

Abstract
Several studies have shown that transforming growth factor β1 (TGF-β1) plays a crucial role in remodeling and proliferation of airway smooth muscle cells (ASMCs). However, its molecular mechanism needs to be further studied. TGF-β1 can up-regulate the level of miR-181a in multiple cells, while miR-181a is expressed in asthma. We asked whether TGF-β1 plays a role in asthma through regulation of miR-181a. For this purpose, ASMCs were stimulated with TGF-β1 and the expression level of miR-181a and extracellular matrix (ECM) protein were measured by quantitative real time polymerase chain reaction (qRT-PCR) and western blotting. The cell proliferation and migration ability of TGF-β1-induced ASMCs were detected by Cell Counting Kit-8 (CCK-8) and transwell method, respectively. Luciferase assay was used to verify whether PTEN is a direct target of miR-181a in ASMCs. miR-181a expression level was increased in TGF-β1-induced ASMCs and miR-181a could inhibit the cell proliferation, migration, and excessive secretion of ECM. The results of the luciferase assay showed that miR-181a plays a role in TGF-β1-induced ASMCs targeting PTEN and the results of western blotting indicated that TGF-β1 could activate Akt/mTOR signaling pathway by up-regulating miR-181a and down-regulating siPTEN. In conclusion, TGF-β1 may induce airway smooth muscle cell proliferation and airway remodeling of asthma by up-regulating miR-181a and suppressing PTEN, and miR-181a inhibitor may function as an inhibitor of ASMCs proliferation through inactivation of the Akt/mTOR pathway.
Keywords: Asthma, TGF-β1, miR-181a, PTEN, Akt/mTOR pathway
Introduction
Asthma is a complex respiratory disease that affects at least 300 million people worldwide, and is characterized by chronic inflammation and remodeling of airway tissues [1]. Airway remodeling is one of the important pathologic features of asthma. Its changes mainly include airway epithelial injury, smooth muscle cell hyperplasia, and hypertrophy [2]. Airway smooth muscle cells (ASMCs) are the main structural components of airway remodeling [3], and several reports have demonstrated that the increase in airway smooth muscle (ASM) mass is correlated with the severity of asthma [4]. Increased cell proliferation, cell size and cell migration could all contribute to the increase in ASM mass, which is a hallmark feature of tissue remodeling [5,6].
Transforming growth factor 1 (TGF-β1) is a key mediator which has been reported to participate in the development of lung fibrosis in severe asthma patients and is involved in airway remodeling in asthma [7,8]. TGF-β1 promotes the differentiation of fibroblasts into myofibroblast cells, and enhances their proliferation [9,10]. Furthermore, TGF-β1 secretion is related to increased extracellular matrix (ECM) protein production, including fibronectin, perlecan, collagen (I-V), versican, elastin, and laminin, leading to enhanced proliferation. ECM is a complex network of large molecules composed of a variety of proteoglycans and fibrous proteins produced and deposited locally in the ASMCs [11]. Changes in ECM protein deposition within and surrounding the smooth muscle bundle have been observed in asthmatic airways [12,13]. Thus, TGF-β1 was reported to play a noteworthy role in airway remodeling due to its effects on airway smooth muscle proliferation and ECM deposition. However, the underlying mechanisms remain unknown.
MicroRNAs (miRNAs) are small noncoding RNAs which control the translation of mRNAs and emerge as post-transcriptional regulators in various biologic processes by mediating the degradation of target mRNAs or preventing their translation [14]. miRNAs were reported to play important roles in lung diseases [15,16], including airway diseases such as asthma [17,18]. Therefore, miRNAs are attractive new drug targets.
miR-181a has been reported to be involved in many cell functions including cell proliferation, migration and to be a proinflammatory factor in asthma [19,20]. Some reports have indicated that ectopic overexpression of miR-181a enhanced cell proliferation through suppressing PTEN expression by directly targeting its 3’UTR, thus resulting in increased AKT phosphorylation [21,22]. It suggests that miR-181a may promote cell proliferation through the PTEN/AKT pathway. Recent studies have shown that miR-181a has a relation with TGF-β1 [23,24].
Considering the noteworthy role that TGF-β1 plays in asthma, this study will elucidate if and how miR-181a is involved in the stage of ASMCs proliferation and airway remodeling to study the mechanism of TGF-β1 in asthma and find a new therapeutic method for asthma.
Materials and methods
Isolation and culture of ASMCs
ASMCs were isolated from trachea and bronchus of BALB/c mice. Briefly, the trachea and main bronchus were dissected from surrounding tissue and the remained tissue was incubated in Hanks’ balanced salt solution (HBSS, Sigma Chemical Co., St. Louis, MO, USA) with 0.1% collagenase solution (Sigma Chemical Co., St. Louis, MO, USA) at 37°C for 20 min and then cut into about 1 cubic millimeter cubes. The cubes were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 20% fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin in a humidified atmosphere of 5% CO2 incubator at 37°C. ASMCs were identified by the typical “hill and valley” growth pattern and immunocytochemistry staining for α-smooth muscle actin. ASMCs were cultured in DMEM with 10% FBS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (all were purchased from Gibco, Waltham, MA, USA). The medium was changed every 2 days. Cells at passage numbers 2-5 were used for the experiments.
Stimulation with TGF-β1
ASMCs were cultured in 0.5% FBS (serum-deprived) for one day and then were treated with different concentrations (0, 0.5, 1, 2, 4 ng/ml) of TGF-β1 (R&D Systems, Minneapolis, MN, USA) for 24 h. 2 ng/ml TGF-β1 was used to stimuli ASMCs for 0, 12, 24, 48 h, respectively. The expression level of miR-181a in TGF-β1 treated ASMCs was detected by quantitative real time polymerase chain reaction (qRT-PCR).
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the ASMCs by using 1 ml Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The purity and concentration of total RNA were determined in an ultraviolet spectrophotometer (Eppendorf, Germany). The expression level of miR-181a, extracellular matrix protein and PTEN were detected by TaqMan microRNA assays (Applied Biosystems, Shanghai, China) with specific primers. qRT-PCR was performed using a 7500 Real-Time PCR System (Applied Biosystems, Shanghai, China). PCR programs were carried out as follows: 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 56°C for 30 s, 72°C for 30 s, and a final extension for 5 min at 72°C. The expression level of glyceraldehyde phosphatedehydrogenase (GAPDH) was used as an internal control for mRNAs, and U6 small nuclear RNA was used as the internal miRNA control. The relative expression level was calculated using the 2-ΔΔCt method and all experiments were run in triplicate. The primers used for detection are listed in Table 1.
Table 1.
List of primers used for qRT-PCR
| Primers | Forward | Reverse |
|---|---|---|
| miR-181a | 5’-GCGGCGAACATTCAACGATG-3’ | 5’-GTGCAGGGTCCGAGG-3’ |
| PTEN | 5’-GTGGTCTGCCAGCTAAAGGT-3’ | 5’-TCACCACACACAGGTAACGG-3’ |
| collagens I | 5’-GACATCCCTGAAGTCAGCTGC-3’ | 5’-TCCCTTGGGTCCCTCGAC-3’ |
| Fibronectin | 5’-ACCAACCTTAATCCGGGCAC-3’ | 5’-TCAGAAACTGTGGCTTGCTGG-3’ |
| GAPDH | 5’-GGGAGCCAAAAGGGTCATCATCTC-3’ | 5’-CCATGCCAGTGAGCTTCCCGTTC-3’ |
| U6 | 5’-CTCGCTTCGGCAGCACA-3’ | 5’-AACGCTTCACGAATTTGCGT-3’ |
Cell proliferation assay
ASMCs were harvested and seeded in flat-bottomed 96-well culture plates (1 × 104 cells/well) and maintained at 37°C in a humidified incubator overnight. The cells were then treated with TGF-β1 (2 ng/ml) or miR-181a inhibitor for 24, 48, 72 and 96 h using three replicates. Then, 10 μl Cell Counting Kit-8 (CCK-8) solution (Dojindo, Tabaru, Japan) were added to 90 μl of culture medium following treatment at the indicated time. After incubation for 2 h, the optical density (OD) of each sample was measured at 450 nm using microplate reader (Spectra Max MD5, Molecular Devices, CA, USA). Culture medium (DMEM) without cells was used as the blank control and each group included at least 6 replicate wells.
Transwell
The migration ability of ASMCs treated with TGF-β1 (2 ng/ml) and miR-181a inhibitor for 24 h was tested in Corning transwell insert chambers. Briefly, after 48 h transfection, ASMCs were resuspended in serum-free DMEM (200 µL) and placed into the upper chamber of the insert without Matrigel. At bottom chamber, the culture medium was added with 20% FBS into the lower chambers as a chemoattractant. Then, the chambers were incubated for 24 h and cells remaining on the upper membrane were carefully removed by cotton swabs. Cells that had migrated through the membrane were manually counted at 200 × magnification from 10 different fields of each filter.
Western blotting
ASMCs were seeded in flat-bottomed 6-well culture plates (1 × 106 cells/well) and maintained at 37°C in a humidified incubator overnight. The cells were then treated with TGF-β1 (2 ng/ml) or miR-181a inhibitor for 24, 48, 72 and 96 h. Total protein was extracted from the cells by using 1 ml Radio Immunoprecipitation Assay (RIPA, Sigma Chemical Co., St. Louis, MO, USA) buffer plus protease inhibitors (Roche, IN, USA). The concentrations of protein were determined with a Pierce BCA Protein Assay Kit (Thermo Scientific, Logan, UT, USA). Equal protein amounts were loaded and separated on 10% SDS-PAGE, and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, MA, USA). After blocking with 5% non-fat milk, the membranes were incubated with primary antibodies: rabbit monoclonal to CDK4 (Cell Signaling Technology, Boston, MA, USA), mouse monoclonal to CDK6 (Cell Signaling Technology, USA), rabbit polyclonal to Cyclin D1 (Cell Signaling Technology, Boston, MA, USA), rabbit polyclonal to PTEN (Abcam, Cambridge, MA, USA), rabbit polyclonal to Akt and Phospho-Akt (Abcam, USA), rabbit polyclonal to mTOR and Phospho-mTOR (Abcam, Cambridge, MA, USA) overnight at 4°C. All first antibodies were diluted by 1:1000. After washing thrice with Tris Buffered Saline with Tween-20 (TBST) buffer to remove unbound antibody, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Inc., CA, USA) at room temperature for 1 h. The protein band was detected using ECL reagent (Millipore, MA, USA). The relative intensities of protein bands were quantified using Image J software. GAPDH was used as an internal control.
Cell transfection
The miR-181a mimics, mimic negative control (NC), miR-181a inhibitor, and inhibitor negative control (inhibitor control) were purchased from Genepharma Inc (Shanghai, China). ASMCs were seeded at a density of 3 × 105 cells per well in 6-well plates for 24 h. Cells were then transfected with oligonucleotides or plasmid by using Lipofectamine 2000 (Invitrogen; Grand Island, NY, USA) based on the manufacturer’s instructions. Cells were harvested at the indicated time for Luciferase assay and protein analysis.
Luciferase assay
Prediction of potential miR-181a target genes in ASMCs was performed using the Dual Luciferase reporter assay System (Promega, Madison, WI, USA). For the luciferase assay, the cells were seeded in 24-well plates for 24 h and transfected with 100 ng luciferase reporter plasmid and 5 ng pRL-TK vector expressing the Renilla luciferase (Promega, Madison, WI, USA) using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s protocol. After 24 h, ASMCs were harvested and lysed and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The firefly luciferase fluorescence was normalized to Renilla, and the relative ratios of firefly to Renilla activity were reported.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software. The values are presented as mean ± standard deviation (SD). The differences between two groups were calculated by Student’s t-test. A P-value < 0.05 was considered a significant difference.
Results
TGF-β1 increased the level of miR-181a expression in ASMCs
To study the mechanism of the TGF-β1 effect on asthma, we first studied the relationship between TGF-β1 and miR-181a in ASMCs. The results showed that the expression level of miR-181a was significantly increased with increasing concentration of TGF-β1 (Figure 1A). When the concentration of TGF-β1 was 2 ng/ml in ASMCs, the expression level of miR-181a was 3.96 times of that of controls (P < 0.01). Figure 1B showed that the expression level of miR-181a increased significantly with the extension of time. When the induced time of TGF-β1 was 24 hours, the expression level of miR-181a was as high as 4.09 (compared with blank space). In conclusion, TGF-β1 is positively correlated with miR-181a within a certain range.
Figure 1.
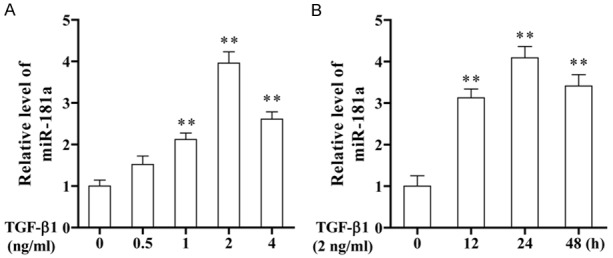
The expression level of miR-181a was increased in TGF-β1-induced ASMCs. A. Cells were treated with different concentrations of TGF-β1 for 24 h, and the expression level of miR-181a was detected by qRT-PCR. B. Cells were treated with 2 ng/ml TGF-β1 for different times. The expression level of miR-181a was detected by qRT-PCR.
miR-181a inhibitor suppressed the proliferation, migration, and extracellular matrix secretion of TGF-β1- induced ASMCs
ASMCs were stimulated by TGF-β1 and miR-181a inhibitor for 24, 48, 72, 96 h. Subsequently the expression levels of miR-181a and cell proliferation were measured by qRT-PCR and CCK-8. The results showed that the level of miR-181a expression is higher in ASMCs in the presence of TGF-β1 than that of the control and inhibitor group (Figure 2A). Additionally, miR-181a inhibitor dramatically reduced the TGF-β1-induced proliferation of ASMCs compared with the miR-181a group (Figure 2B). According to Figure 2C, the levels of proliferation-related proteins including CDK4, CDK6, and Cyclin D1 were significantly higher in TGF-β1 group than in normal and inhibitor group. These data further indicated that TGF-β1 could up-regulate miR-181a in ASMCs and miR-181a inhibitor’s anti-proliferative effects imply a potential value in the treatment of asthma.
Figure 2.

miR-181a inhibitor partially suppressed the proliferation, migration and ECM secretion of ASMCs induced by TGF-β1. A. The expression level of miR-181a in ASMCs stimulated by TGF-β1 and miR-181a inhibitor was determined by qRT-PCR. B. CCK-8 assay for cell proliferation. C. Expression of proliferation-related proteins was determined by western blotting. D. The expression level of ECM protein was determined by qRT-PCR. E. Transwell assay for cell migration.
The expression level of extracellular matrix protein was obtained from qRT-PCR. The results showed that collagen I and fibronectin were both increased obviously in TGF-β1 group when compared with other groups (P < 0.01, Figure 2D). According to Figure 2E, the migration ability of ASMCs was observably increased in the presence of TGF-β1, while the number of cells that migrated in inhibitor group was less than that in TGF-β1 group.
MiR-181a directly targets PTEN in ASMCs
The luciferase reporter assay was performed to verify whether PTEN was a direct target of miR-181a by co-transfection of miR-181a and a luciferase reporter plasmid containing the 3’-UTR of PTEN. The sequence binding sites of miR-181a on 3’-UTR of PTEN gene is shown in Figure 3A. Consistent with binding data, a decrease in relative luciferase activity was shown when the wt PTEN was co-transfected with the miR-181a; however, there was no decrease in relative luciferase activity in ASMCs when transfected with mutant PTEN (Figure 3B). The data showed that both the mRNA and protein expression levels of PTEN in the mimic group were significantly lower than those in the inhibitor group (Figure 3C and 3D).
Figure 3.

miR-181a is direct binding to PTEN in ASMCs. A. The sequence binding sites of miR-181a and PTEN. B. Double luciferase report. C. Expression level of PTEN mRNA. D. The protein expression level of PTEN was determined by western blotting.
miR-181a plays a role in ASMCs by directly targeting PTEN
ASMCs were treated with TGF-β1, miR-181a inhibitor and siPTEN for 24, 48, 72, 96 h, subsequently the expression level of PTEN and cell proliferation were measured by western blotting and CCK-8. The results showed that the levels of PTEN in TGF-β1 and (TGF-β1+miR-181a inhibitor + siPTEN) group were both lower than those in (TGF-β1+miR-181a inhibitor) group (Figure 4A). However, the proliferation rate, relative mRNA expression of collagen I and fibronectin, and the migration ability in TGF-β1 and (TGF-β1+miR-181a inhibitor + siPTEN) group were all higher than the (TGF-β1+miR-181a inhibitor) group (Figure 4B-D). These results suggest that the PTEN 3’-UTR is a functional target site for miR-181a in the ASMCs.
Figure 4.

miR-181a plays a role in ASMCs by targeting PTEN. A. The expression of PTEN was measured by western blotting in different treatment groups. B. CCK-8 assay for cell proliferation. C. Expression level of ECM protein was determined by qRT-PCR. D. Transwell assay for cell migration.
TGF-β1 activates Akt signaling pathway by down-regulating of siPTEN but up-regulating miR-181a
ASMCs were treated with TGF-β1, miR-181a inhibitor and siPTEN for 24 h; subsequently the expression level of p-Akt/Akt and p-mTOR/mTOR were measured by western blotting. The results showed that the level of p-Akt/Akt and p-mTOR/mTOR in TGF-β1 and (TGF-β1+miR-181a inhibitor + siPTEN) group were both higher than (TGF-β1+miR-181a inhibitor) group (Figure 5A). Also, ASMCs were treated with TGF-β1 and Akt pathway inhibitors (MK2206) for 24, 48, 72, 96 h, then the cell proliferation was measured by CCK-8. The results showed that cell proliferation in TGF-β1 group was more than the normal and (TGF-β1+MK2206) group (Figure 5B), which indicated that TGF-β1 plays a role in ASMCs via activating Akt signaling pathway by down-regulating of siPTEN but up-regulating miR-181a.
Figure 5.

TGF-β1 activates Akt signaling pathway by up-regulating siPTEN but down-regulating miR-181a. A. The levels of Akt and mTOR were determined by western blotting in ASMCs. B. CCK-8 assay for ASMCs treated with TGF-β1 and Akt pathway inhibitors.
Discussion
TGF-β1 is a particularly important mediator involved in airway remodeling in asthma [25], and has been shown to induce human ASMCs proliferation and secretion of ECM proteins [26]. Although TGF-β1 has been regarded as having a crucial role in airway remodeling and the proliferation of ASMCs, its molecular mechanism is still unclear. To fulfill this goal in our study, the ASMCs model is essential to study the related molecular changes in airway remodeling. In the present study, we incubated the ASMCs with TGF-β1 to induce the injury leading to airway remodeling.
Since miRNAs can modulate a variety of biological processes including cell proliferation, inflammation, and survival and some miRNAs were reported taking participate in asthma [15,27], we began from the perspective of miRNAs. miR-181a has been reported to be involved in some cell functions in asthma and related to TGF-β1 that made it an important subject of our research. However, it is not clear whether there is a relationship between miR-181a and TGF-β1. Thus, ASMCs were stimulated with TGF-β1 in different concentrations or for different times, and the expression level of miR-181a was determined by qRT-PCR. The results showed that the expression of miR-181a was significantly up-regulated in TGF-β1 induced ASMCs, proving that there was a positive correlation between miR-181a and TGF-β1. We further found that miR-181a inhibitor could inhibit TGF-β1-induced cell proliferation, cell migration, and ECM deposition in ASMCs. Also, miR-181a inhibitor markedly inhibited the levels of collagen I and fibronectin, and suppressed the expression of CDK4, CDK6 and Cyclin D1 in TGF-β1 induced ASMCs. Type I collagen, one of the major ECM proteins in the normal and diseased airway wall, can be synthesized by ASMCs, and affects the proliferation and migration of ASMCs in turn. Fibronectin is a high-molecular weight glycoprotein of the ECM that binds to membrane-spanning receptor proteins called integrins and plays a major role in cell adhesion, growth, and migration [28,29]. CDK4, CDK6 and Cyclin D1 are known to regulate cell proliferation through regulation of the cell cycle [30]. Thus, the results suggested that miR-181a regulation may be deeply involved in the airway remodeling induced by TGF-β1.
To further investigate the specific role of miR-181a, it is important to find the target of miR-181a. The Luciferase report showed that miR-181a was a repressor for PTEN. In the present study, we found that miR-181a directly targets and regulates PTEN expression in ASMCs treated with TGF-β1, miR-181a inhibitor, and siPTEN. Since the target gene PTEN is the key regulator of PI3K/AKT/mTOR pathway [22], the expression levels of p-Akt/Akt and p-mTOR/mTOR were measured by western blotting. The results indicated that TGF-β1 activates the Akt signaling pathway by down-regulating the level of siPTEN but up-regulating miR-181a, which demonstrated a novel link between TGF-β1 and Akt/mTOR signaling in cell proliferation, migration, and ECM deposition.
In summary, this study demonstrated that TGF-β1 could regulate airway remodeling, including decreasing cell proliferation and migration, and reducing ECM deposition by activating the Akt signaling pathway, down-regulating siPTEN but up-regulating miR-181a. miR-181a plays a role in ASMCs by directly targeting PTEN. Thus, miR-181a inhibitor may be a new effective method for the treatment of asthma.
Acknowledgements
This study was supported by the Project of “Discussion on the construction and clinical application of ECG network based on INTERNET” of Qingdao Medical Research Guidance Program in 2015 (Grant No.: 2015-WJZD042).
Disclosure of conflict of interest
None.
References
- 1.McDonald VM, Maltby S, Reddel HK. Severe asthma: current management, targeted therapies and future directions-A roundtable report. Respirology. 2017;22:53–60. doi: 10.1111/resp.12957. [DOI] [PubMed] [Google Scholar]
- 2.Chen M, Lv Z, Huang L, Zhang W, Lin X, Shi J, Zhang W, Liang R, Jiang S. Triptolide inhibits TGF-beta1-induced cell proliferation in rat airway smooth muscle cells by suppressing Smad signaling. Exp Cell Res. 2015;331:362–368. doi: 10.1016/j.yexcr.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Luo L, Gong YQ, Qi X, Lai W, Lan H, Luo Y. Effect of tumor suppressor PTEN gene on apoptosis and cell cycle of human airway smooth muscle cells. Mol Cell Biochem. 2013;375:1–9. doi: 10.1007/s11010-012-1484-7. [DOI] [PubMed] [Google Scholar]
- 4.Martin JG, Ramos-Barbon D. Airway smooth muscle growth from the perspective of animal models. Respir Physiol Neurobiol. 2003;137:251–261. doi: 10.1016/s1569-9048(03)00151-4. [DOI] [PubMed] [Google Scholar]
- 5.Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol. 2013;305:L912–933. doi: 10.1152/ajplung.00259.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goorsenberg AWM, d'Hooghe JNS, de Bruin DM, van den Berk IAH, Annema JT, Bonta PI. Bronchial thermoplasty-induced acute airway effects assessed with optical coherence tomography in severe asthma. Respiration. 2018;96:564–570. doi: 10.1159/000491676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozier A, Allard B, Bara I, Girodet PO, Trian T, Marthan R, Berger P. The pivotal role of airway smooth muscle in asthma pathophysiology. J Allergy (Cairo) 2011;2011:742710. doi: 10.1155/2011/742710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown SD, Baxter KM, Stephenson ST, Esper AM, Brown LA, Fitzpatrick AM. Airway TGF-beta1 and oxidant stress in children with severe asthma: association with airflow limitation. J Allergy Clin Immunol. 2012;129:388–396. doi: 10.1016/j.jaci.2011.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen M, Huang L, Zhang W, Shi J, Lin X, Lv Z, Zhang W, Liang R, Jiang S. MiR-23b controls TGF-beta1 induced airway smooth muscle cell proliferation via TGFbetaR2/p-Smad3 signals. Mol Immunol. 2016;70:84–93. doi: 10.1016/j.molimm.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Michalik M, Pierzchalska M, Legutko A, Ura M, Ostaszewska A, Soja J, Sanak M. Asthmatic bronchial fibroblasts demonstrate enhanced potential to differentiate into myofibroblasts in culture. Med Sci Monit. 2009;15:Br194–201. [PubMed] [Google Scholar]
- 11.Johnson PR, Black JL, Carlin S, Ge Q, Underwood PA. The production of extracellular matrix proteins by human passively sensitized airway smooth-muscle cells in culture: the effect of beclomethasone. Am J Respir Crit Care Med. 2000;162:2145–2151. doi: 10.1164/ajrccm.162.6.9909111. [DOI] [PubMed] [Google Scholar]
- 12.Chen G, Khalil N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir Res. 2006;7:2. doi: 10.1186/1465-9921-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, Gosens R. beta-Catenin signaling is required for TGF-beta1-induced extracellular matrix production by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;301:L956–965. doi: 10.1152/ajplung.00123.2011. [DOI] [PubMed] [Google Scholar]
- 14.Williams M, Cheng YY, Blenkiron C, Reid G. Exploring mechanisms of microRNA downregulation in cancer. Microrna. 2017;6:2–16. doi: 10.2174/2211536605666161208154633. [DOI] [PubMed] [Google Scholar]
- 15.Oglesby IK, McElvaney NG, Greene CM. MicroRNAs in inflammatory lung disease--master regulators or target practice? Respir Res. 2010;11:148. doi: 10.1186/1465-9921-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomankova T, Petrek M, Kriegova E. Involvement of microRNAs in physiological and pathological processes in the lung. Respir Res. 2010;11:159. doi: 10.1186/1465-9921-11-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu TX, Munitz A, Rothenberg ME. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009;182:4994–5002. doi: 10.4049/jimmunol.0803560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3beta. J Biol Chem. 2010;285:29336–29347. doi: 10.1074/jbc.M110.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng MJ, Shi F, Qiu C, Peng WK. MicroRNA-181a, -146a and -146b in spleen CD4+ T lymphocytes play proinflammatory roles in a murine model of asthma. Int Immunopharmacol. 2012;13:347–353. doi: 10.1016/j.intimp.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Lei Z, Ma X, Li H, Zhang Y, Gao Y, Fan Y, Li X, Chen L, Xie Y, Chen J, Wu S, Tang L, Zhang X. Up-regulation of miR-181a in clear cell renal cell carcinoma is associated with lower KLF6 expression, enhanced cell proliferation, accelerated cell cycle transition, and diminished apoptosis. Urol Oncol. 2018;36:93.e23–93.e37. doi: 10.1016/j.urolonc.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Wei Z, Cui L, Mei Z, Liu M, Zhang D. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett. 2014;588:1773–1779. doi: 10.1016/j.febslet.2014.03.037. [DOI] [PubMed] [Google Scholar]
- 22.Geletina NS, Kobelev VS, Babayants EV, Feng L, Pustylnyak VO, Gulyaeva LF. PTEN negative correlates with miR-181a in tumour tissues of non-obese endometrial cancer patients. Gene. 2018;655:20–24. doi: 10.1016/j.gene.2018.02.051. [DOI] [PubMed] [Google Scholar]
- 23.Bhushan R, Grunhagen J, Becker J, Robinson PN, Ott CE, Knaus P. miR-181a promotes osteoblastic differentiation through repression of TGF-beta signaling molecules. Int J Biochem Cell Biol. 2013;45:696–705. doi: 10.1016/j.biocel.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Maity S, Bera A, Ghosh-Choudhury N, Das F, Kasinath BS, Choudhury GG. microRNA-181a downregulates deptor for TGFbeta-induced glomerular mesangial cell hypertrophy and matrix protein expression. Exp Cell Res. 2018;364:5–15. doi: 10.1016/j.yexcr.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK, Chung KF. TGF-beta regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L295–304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jude JA, Dileepan M, Subramanian S, Solway J, Panettieri RA Jr, Walseth TF, Kannan MS. miR-140-3p regulation of TNF-alpha-induced CD38 expression in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;303:L460–468. doi: 10.1152/ajplung.00041.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Araujo BB, Dolhnikoff M, Silva LF, Elliot J, Lindeman JH, Ferreira DS, Mulder A, Gomes HA, Fernezlian SM, James A, Mauad T. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J. 2008;32:61–69. doi: 10.1183/09031936.00147807. [DOI] [PubMed] [Google Scholar]
- 29.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 30.Cheng W, Yan K, Xie LY, Chen F, Yu HC, Huang YX, Dang CX. MiR-143-3p controls TGF-beta1-induced cell proliferation and extracellular matrix production in airway smooth muscle via negative regulation of the nuclear factor of activated T cells 1. Mol Immunol. 2016;78:133–139. doi: 10.1016/j.molimm.2016.09.004. [DOI] [PubMed] [Google Scholar]