

Summary
We characterized the landscape and drug sensitivity of ERBB2 (HER2) mutations in cancers. In eleven datasets (n = 211,726), ERBB2 mutational hotspots varied across 25 tumor types. Common HER2 mutants yielded differential sensitivities to eleven EGFR/HER2 tyrosine kinase inhibitors (TKIs) in vitro, and molecular dynamics simulations revealed that mutants with a reduced drug-binding pocket volume were associated with decreased affinity for larger TKIs. Overall, poziotinib was the most potent HER2 mutant-selective TKI tested. Phase II clinical testing in ERBB2 exon 20-mutant NSCLC resulted in a confirmed objective response rate of 42% in the first twelve evaluable patients. In preclinical models, poziotinib upregulated HER2 cell-surface expression and potentiated the activity of T-DM1, resulting in complete tumor regressions with combination treatment.
Graphical Abstract
Robichaux et al. show that ERBB2 mutation hotspots vary across human tumor types, which affect the volume of the HER2 TKI binding pocket and dictate drug sensitivity. Poziotinib is the most potent HER2 TKI among those tested. Moreover, poziotinib enhances T-DM1 efficacy by increasing the cell surface HER2 level.
Introduction
Amplifications of the Erb-b2 receptor tyrosine kinase 2 gene (ERBB2), also known as human epidermal growth factor receptor 2 gene (HER2), occur in many cancer types and targeted agents such as trastuzumab, trastuzumab emtansine (T-DM1), lapatinib, and neratinib have been shown to improve clinical outcomes compared to chemotherapy alone (Bang et al., 2010; Baselga et al., 2012b; Cameron et al., 2008; Di Leo et al., 2008; Gomez et al., 2008; Park et al., 2016; Slamon et al., 2011; Verma et al., 2012). Activating mutations of ERBB2 have been reported in many cancer types (Connell and Doherty, 2017; Kourie et al., 2016; Kris et al., 2015; Shan et al., 2015). While FDA-approved targeted therapies exist for cancers harboring ERBB2 amplifications, there are no approved targeted therapies for tumors having ERBB2 mutations. However, the National Comprehensive Cancer Network non-small cell lung cancer (NSCLC) guidelines recommend newly diagnosed patients undergo broad molecular profiling to detect ERBB2 mutations (Ettinger et al., 2018).
Recent clinical studies of targeted agents for ERBB2 mutant cancers have focused on covalent tyrosine kinase inhibitors (TKIs), but have shown differential results. Patients with breast cancer treated with neratinib had objective response rates (ORR) of 12.5% - 32%, whereas patients with lung cancer had 0%-4% ORR (Hyman et al., 2018; Ma et al., 2017; Mazieres et al., 2016). Within a single cancer type, HER2 TKIs elicit variant-specific differences. Patients receiving neratinib with ERBB2 kinase domain point mutations had an ORR of 21.4%, whereas patients with exon 20 insertions had an ORR of 7.1% (Hyman et al., 2018). Furthermore, dacomitinib treatment resulted in an ORR of 11.5% for ERBB2 mutant NSCLC but no responses among ERBB2 exon 20 insertion mutation, Y772dupYVMA (Kris et al., 2015). Studies of HER2 monoclonal antibodies and antibody-drug conjugates (ADCs) revealed similar results. The MyPathway study tested the efficacy of the combination of anti-HER2 monoclonal antibodies trastuzumab and pertuzumab in 35 different tumor types and reported an ORR of 11% for all ERBB2 mutations and cancer types, but a 21% ORR for NSCLC patients (Hainsworth et al., 2018). In a pan-HER2 mutant NSCLC study testing the efficacy of T-DM1, patients harboring ERBB2 exon 20 insertion mutations had an ORR of 54.5%, but patients with ERBB2 exon 19 mutations did not have responses (Li et al., 2018). These cancer-specific and variant-specific differences in patient outcomes demonstrate the unmet need for a detailed and systematic understanding of the landscape of ERBB2 mutations across cancer types and the identification of effective therapies for the various ERBB2 mutations identified.
Pre-clinical studies of HER2 activating mutations have also reported differential sensitivity to various TKIs. Studies have shown that HER2 extracellular domain mutants are associated with resistance to non-covalent inhibitors such as lapatinib, yet exhibit robust sensitivity to covalent TKIs (Greulich et al., 2012; Nagano et al., 2018). Exon 19 mutants demonstrate varying sensitivity to lapatinib and covalent inhibitors (Bose et al., 2013; Nagano et al., 2018). Studies have demonstrated that exon 20 mutants have extensive resistance to most non-covalent and covalent TKIs (Nagano et al., 2018; Robichaux et al., 2018), including neratinib, afatinib, and dacomitinib, although some uncommon HER2 exon 20 mutants demonstrated sensitivity (Kosaka et al., 2017). More recently, we reported that poziotinib effectively inhibited HER2 exon 20 insertion mutants at concentrations achievable in patients, and poziotinib treatment induced a radiological response in one patient whose lung cancer harbored an ERBB2 exon 20 mutation (Robichaux et al., 2018).
In the present report, we examined the frequency and drug sensitivity of the most common genomic variants of ERBB2 mutations across various malignancies, and sought to determine a molecular link between the structure and function of HER2 variants and TKI activity. Furthermore, we aimed to determine a rational therapeutic approach for targeting the most common ERBB2 mutations, including the most drug resistant variants.
Results
Cancers of the bladder, stomach, and bile duct have the highest frequency of ERBB2 mutations.
To understand the diversity of ERBB2 mutations across cancer types, we queried several databases, including cohorts from cBioPortal, MD Anderson Cancer Center (MDACC), and Foundation Medicine (FMI), and a circulating free DNA (cfDNA) cohort from Guardant Health (GH). Across all databases, we analyzed all non-synonymous ERBB2 mutations within 25 different cancer types (Table S1). The weighted average frequency for ERBB2 mutations was calculated. Similar to observations in in the AACR GENIE database (Meric-Bernstam et al., 2018), ERBB2 overall mutations occurred most frequently in bladder (8.3%), bile duct (5.3%), and stomach (4.5%) cancers (Figure 1A), whereas ERBB2 exon 20 mutations occurred most frequently in cancers of the small intestine (1.8%), lung (1.5%), and breast (0.9%) (Figure 1B).
Figure 1:

ERBB2 mutations occur in a variety of cancer types with mutational hotspots occurring across the receptor. (A and B) Bar plot of weighted averages of ERBB2 mutations (A) and ERBB2 exon 20 mutation (B) frequency by cancer. Bars are representative of the weighted average ± SEM. Dot sizes are representative of number of cases in each database. Frequency of ERBB2 mutations detected by cfDNA reported by Guardant Health were normalized for clinical sensitivity as reported in Odegaard et al 2018 (Odegaard et al., 2018). See also Table S1.
ERBB2 mutations occur most frequently in the tyrosine kinase domain-coding region and mutational hotspots vary by malignancy.
We analyzed the frequency of mutations within the various regions of HER2 reported in cBioPortal and at MDACC. Across all cancers, ERBB2 mutations occurred most frequently in the tyrosine kinase domain (46%), which included mutations in exon 20 (20%), exon 19 (11%), and exon 21 (9%) (Figure 2A). In addition, 37% of HER2 mutations occur in the extra-cellular domain. Across all cancers, the most common ERBB2 mutations were S310F/Y (11.0%), Y772dupYVMA (5.7%), L755P/S (4.6%), V842I (4.4%), and V777L/M (4.0%) (Figure 2B). In lung cancer, the most frequent ERBB2 mutations occurred in exon 20 (48%), with the Y772dupYVMA mutation comprising 34% of all ERBB2 mutations in lung cancer (Figures 2C, 2D). In breast cancer, ERBB2 mutations occurred most frequently in exon 19 (37%), with L755 mutations being the most prevalent comprising 22% of mutations (Figure 2E). Unlike lung cancer where one variant was dominant among exon 20 mutations, there was more diversity among exon 19 mutations in breast cancer (Figure 2F). In colorectal cancer, ERBB2 mutations occurred most frequently in exon 21 (23%) and the extracellular domain (23%), and the V842I variant in exon 21 was the most prevalent (19%) (Figures 2G, 2H).
Figure 2:

ERBB2 mutation hotspots vary by cancer type. (A-B) Frequency and location of ERBB2 mutations found across all cancers analyzed by protein coding region (A) and specific variant frequency of top ten variants (B) (n = 2338). (C-D) Frequency and location of ERBB2 mutations found in NSCLC (n = 177) analyzed by protein coding region (C) and specific variant (D). (E-F) Frequency and location of ERBB2 mutations found in breast cancer (n = 143) analyzed by protein coding region (E) and specific variant (F). (G-H) Frequency and location of ERBB2 mutations found in colon cancer (n = 219) analyzed by protein coding region (G) and specific variant (H). (B, D, F, and H) Length of bars on lollipop plots are relative to frequency of mutation reported. See also Figure S1.
Y772dupYVMA is the most common ERBB2 exon 20 insertion mutation across cancer types.
Exon 20 mutations are the most common mutations within the tyrosine kinase domain of HER2, and treatment of ERBB2 exon 20 insertion mutations remains a clinical challenge (Connell and Doherty, 2017; Hainsworth et al., 2018; Hartmaier et al., 2017; Hyman et al., 2018; Kosaka et al., 2017). To understand the diversity and prevalence of exon 20 insertions, we analyzed the frequency of ERBB2 exon 20 insertion sequences by cancer type in cBioportal, MDACC, FMI and GH databases. The Y772dupYVMA insertion was the most common ERBB2 exon 20 insertion, comprising 68% of all ERBB2 exon 20 insertions, and the G778dupGSP (14%) and G776del insVC (9%) insertions were the second and third, respectively, most common variants (Figure S1A). Among all cancer types analyzed, NSCLC showed the greatest diversity in exon 20 insertion mutations (Figure S1B) whereas breast cancer showed little diversity, with only four distinct variants reported (Figure S1C). Additional rare insertion mutations were seen across other cancer types, but the duplications at Y772 and G778 occurred most frequently in every cancer type analyzed (Figure S1D).
Frequently detected ERBB2 alterations are activating mutations.
To assess the functional impact of common ERBB2 mutations, we stably expressed each of the 17 most frequently detected HER2 mutants across exons 19, 20, and 21 or the wild-type (WT) HER2 in Ba/F3 cells. All 17 HER2 mutants tested were found to induce IL-3 independent survival of Ba/F3 cells whereas the WT HER2 (blue line) did not (Figure 3A–3C). Using Western blotting and ELISA we validated the expression of total and phosphorylated HER2 in these stable Ba/F3 cell lines and found that all 17 HER2 mutants resulted in phosphorylation of HER2 (Figure 3D–3F, S2) and activation of downstream signal transduction molecules including Akt and p44/42 (ERK1/2), indicating that these mutations result in receptor activation (Warmuth et al., 2007). Interestingly, expression of some mutants such as V773M resulted in lower levels of phosphorylated HER2 and downstream signaling molecules (Figure 3E, S2A) suggesting that some HER2 mutants may not be as activating as others.
Figure 3:

The most common HER2 variants in the tyrosine kinase domain are activating mutations. (A-C) Cell viability of stable Ba/F3 cell lines expressing HER2 exon 19 (A), HER2 exon 20 (B), and HER2 exon 21 (C) mutations grown in IL-3 free conditions for 15 days. Fluorescence values were normalized to day zero, and the mean fold change ± SEM is plotted for each cell line (n = 3). (D-F) Representative Western blot of Ba/F3 cells expressing HER2 exon 19 (D), HER2 exon 20 (E), and HER2 exon 21 mutants (F) or empty vector (n = 3). See also Figure S2.
Poziotinib was the most potent TKI tested and inhibited the most common HER2 mutants in vitro.
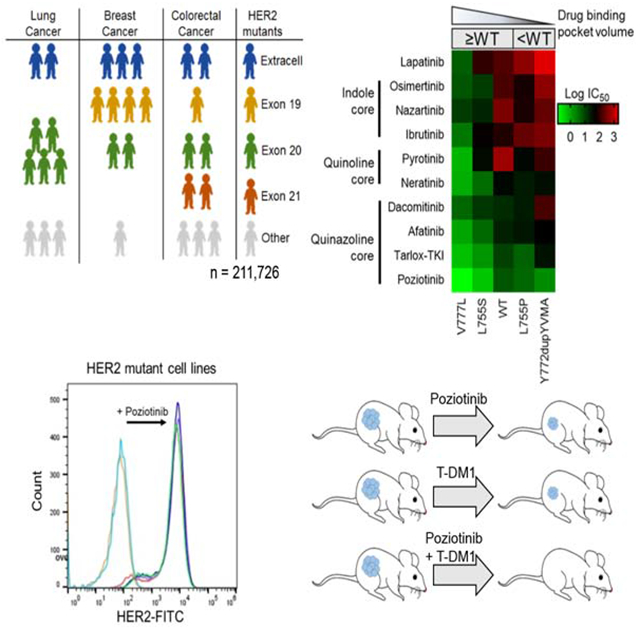
Recent reports highlight the effectiveness of covalent quinazoline and quinoline-based TKIs in pre-clinical models of HER2 mutant diseases (Kosaka et al., 2017; Nagano et al., 2018; Robichaux et al., 2018; Shen et al., 2016; Yang et al., 2015). However, clinical studies of afatinib (Oh et al., 2018; Peters et al., 2018), dacomitinib (Kris et al., 2015), and neratinib (Hyman et al., 2018; Ma et al., 2017) reported low ORR as well as cancer-specific and variant-specific differences in patient outcomes. To systematically evaluate drug sensitivity across the most commonly detected HER2 variants, we screened our panel of HER2 mutant Ba/F3 cells against 11 covalent and non-covalent EGFR and HER2 TKIs. As a control, we included Ba/F3 cells expressing WT EGFR supplemented with 10 ng/ml EGF in the screen (Figure 4A, S3A). HER2 mutants showed robust resistance to the non-covalent inhibitors lapatinib and sapitinib (Figure 4A, S3A). Covalent indole-based TKIs osimertinib, ibrutinib, and nazartinib were not effective in decreasing cell viability in cells expressing exon 20 mutants, however, these TKIs were effective in against cells expressing D769 variants (Figure 4A, S3A). By comparison, the covalent quinazoline and quinoline-based TKIs, afatinib, neratinib, dacomitinib, tarloxotinib-TKI (tarlox-TKI), and poziotinib, had inhibitory activity for HER2 mutants across all three exons (Figure 4A, S3A). Across all HER2 mutants and TKIs tested, poziotinib had the lowest average IC50 (Figure 4B) and was significantly more effective than afatinib, neratinib, or tarlox-TKI for mutations in exons 19 and 20 (Figures 4C–D). However, there was no significant difference in average IC50 for exon 21 mutants (Figure 4E), suggesting that mutation location impacts drug binding. To verify that differential sensitivity of Ba/F3 clones expressing different HER2 mutants to TKIs was not an artifact of varying p-HER2 levels expressed in these cell lines, we compared the IC50 values of each mutant to poziotinib to the p-HER2 level determined by ELISA and found that there was no correlation between p-HER2 level and poziotinib activity (Figure S3B) suggesting that differential sensitivity is driven by mutation effects on drug binding. Since the majority of adverse events (AEs) related to EGFR and HER2 targeted therapy are due to inhibition of WT EGFR, we compared the IC50 values of Ba/F3 cells expressing WT EGFR supplemented with 10 ng/ml EGF to support viability (Figure S3A) to the IC50 values of Ba/F3 cells expressing HER2 mutants. We found that poziotinib and pyrotinib were the most mutant-selective TKIs across all HER2 mutants (Figure 4F, mutant/WT = 0.23), whereas poziotinib was the most mutant-selective TKI across HER2 exon 20 mutants (Figure 4G, mutant/WT = 0.31).
Figure 4:

Potency of TKIs in Ba/F3 cell lines expressing HER2 mutants. (A) Heatmap of log IC50 values calculated in GraphPad for Ba/F3 cells stably expressing the indicated mutants and after 72 hr of drug treatment, (n ≥ 3). (B-E) Average IC50 values for Ba/F3 cell lines expressing all HER2 mutants (B), HER2 exon 19 mutants (C), HER2 exon 20 mutants (D), or HER2 exon 21 mutants (E) after treatment for 72 hr with indicated TKIs. (F and G) Bar plots of ratio of IC50 values of all HER2 mutant (F) or HER2 exon 20 mutant (G) to WT EGFR in the presence of 10 ng/ml EGF. (B-G) Bars are representative of mean ± SEM (n ≥ 3). See also Figure S3.
HER2 mutation location and amino acid change affect drug binding affinity.
To understand further, how the location of mutations and amino acid changes affect drug efficacy, we used molecular dynamics simulations (MDS) to investigate how various mutations impact the structure and motion of the HER2 kinase domain. Molecular models of the L755S, L755P, Y772dupYVMA, and V777L HER2 mutants (Figure S4) were constructed using a reported HER2 X-ray structure (Aertgeerts et al., 2011) (PDB 3PP0) and subjected to accelerated molecular dynamics (Wang et al., 2011). The range of protein conformations sampled varied among the HER2 mutants. Differences were evident even between exon 20 mutants, especially in the αC-helix region, where the duration of the conformation of the αC-helix varied between the “in” (active conformation with smaller binding pocket) and the “out” (inactive conformation with larger binding pocket). The V777L mutant heavily sampled the “out” conformation while the Y772dupYVMA mutant sampled both “in” and “out” conformations (Figure 5A). Overall, these differences in the distribution of conformational states resulted in the Y772dupYVMA mutant sampling the “in” conformation 10-times more often than the V777L mutant (Figure 5B), and, on average, a smaller binding pocket size for Y772dupYVMA compared to V777L (Figures 5C and 5D).
Figure 5:

MDS of HER2 mutants reveal possible mechanisms for decreased drug sensitivity for Y772dupYVMA and L755P mutations. (A) αC-helix positions for the HER2 V777L and Y772dupYVMA exon 20 mutants during the 150 ns accelerated MDS. (B) Fractional population of molecular dynamics snapshots for the HER2 exon 20 mutants in the αC-helix “in” vs. “out” conformations. (C) Molecular dynamics snapshots of the V777L (white backbone, light green P-loop) and Y772dupYVMA (grey backbone, dark green P-loop) mutants. Note minor differences in P-loop and kinase hinge conformations but a significant shift in αC-helix position (“out” position for V777L in blue, “in” position for Y772dupYVMA in purple). (D) Binding pocket volume profiles for the HER2 mutants taken from the accelerated MDS. (E) Molecular dynamics snapshots of L755P (white, backbone; light green, P-loop; yellow, hinge; blue, αC-helix) and L755S (grey, backbone; dark green, P-loop; orange, hinge; purple, αC-helix) HER2 mutants. (F) Bar plots of IC50 values of HER2 mutants with binding pocket volumes ≥ WT HER2 or smaller than WT HER2 treated with quinazoline or indole based TKIs. Bars are representative of mean ± SEM (n ≥ 3). See also Figure S4.
Further analysis of the HER2 mutants by MDS demonstrated that distinct mutations at the same residue can have drastically different effects on protein structure and dynamics. The L755P mutation lacks a hydrogen bond donor and breaks the backbone hydrogen bond between L755 on the β3 strand and V790 on the β5 strand resulting in destabilization of the β-sheet and a structural rearrangement in the kinase hinge region (Figure 5E). In particular, the L800 residue in the hinge of the L755P mutant protruded into the active site during part of the simulation and reduced the pocket size considerably. The changes in hinge mobility may also play a role in kinase activation (Chen et al., 2007; Sours et al., 2014). Changes in the β3 strand conformation also caused the P-loop to collapse inward, further reducing the pocket volume and making this mutant less sensitive to most TKIs. These changes in the L755P mutant contrasted with conformational and pocket volume profiles of the L755S mutant, which are more similar to those of the WT HER2 (Figure 5D, 5E).
The smaller binding pocket of the Y772dupYVMA and L755P mutants may be an important contributor to the weaker potency of larger, covalent, indole-based TKIs compared to V777L and L755S mutants (Yun et al., 2008). To test this hypothesis, we compared the average IC50 values of Ba/F3 cells expressing HER2 mutants with larger binding pocket volumes (V777L and L755S) to the average IC50 values of Ba/F3 cells expressing HER2 mutants with smaller binding pocket volumes (Y772dupYVMA and L755P) treated with either smaller quinazoline-based TKIs (i.e. afatinib, poziotinib, etc.) or larger indole-based TKIs (i.e. osimertinib, nazartinib, etc., Figure 5D). We found that in mutants with larger binding pocket volumes, the average IC50 values were not significantly different between the classes of compounds (Figure 5F). In contrast, in mutants with smaller average binding pocket volumes, the smaller quinazoline-based TKIs had a significantly lower average IC50 value compared to the larger indole-based TKIs (Figure 5F). Furthermore, when comparing average IC50 values of quinazoline-based TKIs across mutation types, HER2 mutants with smaller average binding pocket volumes had significantly higher IC50 values compared to those with larger average binding pocket volumes (Figure 5F). Together, these data suggest that HER2 mutants with smaller drug binding pocket volumes are less sensitive to TKIs, especially larger indole-based TKIs.
HER2 mutant human cell lines showed enhanced sensitivity to poziotinib.
Clinical studies testing HER2 inhibitors have revealed cancer-type specific differences in drug sensitivity (Hainsworth et al., 2018; Hyman et al., 2018). To determine whether covalent, quinazoline-based TKIs have activity in models of HER2 mutant disease, we tested our panel of EGFR/HER2 TKIs in human cell lines. We ectopically expressed HER2 exon 20 mutants in MCF10A mammary epithelial cells and evaluated in vitro sensitivity to 12 EGFR/HER2 TKIs. MCF10A cells expressing G776del insVC, Y772dupYVMA, or G778dupGSP HER2 mutants were most sensitive to poziotinib, with an average IC50 values of 8.2 nM (Figure 6A–6C). In comparison, the average IC50 values of tarlox-TKI and neratinib were 21 nM and 150 nM, respectively (Figure 6A–6C), indicating that poziotinib is 2.6 and 18 times more potent than tarlox-TKI and neratinib, respectively (p < 0.001). Furthermore, Western blotting showed that poziotinib, but not neratinib, completely inhibits p-HER2 at 10 nM in MCF10A cells expressing HER2 G776delinsVC (Figure S5A). Since WT HER2 does not transform Ba/F3 cells to grow without IL-3 (Figure 3A), we used MCF10A cells to determine the HER2 mutant specificity of each compound. To this end, we determined the IC50 of each drug in MCF10A cells expressing WT HER2 (Figure S5B) then calculated the mutant to WT ratio for each inhibitor and found that, similar to the mutant to WT EGFR ratio found in Ba/F3 cells (Figure 4G), poziotinib was the most mutant selective TKI tested in MCF10A cell lines, followed by pyrotinib, and tarlox-TKI (Figure 6D).
Figure 6:

Human cell lines expressing HER2 mutants are also sensitive to poziotinib. (A-C) Dose response curves of MCF10A cells expressing HER2 exon 20 insertion mutants G776delinsVC (A), Y772dupYVMA (B), or G778dupGSP (C), treated with indicated TKIs for 72 hr. (D) Dot plot of ratio of IC50 values comparing MCF10A expressing HER2 exon 20 mutants to MCF10A expressing WT HER2. Dots are representative of mean ± SEM for each cell line and bars are representative of mean ± SEM of all three cell lines (n ≥ 3 for each cell line). (E) Dose response curve of CW-2 colorectal cancer cell line harboring endogenous ERBB2 exon 19 mutation, L755S, treated with indicated inhibitors for 72 hr. (A-C, E) Curves are representative of mean ± SEM, n = 3. (F) Bar graph of CW-2 tumor volume at day 21. Tumors were randomized at 350 mm3, indicated by the dotted line. Dots are representative of individual tumors (n = 5/ group), and bars are representative of mean ± SEM. See also Figure S5.
In a model of colorectal cancer (CW-2) with an endogenous ERBB2 exon 19 point mutation (L755S) (Barretina et al., 2012), differences in sensitivity between poziotinib, tarlox-TKI and neratinib were less dramatic, but still significant (p = 0.02 and p = 0.0004), with average IC50 values of 3.19 nM, 4.24 nM, and 68.8 nM, respectively (Figure 6E). Furthermore, in a xenograft mouse model of CW-2 cells, poziotinib treated animals showed a reduction of 58% in tumor volume compared to the vehicle treated group at day 21 (p = 0.011, Figure 6F). In comparison, neratinib treatment increased (p = 0.023) and afatinib treatment did not significantly affect tumor growth compared to vehicle control (Figures 6F, S5C, S5D).
Poziotinib has anti-tumor activity in patients with NSCLC harboring ERBB2 mutations.
Based on these preclinical data and our previously published work on exon 20 mutations (Robichaux et al., 2018), we initiated an investigator-initiated, phase II clinical trial of poziotinib in NSCLC with EGFR and ERBB2 exon 20 mutations (). Patients with ERBB2 exon 20 mutations detected in tissue and/or plasma were treated with poziotinib 16 mg orally daily until progression, death, or withdrawal. Objective response was evaluated every eight weeks, based on RECIST v1.1. Additional information including eligibility, study objectives and assessments can be found in figures S6A and S6B. Of the first twelve evaluable patients, six (50%) had a best response of partial response (PR) eight weeks after beginning poziotinib treatment. This response was confirmed by a repeat scan two months later (16 weeks after commencing treatment) in 5/12 patients (confirmed ORR = 42%) (Figure 7A). Two patients had progressive disease (PD) at first response evaluation, resulting in a disease control rate of 83%. As of July 2019, ten of the twelve patients had progressed; the median progression free survival (mPFS) was 5.6 months (Figure 7B) and the median duration of response was 4.6 months (Figure S6C). All patients included in the study harbored either the Y772dupYVMA or G778dupGSP insertions (Figure 7A, S6D). Representative images of one NSCLC patient with an Y772dupYVMA mutation pre- and post-treatment showed robust tumor shrinkage in the right lung (Figure 7C).
Figure 7:

Patients with NSCLC harboring ERBB2 mutations had a 42% confirmed response rate to poziotinib. (A) Waterfall plot of the objective responses of patients on clinical trial . Objective PR are shown in dark green, an unconfirmed response is shown in light green, SD is shown in yellow, and PD is shown in red. (B) Kaplan-meier plot of PFS of the 12 patients in (A). (C) Computed tomography (CT) scan of a patient with an ERBB2 Y772dupYVMA mutation one day before and eight weeks after poziotinib treatment. Blue arrow indicates the target lesion and the red arrow indicates the resolved pleural effusion. (D) Positron emission tomography (PET) scans of patient with ERBB2 L755P mutant NSCLC one day before and four weeks after poziotinib treatment. White boxes highlight the lesion of interest. See also Figure S6.
Overall, poziotinib safety and AE profile are similar to those of other FDA- approved quinazoline TKIs, afatinib and dacomitinib (Mok et al., 2018; Sequist et al., 2013). Most patients receiving poziotinib (8/12, 67%) had at least one dose reduction due to AEs (Figure S6E, S6F). Eight patients experienced grade 3-4 AEs, the majority of which were diarrhea (17%) and rash (58%, Figure S6F). No patients discontinued treatment due to AEs (Figure S6E). Additional patient characteristics including the number of previous treatments, dose reductions, treatment related AEs occurring in more than one patient, and co-mutations are in Figures S6D and S6F.
In addition, one patient with NSCLC harboring an ERBB2 exon 19 point mutation, L755P, and a prior history of progression after treatment with multiple regimens including platinum doublet chemotherapy, trastuzumab, afatinib, and TDM-1 was treated on a compassionate care use protocol (C-IND18-0014). The patient was treated with 16 mg poziotinib daily and was evaluated after four weeks of treatment (Figure 7D, white box). The patient had stable disease (SD) per RECIST v1.1 (−12% reduction). The patient remained on poziotinib for more than seven months and proceeded to received further systemic therapy after experiencing disease progression on poziotinib.
Combination of poziotinib and T-DM1 treatment potentiates anti-tumor activity.
Previous studies of the HER2 TKI lapatinib in HER2-positive breast cancer models have shown that TKI treatment results in increased cell surface receptor accumulation, by stabilizing HER2 and reducing ubiquitination of the receptor, and increased cell surface HER2 increased sensitivity to antibody-dependent cellular cytotoxicity (Scaltriti et al., 2009). To determine if poziotinib treatment increases HER2 cell surface expression, we analyzed cell surface HER2 by FACS after 24 hr of low dose poziotinib treatment. We found that, poziotinib treatment increased cell surface HER2 level 2-fold in MCF10A cells expressing mutant, but not WT, HER2 (p<0.0001, Figure 8A). To determine if the increase in cell surface HER2 after low dose poziotinib treatment was related to decreased ubiquitination of the receptor, we performed immunoprecipitation of HER2 from MCF10A cells expressing WT or mutant HER2, and immunoblotted membranes with antibodies against ubiquitin. We found that low dose poziotinib treatment increased ubiquitination of WT HER2 but decreased the ubiquitination of mutant HER2 (Figure 8B), which correlated with the decreased cell surface WT HER2 level but increased cell surface mutant HER2 level. Next, we tested whether the combination of poziotinib and T-DM1 would decrease cell viability in vitro. We found that while T-DM1 alone did not inhibit cell viability of MCF10A expressing mutant or WT HER2, combination with poziotinib significantly lowered IC50 values of T-DM1 in mutant, but not WT, HER2 expressing MCF10A cells (Figure 8C, S7A).
Figure 8:

Poziotinib treatment induces accumulation of HER2 on the cell surface, and combination of poziotinib and T-DM1 treatment potentiates anti-tumor activity. (A) FACS analysis of HER2 expression on MCF10A cells expressing indicated HER2 mutants or WT HER2 after 24 hr of 10 nM poziotinib treatment. (B) Representative western blot of ubiquitin and total HER2 after immunoprecipitation of HER2 in MCF10A cells expressing WT HER2 or indicated HER2 mutants. Molecular weight ladder run between samples was omitted as indicated by the white space between lanes. Ubiquitin level was normalized to HER2 level then to DMSO control to determine fold change in relative ubiquitin expression (C) Bar plot of T-DM1 IC50 values of MCF10A cells expressing indicated HER2 mutants treated with T-DM1 alone or in combination with poziotinib. (n = 2), *, p < 0.0001. (D) Tumor growth curves of ERBB2 Y772dupYVMA NSCLC PDX treated with the indicated inhibitors. (E) Dot plot of percent change in tumor volume of mice treated with indicated inhibitors at day 15. (A-E) Bars and symbols are representative of mean ± SEM. (F) Kaplan-Meier curve of PFS of mice bearing ERBB2 Y772dupYVMA PDX treated with indicated inhibitors. (G) Summary of number of tumor bearing mice in each group at day 15 and day 45. (H) ERBB2 Y772dupYVMA GEMM were treated with indicated inhibitors for four weeks and tumor volume was determined by MRI. Bars are representative of individual mouse tumor volume. (I) Percent change in body weight for ERBB2 GEMMs over the first 14 days of treatment with indicated inhibitors. Symbols are representative of mean body weight ± SEM. See also Figure S7.
To validate these findings in vivo, we tested the combination of low dose poziotinib with a single dose of T-DM1 in an ERBB2 mutant, Y772dupYVMA, NSCLC PDX model (Figure 8D, S7B). At the time of best response, day 15, the combination of low dose poziotinib and a single dose of T-DM1 resulted in complete tumor regression in 20/20 mice, compared to 2/9 mice receiving T-DM1 alone, or 0/12 mice receiving poziotinib (Figure 8E). To further assess response to treatment, we determined mPFS, defined as time to tumor doubling from best response. Mice receiving vehicle control had a mPFS of 3 days, whereas mice receiving low dose poziotinib or T-DM1 had a mPFS of 15 days and 27 days, respectively (Figure 8F). By day 30, tumor growth resumed in all mice receiving T-DM1 alone. However, in 14/20 mice receiving combination treatment there was no evidence of tumor reoccurrence at 45 days (Figure 8G, S7B). To validate these findings in an additional model of ERBB2 mutant NSCLC, we tested low dose poziotinib, T-DM1, or the combination in a genetically engineered mouse model (GEMM) of NSCLC harboring Y772dupYVMA. By four weeks, mice receiving either poziotinib or T-DM1 had on average an 11% increase in tumor growth. In contrast, mice receiving the combination of poziotinib and T-DM1 had an average 47% reduction in tumor burden (Figure 8H). Furthermore, we found that the combination of low dose poziotinib and T-DM1 was tolerable in the GEMMs with a 4% increase in body weight in the combination group at two weeks (Figure 8I).
Discussion
Our pan-cancer analysis reveals that ERBB2 mutational hotspots vary by cancer type and have differential sensitivity to HER2 TKIs in vitro, which likely affect clinical efficacy. In the SUMMIT trial, neratinib was most effective in breast cancer patients, with the majority of responders having tumors with L755S, V777L, or L869R (Hyman et al., 2018). In our in vitro screening, these mutations correlated with low IC50 values. In contrast, patients with colorectal cancer did not respond to neratinib (Hyman et al., 2018). We found that the V842I mutation is the most common ERBB2 mutation in colorectal cancers and this mutant was not sensitive to neratinib in vitro. These data suggest that different TKI sensitivities between malignancies may be, in part, explained by cancer-specific mutational hotspots, which directly impact drug sensitivity. Data from the SUMMIT trial showed that while specific exon 20 insertions were associated with neratinib sensitivity in breast cancer patients, these identical mutations were associated with resistance in other cancer types demonstrating that there may be other mechanisms underlying these tumor-type specific differences that merit further investigation.
Exon 20 insertion mutations and the exon 19 L755P mutation are resistant to most HER2 TKIs. MDS revealed that these mutations induce conformational changes that affect the overall size and shape of the drug binding pocket. Collectively, these in vitro and in silico findings are consistent with the clinical observations that patients with ERBB2 exon 20 insertion mutations historically have had poor responses to TKIs. In NSCLC, where exon 20 insertions frequently occur, patients harboring ERBB2 exon 20 insertion mutations had response rates of 0% (Hyman et al., 2018), 11.5% (Kris et al., 2015), and 18.2% (Mazieres et al., 2016) – 18.8% (Peters et al., 2018) to neratinib, dacomitinib, and afatinib, respectively. Moreover, while L755S mutations have been shown to respond to neratinib, L755P mutations are profoundly resistant to both TKIs and ADCs (Hyman et al., 2018; Li et al., 2018; Mazieres et al., 2016).
Our testing of a panel of covalent and non-covalent EGFR and HER2 TKIs against the most common HER2 mutants revealed that poziotinib has activity against the most common HER2 variants, including exon 19 and 20 mutants that are resistant to other HER2 TKIs. We have previously reported that exon 20 insertions have a sterically hindered drug binding pocket, and that the size and flexibility of poziotinib can overcome this challenge (Robichaux et al., 2018). Likewise, in silico modeling of the L755P variant in the current report indicates that this alteration reduces the volume of the drug binding pocket, which is predicted to limit the binding of drugs with larger terminal groups oriented towards the αC-helix such as neratinib and osimertinib. In contrast, due to the terminal halogenated benzene group of poziotinib, the drug can bind deeply within the hydrophobic cleft created by A751, K753, L796, and T798 in HER2 (Robichaux et al., 2018). This interaction of the terminal benzene group with the hydrophobic cleft results in the alignment of the quinazoline core with residues at the back of the drug binding pocket, maintaining receptor binding.
Previous pre-clinical data in lung cancer (Robichaux et al., 2018) and current pre-clinical models of breast and colon cancer demonstrate that poziotinib has broad anti-tumor effects in multiple ERBB2 mutant cancer types and across exons. In a recent report, two of five (40%) patients with NSCLC harboring ERBB2 exon 20 insertions treated with poziotinib (12 mg or 8 mg) had a partial response (Oh et al., 2018). A recent case study reported that poziotinib induced a radiological response and clinical improvement in a heavily pretreated patient with ERBB2 mutant (G778dupGSP) breast cancer (Pandey and Brufsky, 2018). In the first twelve patients of clinical trial with ERBB2 exon 20 mutant disease, we observed a confirmed ORR of 42% and a mPFS of 5.6 months. In addition, a patient with an L755P mutation, treated on a compassionate care use protocol, demonstrated a durable minor response to poziotinib, lasting more than seven months. Collectively, these data strongly suggest that poziotinib has activity against the most frequent ERBB2 variants and across diverse malignancies harboring ERBB2 mutations. In comparison, previous studies have reported that patients with ERBB2 mutant NSCLC had an ORR of 6% and a mPFS of 1.9 months to second-line immune checkpoint (PD-1/PDL-1) blockade (Negrao et al., 2018); an ORR of 10% and a mPFS of 4.3 months to second-line non-HER2 targeted therapies (Mazieres et al., 2016); or an ORR of 7% and a mPFS of 3.4 months to second-line HER2 targeted agents, neratinib, lapatinib, or afatinib (Mazieres et al., 2016). Furthermore, in the general platinum-refractory NSCLC population, standard second-line chemotherapy, docetaxel, has an ORR of 7-13% and a mPFS of 2-4 months (Hanna et al., 2004; Herbst et al., 2016; Horn et al., 2017; Rittmeyer et al., 2017; Shepherd et al., 2000).
Toxicity of poziotinib was comparable to other quinazoline-based, FDA-approved TKIs afatinib and dacomitinib (Mok et al., 2018; Sequist et al., 2013). In the Phase III study of afatinib for EGFR mutant NSCLC (Lux-lung 3, Sequist et al., 2013) and in the Phase III study of dacomitinib for EGFR mutant NSCLC (Archer1050, Mok et al., 2018), 52% and 67% of patients had dose reductions due to AEs, respectively. Of the first twelve patients in our study receiving poziotinib, 67% of patients received dose reductions. Furthermore, in the Lux-lung 3 study and Archer1050 study, 8% and 10% of patients discontinued treatment due to AEs. In the first twelve patients receiving poziotinib, no patients discontinued treatment due to AEs.
Previous studies of lapatinib in ERBB2 amplified breast cancer showed that lapatinib caused accumulation of HER2 on the cell surface, which enhanced trastuzumab binding and anti-tumor effects (Scaltriti et al., 2009). Furthermore, meta-analysis of several clinical trials found that combination of lapatinib and trastuzumab significantly increases pathological complete responses of HER2-positive breast cancers regardless of hormone receptor status (Baselga et al., 2012a; Xu et al., 2017). We find that poziotinib upregulates mutant, but not WT, HER2 on the cell surface and that combination of poziotinib with T-DM1 decreases cell viability in vitro and causes complete regression of HER2 exon 20 mutant NSCLC tumors in mice. Together, these data suggest that the high affinity and specificity of poziotinib for HER2 mutants make poziotinib a good candidate for combination with T-DM1 by enhancing mutant, but not WT, HER2 on the cell surface for targeting with T-DM1. Furthermore, these data highlight the need for clinical trials testing the efficacy of poziotinib and T-DM1 combination therapy in ERBB2 mutant malignancies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for additional information, regents, and resources should be directed and will be fulfilled by the Lead Contact John V. Heymach (JHeymach@mdanderson.org).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Clinical Trial CIND 18-0014
Clinical study is a single-center investigator-initiated Phase II study of poziotinib conducted at MD Anderson Cancer Center in Houston, TX. The trial protocol was approved by the MD Anderson Cancer Center institutional review board and the FDA. Patient eligibility included histologically or cytologically confirmed recurrent NSCLC not amenable to curative intent therapy or stage IV NSCLC with a documented ERBB2 exon 20 mutation by a CLIA certified laboratory. Additional eligibility criteria included measurable disease by RECIST 1.1 criteria (Eisenhauer et al., 2009), ECOG performance status of 0 or 1, and adequate respiratory, cardiac (>50% cardiac ejection fraction), hepatic, and renal function. Brain metastases were allowed as long as the metastases were stable and the patient did not require treatment with anticonvulsants or escalating doses of steroids. Written and informed consent was obtained from all patients before beginning poziotinib treatment. Eligible patients received daily poziotinib by mouth. Poziotinib was provided by Spectrum Pharmaceuticals. Poziotinib treatment was continued until progression, death or until intolerable AEs. Treatment beyond progression was allowed at the discretion of treating physician if there was evidence of ongoing clinical benefit. Poziotinib starting dose was 16 mg orally daily, and dose reductions to 12 mg daily and 8 mg daily were allowed.
The primary endpoint was investigator-assessed objective response rate (ORR) determined by RECSIT 1.1 criteria. CT and or PET-CT were completed every eight weeks. ORR of 20% was considered to be clinically meaningful, and response rate was defined as the percentage of patients having either a partial (PR) or a complete response (CR). Disease control rate was defined as the percentage of patients has a PR, a CR, or stable disease (SD) lasting at least 8 weeks after initiation of treatment. Secondary objectives included progression free survival, overall survival, disease control rate, duration of response, and safety and toxicity. Toxicities were assessed using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events [NCI CTCAE] version 4.03 (Hay et al., 2014). To analyze efficacy waterfall plots and swimmer plots were created. Progression free survival was calculated from treatment start until disease progression per RECIST 1.1 or death. Duration of response was calculated from date on response per RECIST 1.1 until disease progression or death.
The patient treated on C-IND18-0014 was found to have a ERBB2 exon 19 mutation (L755P) and was not eligible for clinical trial , therefore the patient was enrolled on a compassionate care use protocol approved by both the MD Anderson Cancer Center institutional review board and the FDA. The patient received daily 16 mg poziotinib and was evaluated for response defined by RECIST 1.1 criteria at four weeks.
Human cell lines and Ba/F3 cell lines
MCF10A cells were purchased from ATCC and were cultured in DMEM/F12 media supplemented with 1% penicillin/streptomycin, 5% horse serum (sigma), 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, and 10 μg/ml insulin. Stable cell lines were created by retroviral transduction, and retroviruses were generated by transfecting pBabe-Puro based vectors summarized in Table 1 (Addgene and Bioinnovatise) into Phoenix 293T-ampho cells (Orbigen) using Lipofectamine 2000 (Invitrogen). Two days after transduction, 0.5 μg/ml puromycin (Invitrogen) was added to the RPMI media. After 14 days of selection, cells were tested in cell viability assays as described above. CW-2 cells, which harbor the endogenous ERBB2 L755S mutation (Barretina et al., 2012), were provided by the Riken cell line database under MTA and were maintained in RPMI containing 10% FBS and 1% penicillin/streptomycin. Cell lines are authenticated on an annual basis on all cell lines in active use using a PCR-based DNA fingerprinting assay (PowerPlex 16HS Kit, Promega) that detects genomic DNA sequences specific to each cell line. Cell line mycoplasma-free status is verified using the MycoAlert mycoplasma detection kit (Lonza). Ba/F3 cell lines were established as previously described (Robichaux et al., 2018). Briefly, stable Ba/F3 cell lines were generated by retroviral transduction of Ba/F3 cell line for 12 hr. Retroviruses were generated by transfecting pBabe-Puro based vectors summarized in KEY RESOURCE TABLE (Addgene and Bioinnovatise) into Phoenix 293T-ampho cells (Orbigen) using Lipofectamine 2000 (Invitrogen). Three days after transduction, 2 μg/ml puromycin (Invitrogen) was added to the RPMI media. After 5 days of selection, cells were stained with FITC-HER2 (Biolegend) sorted by FACS.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HER2 | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/her2-erbb2-d8f12-xp-rabbit-mab/4290?site-search-type=Products |
| HER2 | Abcam | https://www.abcam.com/erbb-2-antibody-ep1045y-ab134182.html |
| p-HER2 Tyr1248 | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/phospho-her2-erbb2-tyr1248-antibody/2247?site-search-type=Products |
| pHER2 Tyr1221/1222 | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/phospho-her2-erbb2-tyr1221-1222-6b12-rabbit-mab/2243?site-search-type=Products |
| pAkt | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/phospho-akt-ser473-d9e-xp-rabbit-mab/4060?site-search-type=Products |
| Akt | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/akt-antibody/9272?site-search-type=Products |
| p-p44/42 | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/phospho-p44-42-mapk-erk1-2-thr202-tyr204-d13-14-4e-xp-rabbit-mab/4370?site-search-type=Products |
| P44/42 | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/p44-42-mapk-erk1-2-137f5-rabbit-mab/4695?site-search-type=Products |
| Ubquitin | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/ubiquitin-antibody/3933?site-search-type=Products |
| tubulin | Cell Signaling Technologies | https://www.cellsignal.com/products/primary-antibodies/b-tubulin-antibody/2146?site-search-type=Products |
| Rabbit IgG | Santa Cruz | https://datasheets.scbt.com/sc-2027.pdf |
| Goat Anti-Rabbit IgG (H+L) HRP Conjugate | BioRad | http://www.bio-rad.com/en-us/sku/1706515-goat-anti-rabbit-igg-h-l-hrp-conjugate?ID=1706515 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Erlotinib | Selleck Chem | https://www.selleckchem.com/products/erlotinib.html |
| Lapatinib | Selleck Chem | https://www.selleckchem.com/products/lapatinib.html |
| Afatinib | Selleck Chem | https://www.selleckchem.com/products/BIBW2992.html |
| Dacomitinib | Selleck Chem | https://www.selleckchem.com/products/pf299804.html |
| Neratinib | Selleck Chem | https://www.selleckchem.com/products/Neratinib(HKI-272).html |
| Poziotinib (in vitro / in vivo) | Selleck Chem | https://www.selleckchem.com/products/poziotinib-hm781-36b.html |
| Osimertinib | Selleck Chem | https://www.selleckchem.com/products/azd9291.html |
| Ibrutinib | Selleck Chem | https://www.selleckchem.com/products/pci-32765.html |
| Nazartinib | Selleck Chem | https://www.selleckchem.com/products/nazartinib-egf816-nvs-816.html |
| Olmutinib | Selleck Chem | https://www.selleckchem.com/products/olmutinib-hm61713-bi-1482694.html |
| Rocelitinib | Selleck Chem | https://www.selleckchem.com/products/co-1686.html |
| Tarloxotinib-TKI | Synthesized by IACS | |
| T-DM1 | MD Anderson institutional pharmacy | |
| Poziotinib (clinical trial) | Spectrum Pharmaceuticals | |
| Critical Commercial Assays | ||
| Cell Titer Glo | Promega | https://www.promega.com/products/cell-health-assays/cell-viability-and-cytotoxicity-assays/celltiter_glo-luminescent-cell-viability-assay/?catNum=G7570 |
| ECL | Advansta | https://advansta.com/products/western-blot-substrate-WesternBright-ECL/ |
| Crosslink Immunoprecipitation | Pierce | https://www.thermofisher.com/order/catalog/product/26147 |
| HER2 ELISA | Cell signaling | https://www.cellsignal.com/products/elisa-kits/total-her2-erbb2-sandwich-elisa-kit/7310 |
| Pan-pHER2 ELISA | Cell Signaling | https://www.cellsignal.com/products/elisa-kits/phospho-her2-erbb2-pantyr-sandwich-elisa-kit/7968?_=1563400997725&Ntt=pHER2%20ELISA&tahead=true |
| Experimental Models: Cell Lines | ||
| Ba/F3 | Creative Biogene | https://www.creative-biogene.com/support/ba-f3-cell-line.html |
| MCF10A | ATCC | https://www.atcc.org/products/all/CRL-10317.aspx |
| CW-2 | Riken Cell Bank | http://cellbank.brc.riken.jp/cell_bank/CellInfo/?cellNo=RCB0778 |
| Experimental Models: Organisms/Strains | ||
| HER2 Y772dupYVMA PDX | JAX Labs | http://tumor.informatics.jax.org/mtbwi/pdxDetails.do?modelID=TM01446 |
| Athymic Nude Mice | Harlan Envigo | https://www.envigo.com/products-services/research-models-services/models/research-models/mice/mutant/athymic-nude-mice/ |
| NSG Mice | JAX Labs | https://www.jax.org/strain/005557 |
| Recombinant DNA | ||
|
ERBB2 L755S c.2264T>C |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 D769H c.2305G>A |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 D769N c.2305G>C |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 D769Y c.2305G>T |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 Y772dupYVMA c.2323_2324insTATGTCATGGCT |
Purchased from Addgene (#40982) | |
|
ERBB2 G776delinsVC c.2326_2328insTCT |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 G776del insVV c.2327delinsTTGT |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 G776del insLC c.2326G>TTGT |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 V773M c.2317G>A |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 V777L c.2329G>T |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 G778insLPS c.2332_2333insGGCTCCCCA |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 P780insGSP c.2339_2340insTGGCTCCCC |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 L786V c. 2356C>G |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 V842I c.2524G>A |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
|
ERBB2 L869R c.2606T>G |
Created by Bioinnovatise from pBabe-puro ERBB2 WT from Addgene (#40978) | |
| Other | ||
| HER2 Crystal Structure | PDB | https://www.rcsb.org/structure/3PP0 |
Mouse models
HER2 Y772dupYVMA (A775insYVMA) GEMMs were generated as previously described (Perera et al., 2009). Male and female mice were fed a continuous doxycycline diet from 6 weeks of age. Tumor volume was determined through MRI. Mice were handled in accordance with Good Animal Practices as defined by Laboratory Animal Welfare and were done with approval from the New York University Institutional Animal Care and Use Committee (NY,NY). Six-week old female athymic nude mice (Hsd: Athymic Nud-Foxn1nu) were purchased from Harlan Envigo., and 5- to 6-week old female NSG mice (NOD.Cg-Prkdcscid IL2rtftmWjl/Szj) were purchased from Jax Labs. Mice were maintained in agreement with Good Animal Practices and with approval from MD Anderson Cancer Center Institutional Animal Care and Use Committee (Houston, TX)
METHOD DETAILS
Analysis of ERBB2 variant frequency
To determine the frequencies of each ERBB2 mutation reported in databases from MD Anderson Cancer Center, cBioPortal, Foundation Medicine, or Guardant Health, each database was queried individually, then frequencies were weighted by the total number of patients in each database and are reported as weighted averages. To determine the frequency of ERBB2 mutations across cancer types in cBioPortal, all non-overlapping studies were selected and exported. For overlapping studies, only the largest dataset was used. To determine ERBB2 mutation frequencies at MD Anderson Cancer Center, the Institute for Personalized Cancer Therapy database was queried for all ERBB2 mutations independent of cancer type. To determine the frequency of ERBB2 exon 20 mutations from Foundation Medicine, de-identified data of the number of patients with ERBB2 deletions, frame shifts, insertions, and point mutation were tabulated, and cancer types with less than 5 mutations were excluded. Lastly, to determine the frequency of ERBB2 exon 20 mutations at Guardant Health, the Guardant360 clinical database was queried for samples tested between October 2015 and May 2018 (70 and 73 gene panels) with an ERBB2 exon 20 mutation. Guardant360® is a CLIA - certified, CAP / NYSDOH accredited comprehensive circulating free DNA (cfDNA) NGS test that reports out SNVs, indels, fusions, and SNVs in up to 73 genes. Frequencies reported from Guardant Health were then normalized to correct for clinical sensitivity as reported in Odegaard et al 2018. Specifically, frequencies were divided by the percent clinical sensitivity, 85.9%.
Ba/F3 IL-3 deprivation
Established Ba/F3 cell lines were then grown in the absence of IL-3 for two weeks and cell viability was assessed every three days using the Cell Titer Glo assay (Progema). Relative fluorescence values (RFV) were normalized to day zero by dividing each RFV by the average RFV of three technical replicates at day zero. RFV were determined in both technical and biological triplicate. The average fold change from day zero ±SEM of three biological replicates were graphed using GraphPad prism. Resulting stable cell lines were maintained in RPMI-1640 media containing 10% FBS without IL-3.
Cell viability assay and IC50 estimation
Cell viability was determined using the Cell Titer Glo assay (Promega) as previously described (Robichaux et al., 2018). Briefly, 2000-3000 cells per well were plated in 384-well plates (Greiner Bio-One) in technical triplicate. Cells were treated with seven different concentrations of tyrosine kinase inhibitors or vehicle alone at a final volume of 40 μL per well. After 3 days, 11 μL of Cell Titer Glo was added to each well. Plates were shaken for 15 min, and bioluminescence was determined using a FLUOstar OPTIMA multi-mode micro-plate reader (BMG LABTECH). Bioluminescence values were normalized to DMSO treated cells, and normalized values were plotted in GraphPad Prism using non-linear regression fit to normalized data with a variable slope. IC50 values were calculated by GraphPad Prism at 50% inhibition.
ELISA correlation with IC50 values
Protein was harvested from the parental Ba/F3 cell line and each of the Ba/F3 cell lines expressing HER2 mutants described above. 5 μg/ml of protein was added to each ELISA plate and ELISA was performed as described by the manufacture instructions for phosphorylated HER2 Cell signaling, (#7968) and total HER2 (Cell Signaling, #7310). Relative p-HER2 expression was determined by taking the ratio of p-HER2 over total HER2 as determined by ELISA. The relative level of p-HER2 determined by ELISA was plotted against poziotinib IC50 values calculated as described above. Pearson correlations and p values were determined by GraphPad Prism.
Molecular Dynamics Simulations
Protein structural models of the HER2 mutants were constructed using the MOE computer program (Chemical Computing Group) by introducing in silico mutations to the PDB 3PP0 X-ray structure of HER2 with SYR127063 bound. Coordinates for Asp880 and G881, which were absent from the A-chain of the X-ray structure, were built and subjected to local minimization.
Classical MD (cMD) simulations were performed using the NAMD 2.12 simulation package (Phillips et al., 2005). The LEaP module contained with the AmberTools16 (D.A. Case, 2016) was used to generate the Amber topology files. Hydrogen atoms were added using the Reduce program (Word et al., 1999) and the system was parameterized using the Amber force field 14SB (Maier et al., 2015) in conjunction with the TIP3P water model (Jorgensen et al., 1983). The structures were then solvated in a cubic box, keeping the boundary of the box at least 10.0 Å away from any solute atom. Appropriate number of counter ions were added to neutralize the system. A thorough equilibration scheme was applied to minimize and relax the system. MDS were then performed under periodic boundary conditions, beginning from the energy minimized structures. Bond lengths of hydrogen atoms were constrained using the SHAKE algorithm (Ryckaert et al., 1977) and the integration time step was 2 fs. A distance cutoff of 9.0 Å with PME (Darden et al., 1993) implementation was used compute the long-range electrostatic and short range van der Waals interactions. The system was linearly heated from 0 K to 300 K and held at 300 K for over 200 ps at constant volume (NVT). The system was further equilibrated for 0.5 ns in NPT ensemble to allow temperature and pressure equilibration prior to the NPT production run. A Langevin thermostat (Izaguirre et al., 2001) and a Nosé-Hoover Langevin piston barostat were used to maintain a constant temperature (300 K) and pressure (1 bar). Weak harmonic position restraints were imposed on the protein backbone and maintained for the full heating and equilibration runs. Finally, the equilibrated bimolecular systems were subjected to 50 ns of NPT production simulation using the parallel CUDA version of NAMD.
Starting from the final structure of the classical MD simulations, 100 ns of accelerated MD (aMD) simulations were performed using the ‘dual-boost’ methodology. The parameters for boosting were derived from their corresponding 50 ns classical MD (cMD) simulations, as proposed by Pierce et al (Pierce et al., 2012) and de Oliveira et al (Hamelberg et al., 2007).
aMD simulations were used to identify metastable states that typically occur on the millisecond timescale. Hence, reweighting of the phase space to obtain the canonical ensemble was not undertaken as other equilibrium properties were not considered.
To track αC helixin to αC helixout transition over the course of the simulation, a structural order parameter that measures the distance between the conserved Glu of the αC helix and the Lys of the β3 strand in the N lobe was used (Mobitz, 2015; Sultan et al., 2017).
FACS
MCF10A cells expressing HER2 mutants were plated overnight in a 6-well plate, then treated with 10 nM poziotinib. After 24 hr, cells were washed twice with PBS, and trypsinized. Cells were then resuspended in 0.5% FBS in PBS, and stained with anti-HER2-FITC antibody from Biolegend (#324404) for 45 min on ice. Cells were washed with 0.5% FBS in PBS twice, and analyzed by flow cytometry. IgG and unstained controls were used for gating.
Western Blotting
For Western blotting, cells were washed in PBS and lysed in RIPPA lysis buffer (ThermoFisher) and protease inhibitor cocktail tablets (Roche). Protein (30-40 μg) was loaded into gels purchased from Invitrogen. Overnight, transfers were preformed and blots were probed with antibodies against pHER2, HER2, pPI3K, PI3K, p-Akt, Akt, p-ERK1/2, ERK1/2, or ubquitinib (1:1000; Cell Signaling). Blots were probed with antibodies against vinculin or β-actin (Sigma-Aldrich) as a loading control, and exposed using ECL Western Blotting substrate (Promega).
Immunoprecipitation
MCF10A cells were grown in 10 cm plates and treated with 10 μM MG-132, and DMSO or 10 nM poziotinib for 2 hr. Immunoprecipitations were completed using Pierce Crosslink Immunoprecipitation Kit as described by the manufacture instructions. Briefly, cells were lysed with 500 μl lysis buffer provided from the kit supplemented with cOmplete protease inhibitor tablet (Roche 31075800) and PhosSTOP (Roche 4906845001). HER2 antibody (Abcam EP1045Y) and Rabbit IgG control (Santa Cruz) were cross-linked to the column provided in the kit at 4 C overnight. Protein lysates were pre-cleared and loaded onto the antibody cross-linked columns overnight at 4 C. Protein was eluted, prepared for Western Blotting using a reducing sample buffer, and run on a 4-12% precast gel (Invitrogen NP0321). Western blot was run as described above in western blotting section.
In vivo xenograft studies
CW-2 cell line xenografts were created by injecting 1x106 cells in 50% matrigel into six weeks old female nu/nu nude mice. When tumors reached 300-375 mm3 mice were randomized into 4 groups: 20 mg/kg afatinib, 5 mg/kg poziotinib, 30 mg/kg neratinib, or vehicle control (0.5% Methylcellulose, 2%Tween-80 in dH2O). Tumor volumes were measured three times per week. Mice received drug Monday- Friday (5 days per week), but began dosing on Wednesday allowing for a 2 day holiday after the first 3 days of dosing. ERBB2 Y772dupYVMA PDX mice were purchased from Jax Labs (Model # TM01446). Fragments from tumors expressing ERBB2 Y772dupYVMA were inoculated into 5- to 6-week old female NSG mice (Jax Labs #005557). Mice were measured three times per week, and when tumors reached a volume of 225-325 mm3 mice were randomized into four treatment groups: vehicle control (0.5% Methylcellulose, 0.05% Tween-80 in dH2O), 2.5 mg/kg poziotinib, 10 mg/kg T-DM1, or combination of 2.5 mg/kg poziotinib and 10 mg/kg T-DM1. Tumor volumes and body weight were measured three times per week. Mice treated with 2.5 mg/kg poziotinib received drug orally Monday- Friday (5 days per week). Mice treated with 10 mg/kg T-DM1 received one intravenous (IV) dose of T-DM1 on the day of randomization. Mice treated with combination poziotinib and T-DM1 received one IV dose of T-DM1 and began 2.5 mg/kg poziotinib five days per week, 3 days after the dose of T-DM1. Mice received a holiday from dosing if the mouse dropped in body weight by greater than 10% or if body weight dropped below 20 grams. Progression free survival was defined as tumor doubling from best response for two consecutive measurements. Complete regression was defined as greater than 95% reduction in tumor burden, and for mice with complete regression, tumor doubling was defined greater than 75 mm3 for more than two consecutive measurements. Experiments were completed in agreement with Good Animal Practices and with approval from MD Anderson Cancer Center Institutional Animal Care and Use Committee (Houston, TX).
GEMM study
Mice were fed a continuous doxycycline diet from 6 weeks of age. Tumor volume was determined through MRI. Mice with equal initial tumor volume were non-blindly randomized to four groups: vehicle control (0.5% Methylcellulose, 0.05% Tween-80 in dH2O), 2 mg/kg poziotinib daily (oral), 10 mg/kg T-DM1 (tail vein), or combination of 2 mg/kg poziotinib and 10 mg/kg T-DM1 upon obvious tumor formation as determined through MRI. Mice treated with combination poziotinib and T-DM1 received one IV dose of T-DM1 and began 2 mg/kg poziotinib daily, 3 days after the dose of T-DM1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blotting quantification
Quantification of western blotting was completed in Photoshop and calculated as (background mean intensity – sample mean intensity) × (number of pixels) = band intensity. Samples were normalized first to total HER2 then to DMSO and were graphed in GraphPad Prism.
Statistical analyses
Statistical differences between IC50 values was determined by One-way ANOVA with Dunn’s multiple comparisons test. Statistical differences between tumor volumes in xenografts was determined using a One-way ANOVA with Dunn’s multiple comparison. Differences in cell surface HER2 expression between DMSO and poziotinib treated were determined by a two-tailed student’s t-test for each cell line. Differences in ubiquitin expression between DMSO and poziotinib treated were determined by a two-tailed student’s t-test for WT and for mutant expressing cell lines. Changes in IC50 values with the addition of poziotinib to T-DM1 treatment in vitro was determined by two-way ANOVA and Dunn’s multiple comparison post-hoc for each cell line. Differences in percent change in tumor volume after treatment was determined by two-way ANOVA and Dunn’s multiple comparison post-hoc. Changes in progression free survival were determined by Mantel-Cox Log rank test between groups. Mice were censored at time tumor doubling. Changes in GEMM tumor volume were assessed by a one-way ANOVA. Pearson Correlations between pHER2 expression and poziotinib IC50 were determined by GraphPad Prism. Exact values of experimental replicates or samples (n) are located in the figure legends of each experiment.
DATA AND CODE AVAILABILITY
Public data resources
Data from TCGA/cBioPortal were downloaded from https://www.cbioportal.org/. HER2 crystal structure was downloaded from http://www.rcsb.org/. Data from Guardant Health and Foundation Medicine were available through MTA. For request related to data and code availability please contact the Lead Contact, John V. Heymach (JHeymach@mdanderson.org).
ADDITIONAL RESOURCES
Clinical trial description for :
https://clinicaltrials.gov/ct2/show/NCT03066206?term=Poziotinib&rank=6
Supplementary Material
Highlights.
ERBB2 mutations occur in at least 25 tumor types with varying patterns of mutations.
Mutation-induced changes in drug binding pocket volume dictate drug sensitivity.
Poziotinib inhibits mutant HER2, yielding a 42% response rate in NSCLC patients.
Combination of poziotinib with T-DM1 potentiates antitumor activity of both agents.
Significance.
Although ERBB2 mutations occur across multiple cancer types, there are currently no approved TKIs for these patients. We identified that ERBB2 mutations occurred in at least 25 different tumor types, and that mutation hotspots varied by malignancy. Through an integrative analysis using in vitro testing and molecular modeling, we identified key molecular features impacting the sensitivity to different TKIs. A phase II clinical study of the most potent TKI, poziotinib, confirmed that poziotinib has substantial clinical activity. Finally, we identified that the combination of poziotinib with an antibody-drug conjugate resulted in markedly enhanced efficacy in preclinical models. These findings can guide the development of more effective and targeted treatment regimens across the landscape of ERBB2-mutant cancers.
Acknowledgements
The authors wish to thank the patients, families, and caregivers involved in the clinical trial as well as the Exon 20 Group, Marcia Horn, Robert Hanlon, and Kevin Hanlon.
This work was supported by funds from R01 CA190628-NIH/NCI, the David Bruton, Jr. Endowment, the Rexanna Foundation for Fighting Lung Cancer, CCSG Lung Cancer Program, the Lung Cancer Moon Shot Program, Spectrum Pharmaceuticals, the Kopelman Fund for Lung Cancer Research, the Exon 20 Group, the Lung Cancer Research Fund, the Hallman fund, Lung SPORE grant 5 P50 CA070907, ASCO CDA-57112 and 1U54CA224065-01.
JPR, MBN, & JVH have filed a patent for the use of poziotinib for treating EGFR & HER2 mutant cancers & licensed the technology to Spectrum Pharmaceuticals (SP). JVH also has/had the following disclosures: grant/research support: AstraZeneca (AZ), Bayer, & GlaxoSmithKline (GSK); advisory committees: AZ, Boehringer Ingelheim, Bristol-Myers Squibb (MBS), Exelixis, Hengrui Therapeutics, Genentech, GSK, Guardant Health (GH), Lilly, Novartis, SP, Takeda, & Synta. XL consultant: Lilly & AZ. KKW founder & equity holder: G1 Therapeutics; Consulting/Sponsored Research: AZ, Janssen, Pfizer, Array, Novartis, Merck, Takeda, Ono, Targimmune & BMS. FMB has/had grants, travel/research support: Novartis, AZ, Calithera, Aileron, Bayer, Jounce, CytoMx, eFFECTOR, Zymeworks, PUMA, Curis, Millennium, Daiichi Sankyo, Abbvie, GH, Takeda, GSK, Taiho, Genentech, Debiopharm Group, & Pfizer; consult/advisor: Pieris, Dialectica, Sumitomo Dainippon, Samsung Bioepis, Aduro, OrigiMed, Xencor, JAX Labs, Inflection Biosciences, GRAIL, Darwin Health, Clearlight Diagnostics, SP, & Mersana. JRA reasonable reimbursement for travel: European Journal of Cancer, Vall d’Hebron Institut of Oncology, CU of Hong Kong, SOLTI, Elsevier, GSK; consulting/travel fees: Novartis, Lilly, Orion, Servier, Peptomyc, Merck Sharp & Dohme, Kelun/Klus Pharma, SP, Pfizer, Roche, Ellipses; research funding: Bayer & Novartis; investigator in clinical trials: SP, Tocagen, Symphogen, BioAtla, Pfizer, GenMab, CytomX, KELUN-BIOTECH, Takeda-Millenium, GSK, IPSEN; travel fees: ESMO, US DoD, LSU, Hunstman Cancer Institute, Cancer Core Europe, Karolinska Cancer Institute & King Abdullah International Medical Research Center. VMR & RBL shareholders/full time employees: GH. RBL stockholder and board of directors: Biolase, Inc; stockholder and advisory board: Forward medical. BR & ZY shareholders/full-time employees: SP. LAA, AS, GMF, & VAM shareholders/full-time employees: FMI; stockholders: Roche Holding AG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The research being reported in this publication is research in which The University of Texas MD Anderson Cancer Center has an institutional financial conflict of interest. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’ s conduct of this research.
References
- Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A , et al. (2011). Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem 286, 18756–18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gomez H, Dinh P, Fauria K, Van Dooren V, et al. (2012a). Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 379, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, et al. (2012b). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 366, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, et al. (2013). Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 3, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu CG, Chan S, Jagiello-Gruszfeld A, Kaufman B, Crown J, et al. (2008). A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 112, 533–543. [DOI] [PubMed] [Google Scholar]
- Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, and Mohammadi M (2007). A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell 27, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell CM, and Doherty GJ (2017). Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open 2, e000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, M. B R, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L and Kollman PA (2016). AMBER 2016. In, (San Francisco, California: University of Califorina; ). [Google Scholar]
- Darden T, York D, and Pedersen L (1993). Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Phys 98, 10089–10092. [Google Scholar]
- Di Leo A, Gomez HL, Aziz Z, Zvirbule Z, Bines J, Arbushites MC, Guerrera SF, Koehler M, Oliva C, Stein SH, et al. (2008). Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol 26, 5544–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, Chang JY, Chirieac LR, D’Amico TA, Dilling TJ, Dobelbower M, et al. (2018). NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw 16, 807–821. [DOI] [PubMed] [Google Scholar]
- Gomez HL, Doval DC, Chavez MA, Ang PC, Aziz Z, Nag S, Ng C, Franco SX, Chow LW, Arbushites MC, et al. (2008). Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol 26, 2999–3005. [DOI] [PubMed] [Google Scholar]
- Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, et al. (2012). Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A 109, 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, Burris H, Bose R, Yoo B, Stein A, et al. (2018). Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol 36, 536–542. [DOI] [PubMed] [Google Scholar]
- Hamelberg D, de Oliveira CAF, and McCammon JA (2007). Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J Chem Phys 127. [DOI] [PubMed] [Google Scholar]
- Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, et al. (2004). Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 22, 1589–1597. [DOI] [PubMed] [Google Scholar]
- Hartmaier RJ, Albacker LA, Chmielecki J, Bailey M, He J, Goldberg ME, Ramkissoon S, Suh J, Elvin JA, Chiacchia S, et al. (2017). High-Throughput Genomic Profiling of Adult Solid Tumors Reveals Novel Insights into Cancer Pathogenesis. Cancer Res 77, 2464–2475. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, et al. (2016). Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550. [DOI] [PubMed] [Google Scholar]
- Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M, Poddubskaya E, Borghaei H, Felip E, Paz-Ares L, et al. (2017). Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J Clin Oncol 35, 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, Juric D, Quinn DI, Moreno V, Doger B, et al. (2018). HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaguirre JA, Catarello DP, Wozniak JM, and Skeel RD (2001). Langevin stabilization of molecular dynamics. J Chem Phys 114, 2090–2098. [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 79, 926–935. [Google Scholar]
- Kosaka T, Tanizaki J, Paranal R, Endoh H, Lydon C, Capelletti M, Repellin CE, Choi J, Ogino A, Calles A, et al. (2017). Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourie HR, Chaix M, Gombos A, Aftimos P, and Awada A (2016). Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in HER2-positive breast cancer and breast cancer with HER2 mutations. Expert Opin Drug Metab Toxicol 12, 947–957. [DOI] [PubMed] [Google Scholar]
- Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O’Connell J, Taylor I, Zhang H, Arcila ME, Goldberg Z, and Janne PA (2015). Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 26, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, Ulaner GA, Offin M, Feldman D, Hembrough T, et al. (2018). Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J Clin Oncol 36, 2532–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Bose R, Gao F, Freedman RA, Telli ML, Kimmick G, Winer E, Naughton M, Goetz MP, Russell C, et al. (2017). Neratinib Efficacy and Circulating Tumor DNA Detection of HER2 Mutations in HER2 Nonamplified Metastatic Breast Cancer. Clin Cancer Res 23, 5687–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, Peters S, Dansin E, Fruh M, Pless M, et al. (2016). Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol 27, 281–286. [DOI] [PubMed] [Google Scholar]
- Meric-Bernstam F, Johnson A, Ileana Dumbrava EE, Raghav K, Balaji K, Bhatt M, Murthy RK, Rodon J, and Piha-Paul SA (2018). Advances in HER2-Targeted Therapy: Novel Agents and Opportunities Beyond Breast and Gastric Cancer. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobitz H (2015). The ABC of protein kinase conformations. Biochim Biophys Acta 1854, 1555–1566. [DOI] [PubMed] [Google Scholar]
- Nagano M, Kohsaka S, Ueno T, Kojima S, Saka K, Iwase H, Kawazu M, and Mano H (2018). High-throughput functional evaluation of variants of unknown significance in ERBB2. Clin Cancer Res. [DOI] [PubMed] [Google Scholar]
- Negrao MV, Reuben A, Robichaux JP, Le XN, Nilsson MB, and Wu CJ (2018). Association of EGFR and HER-2 exon 20 mutations with distinct patterns of response to immune checkpoint blockade in non-small cell lung cancer. Journal of Clinical Oncology 36. [Google Scholar]
- Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, Fairclough SR, Zill OA, Sikora M, Mokhtari R, et al. (2018). Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res 24, 3539–3549. [DOI] [PubMed] [Google Scholar]
- Oh IJ, Hur JY, Park CK, Kim YC, Kim SJ, Lee MK, Kim HJ, Lee KY, Lee JC, and Choi CM (2018). Clinical Activity of Pan-HER Inhibitors Against HER2-Mutant Lung Adenocarcinoma. Clin Lung Cancer 19, e775–e781. [DOI] [PubMed] [Google Scholar]
- Pandey A, and Brufsky AM (2018). Metastatic Breast Cancer Patient With Activating HER2 Exon 20 Insertion Mutation With Response to Poziotinib: Case Report of Compassionate Drug Use. Clin Breast Cancer. [DOI] [PubMed] [Google Scholar]
- Park JW, Liu MC, Yee D, Yau C, van ‘t Veer LJ, Symmans WF, Paoloni M, Perlmutter J, Hylton NM, Hogarth M, et al. (2016). Adaptive Randomization of Neratinib in Early Breast Cancer. N Engl J Med 375, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, Borgman CL, Zaghlul S, Brandstetter KA, Kubo S, et al. (2009). HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci U S A 106, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S, Curioni-Fontecedro A, Nechushtan H, Shih JY, Liao WY, Gautschi O, Spataro V, Unk M, Chih-Hsin Yang J, Lorence RM, et al. (2018). Activity of Afatinib in Heavily Pretreated Patients with HER2 Mutation-Positive Advanced NSCLC: Findings from a Global Named Patient Use Program. J Thorac Oncol. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce LCT, Salomon-Ferrer R, de Oliveira CAF, McCammon JA, and Walker RC (2012). Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. Journal of Chemical Theory and Computation 8, 2997–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, et al. (2017). Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, et al. (2018). Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 24, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, and Berendsen HJC (1977). Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys 23, 327–341. [Google Scholar]
- Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S, Arribas J, and Baselga J (2009). Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28, 803–814. [DOI] [PubMed] [Google Scholar]
- Shan L, Qiu T, Ling Y, Guo L, Zheng B, Wang B, Li W, Li L, and Ying J (2015). Prevalence and Clinicopathological Characteristics of HER2 and BRAF Mutation in Chinese Patients with Lung Adenocarcinoma. Plos One 10, e0130447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Chen B, Ma Z, Xie B, Cao X, Yang T, Zhao Y, Qin J, Li J, Cao F, and Chen X (2016). A systematic analysis of the resistance and sensitivity of HER2YVMA receptor tyrosine kinase mutant to tyrosine kinase inhibitors in HER2-positive lung cancer. J Recept Signal Transduct Res 36, 89–97. [DOI] [PubMed] [Google Scholar]
- Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O’Rourke M, Levitan N, Gressot L, Vincent M, Burkes R, et al. (2000). Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18, 2095–2103. [DOI] [PubMed] [Google Scholar]
- Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et al. (2011). Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 365, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sours KM, Xiao Y, and Ahn NG (2014). Extracellular-regulated kinase 2 is activated by the enhancement of hinge flexibility. J Mol Biol 426, 1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan MM, Denny RA, Unwalla R, Lovering F, and Pande VS (2017). Millisecond dynamics of BTK reveal kinome-wide conformational plasticity within the apo kinase domain. Sci Rep 7, 15604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, Guardino E, et al. (2012). Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 367, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Harrison CB, Schulten K, and McCammon JA (2011). Implementation of Accelerated Molecular Dynamics in NAMD. Comput Sci Discov 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmuth M, Kim S, Gu XJ, Xia G, and Adrian F (2007). Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol 19, 55–60. [DOI] [PubMed] [Google Scholar]
- Word JM, Lovell SC, Richardson JS, and Richardson DC (1999). Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285, 1735–1747. [DOI] [PubMed] [Google Scholar]
- Xu ZQ, Zhang Y, Li N, Liu PJ, Gao L, Gao X, and Tie XJ (2017). Efficacy and safety of lapatinib and trastuzumab for HER2-positive breast cancer: a systematic review and meta-analysis of randomised controlled trials. BMJ Open 7, e013053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zhang H, and Wang H (2015). Atomistic insights into the lung cancer-associated L755P mutation in HER2 resistance to lapatinib: a molecular dynamics study. J Mol Model 21, 24. [DOI] [PubMed] [Google Scholar]
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, and Eck MJ (2008). The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 105, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Public data resources
Data from TCGA/cBioPortal were downloaded from https://www.cbioportal.org/. HER2 crystal structure was downloaded from http://www.rcsb.org/. Data from Guardant Health and Foundation Medicine were available through MTA. For request related to data and code availability please contact the Lead Contact, John V. Heymach (JHeymach@mdanderson.org).