

Abstract
Understanding the biosynthesis of cofactors is fundamental to the life sciences, yet to date a few important pathways remain unresolved. One example is the redox cofactor pyrroloquinoline quinone (PQQ) which is critical for C1 metabolism in many microorganisms, a disproportionate number of which are opportunistic human pathogens. While the initial and final steps of PQQ biosynthesis, involving PqqD/E and PqqC, have been elucidated, the precise nature and order of the remaining transformations in the pathway are unknown. Here we show evidence that the remaining essential biosynthetic enzyme PqqB is an iron-dependent hydroxylase catalyzing oxygen insertion reactions that are proposed to produce the quinone moiety of the mature PQQ cofactor. The demonstrated reactions of PqqB are unprecedented within the metallo β-lactamase protein family and expand the catalytic repertoire of non-heme iron hydroxylases. These new findings also generate a nearly complete description of the PQQ biosynthetic pathway.
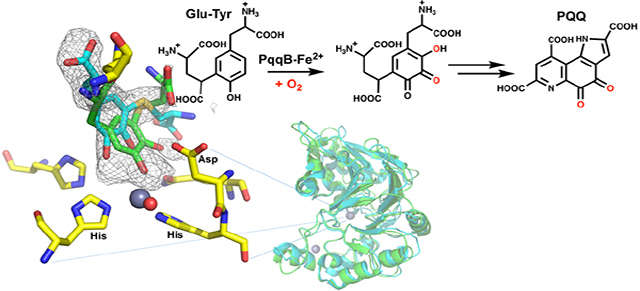
Graphical Abstract
INTRODUCTION
The redox cofactor PQQ is a member of secondary metabolites annotated as ribosomally synthesized and posttranslationally modified peptides (RiPPs) and functions as a prosthetic group of alcohol and/or sugar dehydrogenases.1–5 PQQ is produced by a plethora of gram negative bacteria, a disproportionate number of which are opportunistic human pathogens, making the biosynthetic pathway for PQQ an attractive target for antibiotic development.1, 6–8 Despite being categorized as a third major redox cofactor behind flavins and nicotinamides, a detailed account of PQQ biosynthesis has remained unresolved for over 40 years.4, 6, 8–9
Gene knockout and bioinformatics studies indicate that five genes are required (i.e., pqqA-E), and up to seven genes are implicated (e.g., pqqF and pqqG), in PQQ biosynthesis.8–9 The first step is a C-C crosslinking reaction between the conserved Glu and Tyr residues within the peptide substrate PqqA (Fig. 1a). This is catalyzed by the radical SAM enzyme PqqE and requires the protein PqqD to chaperone PqqA to the active site of PqqE for crosslinking.10–11 The final and most characterized step of PQQ biosynthesis is catalyzed by PqqC, a cofactorless oxidase that carries out a step-wise 8 electron, 8 proton oxidation of the isoquinoline heterocycle AHQQ to PQQ.12–13 However, reactions occurring after the initial crosslinking by the PqqE/D complex and prior to the final reaction catalyzed by PqqC have remained an enigma (Fig. 1a).
Figure 1: PQQ cofactor biosynthesis.

a) A biosynthetic schematic. Steps in blue are derived from the analog studies presented herein. A protease, to convert the peptide-bound cross-linked Glu and Tyr to the substrate of PqqB, is inferred from the present study but not yet characterized. b) Structures of the PqqB substrate analog Cys-Tyr and catalytic intermediate analog Cys-DOPA.
PqqB belongs to the metallo β-lactamase (MBL) family, and contains two metal binding sites per monomer; one of these contains a tightly bound structural Zn2+ that induces correct protein folding and dimerization.14 The second metal binding site, and putative active site, presents a 2-His/1-Asp ligand set for metal coordination (see Supplementary Information Fig. S1). PqqB has been reported to hydrolyze amide and thioester bonds of the substrates of MBL and glyoxalase,15 respectively; however, the turnover rates for these processes are extremely low and suggest residual ancestral activity. Without a conclusive demonstration of robust PqqB activity, proposals of function remain speculative.8, 14, 16 Herein, we present a combination of metal and ligand binding studies, X-ray crystallography and detailed activity assays to show that PqqB is a previously uncharacterized hydroxylase. The aggregate data indicate that PqqB is capable of generating a hydroxyquinone intermediate that possesses the final quinone moiety of the mature PQQ cofactor. The implication of two stepwise insertions of oxygen into the tyrosine ring of a PqqA-derived crosslinked diamino acid substrate, along with a down-pathway reaction also detected herein, permit a close to complete description of this elusive biosynthetic pathway.
RESULTS AND DISCUSSION
Metal and substrate analog binding determinates.
A range of metals (Table S1) and potential substrates (Fig. S2) were tested with the Methylobacterium extorquens PqqB (Me PqqB), which identified Fe2+ as the most probable catalytic metal ion. At the outset, we considered the crosslinked Glu-Tyr a likely substrate (Fig. 1a), and generated the synthetically tractable Cys-Tyr (a putative substrate analog) and Cys-DOPA (a mimic of a potential PqqB catalytic intermediate, discussed below); these analogs replace the methylene and γ–carboxylate of Glu by sulfur (Fig. 1b).17–18 Initial metal binding experiments assayed for the affinity of Cu2+, Zn2+, Fe2+, Fe3+, Mg2+, and Mn2+ within the inferred active site of PqqB (Fig. S3 and S4). The highest affinities detected were for Cu2+ followed by Zn2+ (Fig. S3, Table S1), initially suggesting that PqqB might be a novel copper-dependent enzyme. Additional experiments pursued the extent to which the addition of Cys-Tyr or Cys-DOPA leads to altered metal binding and/or new UV/Vis spectral features. As shown in Fig. S5, aerobic addition of Cu2+, Cu1+ (anaerobic), Zn2+ and Fe3+, but not Mg2+ nor Mn2+, gives rise to a charge transfer band between 300 nm and 350 nm in the presence of Cys-DOPA. The presence of Cys-DOPA also increases the strength of metal binding for Zn2+ and Fe2+ (Fig. 2a and b, Table S1). A similar trend of increased binding for Cu2+ was anticipated, but could not easily be tested due to the very high intrinsic affinity of Cu2+ in the absence of added ligand. The additional feature, of the comparative impact of Cys-Tyr vs Cys-DOPA on the strength of metal ion binding, was then examined. Only a single metal, Fe2+, is observed to undergo increase in affinity to Me PqqB in the presence of both Cys-Tyr (ca. 3-fold) and Cys-DOPA (ca. 10-fold), Fig. 2b. Of potential mechanistic importance, replacement of Fe2+ by Fe3+ obliterated the impact of Cys-DOPA on metal affinity (Fig. S4 and Table S1), which when taken collectively with the results above implicated a functional role for Fe2+.
Figure 2. Substrate and metal binding determination by ICP and ITC.

a) ICP binding titration of ZnCl2 or b) FeSO4 into MePqqB (black), with Cys-DOPA (red) or with substrate analog Cys-Tyr (blue). The subsequent discovery of rapid oxidation of Cys-DOPA by Fe2+-PqqB (Fig. 3 below) indicates that the red trace would have contained a mixture of Cys-DOPA and oxidized product. Nevertheless, a strong impact on Fe2+ binding is observed. c) ITC titration of Cys-DOPA into MePqqB without metal present, or d) with excess ZnCl2. The background traces (buffer titrated into PqqB) are in red.
The implied synergy observed for the binding of either Zn2+ or Fe2+ in the presence of Cys-DOPA led to an examination of the affinity of Cys-DOPA to PqqB in the presence or absence of the non-redox active Zn2+. As assayed by ITC, the interaction of Cys-DOPA with MePqqB is negligible in the absence of Zn2+ (Fig. 2c) but undergoes very tight binding (Kd ~ 200 nM) when excess Zn2+ is included in the titration (Fig. 2d). While a comprehensive analysis of all substrate analogs (cf., Fig. S2) was beyond the scope of this work, an X-ray structure of the Zn2+-PqqB-Cys-DOPA complex provides additional details on the substrate analog-metal interaction (presented below).
Fe2+-dependent hydroxylase activity of PqqB.
Our expectation was that the substrate analog Cys-Tyr might be capable of ring hydroxylation, mimicking an early step in the requisite addition of two addition oxygen atoms into the tyrosine precursor of PQQ. However, all efforts to establish a robust, metal-dependent reaction of PqqB with Cys-Tyr were unsuccessful, with the notable exception of an Fe2+-catalyzed oxidative decarboxylation of alpha-ketoglutarate (α-KG) (Fig. 3a and b and Fig. S6). The suggestion that α-KG, a common co-substrate of non-heme iron hydroxylases,19 could act as a redox co-substrate in PqqB came from the X-ray structure of the Mn2+-reconstituted enzyme from P. putida, which required malic acid to retain significant bound metal in the crystal.14 Addition of Cys-Tyr and α-KG to PqqB does, in fact, give rise to a slow increase in absorbance between 350 and 550 nm (Fig. 3a), though the very low level of product precluded detailed characterization. Reasoning that a highly uncoupled decarboxylation of α-KG might be occurring, we looked for evidence of the accompanying reaction product, succinate. As shown in Fig. 3b, succinate (comparable to a standard Fig. S6) is clearly detected by LC-HRMS in the reaction of Fe2+-PqqB with Cys-Tyr and α-KG, falling to background when Cys-Tyr is excluded from the reaction mixture. The low (5% turnover after 1.5 h) but unambiguous (tens of μM) production of succinate indicates an uncoupling of α-KG decarboxylation from substrate hydroxylation in the presence of Cys-Tyr; this can be attributed to a disruption in the binding geometry as well as altered ring electronics and reactivity relative to a native Glu-Tyr substrate.
Figure 3. Reaction details of Fe2+-dependent PqqB hydroxylation.

a) and b) Reaction with Cys-Tyr: a) UV-visible spectra of PqqB, Fe2+ and Cys-Tyr in solution (black) and after addition of β-KG (blue). b) LC-HRMS extracted ion chromatograms quantifying the succinate fragment ion by tandem MS (m/z = 73.33, [M-H]−) relative to a succinate standard (Fig. S6). Succinate formation is seen for reaction mixtures of PqqB, Fe2+, α-KG and Cys-Tyr (blue), but not in the control in absence of Cys-Tyr (black). c) and d) Reaction with Cys-DOPA: c) Reaction time course showing O2 consumption (red), and 413-nm absorption band formation (blue). d) Absence of hydrogen peroxide production during Fe2+-PqqB hydroxylation of Cys-DOPA. e) UV-visible spectra of anaerobic PqqB and Cys-DOPA (black). Addition of FeSO4 results in charge transfer (gold). Exposure to O2 leads to rapid formation of OH-Cys-DOPA quinone (blue). An analogous reaction is conducted with L-DOPA to give OH-DOPA quinone (magenta).
The final structure of PQQ (Fig. 1a) indicates that one oxygen atom must undergo insertion ortho-to the C4 hydroxyl of tyrosine, pointing toward Glu-DOPA as a reasonable first intermediate generated via a stepwise oxygenation of Glu-Tyr. We proceeded to synthesize the Cys-DOPA analog as a model for the putative Glu-DOPA, testing the former’s activity with PqqB. Remarkably, incubation of Cys-DOPA with Fe2+-reconstituted PqqB leads to an almost instantaneous (< 1 min) oxygen-dependent reaction (Fig. 3c), and occurs without detectable production of hydrogen peroxide (Fig. 3d). This process converts an initially formed charge-transfer complex to a 413 nm species in an O2-dependent process (Fig. 3e). Liquid chromatography-high resolution mass spectrometry (LC-HRMS) analyses showed a new ion of mass-to-charge ratio (m/z) = 331.0596 ([M+H]+) (Fig. 4a and Fig. S7) consistent with OH-Cys-DOPA quinone (+14 Da). The source of the inserted oxygen was then monitored by 18O-isotopic labeling. Reactions of Cys-DOPA and PqqB were carried out either in an atmosphere of 97% 18O2, or with 97% 18OH2 as a solvent, and evaluated by LC-HRMS. The uptake of one oxygen atom during the reaction with 18O2 is shown from the 2 Da change in the product: there is no mass change when the reaction is conducted in 18OH2 (Fig. 4a) indicating that the source of the inserted oxygen is from molecular O2.
Figure 4. High-Resolution Mass Spectrometric (HRMS) analyses of Fe2+-dependent PqqB hydroxylation.

a) Overlaid mass spectra showing isotope 18O-incorporation from 18O2 atmosphere exposure during the Fe2+-PqqB-catalyzed hydroxylation of Cys-DOPA (blue to red). No 18O-atom incorporation was observed when the reaction was conducted in 18OH2 (blue to black). b) MS/MS analyses of the OH-Cys-DOPA quinone and c) 18OH-Cys-DOPA quinone product. Details of the LC-HRMS experiments are provided in Supplementary Information Figs. S6, S7, and S8. The behavior in (c) is ascribed to an equilibration of OH-Cys-DOPA quinone between the species shown in red in (a) and a tautomer bearing the ring hydroxyl group at C-4 of the OH-Cys-DOPA quinone product.
To confirm aromatic ring hydroxylation, and exclude the possibility of sulfur oxidation accompanied by quinol to quinone formation (also predicting a +14 Da product), we performed several essential controls. First, we reacted PqqB with L-DOPA (lacking the Cys moiety all together), observing a UV-Vis absorbance feature similar to the OH-Cys-DOPA quinone product (Fig. 3e); LC-HRMS analyses showed a product mass consistent with OH-DOPA quinone (addition of 14 Da, Fig. S8). Next, tandem MS (MS/MS) analyses were performed on the product of the PqqB reaction with Cys-DOPA under an 16O2 atmosphere. Collision induced dissociation (CID) of the hydroxylated Cys-DOPA (m/z = 331.0596, [M+H]+) produces a major fragment ion at m/z = 313.05 ([M+H]+) ascribed to a neutral loss of a molecule of water (−18 Da) (Fig. 4b). By contrast, CID of the 18O-labelled OH-Cys-DOPA quinone (m/z = 333.0639, [M+H]+) resulted in fragment ions arising from loss of both 16OH2 (−18 Da) and 18OH2 (−20 Da), leading to the distribution of peaks shown in Fig. 4c. While a loss of 18 Da could arise from lactone formation between the inserted oxygen and side chain carboxylate, the loss of 20 Da requires scission of the newly inserted OH. From the near equal distribution of −18 Da and −20 Da fragments we suggest loss of a hydroxyl group from an equilibrium mixture of tautomeric trihydroxyquinone structures (either 18OH from C-2 or 16OH from C-4 (Fig. 4). These results both confirm production of OH-Cys-DOPA quinone and rules out a dominant oxidation of the thioether occurring concomitant with quinol oxidation. These assignments are fully supported by our failure to detect significant peroxide production, indicating that water is the second product derived from O2 reduction (Fig. 3d). While it is conceivable that a net 4-electron oxidation of both sulfur and the quinol of Cys-DOPA could lead to some water production, at least some hydrogen peroxide is expected from two parallel oxidative processes.
The collective results of (i) rapid ring hydroxylation of Cys-DOPA and (ii) conversion of α-KG to succinate in the presence of Cys-Tyr provide strong support for a PqqB-catalyzed hydroxylation of both the C2 and C3 positions of the PqqA-derived diamino acid substrate (Fig. 1a, numbering based on the Tyr ring alone). We note that the above data cannot distinguish between an oxygen addition to Cys-DOPA at the 2-vs. the 6-position of the DOPA ring; however, the fortuitous observation of a downstream product from OH-Cys-DOPA quinone (discussed below) provides experimental support for the regiochemistry presented in Fig. 1a.
X-ray structure of the inactive Zn2+-PpPqqB-Cys-DOPA complex.
To interrogate the structure of a Cys-DOPA complex we soaked this analog into crystals of Pseudomonas putida (Pp) PqqB reconstituted with inactive Zn2+ in the active site. Attempts to obtain a crystalline complex of Cys-DOPA with Zn2+-MePqqB were unsuccessful, but this was achieved using Zn2+-PpPqqB crystals (41.5% sequence identity, Fig. S1)14. The X-ray structure of this complex shows high occupancy of Cys-DOPA at the active site and close contact of the DOPA moiety with the metal ion (Fig. 5, Fig. S9 and Table S2). This is consistent with the metal-dependent tight binding of Cys-DOPA (Fig. 2d) and observed charge-transfer complexation (Fig. S5). These structural studies confirm the location of the active site of PqqB and suggest a functional role for the metal ion.
Figure 5. X-ray crystal structure of the Zn2+-PpPqqB-Cys-DOPA complex.

A close-up of the active site of the crystal structure of the inactive Zn2+-PpPqqB-Cys-DOPA complex (PDB ID 6E13). Two poses of Cys-DOPA are shown of the multiple possible poses that are consistent with the observed electron density. Further structure details are presented in Table S2 and Fig. S9.
Although omit electron density was strong in the active site, the Cys-DOPA could be modelled in multiple binding modes (Fig. S9a). One Cys-DOPA model is ligating the zinc as a monodentate through the O–3 position of the DOPA moiety (Fig. S9b), but there are other off-metal binding modes within the electron density at occupancies similar to the metal-ligated mode (one is shown in Fig. S9c). In one of the possible poses for Cys-DOPA, the electron density also contains difference density consistent with displacement of the metal-coordinating Asp92 ligand (Fig. S9 and S1)14; this may be relevant to the mobility of this Asp ligand in our proposed catalytic mechanism (Fig. 7 below). More than three distinct Cys-DOPA poses could simultaneously fit into the electron density, and although we refined combinations of these with differing relative occupancies, the results were unable to fully account for all the electron density. In light of this low level of data resolution, the deposited structure (PDB ID 6E13) was restricted to the protein without the bound ligand. The presence of multiple binding modes may well be the result of features that include the absence of a gamma-carboxylate in the Cys-DOPA analog as well as a tetrahedral geometry associated with zinc ligation that contrasts with an anticipated octahedral geometry for an active Fe2+ as observed in the Mn2+- reconstituted PpPqqB (PDB ID 4Z67)14. There is also the unresolved issue of whether the cross-linked Glu-Tyr product formed with PqqE/D will undergo full hydrolysis prior to interaction with PqqB. This question is under further investigation in the laboratory.
Figure 7. Proposed reactions of PqqB.

a) A proposed mechanism for the PqqB reaction with the Glu-Tyr diamino acid. b) Our postulated mechanism for the PqqB-catalyzed hydroxylation of Cys-DOPA. The X-ray structure of the Zn2+-PqqB-Cys-DOPA complex (Fig. 5), in which Asp is either on or off metal, is compatible with the illustrated mobility of the Asp ligand within intermediate (2). An alternative mechanism is presented for consideration in Fig. S10.
Identification of a downstream product from OH-Cys-DOPA quinone.
While monitoring the PqqB catalyzed formation of OH-Cys-DOPA quinone from Cys-DOPA, we observed a slower, time dependent, second product (Table S3) that is red-shifted in its absorbance features (Fig. 6a) and detected as an ion of m/z = 313.0490 ([M+H]+) (Fig. 6b). MS/MS analysis (Fig. 6c) and comparison of this products’ λmax at 460 nm to AHQQ13 suggested an isoquinoloid heterocycle derivative that is a structural analog AHQQ (Fig. 1a), the direct precursor to PQQ.13 Importantly, we were able to follow stable isotope incorporation from 18OH-Cys-DOPA quinone (formed in the presence of 18O2) into the 460 nm product, observing an ion of m/z = 315.0528 ([M+H]+) under these reaction conditions (Fig. 6b, inset). Thus, the new product is concluded to derive from OH-Cys-DOPA quinone in a precursor-product fashion.
Figure 6. Characterization of down-pathway reactions.

a) Overlaid UV-Visible spectra of the proposed isoquinoloid (magenta) product that appears after formation of OH-Cys-DOPA quinone (blue). Inset shows a time-course comparing the appearance of the initial 413 nm absorbance band (OH-Cys-DOPA quinone) and the subsequent 460 nm absorbance band (assigned to the putative isoquinoloid). b) A HRMS spectrum of a reaction of PqqB with Cys-DOPA at 1hr shows two predominant products occurring at m/z = 313.0490 ([M+H]+) (isoquinoloid) and m/z = 331.0596 ([M+H]+) (OH-Cys-DOPA quinone). Relative product distribution has been calculated from integrated extracted ion chromatographs (Table S3). Inset is a mass spectrum that shows isotope 18O-incorporation when reactions were conducted under an 18O2 atmosphere. c) MS/MS analysis of the product where the ion occurring at m/z = 313.0490 ([M+H]+) has been fragmented by collision induced dissociation (CID).
We estimate the rate of conversion of OH-Cys-DOPA quinone to the proposed isoquinoloid product to be ca. 10-fold slower than the initial hydroxylation of Cys-DOPA (Fig. 6a inset). Determination of the relative amounts of OH-Cys-DOPA quinone and isoquinoloid by LC-HRMS indicated that up to 15% of the original Cys-DOPA converts to the isoquinoloid within 10 minutes (Table S3), and that this value does not change substantially at longer incubations. Given the evidence for a precursor-product relationship between OH-Cys-DOPA quinone and isoquinoloid, one explanation for a leveling off of the latter at ca. 15% is the presence of competing reactions in which bound OH-Cys-DOPA quinone partitions between further cyclization and dehydration to form an isoquinoloid vs. release to solvent (in a ratio of 85:15). The use of Glu-Tyr as native substrate for PqqB could, in principle slow down dissociation of the hydroxylated reaction intermediate significantly, allowing sufficient time for near complete formation of enzyme-bound isoquinoloid product. However, the available structure of the Cys-DOPA complex with Pp-PqqB does not allow us to conclude whether reorientation and subsequent cyclization of a OH-Cys DOPA quinone to isoquinoloid is feasible since multiple poses of the analog fit the electron density. Consequently, we also considered an alternative possibility in which only a small fraction of the OH-Cys-DOPA quinone dissociates from PqqB during the single turnover reactions, subsequently leading to isoquinoloid formation via a spontaneous reaction in solution.20 The latter is consistent with the observation that rapid acid quenching of the initially formed PqqB/product OH-Cys-DOPA quinone complex and subsequent neutralization leads to detectable amounts of isoquinoloid. Distinguishing between spontaneous isoquinoloid production vs. a guided PqqB reaction will be the subject of future studies.
The MS/MS spectrum of the isoquinoloid is of considerable interest in the context of the position of oxygen insertion during the hydroxylation of Cys-DOPA to OH-Cys-DOPA quinone. Fig. 6c shows an MS/MS spectrum exhibiting a prominent neutral loss of 117.0 Da, ascribed to scission of both the sulfur and nitrogen side chains that are derived from Cys, to generate 2,3-DOPA quinone. Given the anticipated formation of a Schiff’s base between the amine side chain of cysteine and the 4-position of the initial tyrosine ring, the appearance of the 2,3-DOPA quinone fragment in the MS/MS spectrum supports an initial hydroxylation of Cys-DOPA at its C-2 position.
Mechanism of PqqB hydroxylation.
The collective observations in this work strongly implicate PqqB as a novel non-heme hydroxylase. Several mechanisms for aromatic hydroxylation can be envisioned and we propose the simplest model to explain our findings. While aromatic dioxygenases exist that can install two oxygen atoms from one molecule of O2 to produce a catechol moiety,21 to our knowledge an enzyme capable of directly producing hydroxyquinone from tyrosine and one equivalent of O2 is unprecedented. Given the combined experimental observations that first, α-KG is converted to succinate in an O2-dependent reaction and second, that a catechol substrate is rapidly converted to hydroxyquinone in a subsequent O2-dependent reaction giving water and not peroxide as product, we propose a two-step, O2-dependent reaction for PqqB in which α-KG is the two electron donor in the first step and the quinol intermediate provides the two electrons required for hydroxylation in the second step (Fig. 7a). While the nature of this proposed two-step hydroxylation may share consistencies with regard to the mechanism of each hydroxylation, it is likely that these reactions are unique and features fully explaining the multifunctionality of PqqB will emerge in future works.
The unusual properties for the conversion of Cys-DOPA to OH-Cys-DOPA quinone can be rationalized by a mechanism (Fig. 7b) in which initial coordination of the catechol moiety of Cys-DOPA (1) and O2 to the active-site iron produces an activated Fe2+-superoxo (or Fe3+-peroxo) intermediate (2). Radical combination of the metal-bound superoxo- with the catechol-derived semiquinone leads to oxygen insertion at the C2 position of the substrate to give (3). Subsequent breakdown of this intermediate, in a manner reminiscent of Baeyer-Villiger rearrangements,22 yields the 2-hydroxy-3,4-orthoquinone, (4), resetting the active site for another catalytic turnover.
We note that other mechanisms are possible, and introduce one alternative in Fig. S10. In this instance, Cys-DOPA is first oxidized to Cys-DOPA quinone concomitant with formation of iron-hydroperoxide, which then undergoes a nucleophilic attack at C2 of the electrophilic Cys-DOPA quinone. Subsequent proton abstraction from C-2 of the ring would yield 4-hydroxy-2,3-orthoquinone, with cleavage of peroxide to water as a second product (Fig. S10). This mechanism is attractive since it avoids the proposed 1,2-hydride-shift necessary for the breakdown of the intermediate (3) in Fig. 7b. However, we consider this option less likely since it allows for a possible disruption at the Cys-DOPA 3,4-orthoquinone and iron-hydroperoxide step, leading to loss of peroxide from the enzyme; as shown in Fig. 3d virtually no detectable hydroperoxide forms during Cys-DOPA oxidation. Additionally, the oxygen inserted at C2 would be one of the carbonyls of a 2,3-orthoquinone and, as such, may be susceptible to exchange with solvent. However, the product produced in the presence of 18O2 is seen to completely retain its O-18 label (Fig. 4a). While details of the unusual mechanism presented in Figure 7b are expected to emerge on further study, as presented it captures all of the essential experimental features discussed herein.
CONCLUSIONS
The activity of PqqB demonstrated herein is unprecedented and to our knowledge has never been reported for a member of the MBL-fold family. MBL-fold proteins bind various metals,23 and have been shown to catalyze reactions beyond the canonical hydrolysis of β-lactam substrates, such as redox and hydroxylation reactions.24 However, none of the enzymes reported possess substrate, metal, or reaction features similar to PqqB, which appears to more closely resemble other non-heme iron-dependent oxidases such as catechol dioxygenase.25 Importantly, since PqqB is substantially different from MBL-fold proteins and carries out chemistry more akin to non-heme iron enzymes, it presents an unrecognized crossover between these two distinct classes of proteins. An additional product from reaction of Cys-DOPA with PqqB is also detected in this study, shedding light on the down-stream portion of the PQQ pathway and further corroborating the identity of the initial Cys-DOPA hydroxylation product. These findings piece together the long standing mechanistic enigma of PQQ biosynthesis and show how a remarkably complex pathway may be achieved by a small number of gene products that are multifunctional and capable of catalyzing multiple stepwise reactions (e.g., PqqB as well as PqqC).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Prof. Nigel Richards for helpful discussion and the initial suggestion that we explore a molecular oxygen insertion mechanism for PqqB with Cys-DOPA. The work used Advanced Photon Source beamline 19-BM and GM/CA 23-ID-B funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research also used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding Sources
This work was supported by National Institutes of Health grants to J.P.K. (GM118117-015), C.M.W (GM66569) and A.T.I (1S10OD020062-01).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Materials and experimental procedures, synthesis of substrate analogs, PqqB structure comparison for source organisms, ICP binding data, UV-vis data, metal controls, additional LC-HRMS data, MS/MS data and crystallization statistics.
REFERENCES
- (1).Goodwin PM; Anthony C, The Biochemistry, Physiology and Genetics of Pqq and Pqq-Containing Enzymes. Adv. Microb. Physiol 1998, 40, 1–80. [DOI] [PubMed] [Google Scholar]
- (2).Duine JA, The Pqq Story. J. Biosci. Bioeng 1999, 88, 231–236. [DOI] [PubMed] [Google Scholar]
- (3).Anthony C, Pyrroloquinoline Quinone (Pqq) and Quinoprotein Enzymes. Antioxid. Redox Signaling 2001, 3, 757–774. [DOI] [PubMed] [Google Scholar]
- (4).Rucker R; Storms D; Sheets A; Tchaparian E; Fascetti A, Is Pyrroloquinoline Quinone a Vitamin? Nature 2005, 433, E10–E11. [DOI] [PubMed] [Google Scholar]
- (5).Arnison PG; Bibb PG; Bierbaum MG, et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Nature Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gallop PM; Paz MA; Flückiger R; Kagan HM, Pqq, the Elusive Coenzyme. Trends in Biochem. Sci 1989, 14, 343–346. [DOI] [PubMed] [Google Scholar]
- (7).He K; Nukada H; Urakami T; Murphy MP, Antioxidant and Pro-Oxidant Properties of Pyrroloquinoline Quinone (Pqq): Implications for Its Function in Biological Systems. Biochem. Pharmacol 2003, 65, 67–74. [DOI] [PubMed] [Google Scholar]
- (8).Klinman JP; Bonnot F, Intrigues and Intricacies of the Biosynthetic Pathways for the Enzymatic Quinocofactors: Pqq, Ttq, Ctq, Tpq, and Ltq. Chem. Rev 2014, 114, 4343–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shen YQ; Bonnot F; Imsand EM; RoseFigura JM; Sjölander K; Klinman JP, Distribution and Properties of the Genes Encoding the Biosynthesis of the Bacterial Cofactor, Pyrroloquinoline Quinone. Biochemistry 2012, 51, 2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Barr I; Latham JA; Iavarone AT; Chantarojsiri T; Hwang JD; Klinman JP, Demonstration That the Radical S-Adenosylmethionine (Sam) Enzyme Pqqe Catalyzes De Novo Carbon-Carbon Cross-Linking within a Peptide Substrate Pqqa in the Presence of the Peptide Chaperone Pqqd. J. Biol. Chem 2016, 291, 8877–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Latham JA; Iavarone AT; Barr I; Juthani PV; Klinman JP, Pqqd Is a Novel Peptide Chaperone That Forms a Ternary Complex with the Radical S-Adenosylmethionine Protein Pqqe in the Pyrroloquinoline Quinone Biosynthetic Pathway. J. Biol. Chem 2015, 290, 12908–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bonnot F; Iavarone AT; Klinman JP, Multistep, Eight-Electron Oxidation Catalyzed by the Cofactorless Oxidase, PqqC: Identification of Chemical Intermediates and Their Dependence on Molecular Oxygen. Biochemistry 2013, 52, 4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Magnusson OT; Toyama H; Saeki M; Schwarzenbacher R; Klinman JP, The Structure of a Biosynthetic Intermediate of Pyrroloquinoline Quinone (Pqq) and Elucidation of the Final Step of PQQ Biosynthesis. J. Am. Chem. Soc 2004, 126, 5342–5343. [DOI] [PubMed] [Google Scholar]
- (14).Tu X; Latham JA; Klema VJ; Evans RL; Li C; Klinman JP; Wilmot CM, Crystal Structures Reveal Metal-Binding Plasticity at the Metallo-β-Lactamase Active Site of PqqB from Pseudomonas Putida. J. Biol. Inorg. Chem 2017, 22, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baier F; Tokuriki N, Connectivity between Catalytic Landscapes of the Metallo-β-Lactamase Superfamily. J. Mol. Biol 2014, 426, 2442–2456. [DOI] [PubMed] [Google Scholar]
- (16).Wan H; Xia Y; Li J; Kang Z; Zhou J, Identification of Transporter Proteins for PQQ-Secretion Pathways by Transcriptomics and Proteomics Analysis in Gluconobacter Oxydans Wsh-003. Front. Chem. Sci. Eng 2017, 11, 72–88. [Google Scholar]
- (17).Chioccara F; Novellino E, A Convenient One Step Synthesis of 5-Cysteinyl-S-dopa Using Ceric Ammonium Nitrate. Synth. Commun 1986, 16, 967–971. [Google Scholar]
- (18).Ito S; Inoue S; Yamamoto Y; Fujita K, Synthesis and Antitumor Activity of Cysteinyl-3,4-Dihydroxyphenylalanines and Related Compounds. J. Med. Chem 1981, 24, 673–677. [DOI] [PubMed] [Google Scholar]
- (19).Martinez S; Hausinger RP, Catalytic Mechanisms of Fe(II)-and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Deibel RB; Chedekel MR, Biosynthetic and Structural Studies on Pheomelanin. J. Am. Chem. Soc 1982, 104, 7306–7309. [Google Scholar]
- (21).Eby DM; Beharry ZM; Coulter ED; Kurtz DM; Neidle EL, Characterization and Evolution of Anthranilate 1,2-Dioxygenase from Acinetobacter Sp. Strain Adp1. J. Bateriol 2001, 183, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).March J, Advanced Organic Chemistry. 4th ed.; John Wiley and Sons, Inc.: 1992, pp 1099–1111. [Google Scholar]
- (23).Cahill ST; Tarhonskaya H; Rydzik AM; Flashman E; McDonough MA; Schofield CJ; Brem J, Use of Ferrous Iron by Metallo-β-Lactamases. J. Inorg. Biochem 2016, 163, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pettinati I; Brem J; Lee SY; McHugh PJ; Schofield CJ, The Chemical Biology of Human Metallo-β-Lactamase Fold Proteins. Trends in Biochem. Sci 2016, 41, 338–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lipscomb JD, Mechanism of Extradiol Aromatic Ring-Cleaving Dioxygenases. Curr. Opin. Struc. Biol 2008, 18, 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.