

Abstract
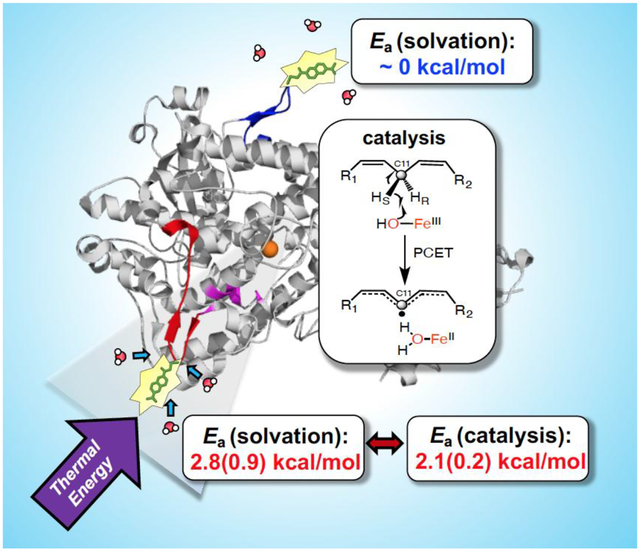
The rate-limiting chemical reaction catalyzed by soybean lipoxygenase (SLO) involves quantum mechanical tunneling of a hydrogen atom from substrate to its active site ferric-hydroxide cofactor. SLO has emerged as a prototypical system for linking the thermal activation of a protein scaffold to the efficiency of active site chemistry. Significantly, hydrogen-deuterium exchange-mass spectrometry (HDX-MS) experiments on wild type and mutant forms of SLO have uncovered trends in the enthalpic barriers for HDX within a solvent-exposed loop (position 317–334) that correlate well with trends in the corresponding enthalpic barriers for kcat. A model for this behavior posits that collisions between water and loop 317–334 initiate thermal activation at the protein surface that is then propagated 15–34 Å inward toward the reactive carbon of substrate in proximity to the iron catalyst. In this study, we have prepared protein samples containing cysteine residues either at the tip of the loop 317–334 (Q322C) or on a control loop, 586–603 (S596C). Chemical modification of cysteines with the fluorophore 6-bromoacetyl-2-dimethylaminonaphthalene (Badan, BD) provides site-specific probes for the measurement of fluorescence relaxation lifetimes and Stokes shift decays as a function of temperature. Computational studies indicate that surface water structure is likely to be largely preserved in each sample. While both loops exhibit temperature-independent fluorescence relaxation lifetimes as do the Stokes shifts for S596C-BD, the activation enthalpy for the nanosecond solvent reorganization at Q322C-BD (Ea(ksolv) = 2.8(0.9) kcal/mol)) approximates the enthalpy of activation for catalytic C-H activation (Ea(kcat) = 2.1(0.2) kcal/mol). This study establishes and validates methodology for measuring rates of rapid local motions at the protein/solvent interface of SLO. These new findings, when combined with previously published correlations between protein motions and the rate limiting hydride transfer in a thermophilic alcohol dehydrogenase, provide experimental evidence for thermally-induced ‘protein quakes’ as the origin of enthalpic barriers in catalysis.
Graphical Abstract
INTRODUCTION
Proteins are inherently “soft” dynamical structures accessing a multitude of sub-states that interconvert on time scales (millisecond to femtosecond, ms-fs) relevant to biological function.1–4 A stochastic search among such sub-states is increasingly implicated in the optimization of enzyme active sites.5 In this context, the empirical temperature dependence6–7 of biological catalysis raises questions regarding the origins of thermal adaptation and rate acceleration. It has long been recognized that the source of thermal activation in enzyme-catalyzed reactions must originate from the solvent/heat bath at the protein-surface interface.8–10 Models describing protein motions during catalysis rarely take into account the contribution of such solvent dynamics,11–13 and there has, at the same time, been debate14–15 regarding the possible role of protein motions in modulating catalysis. One way to identify the role of specific protein/solvent interactions in accelerating active site chemistry is to measure and correlate the thermal activation barriers for these temporally distinct processes to catalysis.11–12, 16–17 The feasibility of performing such studies is greatly aided by the emerging evidence for spatially defined networks that link an enzyme’s active site chemistry to the protein surface.9, 18
Soybean lipoxygenase (SLO) is a non-heme, mononuclear iron enzyme that catalyzes the rate-limiting cleavage of a C-H bond within linoleic acid (LA) (Figure 1a), utilizing a well-defined hydrogen tunneling mechanism that is intrinsically dependent on motions within the protein scaffold.19–22 Recognizing that SLO would provide a robust prototype in which to explore catalysis-linked protein motions, site-specific mutations had been introduced that result in the perturbation of protein packing both proximal and distal from the Fe active site (Figure 1b).20, 23 In particular, reduction in the size of key hydrophobic side chains was found to alter many features that include the rate of C-H activation (kcat), the accompanying energy of activation (Ea), and the donor-acceptor distance for hydrogen transfer as seen in ground state ENDOR-derived structures,24 and in transiently achieved tunneling ready states (from the temperature dependence of kinetic isotope effects25). In the latter case, local increases in protein flexibility most generally alleviate the accompanying unfavorable donor-acceptor distances for C-H activation via increased involvement of distance sampling.26 The impact of these activity-altering mutations has also been examined using hydrogen-deuterium exchange mass spectrometry (HDX-MS), revealing a discrete and spatially resolved segment of the protein that undergoes changes in thermally-activated protein breathing motions that correlate with catalysis.18 The identified network extends from the Fe active site to a surface-exposed loop (residues 317–334) at a distance of 15–34 Å (Figure 1c).
Figure 1.

a) Rate-limiting PCET reaction catalyzed by an FeIII(OH) cofactor of SLO. b) Ground state active site structure of SLO with bound linoleic acid substrate (blue), from QM/MM calculations (ref.69). Flanking side chains L754, I553, and L546 (in gray) have been the subject of various mutational studies. c) HDX-MS identified protein network connecting the surface loop and active site in SLO. Residues L546, I552, I553, S749, S750, and Y317 (shown in pink spheres) line this network (ref. 18).
In this study, we initiate an interrogation of the temporal properties of activity-related protein motions in SLO, choosing to focus on the site of interaction between the remote loop 317–334 and solvent. Following the successful incorporation of the fluorophore 6-bromoacetyl-2-dimethylaminonaphthalene (Badan) at position 322 in SLO (via Q322C), together with a second control loop, 586–603 (via S596C), the temperature dependence of picosecond resolved fluorescence lifetimes and Stokes shifts decays is examined. Our findings show that while both loops exhibit temperature-independent fluorescence relaxation lifetimes, time-dependent Stokes shifts reveal a singular temperature-dependent decay rate for loop 317–334 that mirrors the catalytic barrier for the active site hydrogen tunneling reaction. These results implicate rapid, nanosecond motions, arising from the interplay of a solvent-protein interface, in the thermal activation of H-transfer in SLO. Similar correlations, between a range of rapid protein motions and millisecond catalysis, have been seen for the hydride transfer catalyzed by a thermophilic alcohol dehydrogenase.17, 27–28 A model is described that differentiates protein equilibrations that function to alter the probability of catalysis from ‘protein quakes’ that act to transfer thermal activation from solvent to enzyme active sites via site-specific protein networks.
RESULTS AND DISCUSSION
Insertion of a Fluorescent Probe at the Surface Loops of Lipoxygenase.
Intrinsic tryptophan fluorescence has been widely used to study solvation responses at multiple sites of proteins, owing to their facile insertion or removal by site-specific mutagenesis.29 However, the abundance of tryptophan residues (total of 14) in SLO precluded their use as a site-specific reporter for fluorescence. Site-directed labeling with a solvatochromic fluorescence label such as Badan can be accomplished at different sites of a protein by bioconjugation with a reactive cysteine residue (Figure 2a). In order to assess the solvation dynamics at the surface loop created by residues 317–334, the tip of this loop (residue 322) was mutated to cysteine, and reacted with Badan (cf. Figure 1c and Figure 2b). To deconvolute the specificity of catalysis-related dynamics, a second, control surface loop, for which no correlation could be detected between the thermal barriers for HDX and kcat (residues 586–603),18 was also labeled with Badan (Figure 2c). We note that neither of these loops is conserved in the lipoxygenases from cyanobacteria,30 human,31 and fungi,32–33 and could thus be implicated in the unusually low activation energy (Ea) for lipoxygenases from plants.
Figure 2.

a) Schematic for labeling of SLO mutants Q322C and S596C with Badan. b) Structure of SLO (PDB: 3PZW) labeled with Badan at the tip of the 317–334 surface loop (red) or the tip of the 586–603 surface loop (blue). Badan attached to the Cys-modified locations is depicted in green, and its orientation is arbitrary, for illustration purposes. The proposed thermally activated network is depicted in magenta (encircled), and the active site iron is shown as an orange sphere.
Wild-type (WT) SLO has four native cysteine residues (C127, C357, C492, and C679) that, although buried in the hydrophobic pockets of the protein, could potentially introduce non-specific labeling of Badan. We therefore incubated WT SLO with Badan (2 equiv) for 3 h at 4 °C, and re-isolated the protein by size exclusion chromatography. Analysis of the incubation product by UV-visible spectroscopy showed that the buried cysteine residues are not reactive under the specified labeling conditions (labeling efficiency < 5%). In contrast, incubation of Q322C and S596C, variants with cysteine introduced onto the surface loops, with Badan under similar conditions showed ca. 100% labeling efficiency (Table S1). The Badan-SLO conjugates Q322C-BD and S596C-BD were characterized by electrospray ionization mass spectrometry (ESI-MS) (Table 1), confirming the 1:1 Badan/SLO adduct. Liquid chromatography – tandem mass spectrometry (LC-MS/MS) analysis of pepsin digests of Q322C-BD and S596C-BD further showed that only positions 322C and 596C were modified, with a mass addition corresponding to the 6-acetyl-2-diaminonaphthalene group (+211 Da); no further cysteine modifications were detected (Figure S1).
Table 1.
Calculated and measured molecular mass for labeled and unlabeled SLO constructs.
| (–) Badan | (+) Badan | |||
|---|---|---|---|---|
| Calcd. Mass (Da) | Measured Mass (Da) | Calcd. Mass (Da) | Measured Mass (Da) | |
| Q322C | 94,386 | 94,387 | 94,597 | 94,599 |
| S596C | 94,427 | 94,429 | 94,638 | 94,639 |
| Glutathionea | 308.09 | - | 518.18 | 518.18 |
Monoisotopic mass-to-charge ratio (m/z) for the [M+H]+ ion.
The fluorescence probe does not affect the steady-state kinetics of the Q322C-BD and S596C-BD variants, having similar kcat, KM, and Ea parameters with unlabeled WT SLO at 30 °C in pH 9.0 borate buffer (Table 2). The enzyme Fe content is also unaffected by the cysteine replacements and Badan-labeling process (Table S1). Taken together, the data show that the Badan-SLO conjugates Q322C-BD and S596C-BD introduce catalytically faithful, site-specific reporters of relaxation lifetimes and solvation dynamics at targeted surface loops in SLO.
Table 2.
Kinetic parameters for WT and Badan-labeled SLO.
| kcat (s−1)a | KM (μM) | Ea (kcal/mol) | |
|---|---|---|---|
| WTb | 297(12) | 27(2.7) | 2.1(0.2) |
| Q322C-BD | 252(13) | 34(4) | 2.3(0.4) |
| S596C-BD | 239(17) | 38(5) | 2.2(0.5) |
Corrected for Fe content.
From Ref 20.
Steady-State Fluorescence.
The 6-acetyl-2-dimethylaminonaphthalene (DAN) moiety of Badan undergoes a large charge redistribution upon excitation, making it an ideal polarity sensor.34–37 Further, the spectral emission response for DAN probes upon changes in solvent polarity allows for sensing solvation at different protein sites. For example, N-acetyl-aladanamide, an alanine derivative of DAN, exhibits a large change in emission maximum from 409 nm in heptane, to 542 nm in water.38 Fluorescence experiments were conducted with the Fe-containing SLO in the absence of linoleic acid (LA) substrate, to avoid possible secondary effects of LA on the protein structure and flexibility arising from substrate binding or catalytic turnover. Previous HDX-MS measurements of SLO, uncovering a mutation-dependent flexibility at the 317–334 loop, were similarly performed in the absence of substrate.18 The steady-state emission spectra of Q322C-BD and S596C-BD were collected under the conditions in which kinetic data (Table 2) were previously obtained. As shown in Figure 3a, the Q322C-BD peak emission occurs at 485 ± 2 nm, while S596C-BD displays a peak emission at 491 ± 2 nm (Figure S2).
Figure 3.

Temperature dependence of steady-state fluorescence emission spectra (λexc = 373 nm) of a) Q322C-BD and b) glutathione-BD in 0.1 M pH 9.0 borate at 10 – 40 °C. Inset: changes in relative intensity at λmax emission as a function of temperature.
As a mimic for a fully exposed DAN probe, Badan was also attached to a small peptide, glutathione. Glutathione-BD has a peak emission of 548 ± 1 nm, across all temperatures studied (Figure 3b). While both Q322C-BD and S596C-BD are at surface loops, the λmax of S596C-BD is red-shifted by ~6 nm relative to Q322C-BD, indicating a somewhat more solvent-exposed location. For glutathione-BD, the red-shifted λmax agrees with the expectation of the probe being fully solvated. Another interesting feature is the temperature dependence of the emission peak intensity of Badan-labeled SLO vs. glutathione. From 10 to 40 °C, the peak intensity for SLO-BD decreases to about 75%, while glutathione-BD undergoes a decrease to about 55% of the original peak intensity (Figure 3 and Figure S2, inset).
Temperature Dependence of Time-Resolved Fluorescence Lifetime Decays.
Figure 4a shows the representative lifetime decays at the respective wavelength of maximum emission at 30 °C. For Q322C-BD and S596C-BD, the fluorescence decays were essentially identical, and both could be fit to a three-exponential function, while the glutathione-BD fluorescence decay is best described by a bi-exponential function (Table S2). The average relaxation lifetimes at all temperatures for Q322C-BD and S596C-BD are almost constant at ca. 3 ns, whereas the relaxation lifetime of glutathione-BD is much shorter, and varies from ca. 0.5 – 0.8 ns. Arrhenius plots of the relaxation lifetimes were constructed, and the magnitude of the activation energies (Ea) are summarized in Table S3. The lifetime measurements show temperature-independent relaxation lifetimes (Ea < 1 kcal/mol) for both Q322C-BD (Figure 4b) and S596C-BD (Figure S3) while the exposed probe in glutathione-BD (Figure 4c) has lifetimes that are more temperature dependent (Ea = 3–5 kcal/mol). Overall, the differences in both steady-state and time-resolved fluorescence of the surface loops of SLO vs. glutathione reflect the partial shielding of the probe by surface side chains in SLO and the dielectric differences between bulk water (glutathione-BD) and the protein’s hydration shell (SLO-BD).
Figure 4.

a) Time-resolved fluorescence of Q322C-BD, S596C-BD, and glutathione-BD at 30 °C in 0.1 M borate buffer pH 9.0, λexc = 373 nm. Gray line shows the instrument response function (IRF). b, c) Arrhenius plots for fluorescence relaxation lifetime decays for Q322C-BD and glutathione-BD, respectively.
Temperature Dependence of Time-Dependent Stokes Shifts for Q322C-BD and S596C-BD.
The fluorescence lifetimes were then recorded at different emission wavelengths and temperatures, and the time resolved emission spectra (TRES) were constructed from the steady-state and time-resolved fluorescence decay data (see Experimental Section for details). The time-dependent Stokes shift reports on the evolution of the solvent’s electrostatic environment during the excited state of a fluorophore. Following excitation, the solvent reorients around the excited state dipole moment, which occurs with a solvent relaxation rate ksolv. This solvent reorganization is manifested by a shift of the TRES to longer wavelengths, until the steady-state emission wavelength is reached.39–41
The time-resolved emission spectra (TRES) at 30 °C for Q322C-BD, S596C-BD, and GS-BD (Figure 5a–c) were reconstructed from the TRES peak emission. At 30 °C, the magnitude of total Stokes shift is larger for Q322C-BD (Δν = 621 cm−1; τ1 = 0.49 ns, τ2 = 7.42 ns) than S596C-BD (Δν = 434 cm−1; τ1 = 0.49 ns, τ2 = 4.83 ns), whereas GS-BD shows a much smaller Stokes shift (Δν = 215 cm−1, τ = 0.52 ns) in comparison to the BD-labeled SLO constructs (Table S4). At all temperatures, Q322C-BD and S596C-BD were best fit to a bi-exponential decay function (Figure 5d–e), whereas glutathione-BD was fit to a single-exponential function (Figure 5f). In the case of S596C-BD the time traces appear independent of temperature (Figure 5e), whereas these are dependent on temperature for both Q322C-BD (Figure 5d) and GS-BD (Figure 5f) between 10 and 40 °C.
Figure 5.

a-c) Selected time-resolved emission spectra for Q322C-BD (a), S596C-BD (b), and GS-BD (c) in 0.1 M borate pH 9.0 at 30 °C. d-f) Temperature dependence of Stokes shift decay rates for Q322C-BD (d), S596C-BD (e), and GS-BD (f) normalized in terms of solvation correlation function C(t). Decays are shown at 10 – 40 °C and were fit to either a bi-exponential (for Q322C-BD and S596C-BD) or a single exponential (for GS-BD) decay function. g-h) Arrhenius plots of 1/τ2 for Q322C-BD and S596C-BD (g), and 1/τ for GS-BD (h).
Arrhenius plots of the Stokes shift decay rates for Q322C-BD reveal measurable temperature dependences for both its more rapid (τ1) and slower component (τ2) (Figure 5g, Figure S4, and Table 3). The Stokes shift decay for glutathione-BD has a temperature dependence (Ea = 4.3(0.8) kcal/mol) that is similar to the fast relaxation of Q322C-BD (Ea(τ1) = 3.6(0.5) kcal/mol) (Table 3). Of considerable interest, the activation energy of τ2 for Q322C-BD (Ea(τ2) = 2.8(0.9) kcal/mol) (Figure 5e) is almost identical to the reported Ea for kcat. As anticipated from Figure 5g, solvation times are independent of temperature for S596C-BD, with Ea(τ1) = −0.3(0.4) kcal/mol and Ea(τ2) = 0.5(0.7) kcal/mol).
Table 3.
Comparison of activation energies (Ea)a of Stokes shift decays for SLO surface loops and C-H activation.
| Ea (kcat) | Ea (1/τ1) | Ea (1/τ2) | |
|---|---|---|---|
| Q322C-BD | 2.3(0.4) | 3.6(0.5) | 2.8(0.9) |
| S596C-BD | 2.2(0.5) | −0.3(0.4) | 0.5(0.7) |
| GS-BD | - | 4.3(0.8) | - |
Ea values are in kcal/mol.
An early model proposed by Nandi and Bagchi defined water in the vicinity of the protein as “biological water” comprised of bound and free water as differentiated from bulk water (Figure 6a),42 with the biological water and bulk water expected to give rise to distinct time scales of water relaxation in proteins. Femtosecond up-conversion measurements on solvation decays of solvent-exposed DAN probes have been reported to have large Stokes shifts (~2000 cm−1) with decay lifetimes of 0.1–10 ps that are generally ascribed to bulk water reorganization.43 In this study we are focused on the sub-ns regime (~120 ps of instrumental resolution), as a reporter of the reorganization of water within the protein hydration shell (typical Stokes shift decay lifetimes of > 150 ps)42. As indicated, the measured Stokes shift decays for Q322C-BD and S596C-BD are bi-exponential with both a fast (τ1 ~ 0.5 ns) component and a slower (τ2 ~ 5 – 8 ns) component. For comparison, the fully exposed probe, glutathione-BD, shows a singular, fast, red-shifted decay lifetime of 0.3 – 0.9 ns. The longer τ2 for the enzyme constructs is assigned to the reorganization of the more constrained, bound water at the protein interface (Figure 6a). Slow, nanosecond Stokes shift decays have also been observed for a probe attached to a tunnel mouth at the protein surface of haloalkane dehalogenase,44 as well as for a probe attached to the active site of glutaminyl-tRNA synthetase.45 The slow nanosecond component on these fluorescence measurements have been ascribed to either residue side chain motions or the restricted water mobility. Similarly, molecular dynamics simulations have revealed that water molecules hydrating the protein surface exhibit restricted translational and rotational motions.46–47 As shown in Figure 6a, the water molecules described in the biological water layer also includes free water, which is anticipated to be the primary agent in the thermal transfer process. Thus, while Stokes shift decays measure the solvent reorganization around the excited state probe, protein-water interactions and the coupling of their motions makes it non-trivial to separate and identify the contributions originating from different components in the dynamical response of a solvated protein.
Figure 6.

a) Schematic representation of different hydration layers, with the dynamic exchange of free and protein-bound water, the two components of the “biological water”. b, c) Residues surrounding Q322 and S596. In b), Q322 is in close contact (~3 Å) with R301 in the adjacent helix. A longer range but similar interaction is possible between R301 and the carbonyl of the attached probe. In c), while the S596 interaction with D594 is lost after addition of BD, other interactions remain possible.
The structural origin of the respective solvation enthalpic barriers in Q322C-BD and S596C-BD was pursued by inspection of the residues surrounding the probe positions in WT (left) vs. labeled (right) proteins (Figure 6b and c). The optimized structures of the BD-labeled Q322C and S596C constructs were determined by energy minimization (see Experimental Section for details). In WT-SLO, the Q322 has close polar contacts to D320 and H323, together with a primary H-bonding distance to R301 found on an adjacent helix. The carbonyl introduced with BD may, thus, approximate the interaction seen with Q322 (Figure 6b). In the case of the replacement of S596 by Cys and subsequent labeling, there is a greater structural change than that observed with Q322 replacement, yet also a likely retention of nearby H-bonding interactions near C596 (Figure 6c). Coupled with nearly identical kinetic parameters in Q322C-BD and S596C relative to WT (Table 2), the solvation environment is not likely to be greatly altered after addition of BD. The data and modeling argue that thermally activated water restructuring reflected in the Stokes shifts of BD-labeled SLO provides a reasonable surrogate for any impact of thermal water restructuring on the Ea (kcat). Analysis of the local structure around the probe at the 596C position indicates only one polar interaction of 596C-BD with the adjacent proline (P595) (Figure 6C). The scarce number of polar interactions at this site supports a more facile solvent reorganization due the protein-water hydrogen bonds that have to be broken to enable solvent reorganization around the probe. In contrast, the probe environment around 322 site has more interactions with polar sidechains than the 596 position, which is reflected by the increased temperature dependence of the solvent reorganization process.
Relating Solvent Reorganization Kinetics at the Surface Loop of SLO to Active Site Hydrogen Transfer.
Extensive kinetic data have shown that the rate-limiting hydrogen transfer step in SLO (Fig. 1b) occurs via an intrinsically temperature independent H-tunneling process that is dependent on the thermal activation of the heavy atoms of the protein; the latter plays an essential role in creating a transient alignment and compaction that is compatible with efficient wavefunction overlap between the H-donor and acceptor sites. In general, the origin of thermal activation for chemical reactions in condensed phase can be ascribed to either molecular collisions between reactants and/or to collisions between the reacting molecule and the solvent bath.8 Extension of this reasoning to enzyme-catalyzed reactions emphasizes the importance of solvent-exposed surfaces in heat transfer to the active site. Given the anisotropic nature of globular proteins, and the accompanying differences in local heat capacity, specific regions of a protein’s surface are anticipated to play dominant roles in overcoming the enthalpic barriers to catalysis.9, 20, 48
Full understanding of the molecular origins of enzymatic thermal activation will involve first, identification of the preferred protein network(s) for solvent-to-protein heat transfer and second, a deconvolution of the time constants that contribute to this process. One enzyme system that has offered a model for this type of analysis is the thermophilic alcohol dehydrogenase from B. stearothermophilus (ht-ADH). Early studies of ht-ADH had demonstrated a parallel break in both catalytic behavior49 and regional HDX-MS within the substrate binding domain.50 Subsequent spectroscopic probes provided kinetic constants for physical processes within the same (substrate) domain, that include ns fluorescence lifetime measurements27 and μs temperature-jump Förster resonance energy transfer (FRET) measurements.17 In each instance, these studies were aided by the ability to prepare protein constructs that contained only a single or two Trp side chains and retained catalytic activity similar to WT.27–28
In the context of the present study, we have compiled the body of published data for the ht-ADH constructs W87in, W167in and W49in/W167in (see Figure S5 for positions of Trp residues and NADH cofactor in ht-ADH). W167in contains a single Trp near a solvent interface of the cofactor binding domain. This is an important control for the behavior of W87in and W49in/W167in, both of which are situated in the substrate binding domain that has been shown to rigidify at reduced temperatures.50 The construct W167in showed virtually no sensitivity of its Stokes shifts to either the introduction of activity altering mutations51 or to temperature (Table 4). This is in marked contrast to the Stokes shifts behavior for W87in, where the value of ΔH‡ is large and close to that for kcat. We note that neither W167in nor W87in displayed a 30 °C break in the magnitude of their Stokes shifts, suggesting that the protein’s accommodations to changes in charge within the buried excited state Trp dipole may be quite local and, thus, only weakly sensitive to the enhanced rigidification shown to occur in ht-ADH at reduced temperatures. The remainder of the data in Table 4 indicate a remarkable and initially unexpected trend. In the case of both W87in or W49in/W167in, it can be seen that under physiological conditions (>30 °C) the enthalpic barrier for protein motions that control either the lifetime of fluorescence at position W87 or FRET from W49 to the nicotinamide ring of the cofactor is virtually identical to the ΔH‡ measured for the rate-limiting hydride transfer step. These trends are, in fact, mirrored in the activation energy behavior of SLO (Table 3), with the exception that the measurements for SLO were conducted using an extrinsic probe appended either to the loop of the dynamical network (Q322C) or a control loop (S596C).
Table 4.
Summary of enthalpic barriers for catalysis and protein relaxation kinetics in ht-ADHa
| Parameter | ΔH‡b (kcal/mol) | Time scale | Break > 30 °C |
|---|---|---|---|
| k(cat, WT) | 14.6(0.3)e | ms (10−3 s) | Yc |
| k(cat, W87in) | 13.5(0.4)f | ms (10−3 s) | Yc |
| k(fl, W87in) | 12.7(2.9)f | ns (10−9 s) | Yc |
| k(ss, W87in) | 9.4(0.3)f | ns (10−9 s) | Nd |
| k(cat, W49in, W167in) | 11.0(0.6)g | ms (10−3 s) | Yc |
| k(FRET, W49in, W167in) | 10.4(1.3)h | μs (10−6 s) | Yc |
| k(cat, W167in) | 13.3(0.4)f | ms (10−3 s) | Yc |
| k(ss, W167in) | −0.2(0.2)f | ns (10−9 s) | Nd |
For the definition of rate constants, fl refers to fluorescence relaxation lifetimes, ss refers to Stokes shifts and FRET refers to fluorescence resonance energy transfer.
Ea = ΔH‡ + RT
The enthalpies are above the temperature break (physiological conditions)
No observable temperature break.
Ref.49.
Ref.27.
Ref.28.
The well-developed description of the catalytic behavior of the hydrogen transfer step in SLO as non-adiabatic PCET52–54 adds another dimension to our efforts to link specific networks of protein motions to the barriers controlling catalysis. As previously formulated, the observed PCET rate in SLO is well-described by two terms that represent the fraction of enzyme capable of achieving tunneling ready states, Fconf, multiplied by the tunneling rate constant, ktun, that contains the kinetic barrier to catalysis (equation 1).
| (1) |
According to the formalism in equation (1), the enthalpic barrier will be the sum of multiple terms that contain both thermodynamic (ΔH°conf) and kinetic (ΔH‡tun) contributions, with the latter arising primarily from the environmental reorganization term (λ, the sum of inner-sphere λin and outer-sphere λout contributions). We note that, according to Marcus theory, a contribution from ΔH° can also arise from the reaction driving force component of ktun. However, in general, the magnitude of the reorganization energy λ in enzyme reactions greatly exceeds the magnitude of ΔG°.55
Turning to Fconf, this represents the reversible samplings among multiple protein sub-states that constitute the equilibrated conformational landscape. These samplings play an important role in generating the catalytically competent active site configurations that dominate barrier crossing. The actual number of productive states within Fconf can be quite small in relation to the total enzyme concentration, reducing the observed rate of catalysis from the faster time frames of experimentally detected ns and μs protein motions to the typically observed millisecond regime for kcat. The generation of the individual sub-states within Fconf is expected, at least in part, to be initiated via surface unfolding events that reversibly expose buried hydrophobic regions, leading to ΔS°conf as a major component of ΔG°conf.56–57 The type of physical model describing conformational sampling is fundamentally distinct from the kinetic contribution to catalysis that, as noted above, arises from the time and temperature dependent λ term in the Marcus theory of electron and hydrogen tunneling. Very significantly, the data provided (Table 3 for SLO and Table 4 for ht-ADH) indicate almost identical values for the enthalpic barriers that control kcat and the enthalpic barriers that emerge from time and temperature dependent changes in spectroscopic probes of protein motion. While a single correlation between the magnitude of ΔH‡ for kcat and an isolated protein motion could be fortuitous (cf. the discussion in Ref. 17), the aggregate data analyzed herein implicate a hierarchy of detectable protein motions as the origin of the enthalpic barrier to catalysis. The term ‘protein quake’ has been previously coined to describe how proteins undergo a transfer of thermal energy from their interior to the solvent via a range of correlated motions that occur on a hierarchy of scales.58–59 The molecular basis of the behavior described herein can, similarly, be described as the result of ‘protein quakes’ that result from solvent-dependent, collisions within spatially resolved protein networks (Figure 7), serving as the source of the thermal activation for active site chemistry. This behavior is quite distinct from thermodynamically driven conformational changes or landscapes, that involve stochastic sampling among equilibrated ground state protein structures.
Figure 7.

Depiction of solvent-dependent, collision-induced protein quakes as a source for the thermal activation of active site chemistry.
SUMMARY AND CONCLUSIONS
The work discussed herein introduces a fundamental distinction between specific protein networks that serve as functional heat transfer conduits from solvent to active site and the types of small motions that contribute to the equilibration of a given protein among multiple protein sub-states. The latter are sometimes discussed and analyzed in the context of possible dynamical networks; however, these are expected to be thermally equilibrated, primarily entropic in nature and may be distributed across large regions of the protein. We posit that the relatively slow (ms) time scales characteristic of enzymatic catalytic turnover are, in part, be a consequence of the additional rate constraint imposed by Fconf, (equation 1); the latter reduces kobs, but may contribute little to the enthalpic barriers accompanying catalysis. This is in contrast to the motional-directed networks in proteins that play a fundamental role in transmission of thermal energy from the solvent heat bath to the active site. While the model described here for dynamical and site specific heat transfer is generated in the context of hydrogen transfer reactions, where the rate-limiting primary chemical coordinate occurs via wave function overlap, the concepts are expected to be generally applicable to the full range of enzymatic reactions.
To conclude, this study first establishes and validates the methodology for measuring the rates of rapid local motions at the protein/solvent interface of SLO. The experimental findings, when combined with previously published kinetic data for a thermophilic alcohol dehydrogenase, implicate site selective protein motions in the generation of thermally-induced and kinetically controlled ‘protein quakes’. The developed model differentiates the impact of global protein equilibrations that function to alter the probability of catalysis from the role of discrete networks that facilitate thermal transfer to initiate catalysis within pre-formed enzyme-substrate complexes.
EXPERIMENTAL SECTION
General.
All reagents were purchased from commercial sources at the highest grade available. Water was purified to a resistivity of 18.2 MΩ·cm (at 25°C) using a Milli-Q Gradient ultrapure water purification system (Millipore, Billerica, MA). DNA sequencing was performed at the UC Berkeley DNA Sequencing Facility. Mass spectrometric analysis of intact and pepsin-digested proteins was performed at the UC Berkeley QB3/Chemistry Mass Spectrometry Facility.
Site-Directed Mutagenesis.
Site-directed mutation of either the tip of the dynamic loop (Q322C) or the control loop (S596C) to cysteine was performed following the Stratagene QuikChange Lightning protocol starting from the WT SLO-1 gene contained in a pT7–7 E. coli expression vector.60 The mutant plasmids were generated and amplified using a PTC-2000 Peltier Thermal Cycler (MJ Research). The following primers (Eurofins Genomics) and their respective reverse complements (not shown) were used to generate the cysteine variants:
Q322C: 5’-TATCGTACCGATGGTTGCCACATCCTCAAGTTT-3’
S596C: 5’-GCAATTAAGGATCCATGCACCCCACATGGAGTT-3’
Plasmid purification for gene sequencing was performed using the QIAprep spin Miniprep kit (QIAGEN). Mutations were confirmed by sequencing with three different primers that targeted different regions of the gene: the beginning of the gene, a 500-bp region of the gene containing the mutation site, and a region that covers 500-bp up to the end of the gene.
Protein Expression.
SLO-1 was expressed using the pT7–7 vector in E. coli BL21(DE3) Codon Plus RIL cells (Agilent) as described previously with minor modifications.60 Briefly, the expression culture was grown in 2xYT broth, 3% ethanol, and 0.05 mg/mL ampicillin. The culture was grown with shaking (220 RPM) at 37°C to an OD600 of 0.6–0.8, at which time the temperature of the incubator was lowered rapidly to 15 °C. After 48 h, cells were harvested and stored at −80 °C until purification.
Protein Purification.
SLO-1 was purified following a previously described procedure with minor modifications.61 Cell paste from a 3 L cell culture was resuspended in ca. 30 mL lysis buffer (containing 50 mM pH 7.5 phosphate buffer, 50 mM KCl, 10% glycerol, 0.1% Tween 20, 0.1 mM MgSO4, 1 mM aminoethylbenzenesulfonyl fluoride (Sigma), 15 U/mL of benzonase (Novagen), 0.2 kU/mL of lysozyme (Sigma), and 0.1 mM pH 7.0 TCEP). The lysis reaction was allowed to proceed for 20 min at 23 °C. The rest of the purification procedure was performed at 4 °C, in the presence of 0.1 mM TCEP to avoid oxidation of the surface cysteine. The lysis solution was sonicated with three (3x) 5-min on-off cycles, and then centrifuged at 20,000 RPM for 20 min to pellet the insoluble cellular debris. The soluble supernatant was dialyzed in 20 mM Bis-Tris (pH 5.8) for 3–4 h, centrifuged at 20,000 RPM for 20 min, and then loaded onto a column packed with 70 mL SP Sepharose Fast Flow (GE) and pre-equilibrated with 20 mM Bis-Tris (pH 5.8). The column was then washed with 500 mL of 20 mM Bis-Tris (pH 5.8). The bound protein was eluted with a 600-mL linear gradient of 0 to 500 mM KCl (Buffer A: 20 mM Bis-Tris (pH 5.8); Buffer B: 20 mM Bis-Tris (pH 5.8), 500 mM KCl). SLO typically co-elutes with white precipitates in this step, as confirmed by Bradford reagent and SDS-PAGE analysis. Fractions containing SLO were pooled and further dialyzed in 20 mM Bis-Tris (pH 5.8).
A second purification step using a fast protein liquid chromatography (FPLC) (AKTA) was performed to further separate protein impurities that co-elute with SLO. The protein solution (~70 mL) was centrifuged at 20,000 RPM for 20 min and passed through a 0.45 μm filter to remove insoluble debris, and then loaded to an UNO S6 column (Bio-Rad) using a 150-mL Superloop (GE Healthcare). The column was washed with 20 mM Bis-Tris (pH 5.8) until no absorbance at 280 nm was observed. SLO was eluted with a 210 min stepwise gradient of 0 to 500 mM KCl in 20 mM Bis-Tris (pH 5.8). SLO-containing fractions were pooled, buffer-exchanged in 0.5 mM TCEP, 50 mM HEPES (pH 7.0) and concentrated to minimal volume using an Amicon spin filter with a 30-kDa cut-off (Millipore-Sigma) in preparation for labeling with the fluorophore Badan. The purity of the enzymes was greater than 95% as assessed by SDS-PAGE. Typical yields were 10–15 mg SLO per liter of culture.
Labeling of Surface Cys-modified SLO and Glutathione with Badan.
Prior to labeling, TCEP was removed from solution by buffer exchange with deoxygenated 50 mM pH 7.0 HEPES. A solution of SLO (200 μM) was incubated in the presence of 2-fold excess of Badan (in DMSO) at 4 °C in 50 mM HEPES (pH 7.0) for 3 h. The volume of DMSO was kept to <10% of the total buffer volume. Excess unreacted Badan was removed by gel filtration by passing the reaction solution through a PD-10 column (GE Healthcare). Labeling efficiency was measured by UV-visible spectroscopy based on extinction coefficients of SLO (ε280 = 132 mM−1 cm−1)18 and Badan (ε387 = 21,000 M−1 cm−1).62 Badan-labeled SLO was stored in 50–150 μM, 50 μL aliquots at −80 °C until further use.
Mass Spectrometry Analysis.
Badan-labeled SLO was analyzed by mass spectrometry to confirm site-specific cysteine bioconjugation with Badan. A solution of Badan-labeled SLO was acid-quenched with citric acid (pH 2.4) and guanidine HCl. SLO samples were digested with pre-washed immobilized pepsin for 2.5 min. The peptide fragments were filtered from the immobilized pepsin using spin cups (cellulose acetate) and by centrifugation at 8000 RPM for 10 seconds at 4 °C. Samples were flash frozen immediately in liquid N2 and stored at −80 °C until data collection. Table 1 summarizes all mass spectrometry data, and Figure S1 shows the sequence coverage maps obtained from the pepsin digests.
Iron Quantitation.
Iron content for all Badan-labeled SLO variants was measured using both inductively coupled plasma-optical emission spectrometry (ICP-OES) and the ferrozine assay. Table S1 summarizes the amount of Fe ion per SLO monomer. Data represent the mean ± standard deviation of three independent measurements.
Enzyme Kinetics.
Steady-state kinetics was performed on a Cary 50 UV/vis spectrophotometer in the single-wavelength mode. The reaction progress was monitored by following the production of 13-(S)-hydroperoxy-9,11-(Z,E)-octadecadienoic acid [13-(S)-HPOD] (ε234 = 23,600 M−1 cm−1). All assays were performed in 100 mM borate (pH 9.0) under an ambient atmosphere at 10 – 40°C, regulated by a water-jacketed cuvette holder attached to a NESLAB RTE-111 circulating water bath (temperature stability is ± 0.1 °C). The substrate concentrations for the kinetics vary in the range of 1–220 μM. The kinetic parameters kcat and KM were determined from the Michaelis-Menten non-linear fits in KaleidaGraph 4.5 (Synergy) and represent the mean ± standard error from the mean from three independent measurements for each protein variant.
Steady-State Fluorescence.
Fluorescence emission spectra of the Badan-labeled mutants were collected on a custom-built Fluorolog-3 spectrofluorometer (Horiba Jobin-Yvon). Excitation was achieved with a 450 W Xenon lamp. The light was focused using a double Czerny-Turner excitation monochromator (1 nm bandpass) with 1200 grooves/mm blazed at 330 nm. Photons from sample emission were focused using a single Czerny-Turner monochromator (10 nm bandpass) with 1200 grooves/mm blazed at 500 nm. The excitation and emission optics were calibrated using the lamp spectral maximum at 467 nm and the water Raman scattering band at 397 nm, respectively, using HPLC-grade water in a quartz cuvette.
For Badan-labeled SLO emission spectra, the excitation wavelength was set at 373 nm, and the emission spectra were collected in 0.1 M pH 9.0 borate buffer, from wavelengths spanning 400–650 nm at 0.5 nm increments. Spectra were collected over temperatures ranging from 10 to 40 °C at 5 °C intervals with a water bath (NESLAB RTE-111) controlling the cell temperature. Samples were equilibrated for 5 min at each respective temperature in a quartz cuvette (Starna Cells) before collecting emission spectra. Peak emission wavelengths were determined by fitting the corrected spectra to a Gaussian curve using KaleidaGraph 4.5 (Synergy).
Picosecond-Resolved Fluorescence Spectroscopy.
The time-correlated single photon counting technique (TCSPC) was used to obtain fluorescence decays. Selective excitation of Badan in each mutant was achieved using a Nano-LED N370 source having a typical full-width at half-maximum pulse of <1.2 ns with a 1-MHz repetition rate. The Nano-LED was powered by a FluoroHub photon-counting controller. Single-photon signals were detected by a TBX-04 photomultiplier tube detection module. The photon-counting hub window contained 1024 channels with a time-to-amplitude conversion range of 56 ps/channel. Lifetimes were measured using reverse mode counting with a 75-ns coaxial delay and 0 ns sync delay. Magic angle (55°) conditions were employed to eliminate lifetime distortions resulting from rotational motion. Fluorescence decays of the Badan-labeled SLO variants (0.1 μM) were obtained from 450–520 nm in 10 nm intervals with a 10 nm emission bandpass. Instrument response functions were collected once at each temperature using only 0.1 M borate (pH 9.0), with the emission monochromator set at 373 nm. All optical settings and delays were kept constant when acquiring the instrument response function. All decays were collected with a peak preset of 10,000 counts. The time resolution of the instrument was calculated to be ~120 ps. Procedures for sample incubation during data acquisition were performed in the same manner as previously described and executed at the temperatures ranging from 10 to 40 °C in 5 °C intervals. Time-resolved fluorescence measurements were typically performed in triplicate.
Enzyme Stability during Time-Resolved Fluorescence Measurements.
Previous circular dichroism measurements at different temperatures show that SLO has a melting temperature of ca. 55 °C at pH 9.0. The enzyme is also shown to be stable at 40 °C for up to 4 h.18 To ensure that the enzyme is active during acquisition of time-resolved fluorescence data (typically ~10–15 min of incubation), protein samples are replaced after every 30 min of data collection.
Construction of Time-Dependent Stokes Shift Spectra.
Time-resolved emission spectra (TRES) were constructed from fluorescence decay data by methods previously described in more thorough detail.39–41 Briefly, the function describing the fluorescence decay was determined by deconvoluting the instrument response function (IRF) from a sum of exponential decays at each respective wavelength, λ using Decay Analysis Software v6.8 (DAS6) as represented by equation 2:
| (2) |
where I(t) is the IRF, n represents the number of exponential components needed for fitting the decay data, αi represents the weighted amplitudes such that ∑iαi(λ) = 1, and τi represents the fluorescence decay constant for the ith component. One exponential was always used to begin fitting the decays, and additional exponentials were iteratively added until a reduced Γ2 < 1.25 was achieved for the residuals. After obtaining satisfactory fitting parameters, an H(λ) that is linearly proportional to the steady-state emission spectrum is calculated using the steady-state emission, F(λ) at a particular temperature (equation 3):
| (3) |
Equation 4 represents the points Γ(λ,t) that comprise the TRES, which are calculated at arbitrary time points t (0 to 10 ns, in 0.01 increments) by multiplying H(λ) by the deconvoluted decay parameters extracted from the raw data (τi, αi, at a certain λ) represented by equation 2.
| (4) |
Equations 4 and 5 are used to fit the experimental time points at arbitrary times (t) to a log-normal line shape63–64 as a function of frequency (ν) in cm−1:
| (5) |
| (6) |
where νp is peak frequency that is time-dependent, h and Δ represent the respective peak heights and width, and γ is the asymmetry parameter. These parameters were allowed to float freely until a best fit was obtained. Fitting of Γ(λ,t) vs ν to a log-normal line shape was performed from t = 0 to 10 ns in 0.01 increments (1001 datasets) using the software MATLAB. At a time point t, a single value of νp, h, Δ, and γ was obtained. For illustration purposes, the intensity Γ(λ,t) of the time-resolved emission spectra (TRES) was normalized by dividing Γ(λ,t) by the calculated peak height h at time t from the log-normal line fits.
Analysis of Stokes Shift Decay Rates.
The Stokes shift decay rates were calculated by plotting the calculated peak frequency νp as a function of time t. The data for Badan-labeled SLO generally follow a bi-exponential decay behavior, whereas a single-exponential fit is appropriate for glutathione-Badan. The values for the frequencies at time 0 (ν0) and time ∞ (ν∞), the decay lifetimes τi, and the relative amplitudes αi were obtained from fitting the data to equation 7.
| (7) |
The Stokes shift was calculated as the difference between ν(∞) and ν(0). As the instrument resolution is limited (~120 ps), only solvation events that are longer than this time are detected. For illustration purposes, the solvation correlation function C(t) (eq. 8) was plotted vs. time t. As shown in equation 8, C(t) is related to νp(t) in that νp(t) is normalized with respect to the total Stokes shift.
| (8) |
Analysis of Temperature Dependence of Fluorescence Relaxation Lifetimes and Stokes Shift Decay Rates.
The fluorescence relaxation lifetimes (τr) and Stokes shift decay rates (τs) (in seconds) were related to temperature T (in K) using the Arrhenius equation (eq. 9). The activation enthalpy (Ea) was calculated and is related to the Ea values from active site hydrogen transfer rates.
| (9) |
Structure Preparation and Energy Minimization of Badan-labeled SLO Structures.
The initial structure was taken from the RCSB Protein Data Bank (PDB: 3PZW).65 Where alternate amino acid orientations were available, the position with the higher occupancy was chosen. Missing heavy atoms were filled in with Prime,66 and hydrogens were added with Maestro’s Protein Preparation Wizard.67 Waters beyond 3 Å of added heteroatom groups were deleted, as were acetate and ethylene glycol molecules which were artifacts of crystallization. The active site iron is in its ferrous state with a water as a coordination oxygen. The Q322 and S596 positions were mutated to cysteine and Badan was added to the thiol group to generate the two labeled mutants. A hydrogen-bond optimization was run to flip waters and polar groups to maximize hydrogen-bonding, followed by a restrained minimization for hydrogens only. A further minimization was performed with Macromodel.68 A PRCG algorithm was performed for 500 iterations. An OPLS3e force field was used in water, and the convergence threshold was 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the National Institutes of Health grants to JPK (GM118117), ARO (GM113432 (F32)), and ATI (1S10OD020062-01). JPZ thanks the University of California President’s Postdoctoral Program for a fellowship. AN was supported by a Haas Fellowship. We thank Corey Meadows, Shenshen Hu, and members of the Klinman lab for helpful discussions. We also thank Wenju Zhang for developing the MATLAB script used in the calculation of Stokes shift decay curves.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.9b07228.
Mass Spectrometry, Fluorescence, and Enzyme Kinetics data.
REFERENCES
- (1).Henzler-Wildman KA; Lei M; Thai V; Kerns SJ; Karplus M; Kern D, A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [DOI] [PubMed] [Google Scholar]
- (2).Eisenmesser EZ; Millet O; Labeikovsky W; Korzhnev DM; Wolf-Watz M; Bosco DA; Skalicky JJ; Kay LE; Kern D, Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [DOI] [PubMed] [Google Scholar]
- (3).Schwartz SD; Schramm VL, Enzymatic transition states and dynamic motion in barrier crossing. Nat. Chem. Biol 2009, 5, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Daniel RM; Dunn RV; Finney JL; Smith JC, The Role of Dynamics in Enzyme Activity. Annu. Rev. Biophys. Biomol. Struct 2003, 32, 69–92. [DOI] [PubMed] [Google Scholar]
- (5).Benkovic SJ; Hammes GG; Hammes-Schiffer S, Free-Energy Landscape of Enzyme Catalysis. Biochemistry 2008, 47, 3317–3321. [DOI] [PubMed] [Google Scholar]
- (6).Arrhenius S, Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. In Z. Phys. Chem 1889, 4U, 226–248. [Google Scholar]
- (7).Eyring H, The Activated Complex in Chemical Reactions. J. Chem. Phys 1935, 3, 107–115. [Google Scholar]
- (8).Tolman RC, Statistical Merchanics Applied to Chemical Kinetics. J. Am. Chem. Soc 1920, 42, 2506–2528. [Google Scholar]
- (9).Agarwal PK, A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2019, 58, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Agarwal PK, Role of Protein Dynamics in Reaction Rate Enhancement by Enzymes. J. Am. Chem. Soc 2005, 127, 15248–15256. [DOI] [PubMed] [Google Scholar]
- (11).Fenimore PW; Frauenfelder H; McMahon BH; Young RD, Bulk-solvent and hydration-shell fluctuations, similar to α- and β-fluctuations in glasses, control protein motions and functions. Proc. Natl. Acad. Sci 2004, 101, 14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fenimore PW; Frauenfelder H; McMahon BH; Parak FG, Slaving: Solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci 2002, 99, 16047–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Grossman M; Born B; Heyden M; Tworowski D; Fields GB; Sagi I; Havenith M, Correlated structural kinetics and retarded solvent dynamics at the metalloprotease active site. Nat. Struct. Mol. Biol 2011, 18, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kamerlin SCL; Warshel A, At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins 2010, 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nashine VC; Hammes-Schiffer S; Benkovic SJ, Coupled motions in enzyme catalysis. Curr. Opin. Chem. Biol 2010, 14, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Frauenfelder H; Chen G; Berendzen J; Fenimore PW; Jansson H; McMahon BH; Stroe IR; Swenson J; Young RD, A unified model of protein dynamics. Proc. Natl. Acad. Sci 2009, 106, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vaughn MB; Zhang J; Spiro TG; Dyer RB; Klinman JP, Activity-Related Microsecond Dynamics Revealed by Temperature-Jump Förster Resonance Energy Transfer Measurements on Thermophilic Alcohol Dehydrogenase. J. Am. Chem. Soc 2018, 140, 900–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Offenbacher AR; Hu S; Poss EM; Carr CAM; Scouras AD; Prigozhin DM; Iavarone AT; Palla A; Alber T; Fraser JS; Klinman JP, Hydrogen–Deuterium Exchange of Lipoxygenase Uncovers a Relationship between Distal, Solvent Exposed Protein Motions and the Thermal Activation Barrier for Catalytic Proton-Coupled Electron Tunneling. ACS Cent. Sci 2017, 3, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nagel ZD; Klinman JP, A 21st century revisionist’s view at a turning point in enzymology. Nat. Chem. Biol 2009, 5, 543–550. [DOI] [PubMed] [Google Scholar]
- (20).Klinman JP; Offenbacher AR; Hu S, Origins of Enzyme Catalysis: Experimental Findings for C–H Activation, New Models, and Their Relevance to Prevailing Theoretical Constructs. J. Am. Chem. Soc 2017, 139, 18409–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Klinman JP, Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc. Chem. Res 2015, 48, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Klinman JP, Importance of Protein Dynamics during Enzymatic C–H Bond Cleavage Catalysis. Biochemistry 2013, 52, 2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Meyer MP; Tomchick DR; Klinman JP, Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl. Acad. Sci 2008, 105, 1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Horitani M; Offenbacher AR; Carr CAM; Yu T; Hoeke V; Cutsail GE; Hammes-Schiffer S; Klinman JP; Hoffman BM, 13C ENDOR Spectroscopy of Lipoxygenase–Substrate Complexes Reveals the Structural Basis for C–H Activation by Tunneling. J. Am. Chem. Soc 2017, 139, 1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Klinman JP; Offenbacher AR, Understanding Biological Hydrogen Transfer Through the Lens of Temperature Dependent Kinetic Isotope Effects. Acc. Chem. Res 2018, 51, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hu S; Soudackov AV; Hammes-Schiffer S; Klinman JP, Enhanced Rigidification within a Double Mutant of Soybean Lipoxygenase Provides Experimental Support for Vibronically Nonadiabatic Proton-Coupled Electron Transfer Models. ACS Catal. 2017, 7, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meadows CW; Ou R; Klinman JP, Picosecond-Resolved Fluorescent Probes at Functionally Distinct Tryptophans within a Thermophilic Alcohol Dehydrogenase: Relationship of Temperature-Dependent Changes in Fluorescence to Catalysis. J. Phys. Chem. B 2014, 118, 6049–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meadows CW; Balakrishnan G; Kier BL; Spiro TG; Klinman JP, Temperature-Jump Fluorescence Provides Evidence for Fully Reversible Microsecond Dynamics in a Thermophilic Alcohol Dehydrogenase. J. Am. Chem. Soc 2015, 137, 10060–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Abbyad P; Shi X; Childs W; McAnaney TB; Cohen BE; Boxer SG, Measurement of Solvation Responses at Multiple Sites in a Globular Protein. J. Phys. Chem. B 2007, 111, 8269–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Carr CAM; Klinman JP, Hydrogen Tunneling in a Prokaryotic Lipoxygenase. Biochemistry 2014, 53, 2212–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Segraves EN; Holman TR, Kinetic Investigations of the Rate-Limiting Step in Human 12- and 15-Lipoxygenase. Biochemistry 2003, 42, 5236–5243. [DOI] [PubMed] [Google Scholar]
- (32).Su C; Oliw EH, Manganese Lipoxygenase: Purification and Characterization. J. Biol. Chem 1998, 273, 13072–13079. [DOI] [PubMed] [Google Scholar]
- (33).Kostenko A; Ray K; Iavarone AT; Offenbacher AR, Kinetic Characterization of the C–H Activation Step for the Lipoxygenase from the Pathogenic Fungus Magnaporthe oryzae: Impact of N-Linked Glycosylation. Biochemistry 2019, 58, 3193–3203. [DOI] [PubMed] [Google Scholar]
- (34).Macgregor RB; Weber G, Estimation of the polarity of the protein interior by optical spectroscopy. Nature 1986, 319, 70–73. [DOI] [PubMed] [Google Scholar]
- (35).Pierce DW; Boxer SG, Dielectric relaxation in a protein matrix. J. Phys. Chem 1992, 96, 5560–5566. [Google Scholar]
- (36).Lakowicz JR, Principles of Fluorescence Spectroscopy. 3rd ed.; Springer: New York, 2006 [Google Scholar]
- (37).Weber G; Farris FJ, Synthesis and spectral properties of a hydrophobic fluorescent probe: 6-propionyl-2-(dimethylamino)naphthalene. Biochemistry 1979, 18, 3075–3078. [DOI] [PubMed] [Google Scholar]
- (38).Cohen BE; McAnaney TB; Park ES; Jan YN; Boxer SG; Jan LY, Probing Protein Electrostatics with a Synthetic Fluorescent Amino Acid. Science 2002, 296, 1700–1703. [DOI] [PubMed] [Google Scholar]
- (39).Badea MG; Brand L, Time-resolved fluorescence measurements In Methods Enzymol., Academic Press: 1979; Vol. 61, pp 378–425. [DOI] [PubMed] [Google Scholar]
- (40).Lakowicz JR; Cherek H; Laczko G; Gratton E, Time-resolved fluorescence emission spectra of labeled phospholipid vesicles, as observed using multi-frequency phase-modulation fluorometry. Biochim. Biophys. Acta - Biomembranes 1984, 777, 183–193. [Google Scholar]
- (41).Horng ML; Gardecki JA; Papazyan A; Maroncelli M, Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited. J. Phys. Chem 1995, 99, 17311–17337. [Google Scholar]
- (42).Nandi N; Bagchi B, Dielectric Relaxation of Biological Water. J. Phys. Chem. B 1997, 101, 10954–10961. [Google Scholar]
- (43).Arya S; Singh AK; Bhasne K; Dogra P; Datta A; Das P; Mukhopadhyay S, Femtosecond Hydration Map of Intrinsically Disordered α-Synuclein. Biophys. J 2018, 114, 2540–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jesenská A; Sýkora J; Olżyńska A; Brezovský J; Zdráhal Z; Damborský J; Hof M, Nanosecond Time-Dependent Stokes Shift at the Tunnel Mouth of Haloalkane Dehalogenases. J. Am. Chem. Soc 2009, 131, 494–501. [DOI] [PubMed] [Google Scholar]
- (45).Guha S; Sahu K; Roy D; Mondal SK; Roy S; Bhattacharyya K, Slow Solvation Dynamics at the Active Site of an Enzyme: Implications for Catalysis. Biochemistry 2005, 44, 8940–8947. [DOI] [PubMed] [Google Scholar]
- (46).Pal S; Bandyopadhyay S, Importance of Protein Conformational Motions and Electrostatic Anchoring Sites on the Dynamics and Hydrogen Bond Properties of Hydration Water. Langmuir 2013, 29, 1162–1173. [DOI] [PubMed] [Google Scholar]
- (47).Makarov V; Pettitt BM; Feig M, Solvation and Hydration of Proteins and Nucleic Acids: A Theoretical View of Simulation and Experiment. Acc. Chem. Res 2002, 35, 376–384. [DOI] [PubMed] [Google Scholar]
- (48).van der Kamp MW; Prentice EJ; Kraakman KL; Connolly M; Mulholland AJ; Arcus VL, Dynamical origins of heat capacity changes in enzyme-catalysed reactions. Nat. Commun 2018, 9, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kohen A; Cannio R; Bartolucci S; Klinman JP; Klinman JP, Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 1999, 399, 496–499. [DOI] [PubMed] [Google Scholar]
- (50).Liang Z-X; Lee T; Resing KA; Ahn NG; Klinman JP, Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl. Acad. Sci 2004, 101, 9556–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Meadows CW; Tsang JE; Klinman JP, Picosecond-Resolved Fluorescence Studies of Substrate and Cofactor-Binding Domain Mutants in a Thermophilic Alcohol Dehydrogenase Uncover an Extended Network of Communication. J. Am. Chem. Soc 2014, 136, 14821–14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hatcher E; Soudackov AV; Hammes-Schiffer S, Proton-Coupled Electron Transfer in Soybean Lipoxygenase. J. Am. Chem. Soc 2004, 126, 5763–5775. [DOI] [PubMed] [Google Scholar]
- (53).Hammes-Schiffer S, Hydrogen Tunneling and Protein Motion in Enzyme Reactions. Acc. Chem. Res 2006, 39, 93–100. [DOI] [PubMed] [Google Scholar]
- (54).Knapp MJ; Klinman JP, Environmentally coupled hydrogen tunneling. Eur. J. Biochem 2002, 269, 3113–3121. [DOI] [PubMed] [Google Scholar]
- (55).Marcus RA; Sutin N, Electron transfers in chemistry and biology. Biochim. Biophys. Acta, Rev. Bioenerg 1985, 811, 265–322. [Google Scholar]
- (56).Dunitz JD, Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol 1995, 2, 709–712. [DOI] [PubMed] [Google Scholar]
- (57).Breiten B; Lockett MR; Sherman W; Fujita S; Al-Sayah M; Lange H; Bowers CM; Heroux A; Krilov G; Whitesides GM, Water Networks Contribute to Enthalpy/Entropy Compensation in Protein–Ligand Binding. J. Am. Chem. Soc 2013, 135, 15579–15584. [DOI] [PubMed] [Google Scholar]
- (58).Miyashita O; Onuchic JN; Wolynes PG, Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci 2003, 100, 12570–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ansari A; Berendzen J; Bowne SF; Frauenfelder H; Iben IE; Sauke TB; Shyamsunder E; Young RD, Protein states and proteinquakes. Proc. Natl. Acad. Sci 1985, 82, 5000–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Steczko J; Donoho GA; Dixon JE; Sugimoto T; Axelrod B, Effect of ethanol and low-temperature culture on expression of soybean lipoxygenase L-1 in Escherichia coli. Protein Expr. Purif 1991, 2, 221–227. [DOI] [PubMed] [Google Scholar]
- (61).Hu S; Sharma SC; Scouras AD; Soudackov AV; Carr CAM; Hammes-Schiffer S; Alber T; Klinman JP, Extremely Elevated Room-Temperature Kinetic Isotope Effects Quantify the Critical Role of Barrier Width in Enzymatic C–H Activation. J. Am. Chem. Soc 2014, 136, 8157–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Owenius R; Österlund M; Lindgren M; Svensson M; Olsen OH; Persson E; Freskgård P-O; Carlsson U, Properties of Spin and Fluorescent Labels at a Receptor-Ligand Interface. Biophys. J 1999, 77, 2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Strambini GB; Gonnelli M, Fluorescence Quenching of Buried Trp Residues by Acrylamide Does Not Require Penetration of the Protein Fold. J. Phys. Chem. B 2010, 114, 1089–1093. [DOI] [PubMed] [Google Scholar]
- (64).Eftink MR; Ghiron CA, Exposure of tryptophanyl residues and protein dynamics. Biochemistry 1977, 16, 5546–5551. [DOI] [PubMed] [Google Scholar]
- (65).Minor W; Steczko J; Stec B; Otwinowski Z; Bolin JT; Walter R; Axelrod B, Crystal Structure of Soybean Lipoxygenase L-1 at 1.4 Å Resolution. Biochemistry 1996, 35, 10687–10701. [DOI] [PubMed] [Google Scholar]
- (66).Jacobson MP; Pincus DL; Rapp CS; Day TJF; Honig B; Shaw DE; Friesner RA, A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- (67).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (68).MacroModel, Schrödinger, LLC: New York, NY, 2019. [Google Scholar]
- (69).Li P; Soudackov AV; Hammes-Schiffer S, Fundamental Insights into Proton-Coupled Electron Transfer in Soybean Lipoxygenase from Quantum Mechanical/Molecular Mechanical Free Energy Simulations. J. Am. Chem. Soc 2018, 140, 3068–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.