

Abstract
Plant mitogenomes can be difficult to assemble because they are structurally dynamic and prone to intergenomic DNA transfers, leading to the unusual situation where an organelle genome is far outnumbered by its nuclear counterparts. As a result, comparative mitogenome studies are in their infancy and some key aspects of genome evolution are still known mainly from pregenomic, qualitative methods. To help address these limitations, we combined machine learning and in silico enrichment of mitochondrial-like long reads to assemble the bacterial-sized mitogenome of Norway spruce (Pinaceae: Picea abies). We conducted comparative analyses of repeat abundance, intergenomic transfers, substitution and rearrangement rates, and estimated repeat-by-repeat homologous recombination rates. Prompted by our discovery of highly recombinogenic small repeats in P. abies, we assessed the genomic support for the prevailing hypothesis that intramolecular recombination is predominantly driven by repeat length, with larger repeats facilitating DNA exchange more readily. Overall, we found mixed support for this view: Recombination dynamics were heterogeneous across vascular plants and highly active small repeats (ca. 200 bp) were present in about one-third of studied mitogenomes. As in previous studies, we did not observe any robust relationships among commonly studied genome attributes, but we identify variation in recombination rates as a underinvestigated source of plant mitogenome diversity.
Keywords: mitogenome, repeats, recombination, rearrangement rates, structural variation
Introduction
Mitochondria share an α-proteobacterium ancestor, a conserved core proteome, and an almost universal function as the site of cellular energy production (Gray 2014). Despite the broad similarity of mitochondria across eukaryotes, the vestigial mitogenome is remarkably diverse in size, content, and architecture (Burger et al. 2003). Nowhere is this heterogeneity showcased more clearly than in plants: Closely related mitogenomes can vary 100-fold or more in size (Sloan et al. 2012), substitution rates (Cho et al. 2004), and rearrangement rates (Cole et al. 2018). Gene repertoires are fluid due to recurrent horizontal (Rice et al. 2013; Sanchez-Puerta 2014) and intergenomic transfers (Adams and Palmer 2003). Multichromosomal architectures (Alverson, Rice, et al. 2011; Sloan et al. 2012) have also been reported from the relatively few sequenced plant mitogenomes. Underlying the dynamism of plant mitogenomes appears to be the evolution of pervasive homologous recombination (Palmer and Herbon 1988; Gray et al. 1999).
Recombination is the predominant mode of double-strand break repair in plant mitogenomes (Maréchal and Brisson 2010; Gualberto and Newton 2017). In contrast, animal mitogenomes tend to use nonhomologous end joining pathways or may simply degrade damaged molecules (Alexeyev et al. 2013). Recombination can preserve sequence identity, but reliance on this pathway may lead to unsuppressed intramolecular recombination at dispersed repeats (Maréchal and Brisson 2010). While these differences in repair pathways likely contribute to the broad mitogenome differences among eukaryotic kingdoms, the importance of recombination and other mechanisms in generating the diversity within plants is unclear. Our limited understanding stems, in part, from the few available assembled mitogenomes.
While sequencing plant genomes is routine, only one mitogenome is published for every four nuclear genomes (NCBI Genome Resource; accessed March 11, 2019). Often, mitogenome assembly requires isolating DNA from intact mitochondria because frequent intergenomic transfers (e.g., Alverson, Rice, et al. 2011), the rapid decay of intergenic sequence homology (Guo et al. 2016), and the unclear physical organization of the mitogenome (Sloan 2013) can preclude identifying mitochondrial sequences from whole-genome read data (e.g., Nystedt et al. 2013). Strategies to identify mitogenomic scaffolds using GC-content and genome copy number can be effective (e.g., Naito et al. 2013) but become prone to false positives and low recovery rates with increasingly large, complex genomes (Eldfjell 2018).
We used machine learning to assemble the bacterial-sized mitogenome of a coniferous forest tree, Norway spruce (Pinaceae: Picea abies), from single molecule whole-genome shotgun sequencing reads. The P. abies mitogenome helps to fill a phylogenetic gap in comparative analyses, and to that end we analyzed gene repertoires; sources of genome size heterogeneity; intraspecific variation; and substitution, recombination, and rearrangement rates in gymnosperms. Prompted by the detection of highly recombinogenic small repeats in P. abies, we reevaluated published recombination rates in vascular plants. Despite early recognition of recombination as a factor in generating the diversity of eukaryotic mitogenomes (Palmer and Herbon 1988; Gray et al. 1999), surprisingly little attention has been given to recombinational dynamics as a source of mitogenomic diversity within plants. As in previous studies, we found no clear relationship between mitogenome traits and potential mechanisms—for example, genome size and the proportion of intergenomically transferred DNA—but the role of recombination as a driver of mitogenomic diversity within plants merits further scrutiny.
Materials and Methods
Machine Learning Classification of Genomic Scaffolds
Developing the support vector machine (SVM) involved four steps: 1) identification of high-quality training data, 2) training the classifier using this data and pre-determined genomic features, 3) evaluation of the model performance, and 4) application of the classifier to the P. abies v. 1.0 genome assembly (Nystedt et al. 2013) retrieved from ConGenIE.org (Sundell et al. 2015). To curate a reliable set of positive and negative training data from P. abies, we first discarded scaffolds <500 bp and masked repetitive elements using RepeatMasker v. open-4.0.1 (Smit et al. 2013). We aligned the remaining scaffolds to a set of relatively well-annotated genomes, including Arabidopsis thaliana and Populus trichocarpa and 18 additional organelle genomes (supplementary table S8, Supplementary Material online), using BlastN and TBlastX in BLAST version 2.2.29+ with default parameters except for an e-value cutoff of 1.0 × 10−20. Known numts (nuclear mitochondrial DNA sequences) were masked from the Arabidopsis assembly. Piceaabies sequences were considered robustly assigned if they matched an annotated gene region on the subject genome and 1) the top bitscore was >100; 2) the quotient between the top bitscore and next-best hit was >1.2; 3) alignments spanned at least 150 bp for BlastN and 100 aa for TBlastX; 4) BlastN and TBlastX annotations were identical if both existed; and 5) at least two different reference species contained the BLAST hit. Scaffolds meeting all these criteria were then classified as mitochondrial or nonmitochondrial training data for the SVM classifier. This yielded 2.0 and 51.1 Mb of positive and negative data.
After identifying robust training data sets, we trained the SVM classifier to distinguish between them using predetermined features. Following previous studies (Nystedt et al. 2013; Jackman et al. 2016), we used the standardized values of the natural logarithm of mean depth of coverage, the proportion of ambiguous nucleotides (%N), and the proportion of guanine and cytosine (%GC) for each scaffold, and a k-mer-based score based on the frequency of nonredundant “mitochondrial-like” or “nonmitochondrial-like” oligonucleotide sequences in a scaffold. We calculated probability tables for occurring k-mers based on positive and negative training sequences and calculated a score for each accordingly:
where c is the class, n is the number of k-mers in the scaffold, and is the probability of k-mer k occurring in a sequence of class c. The final k-mer classifier score for each scaffold was calculated as:
where L is the scaffold length and and are the positive and negative scores as defined above. Thus, smaller scores are consistent with plastid or nuclear sequence whereas larger scores indicate a scaffold comprising more mitochondrial-like k-mers. Optimal k-mer size for the classifier (k = 7, here) was assessed using test and training data from A. thaliana and visual inspection of the receiver-operating characteristic curves. After defining these features, we used SVMlight (Joachims 1998) with a Gaussian kernel to conduct the training step and to apply model to the set of P. abies scaffolds with length >500 bp and coverage >100×.
We assessed the SVM performance using crossvalidation. The initial training data were divided randomly into nine trials varying in training subsets from 10% to 90%, each consisting of 100 crossvalidation tests. K-mer scores, SVM training, and classification were carried out as described above. For each trial, we calculated the mean false discovery rate (FDR) and recall.
Mitogenome Assembly
We screened subreads from 77 Pacific Biosciences (PacBio) Sequel SMRT cells generated from genomic DNA (Street et al., unpublished data) for 27-mer matches to any of the SVM-classified scaffolds using BBDuk v. 35.14 (http://jgi.doe.gov/data-and-tools/bb-tools; last accessed December 12, 2019). Enriched reads were assembled using canu v. 1.7 (Koren et al. 2017), MECAT v. 1.3 (Xiao et al. 2017), and SMARTdenovo (https://github.com/ruanjue/smartdenovo; last accessed December 12, 2019). Canu was run using default parameters, except corMaxEvidenceErate was set to 0.15 as recommended in the manual for repetitive and GC-skewed genomes. MECAT was also run using default parameters using a target of 45×, 55×, and 65× coverage of the longest corrected reads. For MECAT and canu, we specified a genome size of ∼5.0 Mb, which is used to estimate the coverage of the input reads. Because SMARTdenovo does not include a preassembly read correction step, we used the 45×, 55×, and 65× corrected reads from MECAT as input and the default parameters were then used for assembly.
We selected the most contiguous assembly from each assembler for further refinement. We passed these assemblies and their constituent reads to FinisherSC to reconstruct the overlap graph and identify contigs that can be robustly merged (Lam et al. 2015). Next, we used TBlastX to identify contigs in each upgraded assembly containing the 41 protein-coding genes in the Cycas mitogenome (Chaw et al. 2008), which we retained as “high-confidence” assemblies. Then, we used pairwise alignments from NUCmer (Kurtz et al. 2004) to break the three assemblies at major disagreements. The resulting contigs were put through the assembly reconciliation pipeline implemented in CISA v. 1.3 (Lin and Liao 2013) to reassemble them into a single draft. After checking for circular contigs with dot plots, we evaluated the quality of the assembly by aligning the corrected reads with minimap2 (Li 2018) to the draft and visually inspected their congruency in IGV v. 2.4.14. (Thorvaldsdóttir et al. 2012). Finally, we used the partial order alignment graph approach implemented in Racon to call the consensus sequence from the minimap2 alignment (Vaser et al. 2017).
Genome Annotation
We reused repeat libraries curated for the P. abies v. 1.0 assembly (Nystedt et al. 2013) as input for RepeatMasker v. 4.0.7. (Smit et al. 2013) to identify TEs including long retrotransposons (long-terminal repeat [LTRs]), transposable elements (TEs), and other interspersed elements (LINEs and SINES). We additionally used RepeatModeler v. 1.0.8. to identify potential de novo repeats with significant similarity to the RepBase and Dfam databases (Smit et al. 2013). Direct and inverted repeats were identified with self versus self BlastN searches following Guo et al. (2016). Mitochondrial DNA of plastid origin (MIPTs) were identified following the methods of Guo et al. (2016) and shared mitochondrial-nuclear DNA following the methods of Alverson, Rice, et al. (2011).
We used MAKER v. 3.01.2 to annotate protein-coding genes (Cantarel et al. 2007). Complex repeats identified by RepeatMasker and RepeatModeler were hard-masked prior to annotation, whereas simple repeats were reannotated and soft masked by MAKER. As empirical gene evidence, we used the transcriptomes of P. abies, P. glauca, P. sitchensis, Pinus pinaster, Pinus sylvestris, and Pinus taeda from the PLAZA 3.0 database (Proost et al. 2015) and all curated plant sequences (Swiss-Prot) from UniProt as protein evidence. This provided 312,953 ESTs and 37,954 proteins for use as evidence alignments. Although we initially attempted to use the SNAP and AUGUSTUS ab initio gene predictors, we found the number of high-confidence genes was insufficient for training. Therefore, annotations are based on empirical evidence alone. Coding sequences were manually curated to improve identification of reading-frames and gene structure based on sequence homology. Hypothetical proteins were compared with an RNA-Seq data set obtained from needles (PRJEB26398, Schneider et al., in preparation), in which transcripts were extracted using a ribominus kit and subjected to de novo transcriptome assembly using Trinity v. 2.8.4 using default parameters (Grabherr et al. 2011). The de novo reconstructed transcripts were aligned to the P. abies v. 1.0 assembly (Nystedt et al. 2013) using GMAP v. 2015.11.20 (Wu and Watanabe 2005). The alignment coordinates were intersected with the ab initio prediction to validate the predicted hypothetical proteins using an ad hoc R script.
Identification of Repeat-Mediated Recombination and Rearrangements
For each inverted repeat pair, we extracted the ±2,000 bp single-copy flanking regions and constructed their expected recombination products for use as an alternative genome reference (Day and Madesis 2007) (supplementary fig. S3, Supplementary Material online). We used minimap2 to align the enriched PacBio reads against these four sequences, with the expectation that reads spanning the repeat and flanking regions of the alternative configurations with robust mapping quality (MQ >50) represent actual structural variations present within the sequenced individual (e.g., Park et al. 2014; Skippington et al. 2015; Cole et al. 2018; Dong et al. 2018).
We aligned the mitogenomes of P. abies and P. glauca using the rearrangement-aware progressiveMAUVE aligner (Darling et al. 2010), which identifies locally collinear blocks (LCBs) internally free of recombination and reorders them against a selected genome. Then, we used GRIMM 2.01 (Tesler 2002) to infer a minimum, optimum rearrangement history assuming signed undirected chromosomes. Apparent rearrangements introduced by the draft state of the genomes were corrected following Muñoz and Sankoff (2010) and Cole et al. (2018). Shared sequence between P. abies and P. glauca was estimated using BlastN with the same parameters as Guo et al. (2016) for consistency. To put the rearrangement rates inferred in Picea into a broader phylogenetic context, we realigned and estimated minimum numbers of rearrangements from the species pairs in Guo et al. (2016) with similar levels of divergence (supplementary table S6, Supplementary Material online).
We searched manuscripts associated with all 127 tracheophyte mitogenomes in the NCBI Genome resource (accessed March 11, 2019) for estimations of recombination rates. In addition, we exhaustively searched Google Scholar for “plant mitochondrial genomes” for publications since 2011, the year of the last extensive review of plant mitogenome recombination (Alverson, Zhuo, et al. 2011). To be included in our analysis, we required 1) genomes to be assembled using high-throughput sequencing at a depth of coverage sufficient to allow detection of alternative genome configurations (AGCs) comprising ≥1.6% of the read pool, 2) estimates of repeat-by-repeat recombination rates inferred from mapping statistics or alignment rates to be clearly reported, either tabulated or in figures sufficiently detailed to allow precise interpolation from vectorized figures using Inkscape v. 0.91 (e.g., Sloan et al. 2012, their fig. S6). Wherever possible, we limited the analyses to repeats ≥50 bp with ≥80% identity, although some studies employed more stringent cutoffs. Relevant details for each genome, including the sequencing depth of coverage, repeat number, range of repeat sizes and identities, and other analytical details are summarized in supplementary table S5, Supplementary Material online.
Comparative Analysis of Gymnosperm Mitogenomes
We downloaded the mitogenomes of Amborella trichopoda, Cycas taitungensis, Ginkgo biloba, Welwitschia mirabilis, P. glauca, and Pinustaeda from GenBank to compare substitution rates, gene and repeat content, and genomic structure. For analyses of nucleotide substitution rates, we also used the mitogenome coding sequences of Gnetum gnemon, Pinus sylvestris, and Araucaria heterophylla from NCBI GenBank. Given the draft state of the P. glauca mitogenome, we retained only contigs with conserved mitochondrial genes, which resulted in a 5.2 Mb assembly. We reannotated repeats and intergenome sequence transfers as described for P. abies for consistency.
Nucleotide substitution rates were estimated from the 41 protein-coding genes in Pinaceae. Coding sequences were extracted, aligned with MUSCLE v. 3.8.42 (Edgar 2004), and manually verified to ensure correct reading frames and complete codons. RNA editing sites were predicted using the PREP-mt webserver (Mower 2005) using a cutoff value of 0.2 as in Guo et al. (2016). Edited nucleotide sequences were realigned using the translation alignment option in Geneious v. 11.1.4 and poorly aligned blocks of codons were removed using Gblocks v. 0.91b webserver (Castresana 2000). First and second codon positions were extracted using DnaSP v. 6 (Rozas et al. 2017), and the maximum likelihood phylogeny of the concatenated alignment was inferred using RAxML v. 8.2.12 (Stamatakis 2014) under the GTR-GAMMA model of nucleotide evolution. This phylogeny agreed with the prevailing consensus of gymnosperm evolution (Lu et al. 2014), so we estimated branch lengths in units of synonymous (dS) and nonsynonymous (dN) substitution rates under the free-ratio branch model in PAML on this constrained topology (Yang 2007).
Intraspecific Mitogenome Variation in Picea abies
We selected two putatively ancient trees from Kullman (2008) and collected newly flushed buds. Buds were immediately placed in a solution comprising 10 mM MOPS, 5% (v/v) dimethylsulfoxide, and 5% (w/v) glycerol at pH 7.3 and stored on dry ice and then at −80 °C until isolation. In brief, intact mitochondria were isolated using centrifugation and extraneous DNA was degraded using DNase, and then mitochondrial DNA was isolated using a Gram-negative bacteria genomic DNA purification kit (supplementary methods 1, Supplementary Material online). DNA library construction and PacBio RS II sequencing were performed at the Duke Center for Genomic and Computational Biology according to the manufacturer’s instructions.
PacBio subreads were checked for adapter contamination and then aligned to the whole P. abies v. 1.0 assembly containing the new de novo mitogenome using minimap2 (Li 2018). Scaffolds identified as mitochondrial-like by the SVM in the P. abies v. 1.0 assembly but were not contained within the de novo assembly were left in place, as they may represent numts. Bases were called using GATK v. 3.8.0 Haplotype Caller in gvcf mode followed by joint-genotyping in includeNonVariantSites mode (Van der Auwera et al. 2013). As GATK does not assign confidence metrics to nonvariant sites, we considered those covered by 10–35 reads to be represented. Sites with higher coverage represent unresolved repeats. Structural variants were called with Sniffles v. 1.0.11 using default filtering settings (Sedlazeck et al. 2018). Each structural variant was then visually validated in IGV v. 2.4.14. (Thorvaldsdóttir et al. 2012).
Results and Discussion
Mitogenome Assembly
We combined machine learning, in silico enrichment of long sequencing reads, and assembly reconciliation to produce a highly contiguous P. abies draft mitogenome (Fig. 1). First, we trained a SVM using high-confidence positive and negative sequence data to identify mitochondrial-like scaffolds from whole genome assemblies. We applied the SVM to a reduced set of the P. abies v. 1.0 assembly (Nystedt et al. 2013) comprising 49,500 scaffolds. Of these, the SVM identified 301 scaffolds totaling 4.69 Mb as potentially mitochondrial in origin. While the SVM achieved higher resolution than simple coverage versus GC-content plots (Eldfjell 2018; supplementary fig. S1, Supplementary Material online), the recall and FDR indicated this classification alone would be insufficient to identify the P. abies mitogenome from the P. abies whole genome assembly. Recall ranged from 0.53 to 0.92 and the FDR from 0.13 to 0.22 with varying test-to-training ratios (supplementary table S1, Supplementary Material online), although SVMs achieved more accurate classifications in our tests with other draft gymnosperm assemblies (Eldfjell 2018).
Fig. 1.
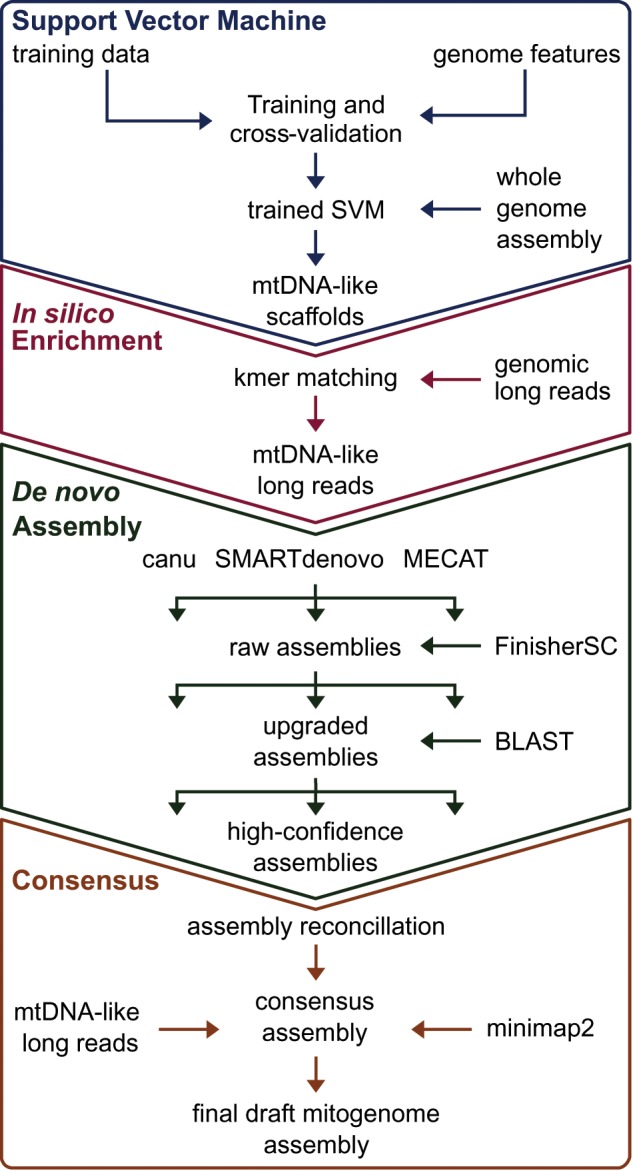
—Strategy used to assemble the mitogenome of Picea abies. First, a support vector machine (SVM) was trained to identify mitochondrial-like scaffolds from the P. abies genome assembly. We used the classified scaffolds to identify PacBio Sequel subreads containing mitogenome-like 27-mers. These enriched reads were then assembled using three different pipelines. Scaffolds from each assembler with at least one mitochondrial protein coding gene were retained for assembly reconciliation and base-pair correction, thus yielding the final mitogenome draft.
We then used the SVM classified scaffolds as “bait” to enrich genomic PacBio reads for mitochondrial-like sequences through k-mer matching (fig. 1). We screened ca. 35.5 million subreads totaling 244 Gb for 27-mer matches to any of the SVM-classified scaffolds, which yielded 3.23 million reads totaling 4.1 Gb, with an average read length of 12,041 bp. We reasoned that the long read length could enable us to recover a greater proportion of the mitogenome, as only a single 27-mer match is required, while the improved contiguity of the assembly could allow removal of dubious contigs postassembly.
We assembled the enriched reads using three assemblers selected based on their prior performance (Jayakumar and Sakakibara 2017; Giordano et al. 2017; fig. 1). The most contiguous results per assembler are summarized in table 1. Upgrading the initial assemblies with unused overlap information in the corrected reads with FinisherSC (Lam et al. 2015) only appreciably improved the N50 of the canu (Koren et al. 2017) assembly (table 1). Despite initial differences in size and contiguity, the assemblies were similar when considering only contigs containing at least 1 of the 41 mitochondrial protein-coding genes conserved in Gingko and Cycas, which we term high-confidence assemblies (table 1). Finally, reconciliation using the Contig Integrator for Sequence Assembly (CISA) pipeline (Lin and Liao 2013) produced a final draft assembly measuring 4.9 Mb over four contigs with a mean depth of coverage of 284×. This project has been deposited in Genbank under the accessions MN642623-MN642626 and at the Plant Genome Integrative Explorer's (Sundell et al. 2015) ftp resource (ftp://plantgenie.org/Publications/Sullivan2019/, last accessed December 12, 2019).
Table 1.
Summary Statistics for Assemblies of Pacific Biosciences Sequel Subreads Enriched In silico for Mitogenome-Like k-mers
| Assembler | No. Contigs | N50 (Mb) | L50 | Longest Contig (Mb) | Assembly Size (Mb) |
|---|---|---|---|---|---|
| canu | |||||
| Raw | 383 | 0.06 | 18 | 1.10 | 10.25 |
| Upgraded | 276 | 0.32 | 4 | 2.36 | 9.76 |
| High conf. | 4 | 1.28 | 2 | 2.56 | 5.29 |
| MECAT | |||||
| Raw | 104 | 0.43 | 5 | 2.29 | 9.24 |
| Upgraded | 95 | 0.43 | 5 | 2.29 | 9.22 |
| High conf. | 6 | 0.70 | 2 | 2.29 | 5.10 |
| SMARTdenovo | |||||
| Raw | 59 | 0.76 | 2 | 3.60 | 7.31 |
| Upgraded | 55 | 0.76 | 2 | 3.60 | 7.32 |
| High conf. | 4 | 3.60 | 1 | 3.60 | 5.13 |
| Final draft | 4 | 3.42 | 1 | 3.42 | 4.90 |
Note.—“Raw” refers to the full contig output produced by each assembler. Upgraded assemblies have been processed with FinisherSC. High-confidence assemblies contain only contigs with at least one protein-coding mitochondrial gene. N50 and L50 are calculated from contig lengths.
Genome Annotation and Gene Repertoires in Gymnosperms
General features of the P. abies mitogenome are summarized in table 2. All 41 protein-coding genes inferred to be present in the last common ancestor of angiosperms were also found in P. abies and, after reannotation, also in P. glauca and Pinustaeda. This pattern of conservation in Pinaceae is consistent with the early-diverging gymnosperms Gingko and Cycas, which may suggest a less dynamic gene repertoire than in angiosperms, although Welwitschia has undergone extensive gene loss (Guo et al. 2016). In addition, the P. abies mitogenome acquired complete copies of four plastid genes, psaB, psbH, psbN, and psbT, although the functionality of these genes, if any, cannot be inferred from the data here. We also identified 20 transcribed open-reading frames: 14 had uniquely mapping transcripts of ≥99% coverage and identity (high confidence), 2 were of medium confidence, where either identity or coverage was <99% but >97%, and 4 low-confidence genes had <97% support (supplementary table S2, Supplementary Material online). Intron and tRNA content were similar in P. abies and P. glauca (Jackman et al. 2016), but both have apparently undergone losses compared with the other gymnosperms, including the Pinaceae conifer Pinustaeda.
Table 2.
Characteristics of the Picea abies Mitogenome
| Genome | |
| Size (Mb) | ∼4.90 |
| GC content | 44.7% |
| Annotation | |
| Repeat content | 15.15% |
| Direct and inverted | 14.25% |
| Tandem | 1.12% |
| Nuclear-mitochondrial DNA | 28.89% |
| Transposable elements | 7.72% |
| Plastid-derived DNA | 0.34% |
| Genes | 1.00% |
| Protein coding genes | 41 |
| Hypothetical proteins high/medium/low confidence | 14/2/4 |
| tRNAs | 17 |
| rRNAs | 3 |
The repetitive fraction of the P. abies mitogenome is larger than the entire mitogenome of most plants (table 2; supplementary table S3, Supplementary Material online). Relative to genome size, however, repeat content in P. abies is unremarkable. Dispersed repeats in an analysis of 82 angiosperms comprised 14% of the mitogenome on average (Dong et al. 2018), identical to the proportion in P. abies (table 2). However, dispersed repeats in P. abies tended to be about half the size of a typical tracheophyte, at 312 versus the 641 bp average (Wynn and Christensen 2019), indicating a relative enrichment of smaller repeats (supplementary fig. S2, Supplementary Material online). At the same time, P. abies also had eight pairs of repeats ≥10 kb, more than all but 2 of the 79 land plants analyzed by Wynn and Christensen (2019).
Repeats and DNA T ransfers D o N ot E xplain Picea Mitogenome Expansion
Extraordinary mitogenome size heterogeneity among plants has long been recognized (Ward et al. 1981), but the sources appear to vary among species. We analyzed intergenomic transfers and the proliferation of repetitive sequences as potential causes of genome size variation in gymnosperms. Transfer of plastid DNA made a negligible contribution to the genome size of the three Pinaceae conifers (table 3), in contrast to some angiosperms, such as cucurbits (Alverson, Rice, et al. 2011; Alverson et al. 2010). Similarly, no relationship between genome size and repeat proliferation was evident: The 410 kb Cycas mitogenome was proportionally the most repetitive (Chaw et al. 2008), and relative repeat content was similar in Picea and Ginkgo despite their >10× size differences (table 3). An ambiguous correlation between genome size and repeat proliferation was also observed in Silene species (Sloan et al. 2012), whereas larger cucurbit genomes tend to contain proportionally more repeats (Alverson et al. 2010; Alverson, Rice, et al. 2011; Rodríguez-Moreno et al. 2011).
Table 3.
Potential Sources of Mitogenome Size Variation among Gymnosperms
| Cycas | Ginkgo | Picea abies | Picea glauca | Pinus taeda | Welwitschia | |
|---|---|---|---|---|---|---|
| Genome size (Mb) | 0.41 | 0.35 | 4.90 | 5.20a | 1.19 | 0.98 |
| Plastid-derived DNA (kb) | 18 (4) | 0 (0) | 17 (0) | 18 (0) | 3 (0) | 9 (9) |
| Dispersed repeats (kb) | 109 (26) | 51 (15) | 699 (14) | 885 (17) | 83 (7) | 42 (4) |
| Nuclear-mtDNA (Mb) | – | 0.35 (100) | 1.40 (29) | 2.29 (44) | 0.60 (50) | – |
| Transposable elements (kb) | 6 (1) | 3 (1) | 482 (10) | 386 (7) | 129 (10) | 15 (2) |
Note.—Dispersed repeats include those in the inverted and direct orientation ≥50 bp and with ≥80% identity. Nuclear-mitochondrial DNA are shared sequences with no direction of transfer inferred. Transposable elements comprises long-terminal repeats (LTR) and non-LTR retrotransposons. Numbers in parenthesis indicate percent coverage of the mitogenome.
Scaffolds containing protein-coding mitochondrial genes extracted from the 5.9 Mb assembly.
Interpreting shared nuclear-mitochondrial content is difficult because 1) the nuclear genomes of Cycas and Welwitschia are not sequenced; 2) the direction of transfer can rarely be determined with confidence under the simplest circumstances (Alverson et al. 2010); and 3) the Cycas and Ginkgo mitogenomes are highly conserved, which implies that any import from the nucleus predate their divergence ∼354 Mya (Lu et al. 2014) and may no longer be detectable in either nuclear genome (Guo et al. 2016). Therefore, it is unsurprising that the four species with nuclear reference genomes show an equivocal relationship between mitogenome size and shared nuclear-mitochondrial sequence (table 3). Remnants of nuclear TEs can potentially act as markers of sequence import because they have an unambiguous origin and generally do not proliferate after transfer (Knoop et al. 1996; Goremykin et al. 2012). All three Pinaceae species showed an increase of TE remnants, but relative content was similar among the two Picea species and the 4-fold smaller Pinus taeda mitogenome (table 3). Similarly, Welwitschia was relatively depauperate in TE elements, despite its large mitogenome size (table 3). Increased taxon sampling may help to clarify this ambiguous relationship between TE import and genome size, but a similarly unclear relationship has also been reported among angiosperms (Alverson et al. 2010; Alverson, Rice, et al. 2011; Rodríguez-Moreno et al. 2011, Goremykin et al. 2012). Overall, gymnosperms tend to reinforce observations in angiosperms: Repeat proliferation and DNA imports do not broadly explain mitogenome size heterogeneity (Alverson et al. 2010; Alverson, Rice, et al. 2011; Sloan et al. 2012; Dong et al. 2018).
Abundant Repeat-Mediated Recombination at Small Repeats
Homologous intermolecular recombination is used to repair double-stranded breaks and recover stalled replication forks (Maréchal and Brisson 2010). In many plants, intramolecular recombination also occurs at dispersed repeats and results in an individual harboring genomes that differ in structure but are identical in sequence. Intramolecular recombination dynamics range from completely inert (Alverson, Rice, et al. 2011; Dong et al. 2018) to astonishingly friable (Skippington et al. 2015), yet recombination rates are not widely estimated (see next section). However, recombination resulting in DNA exchange produces predictable AGCs that are directly observable in a pool of single molecule sequencing reads (e.g., Dong et al. 2018; Wang et al. 2018; supplementary fig. S3, Supplementary Material online). This allowed us to estimate the AGC frequency associated with each pair of inverted repeats in the P. abies mitogenome, which provides a quantitative estimate of recombination rates.
Genome rearrangements consistent with the products of intramolecular recombination (i.e., AGCs) comprised ∼1% of the pool of mapped reads. Half of the 598 repeats showed no evidence of recombination, but AGC frequency averaged 2% at recombinogenic repeats (fig. 2A;supplementary table S4, Supplementary Material online). Recombination was asymmetric in most cases, a result consistent with the repair of stalled replication forks through break-induced replication (supplementary table S4, Supplementary Material online; Maréchal and Brisson 2010). AGC abundance reached a maximum of 32% at a 186 bp repeat, with the 2 possible isomers comprising 12% and 20% of the reads, respectively. Surprisingly, the most recombinogenic repeats (AGCs ≥10%) ranged in size from 50 bp, the minimum size evaluated, to 948 bp (fig. 2A;supplementary table S4, Supplementary Material online). Overall, we found negligible correlation between repeat length and AGC frequency (fig. 2B;r2 = 0.03, P < 0.001), in contrast to some well-characterized examples (e.g., Mower, Floro, et al. 2012; Skippington et al. 2015; Guo et al. 2016) and contrary to the general expectation that, in the absence of other factors, recombination should scale positively with repeat length (Arrieta-Montiel and Mackenzie 2011).
Fig. 2.

—Recombination frequency at inverted repeats ≥50 bp with ≥80% pairwise identity as inferred from long reads mapping to expected recombination products (alternative genome configurations; AGCs). (A) Most repeat pairs have little or no evidence of recombination, but a minority are highly active. (B) Repeat length explains very little of the variation in recombination frequency (r2 = 0.03).
A Reappraisal of Intramolecular Mitogenome Recombination
Plant mitogenomes are widely cited to undergo frequent, reciprocal recombination at large repeats and only rarely elsewhere based on the results of seminal pregenomic studies (Lonsdale et al. 1984; Palmer and Shields 1984; Arrieta-Montiel and Mackenzie 2011). As our results suggest P. abies deviates from this pattern, we tested if this canonical model of mitogenome evolution is still supported by modern sequencing data. We searched the ∼200 published tracheophyte mitogenomes and identified those that 1) analyzed and reported repeat-by-repeat recombination dynamics and 2) achieved at least a 60× average depth of coverage. This coverage allows identification of AGCs comprising ≥1.6% of the read pool and establishes a baseline for comparing genomes sequenced to varying depths (e.g., 60 to over 1,000×), although confirmed AGCs have been documented at much lower frequencies (Woloszynska 2010; Arrieta-Montiel and Mackenzie 2011). Only 18 mitogenomes, representing ferns, gymnosperms, and 4 major angiosperm lineages, met both criteria.
Recombination patterns across tracheophytes provided mixed support for the canonical model of mitogenome evolution (table 4; supplementary table S5, Supplementary Material online). Results are summarized as proportions in table 4 to account for differences in repeat counts across species but are presented in absolute terms in supplementary table S5, Supplementary Material online. Consistent with the canonical model, large repeats tended to be more active than their smaller counterparts (table 4). However, AGC abundances were not at equilibrium with their reciprocal configurations in most species (table 4), indicating a lower recombination rate than anticipated from Southern blots (Maréchal and Brisson 2010). Although small repeats were overall less recombinogenic, they produced AGCs at similar frequencies as their larger counterparts in 33% of species, including P. abies (table 4). All species with active small repeats except for Silene conica showed high AGC frequencies at repeats measuring just ∼200 bp in length (Sloan et al. 2012; Mower, Floro, et al. 2012; Naito et al. 2013; Skippington et al. 2015; Pinard et al. 2019). While references to recombination rates are often necessarily vague, the 10%–50% AGC abundances in these species exceed any reasonable interpretation of phrases such as “highly substoichiometric” that are frequently used to describe the activity of small repeats (Woloszynska 2010; Arrieta-Montiel and Mackenzie 2011). Together, these studies point to the importance of small repeats as drivers of mitogenome evolution, a long anticipated (André et al. 1992) but rarely quantified phenomenon.
Table 4.
Recombination Patterns Summarized from 18 Published Vascular Plant Mitogenomes
| Species | Repeats ≥1,000 bp |
Repeats <1,000 bp |
Study | ||
|---|---|---|---|---|---|
| Proportion Active | Max AGC % | Proportion Active | Max AGC % | ||
| Chrysanthemum nankingense | na | na | 0.17 | 4 | Wang et al. (2018) |
| Cucumis sativus | 1 | 50 | 0.08 | 5 | Alverson, Rice, et al. (2011) |
| Daucus carota | 0.25 | 50 | 0.00 | 0 | Iorizzo et al. (2012) |
| Eucalyptus grandis | 0.40 | 31 | 0.04 | 23 | Pinard et al. (2019) |
| Ginkgo biloba | 1.00 | 50 | 0.00 | 0 | Guo et al. (2016) |
| Mimulus guttatus b | 1.00 | 50 | 0.38a | 50 | Mower, Floro, et al. (2012) |
| Monsonia ciliate | — | — | 0.00 | 0 | Cole et al. (2018) |
| Monsonia herrei | na | Na | 0.00 | 0 | Cole et al. (2018) |
| Nymphaea colorata | 0.00 | 8 | 0.00 | 0 | Dong et al. (2018) |
| Ophioglossum californicum | 0.50 | 25 | 0.00 | 0 | Guo et al. (2016) |
| Picea abies | 0.50 | 6 | 0.13 | 31 | This study |
| Psilotum nudum | 0.00 | 0 | 0.03 | 2 | Guo et al. (2016) |
| Silene conica | 0.48 | 13 | 0.06 | 15 | Sloan et al. (2012) |
| Silene noctiflora | 0.78 | 10 | 0.00 | 0 | Sloan et al. (2012) |
| Silene vulgaris | 1.00 | 50 | 0.14 | 10 | Sloan et al. (2012) |
| Vigna angularis | 1.00 | 33 | 0.26 | 24 | Naito et al. (2013) |
| Viscum scurruloideum | — | — | 0.66 | 50 | Skippington et al. (2015) |
| Welwitschia mirabilis | na | na | 0.00 | 0 | Guo et al. (2016) |
Note.—“Proportion active” refers to the fraction of repeats producing alternative genome configurations (AGCs) inferred to be the product of recombination in frequencies ≥1.6% of the parent molecule. “Max AGC” denotes the maximum frequency obtained by any AGC in the given repeat size class. Missing data because repeats of a size class do not exist in a given genome are listed as “na”, whereas “–” indicates missing data due to study limitations.
Minimum detection threshold is ∼4%, thus this proportion is underestimated.
Only inverted repeats analyzed.
Differences in nuclear-encoded repair pathways may be the mechanism underlying heterogeneity in recombination rates (Maréchal and Brisson 2010; Gualberto and Newton 2017). For example, Arabidopsis mutants for mitochondrial repair and surveillance genes recombine more readily at small repeats than their wild-type counterparts (Zaegel et al. 2006; Shedge et al. 2007; Miller-Messmer et al. 2012; Wallet et al. 2015). Although mutations associated with these or undiscovered genes could contribute to the diversity of recombination dynamics, the results in table 4 do not show any discernable phylogenetic signal. If nuclear-encoded repair genes directly explain most of the observed differences in recombination dynamics, then this lack of signal implies repeated, independent evolution. Identifying factors influencing recombination rates, and in turn how—or if—their variation explains facets of genome evolution such as genome size, repeat content, and mutation rate would be aided by more consistent reporting of recombination rates at a minimum.
Rampant M itogenome R earrangements in Picea
Rearrangements between the P. abies and P. glauca mitogenomes occurred an average of every 1,540 bp and blocks of synteny rarely extended beyond gene boundaries (fig. 3). After accounting for the draft state of the genomes (Muñoz and Sankoff 2010), a parsimonious rearrangement scenario (Tesler 2002) required 1,292 events to explain the size and distribution of synteny blocks between the Picea species (1,310 events, unadjusted for assembly scaffold number). Assuming a divergence time of ∼15 Ma (95% CI: 10–18 Myr; Feng et al. 2019) results in an absolute rearrangement rate of around 36–65 rearrangements/Myr. This rate is similar to those observed in Silene vulgaris and some closely related Monsonia species (Cole et al. 2018), which appear exceptionally rearranged relative to other eukaryotes. Rearrangement inference methods used here and by Cole et al. (2018) do not correct for events that have been lost due to the erosion of shared sequence, which should underestimate rearrangements given increasing divergence. When comparing similarly diverged mitogenomes (ca. 50% shared sequence) to mitigate this bias, the high rearrangement rate in Picea is even clearer: 43 versus an average of 1 rearrangement/Myr (SD 0.20) for 4 other species pairs (supplementary table S6, Supplementary Material online).
Fig. 3.

—The mitogenomes of Picea abies and P. glauca are extensively rearranged and collinear regions are limited to genic regions. An absolute rearrangement rate of 36–65/Myr is needed to explain this level of structural divergence. (A) Simplified diagram of the P. abies–P. glauca mitogenome alignment, where colored blocks represent corresponding homologous regions free of internal rearrangements (locally collinear blocks; LCBs) and heights are proportional to pairwise sequence identity. LCBs below the center line in P. glauca are inverted with respect to P. abies. White space indicates regions with no homology. Only LCBs longer than 2,000 bp are shown. (B) Two gene clusters widely conserved in plant mitogenomes are also found in Picea within a 9-kb block, which also serves to illustrate the typical extent of synteny beyond genic regions. Gene structures are indicated by yellow boxes and introns by black lines.
Mitogenome shuffling in Picea contrasts with the remarkably static Pinaceae nuclear genome, where a high degree of synteny has persisted among genera after ≥140 Myr of divergence (Krutovsky et al. 2004; Pelgas et al. 2006; Pavy et al. 2012). Even the nuclear genomes of Cupressaceae and Pinaceae, which last shared a common ancestor in the Carboniferous (Leslie et al. 2018), are more collinear than the P. abies and P. glauca mitogenomes (Ehrenmann et al. 2015). Despite their extensive rearrangements, two of the three gene clusters widely preserved in tracheophytes (Dong et al. 2018) were also maintained in Picea within the same 9 kb block: rpl2-rps19-rps3-rpl16 and nad3-rps12 (fig. 3B). The third widely conserved gene cluster—rrn5-rrn18—has not been preserved in Picea, Pinustaeda, or Welwitschia but persists in Cycas and Ginkgo (Guo et al. 2016), suggesting it was lost in the common ancestor of conifers and gnetophytes.
Intramolecular recombination creates AGCs that can potentially be transmitted as rearrangements (Gualberto and Newton 2017; Cole et al. 2018). However, the path from isomer to rearrangement is not straightforward because mitogenomes are subject to genetic drift within an individual and within a population (Gualberto and Newton 2017). After an AGC is produced, it must survive multiple rounds of cell division, be recruited into the germline, and persist through potentially multiple generations before the rearrangement becomes firmly established within an individual (Davila et al. 2011). Proliferation throughout the population could then occur as other polymorphisms, probably predominately through stochastic demographic forces but possibly also through natural selection (Shedge et al. 2010). Several aspects of this process are unknown, including how mitogenomes are replicated (Gualberto and Newton 2017), the timing of germline segregation (Lanfear 2018), and when, where, and under what circumstances AGCs arise. For these reasons, the relationship between recombination within individuals summarized in table 4 and rearrangement rates among species is unclear. For example, Monsonia and S.vulgaris have markedly different levels of intramolecular recombination (table 4), yet have similar rearrangement rates (Cole et al. 2018).
Sequence D ivergence in Picea and among Gymnosperms
A strong dichotomy between rates of sequence and structural evolution is a well-known feature of plant mitogenomes (Palmer and Herbon 1988) and is also the dynamic found in Picea and other Pinaceae conifers (fig. 4). Substitution rates across all protein-coding genes averaged 0.0023 for synonymous and 0.0012 for nonsynonymous sites. These rates are about 1/2 of those in the plastid genome (Sullivan et al. 2017) and 1/4 of the nuclear genome (Buschiazzo et al. 2012; Chen et al. 2012). Assuming a divergence time of 10–18 Myr between P. abies and P. glauca (Feng et al. 2019), mitochondrial substitution rates in Picea fall within the lower end of absolute rates observed in angiosperms ( = 7.65 ×10−11; = 4.00×10−11 given a 15 Myr divergence, cf. Mower et al. 2007) and are slightly higher than in Cycas and Ginkgo (Guo et al. 2016). Absolute substitution rates in Pinus, however, include the lowest rates reported so far in vascular plants (Richardson et al. 2013). Using divergence times from Saladin et al. (2017), rates ranged from dS = 0.60×10−11 and dN = 0.30×10−11 site/year in Pinustaeda to dS = 3.15×10−11 and dN = 0.17×10−11 in Pinus strobus. Previous work with more taxa, but fewer loci, similarly inferred exceptionally low substitution rates in some Pinus species (Wang and Wang 2014). At the other extreme, absolute dS measured ∼30.0×10−11 site/year in Welwitschia and A.heterophylla when assuming a divergence from Pinaceae around 342 Ma (Lu et al. 2014). Relative substitution rates were consistent with previous studies analyzing fewer genes or species (e.g., Mower et al. 2007; Guo et al. 2016) and are reported in supplementary table S7, Supplementary Material online.
Fig. 4.

—Mean substitution rates at synonymous (dS) and nonsynonymous (dN) sites vary 100-fold among gymnosperms. Rates within Pinaceae, in gray, are more consistent but are about 6-fold higher in Picea than in the 75% smaller Pinus mitogenomes.
Intraspecific Variation
We used the PacBio Sequel system to sequence partial mitogenomes from two P. abies on the alpine tundra in central Sweden, about 400 km southwest from the reference tree. Picea megafossil remains in this region date to ∼11,000 14C years ago, which suggests the presence of high-latitude glacial refugia in Scandinavia (Kullman 2008). Both formed small clonal groups above the modern tree line and are spatially associated with megafossils dated to 5,120 and 4,820 14C years ago, respectively, raising the possibility that these clonal groups are extremely ancient (Kullman 2001). Thus, the mitogenomes of these two trees is relevant to postglacial recolonization history and to the study of mitochondrial repair and recombination dynamics in long-lived organisms. In total, we recovered 499,180 and 553,660 bp, respectively, of which 82,443 was shared between the 2 trees. Nine variants were shared by the two trees relative to the reference tree. In contrast, 58 structural variations were found between these putatively ancient trees and the reference individual: 29 insertions, 17 deletions, 13 translocations, 9 duplications, and 7 inversions. More data are needed to address ecological and molecular hypotheses, but these sequences support a high rate of structural evolution in Picea.
Conclusion
Plant mitogenomes are highly variable in size, repeat and gene content, and substitution and rearrangement rates. The underlying processes generating this heterogeneity are largely unclear: For each mitogenome supporting a given mechanistic hypothesis, another often suggests the opposite, such as in the relationship between mutation rate and genome size (cf. Sloan et al. 2012; Skippington et al. 2015, Christensen 2018). The P. abies mitogenome and our comparative analyses may lend support to the view of plant mitogenomes as highly idiosyncratic and driven mainly by rapid evolution and/or considerable genetic drift. As in previous studies, we found no clear relationship between genome size, repeat content, intergenomic transfer, or substitution rate. However, we identified recombination as an underinvestigated mechanism of plant mitogenome evolution, despite being recognized as a likely source of the differences among eukaryotes (Palmer and Herbon 1988; Gray et al. 1999). Recombination rates vary extensively among plants and small repeats (<1,000 bp) are highly active in one-third of the reported species. Recombination affects the accumulation of mutations (Maréchal and Brisson 2010; Christensen 2018), influences genome size (Christensen 2018), and induces structural rearrangements (Palmer and Herbon 1988), making this variation a potential but understudied contributor to the diversity of plant mitogenomes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors acknowledge support from the National Genomics Infrastructure in Uppsala funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council, and SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure, and the Danish Council for Independent Research – Natural Science (to I.M.M.). N.R.S. is supported by the Trees and Crops for the Future (TC4F) project. Three anonymous reviewers provided helpful comments that improved the clarity and accuracy of the manuscript.
Data deposition: The authors received GenBank accession numbers MN642623–MN642626 for the assembly and PRJEB26398 for the RNAseq reads.
Literature Cited
- Adams KL, Palmer JD. 2003. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 29(3):380–395. [DOI] [PubMed] [Google Scholar]
- Alexeyev M, Shokolenko I, Wilson G, LeDoux S. 2013. The maintenance of mitochondrial DNA integrity – critical analysis and update. Cold Spring Harb Perspect Biol. 5:a01264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, et al. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27(6):1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD. 2011. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 23(7):2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD. 2011. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One 6(1):e16404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André C, Levy A, Walbot V. 1992. Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 8(4):128–132. [DOI] [PubMed] [Google Scholar]
- Arrieta-Montiel MP, Mackenzie SA. 2011. Plant mitochondrial genomes and recombination In: Kempken F, editor. Plant mitochondria. Advances in plant biology. Vol. 1 New York: Springer; p. 65–82. [Google Scholar]
- Van der Auwera GA, et al. 2013. From fastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Franz Lang B. 2003. Mitochondrial genomes: anything goes. Trends Genet. 19(12):709–716. [DOI] [PubMed] [Google Scholar]
- Buschiazzo E, Ritland C, Bohlmann J, Ritland K. 2012. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, et al. 2007. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18(1):188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Chaw SM, et al. 2008. The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol Biol Evol. 25(3):603–615. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. 2012. Sequencing of the needle transcriptome from Norway spruce (Picea abies Karst L.) reveals lower substitution rates, but similar selective constraints in gymnosperms and angiosperms. BMC Genomics 13(1):589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Mower JP, Qiu Y-L, Palmer JD. 2004. Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci U S A. 101(51):17741–17746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AC. 2018. Mitochondrial DNA repair and genome evolution. In: Logan DC, editor. Annual Plant Reviews, Plant Mitochondria. 2nd Ed. New York (USA): Wiley-Blackwell p. 11–31. doi:10.1002/9781119312994.apr0544 [Google Scholar]
- Cole LW, Guo W, Mower JP, Palmer JD. 2018. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol Biol Evol. 35(11):2773–2785. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila JI, et al. 2011. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 9(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day A, Madesis P. 2007. DNA replication, recombination, and repair in plastids In: Bock R, editor. Cell and molecular biology of plastids. Berlin, Heidelberg (Germany: ): Springer; p. 65–119. [Google Scholar]
- Eldfjell Y. 2018. Identifying mitochondrial genomes in draft whole-genome shotgun assemblies of six gymnosperm species [Bachelor’s thesis]. [Stockholm (Sweden)]: Stockholm University. Available from: http://urn.kb.se/resolve?urn=urn:nbn:se:su:diva-175410; last accessed December 13, 2019.
- Dong S, et al. 2018. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19(1):614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenmann F, et al. 2015. Evidence of intense chromosomal shuffling during conifer evolution. Genome Biol Evol. 7(10):2799–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, et al. 2019. Trans-lineage polymorphism and nonbifurcating diversification of the genus Picea. New Phytol. 222(1):576–587. [DOI] [PubMed] [Google Scholar]
- Giordano F, et al. 2017. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci Rep. 7(1):3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goremykin VV, Lockhart PJ, Viola R, Velasco R. 2012. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71(4):615–626. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW. 2014. The pre-endosymbiont hypothesis: a new perspective on the origin and evolution of mitochondria. Cold Spring Harb Perspect Biol. 6:a016097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. 1999. Mitochondrial evolution. Science 283(5407):1476–1481. [DOI] [PubMed] [Google Scholar]
- Gualberto JM, Newton KJ. 2017. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 68(1):225–252. [DOI] [PubMed] [Google Scholar]
- Guo W, et al. 2016. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol Biol Evol. 33(6):1448–1460. [DOI] [PubMed] [Google Scholar]
- Iorizzo M, et al. 2012. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SD, et al. 2016. Organellar genomes of white spruce (Picea glauca): assembly and annotation. Genome Biol Evol. 8(1):29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar V, Sakakibara Y. 2017. Comprehensive evaluation of non-hybrid genome assembly tools for third-generation PacBio long-read sequence data. Brief Bioinform. 20:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. 1998. Making large-scale support vector machine learning practical In: Schölkopf B, Burges C, Smola A, editors. Advances in kernel methods: support vector machines. Cambridge (MA: ): MIT Press; p. 169–184. [Google Scholar]
- Knoop V, et al. 1996. copia-, gypsy-, and LINE-Like retrotransposon fragments in the mitochondrial genome of Arabidopsis thaliana. Genetics 142(2):579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27(5):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovsky KV, Troggio M, Brown GR, Jermstad KD, Neale DB. 2004. Comparative mapping in the Pinaceae. Genetics 168(1):447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullman L. 2008. Early postglacial appearance of tree species in northern Scandinavia: review and perspective. Quat Sci Rev. 27(27–28):2467–2472. [Google Scholar]
- Kullman L. 2001. Immigration of Picea abies into north-central Sweden. New evidence of regional expansion and tree-limit evolution. Nord J Bot. 21(1):39–54. [Google Scholar]
- Kurtz S, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K-K, Tse D, LaButti K, Khalak A. 2015. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31(19):3207–3209. [DOI] [PubMed] [Google Scholar]
- Lanfear R. 2018. Do plants have a segregated germline? PLoS Biol. 16(5):e2005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AB, et al. 2018. An overview of extant conifer evolution from the perspective of the fossil record. Am J Bot. 105(9):1531–1544. [DOI] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18):3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-H, Liao Y-C. 2013. CISA: contig integrator for sequence assembly of bacterial genomes. PLoS One. 8(3):e60843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale DM, Hodge TP, Fauron CM. 1984. The physical map and organisation of the mitochondrial genome from the fertile cytoplasm of maize. Nucleic Acids Res. 12(24):9249–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Ran J-H, Guo D-M, Yang Z-Y, Wang X-Q. 2014. Phylogeny and divergence times of gymnosperms inferred from single-copy nuclear genes. PLoS One 9(9):e107679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, Brisson N. 2010. Recombination and the maintenance of plant organelle genome stability. New Phytol. 186(2):299–317. [DOI] [PubMed] [Google Scholar]
- Miller-Messmer M, et al. 2012. RecA-dependent DNA repair results in increased heteroplasmy of the Arabidopsis mitochondrial genome. Plant Physiol. 159(1):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP. 2005. PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinformatics 6(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP, Floro ER, Case AL, Willis JH. 2012. Evidence against equimolarity of large repeat arrangements and a predominant master circle structure of the mitochondrial genome from a monkeyflower (Mimulus guttatus) mineage with cryptic CMS. Genome Biol Evol. 4(5):670–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD. 2007. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 7(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz A, Sankoff D. 2010. Rearrangement phylogeny of genomes in contig form. IEEE/ACM Trans Comput Biol and Bioinf. 7:579–587. [DOI] [PubMed] [Google Scholar]
- Naito K, Kaga A, Tomooka N, Kawase M. 2013. De novo assembly of the complete organelle genome sequences of azuki bean (Vigna angularis) using next-generation sequencers. Breed Sci. 63(2):176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, et al. 2013. The Norway spruce genome sequence and conifer genome evolution. Nature 497(7451):579–584. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Herbon LA. 1988. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 28(1–2):87–97. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Shields CR. 1984. Tripartite structure of the Brassica campestris mitochondrial genome. Nature 307(5950):437–440. [Google Scholar]
- Park S, et al. 2014. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics 15(1):405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, et al. 2012. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 10(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelgas B, et al. 2006. Comparative genome mapping among Picea glauca, P. mariana x P. rubens and P. abies, and correspondence with other Pinaceae. Theor Appl Genet. 113(8):1371–1393. [DOI] [PubMed] [Google Scholar]
- Pinard D, Myburg AA, Mizrachi E. 2019. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genomics 20(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost S, et al. 2015. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 43(D1):D974–D981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DW, et al. 2013. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342(6165):1468–1473. [DOI] [PubMed] [Google Scholar]
- Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD. 2013. The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno L, et al. 2011. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genomics 12(1):424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, et al. 2017. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 34(12):3299–3302. [DOI] [PubMed] [Google Scholar]
- Saladin B, et al. 2017. Fossils matter: improved estimates of divergence times in Pinus reveal older diversification. BMC Evol Biol. 17(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Puerta MV. 2014. Involvement of plastid, mitochondrial and nuclear genomes in plant-to-plant horizontal gene transfer. Acta Soc Bot Pol. 83(4):317–323. [Google Scholar]
- Sedlazeck FJ, et al. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15(6):461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedge V, Arrieta-Montiel M, Christensen AC, Mackenzie SA. 2007. Plant mitochondrial recombination surveillance requires unusual RecA and MutS homologs. Plant Cell 19(4):1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedge V, Davila J, Arrieta-Montiel MP, Mohammed S, Mackenzie SA. 2010. Extensive rearrangement of the Arabidopsis mitochondrial genome elicits cellular conditions for thermotolerance. Plant Physiol. 152(4):1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skippington E, Barkman TJ, Rice DW, Palmer JD. 2015. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112(27):E3515–E3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB. 2013. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200(4):978–985. [DOI] [PubMed] [Google Scholar]
- Sloan DB, et al. 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10(1):e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013. RepeatMasker Open 4. Available from: http://www.repeatmasker.org; last accessed December 13, 2019.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan AR, Schiffthaler B, Thompson SL, Street NR, Wang X-R. 2017. Interspecific plastome recombination reflects ancient reticulate evolution in Picea (Pinaceae). Mol Biol Evol. 34(7):1689–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundell D, et al. 2015. The Plant Genome Integrative Explorer resource: plantGenIE.org. New Phytol. 208(4):1149–1156. [DOI] [PubMed] [Google Scholar]
- Tesler G. 2002. GRIMM: genome rearrangements web server. Bioinformatics 18(3):492–493. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2012. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Sovic I, Nagarajan N, Sikic M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27(5):737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallet C, et al. 2015. The RECG1 DNA translocase is a key factor in recombination surveillance, repair, and segregation of the mitochondrial DNA in Arabidopsis. Plant Cell. 27(10):2907–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wang X-R. 2014. Mitochondrial DNA capture and divergence in Pinus provide new insights into the evolution of the genus. Mol Phylogenet Evol. 80:20–30. [DOI] [PubMed] [Google Scholar]
- Wang S, et al. 2018. Assembly of a complete mitogenome of Chrysanthemum nankingense using Oxford Nanopore long reads and the diversity and evolution of Asteraceae mitogenomes. Genes 9(11):547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BL, Anderson RS, Bendich AJ. 1981. The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 25(3):793–803. [DOI] [PubMed] [Google Scholar]
- Woloszynska M. 2010. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes—though this be madness, yet there’s method in’t. J Exp Bot. 61(3):657–671. [DOI] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. 2005. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21(9):1859–1875. [DOI] [PubMed] [Google Scholar]
- Wynn EL, Christensen AC. 2019. Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 (Bethesda) 9(2):549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao CL, et al. 2017. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat Methods 14(11):1072–1074. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Zaegel V, et al. 2006. The plant-specific ssDNA binding protein OSB1 is involved in the stoichiometric transmission of mitochondrial DNA in Arabidopsis. Plant Cell 18(12):3548–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.