

Immune cell function can be modulated by changes in lipid metabolism. Our studies indicate that cholesterol and fatty acid synthesis increases in macrophages between 12 and 18 h after the activation of Toll-like receptors with proinflammatory stimuli and that the upregulation of lipogenesis may contribute to the resolution of inflammation.
KEYWORDS: inflammation, lipids, transcription, macrophage, nuclear receptor, lipid synthesis, transcriptional regulation
ABSTRACT
Immune cell function can be modulated by changes in lipid metabolism. Our studies indicate that cholesterol and fatty acid synthesis increases in macrophages between 12 and 18 h after the activation of Toll-like receptors with proinflammatory stimuli and that the upregulation of lipogenesis may contribute to the resolution of inflammation. The inflammation-dependent increase in lipogenesis requires the induction of the liver X receptors, members of the nuclear receptor superfamily of transcription factors, by type I interferons in response to inflammatory signals. Instead of the well-established role for liver X receptors in stimulating cholesterol efflux, we demonstrate that liver X receptors are necessary for the proper resumption of cholesterol synthesis in response to inflammatory signals. Thus, liver X receptors function as bidirectional regulators of cholesterol homeostasis, driving efflux when cholesterol levels are high and facilitating synthesis in response to inflammatory signals. Liver X receptor activity is also required for the proper shutdown of a subset of type I interferon-stimulated genes as inflammation subsides, placing the receptors in a negative-feedback loop that may contribute to the resolution of the inflammatory response.
INTRODUCTION
Recent studies indicate that the response of immune cells to pro- and anti-inflammatory signals is associated with changes in lipid metabolism (1–6). Activation of Toll-like receptor 4 (TLR4) signaling in macrophages leads to the rapid and transient inhibition of fatty acid synthesis that is followed by a later increase in the synthesis of long-chain unsaturated fatty acids (6, 7). Importantly, fatty acid synthesis at later stages of the inflammatory response has been suggested to play a role in the resolution of inflammation (7). Proinflammatory signals have also been shown to modulate cholesterol synthesis (6, 8–10), and simply repressing cholesterol synthesis in macrophages, independent of other proinflammatory signals, engages a type I interferon (IFN) response (6, 9). TLR4 activation also promotes the enzymatic conversion of cholesterol to 25-hydroxycholesterol, a cholesterol derivative with anti-inflammatory activity (1, 4, 5). As these studies illustrate, the temporal control of lipid metabolism plays an important role in modulating the inflammatory response. The signaling pathways that couple lipid metabolism to inflammation, however, are still being defined.
The liver X receptors (LXRs) LXRα (NR1H3) and LXRβ (NR1H2) are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors that control genetic networks involved in fatty acid and cholesterol metabolism (11, 12). In response to the direct binding of cholesterol derivatives, LXRs regulate the expression of genes encoding proteins such as the ATP binding cassette transporters ABCA1, ABCG5, and ABCG8, which mediate the transport of cholesterol out of peripheral cells (cholesterol efflux) to the liver and ultimately out of the body. Since LXR transcriptional activity can be increased by the direct binding of cholesterol-derived molecules, it has been suggested that LXRs act as cholesterol sensors that function to maintain whole-body cholesterol homeostasis (12, 13). The positive regulation of cholesterol efflux by LXRs complements the classic negative-feedback control of cholesterol biosynthesis mediated at least in part by inhibiting the transcriptional activity of the sterol regulatory element binding proteins (SREBPs) (SREBP1a and SREBP2) (14, 15). Thus, when intracellular cholesterol levels rise, synthesis is inhibited, and efflux/excretion is stimulated. LXRs also directly control the expression of the genes encoding enzymes required for fatty acid and triglyceride synthesis, including SREBP1c, considered the master transcriptional regulator of fatty acid synthesis (11, 12). Indeed, treatment with potent synthetic LXR agonists increases fatty acid synthesis and plasma triglyceride levels (16–18). Similarly, LXR activation in macrophages results in the increased production of long-chain unsaturated fatty acids (3, 19–21). Along with effects on cholesterol efflux and fatty acid synthesis, pretreatment of cells or animals with these same synthetic LXR agonists can also limit the subsequent response to proinflammatory challenges (22–24). A number of mechanisms have been suggested to underlie LXR anti-inflammatory activity, including increased production of anti-inflammatory fatty acids, direct repression of proinflammatory gene expression, and upregulation of cholesterol efflux from cells (12). Most studies exploring LXR anti-inflammatory activity, however, have utilized synthetic ligands, and it is not clear if there are physiological (as opposed to pharmacological) functions for LXRs in regulating the inflammatory response. Importantly, the LXR transcriptional responses to potent synthetic ligands and to endogenous cholesterol-derived ligands differ dramatically (25, 26).
We have now uncovered a previously unexplored link between LXR transcriptional activity and inflammatory signaling. TLR activation leads to the upregulation of LXR expression in a type I interferon-dependent manner at relatively late stages of the inflammatory response. Concurrently, there is an LXR-dependent and selective increase in gene expression associated with the generation of long-chain unsaturated fatty acids. Well-characterized LXR target genes involved in cholesterol efflux, however, are not induced by TLR activation. Instead of the established role for LXRs in stimulating cholesterol efflux, during the later stages of the TLR response, LXRs are necessary for an increase in sterol synthesis. Thus, LXRs can function as bidirectional regulators of cholesterol homeostasis, promoting efflux when cholesterol levels are high and promoting synthesis in response to proinflammatory signals. Interestingly, the LXR-dependent regulation of fatty acid and sterol synthesis that occurs after TLR activation intersects with the control of lipogenesis by the mammalian target of rapamycin (mTOR). In particular, both LXR and mTOR activities are required for the lipogenic response to TLR activation. Type I interferon treatment has been shown to inhibit lipid synthesis and promote lipid uptake (6, 9). Our results suggest that LXRs play a role in the reestablishment of lipid synthesis at late stages of inflammatory signaling. Consistent with a role for LXRs in counteracting the interferon response, LXR activity is also required for the proper shutdown of a subset of type I interferon-stimulated genes (ISGs). Thus, LXRs can be placed in a negative-feedback loop that may play a role in the resolution of the inflammatory response.
RESULTS
TLR activation induces LXR expression.
A number of studies have linked immune cell function to changes in metabolism. In particular, fatty acid synthesis has been suggested to play roles in both proinflammatory signaling as well as the resolution of inflammation (3, 7, 19, 21, 27–30). To explore the regulation of fatty acid synthesis during an inflammatory response, bone marrow-derived macrophages (BMDM) from C57BL/6J mice were treated with the TLR4 ligand Kdo2-lipid A (KLA) (the lipid A component of lipopolysaccharide), and the expression of Srebf1, encoding a master transcriptional regulator of fatty acid synthesis, was quantified by real-time PCR. There are two Srebf1 isoforms derived from differential promoter usage (15). Srebf1a, which has been shown to be highly expressed in macrophages (31), is modestly induced after 4 h of treatment and remains unchanged throughout the rest of the time course (Fig. 1A). In contrast, mRNA levels of Srebf1c, the second isoform, which is expressed at relatively low levels in resting macrophages (31), decrease at 4 h and subsequently increase, peaking at 16 h (Fig. 1B). Increased SREBP1 protein (Fig. 1C) and elevated fatty acid synthesis measured by the incorporation of [14C]acetate into fatty acid are also detected at between 13 and 18 h (Fig. 1D). Since LXRs bind to the Srebf1c promoter and directly regulate the transcription of this gene (17, 18), the same KLA time course was carried out in BMDM from Lxrα−/−/Lxrβ−/− mice (in the C57BL/6J background). In vehicle-treated cells, Srebf1c levels are increased in Lxrα−/−/Lxrβ−/− BMDM relative to C57BL/6J controls (Fig. 1B, compare white bars), a result consistent with the ability of LXRs to repress transcription via the recruitment of corepressors when agonist ligands are absent (3, 32). Nevertheless, decreased Srebf1c expression is still observed in Lxrα−/−/Lxrβ−/− BMDM 4 h after KLA treatment, indicating that the transient repression of Srebf1c is LXR independent (Fig. 1B). In contrast, the subsequent increases in Srebf1c expression and fatty synthesis between 8 and 16 h are dependent on LXR activity (Fig. 1B and D). Treatment of LXR-positive BMDM with the LXR antagonist GSK2033 also blocks the KLA-dependent induction of Srebf1c (Fig. 1E). LXR deletion, however, has no effect on the regulation of well-characterized proinflammatory genes such as the tumor necrosis factor (TNF) gene (Tnf) (Fig. 1F) and the interferon beta (IFN-β) gene (Ifnb) (Fig. 1G). Consistent with a role for LXRs in regulating the KLA-dependent induction of Srebf1c, the expression of Lxrα is induced by KLA with kinetics that precedes the Srebf1c response (Fig. 2A), and an increase in LXRα protein levels is observed by 16 h posttreatment (Fig. 2B). Additionally, increased binding of LXRα to the promoters of known LXR target genes such as Srebf1c and Abca1 is detected by chromatin immunoprecipitation (ChIP) assays in KLA-treated cells at 16 h (Fig. 2C). Increased mRNA levels of both Lxrα and Lxrβ can be detected 8 h after KLA treatment in BMDM as well as in human THP-1 cells; however, we have not been able to detect an increase in LXRβ protein in either cell type (data not shown). The regulation of Lxrα and Srebf1c by inflammatory signals is not limited to TLR4 activation, as activators of TLR1, -2, -3, -6, -7, and -9 also increase the expression of both genes at 16 h posttreatment (data not shown).
FIG 1.

TLR4 activation induces Srebf1c and Lxrα. (A, B, F, and G) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle or 100 ng/ml KLA for the times shown. Following treatment, mRNA levels of Srebf1a (A), Srebf1c (B), Tnf (F), and Ifnb (G) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 10). (C) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 16 h. Following treatment, nuclear extracts were prepared. SREBP1 and actin levels were examined by Western blotting as described in Materials and Methods. (D) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle or 100 ng/ml KLA for 13 h and then labeled with [14C]acetate in the continued presence or absence of KLA for an additional 5 h. Fatty acids were extracted and quantified as described in Materials and Methods. *, statistically significant difference from the vehicle control within the same genotype determined by 2-way ANOVA (P ≤ 0.05; n = 8). (E) C57BL/6 BMDM were treated with the vehicle, 100 ng/ml KLA, or 100 nM GW3965 for 8 h and then treated for an additional 8 h with 1.0 μM GSK2033 in the continued presence of KLA or GW3965. Following treatment, mRNA levels of Srebf1c were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference between the vehicle control and treated groups determined by 2-way ANOVA (P ≤ 0.05; n = 8).
FIG 2.
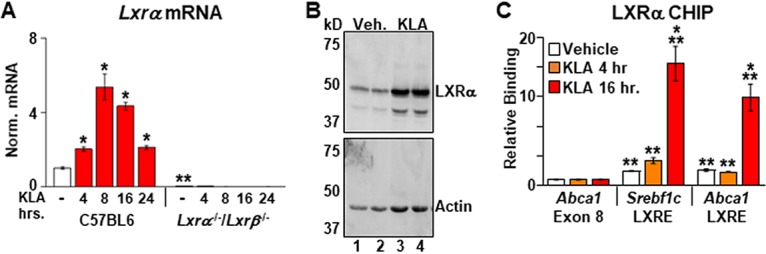
Inflammatory signals induce Lxrα expression. (A) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle or 100 ng/ml KLA for the times shown. Following treatment, mRNA levels of Lxrα were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 10). (B) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 16 h. Following treatment, nuclear extracts were prepared. LXRα and actin levels were examined by Western blotting as described in Materials and Methods. (C) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 4 or 16 h. ChIP assays were performed with an LXRα antibody, and real-time PCR was used to measure binding at the Srebf1c and Abca1 LXR response elements (LXREs) present in the promoters of each gene. A nonspecific site in exon 8 of the Abca1 gene was used as a control. Data are expressed relative to the value for the nonspecific site. *, statistically significant difference from the vehicle control within each group; **, statistically significant difference between specific and nonspecific binding within each treatment group determined by 1-way ANOVA (P ≤ 0.05; n = 8). Data are means ± standard errors.
Type I interferon induction of LXR expression.
The relatively late induction of Lxrα by TLR activation relative to classical proinflammatory genes such as Tnf suggests that LXR gene activation may be associated with a secondary wave of the inflammatory response. Comparison of the mouse and human LXRα promoters using the National Center for Biotechnology Information ECR browser identifies 2 highly conserved blocks of sequences, each of which contains conserved binding sites for STAT1, an interferon-inducible transcription factor (33). Furthermore, analyses of published chromatin immunoprecipitation sequencing (ChIP-Seq) data indicate that these sites are occupied by STAT1 in BMDM and that STAT1 binding increases after treatment with IFN-β (Fig. 3A) (34). TLR activation is known to induce the expression of IFN-β, which can subsequently function in an autocrine or paracrine fashion to induce the expression of ISGs (33). IFN-β treatment alone induces Lxrα in mouse BMDM, and the induction is lost in BMDM isolated from Stat1−/− mice (Fig. 3B). IFN-γ, on the other hand, has no effect on Lxrα expression (data not shown). The KLA-dependent induction of LXRα is also reduced when THP-1 cells are pretreated with a neutralizing antibody to IFN-β (Fig. 3C). Binding of IFN-β to type I interferon receptors leads to the activation of the Janus kinase (JAK)-STAT signaling pathway. Consistent with a role for IFN-β in regulating LXRα expression, treatment with the JAK inhibitor ruxolitinib blocks the KLA-dependent upregulation of LXRα mRNA (Fig. 3D). Finally, as shown in Fig. 3E intraperitoneal (i.p.) injection of recombinant IFN-β into mice leads to increased Lxrα levels in cells recovered from the peritoneal space at 8 h postinjection, indicating that IFN-β can regulate Lxrα expression in vivo.
FIG 3.
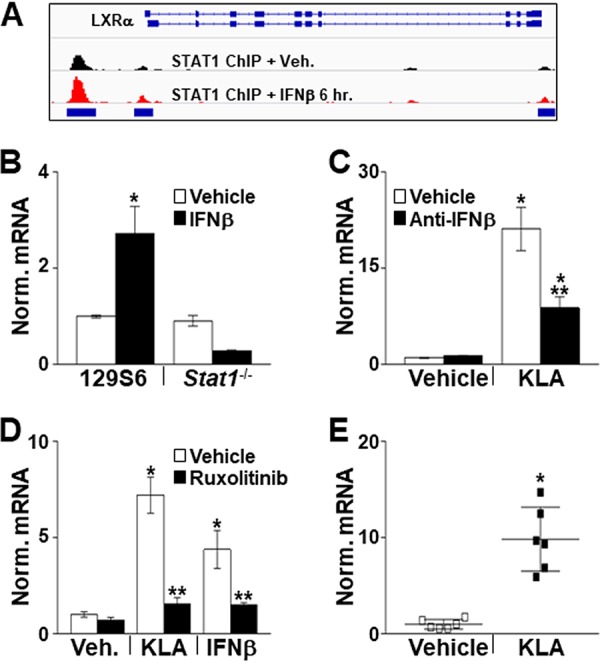
IFN-β induces Lxrα expression. (A) Integrative Genomics Viewer (IGV) screenshot demonstrating binding of STAT1 to the Lxrα promoter in vehicle- and IFN-β-treated BMDM. STAT1 ChIP-Seq data are derived from data reported under GEO accession number GSE33913 (34). (B) Control and Stat1−/− BMDM were treated with the vehicle or 100 ng/ml KLA for 8 h. Following treatment, Lxrα mRNA levels were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control determined by 2-way ANOVA (P ≤ 0.05; n = 8). (C) THP-1 macrophages were treated with the vehicle or 10 ng/ml KLA for 8 h in the presence or absence of a neutralizing antibody against IFN-β. Following treatment, LXRα mRNA levels were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control; **, statistically significant difference between KLA with and without anti-IFN-β determined by 2-way ANOVA (P ≤ 0.05; n = 8). (D) THP-1 macrophages were treated with the vehicle, 10 ng/ml KLA, or 20 ng/ml IFN-β for 8 h in the presence or absence of 1 μM ruxolitinib. Following treatment, LXRα mRNA levels were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control; **, statistically significant difference between KLA or IFN-β with and without ruxolitinib determined by 2-way ANOVA (P ≤ 0.05; n = 8). (E) Male 8-week-old C57BL/6J mice were injected i.p. with the vehicle or 10,000 U of IFN-β. After 8 h, cells were recovered from the peritoneal cavity, RNA was isolated, and Lxrα mRNA levels were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control determined by a Mann-Whitney test (P ≤ 0.05; n = 6). Data are means ± standard errors.
LXR-dependent induction of fatty acid synthesis.
To further define the contribution of LXRs to gene expression late in the inflammatory response, transcriptome sequencing (RNA-Seq) was performed using C57BL/6J and Lxrα−/−/Lxrβ−/− BMDM treated for 17 h with KLA, the LXR agonist GW3965, or acetylated low-density lipoprotein (AcLDL) particles to increase intracellular cholesterol levels. Interestingly, there is little overlap in the LXR-dependent responses to the 3 treatments (Fig. 4A), an observation consistent with previous studies demonstrating that synthetic agonists and cholesterol-derived LXR ligands generate distinct gene expression responses (25, 26). In support of the results in Fig. 1, a number of well-characterized LXR target genes encoding enzymes involved in long-chain unsaturated fatty acid synthesis, including Srebf1c, the stearoyl coenzyme A (stearoyl-CoA) desaturase 1 gene (Scd1), Scd2, and the fatty acid desaturase 2 gene (Fads2), are induced by KLA treatment in an LXR-dependent fashion (Fig. 4B to E). Unbiased pathway analysis using Metascape (35) also identifies genes associated with fatty acid biosynthesis as being significantly enriched in the KLA-induced, LXR-dependent class (q = 2 × 10−9). In contrast, KLA treatment does not alter the expression of well-characterized LXR target genes required for the efflux of cholesterol, including Abca1 and Abcg1, although both genes are induced by GW3965 (Fig. 4B and F).
FIG 4.

LXR-dependent regulation of fatty acid synthetic genes. (A) Genes upregulated ≥2-fold or downregulated ≥50% (P ≤ 0.05) in an LXR-dependent manner in RNA-Seq data from C57BL/6J and Lxrα−/−/Lxrβ−/− BMDM treated with the vehicle, 100 ng/ml KLA, 1.0 μM GW3965, or 50 μg/ml AcLDL for 17 h. (B) Heat map of RNA-Seq data (n = 3/group) demonstrating KLA-dependent induction of fatty acid synthesis genes. (C to F) Validation of RNA-Seq results in independent mice. C57BL/6J and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle, 100 ng/ml KLA, or 1.0 μM GW3965 for 16 h. (C, E, and F) Following treatment, mRNA levels of Scd2 (C), Fads2 (E), and Abca1 (F) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 8 to 16). Data are means ± standard errors. (D) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 16 h, and the protein levels of SCD2 and actin were examined by Western blotting as described in Materials and Methods.
TLR4 activation selectively increases the expression of fatty acid synthetic genes, even though increased binding of LXRα to the promoters of cholesterol efflux genes such as Abca1 is detected in KLA-treated cells (Fig. 2C). Along with the LXRs, the mTOR signaling pathway plays an important role in the regulation of lipogenesis (36, 37). Interestingly, mTOR activity can be induced by TLR4 activation and by type I interferon signaling (38–40). To determine if mTOR participates in the induction of lipogenesis by TLR4 activation, BMDM were treated with KLA as described above. Torin1, an inhibitor of both mTORC1 and mTORC2 (41), was then added beginning 8 h after KLA treatment. As shown in Fig. 5, mTOR inhibition impairs the KLA-dependent induction of fatty acid synthetic gene expression. mTOR modulates lipogenesis at least in part by controlling the transcriptional activity of the SREBPs through multiple mechanisms (36). To further explore a role for SREBPs in the TLR-dependent lipogenic response, BMDM were treated with the site 1 protease inhibitor PF-429242 (42) beginning 8 h after KLA administration to block the processing of SREBPs into mature, transcriptionally active forms. Consistent with a requirement for SREBP activity, PF-429242 also blocks the KLA-dependent induction of fatty acid synthetic genes (Fig. 5A to C). In contrast, Torin1 and PF-429242 have little or no effect on the KLA-dependent induction of Lxrα (Fig. 5D). Srebf1a has been reported to be highly expressed in BMDM (31), while Srebf1c is induced by KLA in an LXR-dependent fashion. To examine the contribution of each SREBP1 isoform, gene expression was measured in BMDM from mice selectively deficient in either SREBP1a or SREBP1c (Fig. 6). Consistent with a dominant role for SREBP1a in macrophages, KLA-dependent regulation of fatty acid synthetic genes is strongly reduced in SREBP1a-deficient cells. Decreased KLA-dependent gene expression is also observed in SREBP1c knockout BMDM, suggesting that both isoforms are necessary for the full response. Taken together, the genetic and pharmacological data in Fig. 4 to 6 indicate that the combined activity of LXR, mTOR, and SREBP is required for TLR4-dependent fatty acid synthesis.
FIG 5.
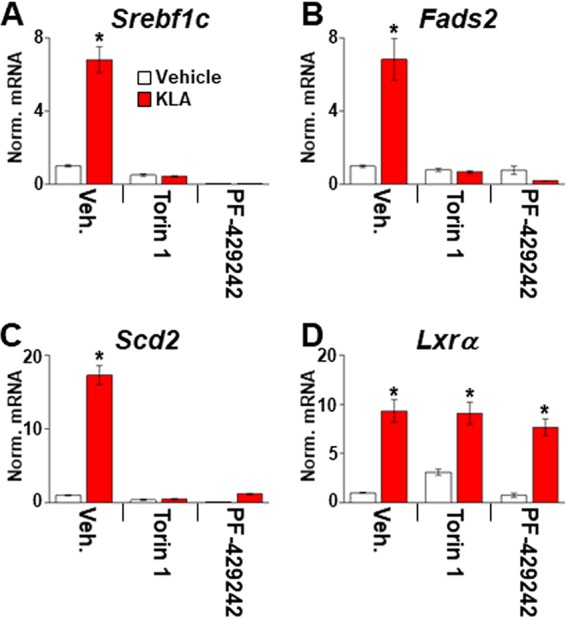
mTOR and SREBP activity is required for TLR4-dependent upregulation of fatty acid synthesis. C57BL/6J BMDM were treated with the vehicle or 100 ng/ml KLA for 8 h and then treated for an additional 8 h with 250 nM Torin1 or 10 μM PF-429242 in the continued presence of KLA. Following treatment, mRNA levels of Srebf1c (A), Fads2 (B), Scd2 (C), and Lxrα (D) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference between the vehicle and KLA within each pair determined by 2-way ANOVA (P ≤ 0.05; n = 8). Data are means ± standard errors.
FIG 6.

Role of SREBP1 isoforms in TLR4-dependent regulation of fatty acid synthesis. BMDM from C57BL/6J, SREBP1a-deficient (SREBP1a-DF), and SREBP1c knockout (SREBP1c-KO) mice were treated with the vehicle, 100 ng/ml KLA, or 1.0 μM GW3965 for 16 h. Following treatment, mRNA levels of Srebf1a (A), Srebf1c (B), Fads2 (C), Scd1 (D), Scd2 (E), and Lxrα (F) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Srebp1 knockouts determined by 2-way ANOVA (P ≤ 0.05; data derived from 2 mice/genotype and 4 replicates/mouse).
Regulation of sterol synthesis.
Surprisingly, the most significantly enriched pathway selectively upregulated by TLR4 activation in an LXR-dependent manner identified by Metascape was cholesterol biosynthesis (Fig. 7A) (q = 4 × 10−28). We confirmed a significant KLA-dependent induction of several genes in this group, including 24-dehydrocholesterol reductase (Dhcr24), farnesyl diphosphate farnesyltransferase 1 (Fdft1) (squalene synthase), and squalene epoxidase (Sqle), in BMDM from independent sets of mice (Fig. 7B to D). A KLA-dependent increase in sterol synthesis is also detected by measuring the 3-hydroxy-3-methyl-glutaryl (HMG)-CoA) reductase-dependent incorporation of [14C]acetate into neutral lipid between 13 and 18 h after KLA treatment (Fig. 7E). Fatty acid synthesis genes are well-established LXR targets, and high-affinity LXR binding sites have been identified in their promoter regions (17, 18, 43, 44). LXR-dependent regulation of cholesterol biosynthesis, however, has not been well characterized. Importantly, genetic deletion of LXR activity in vehicle-treated cells mimics the KLA-dependent increase in cholesterol biosynthesis measured in LXR-positive cells (Fig. 7B to E, compare red bars [C57BL/6 mice] to white bars [Lxrα−/−/Lxrβ−/− mice]). Thus, LXRs appear to function in a repressive pathway that restrains cholesterol biosynthesis in the absence of inflammatory signals. As shown in Fig. 7F and G, mTOR and SREBP activities are also required for the induction of cholesterol biosynthetic genes. Importantly, deletion of LXRs in BMDM derepresses genes involved in cholesterol transport, such as Abca1 and Abcg1 (Fig. 4B and F), leading to increased cholesterol efflux (Fig. 7H and I) (45). Increased cholesterol biosynthesis in LXR knockout cells may therefore represent an indirect response that functions to maintain overall cholesterol levels in the face of elevated cholesterol efflux.
FIG 7.

LXR-dependent regulation of cholesterol synthesis. (A) Heat map of RNA-Seq data (n = 3/group) demonstrating KLA-dependent induction of cholesterol synthesis genes. (B to D) Validation of RNA-Seq results in independent mice. C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle, 100 ng/ml KLA, or 1.0 μM GW3965 for 16 h. Following treatment, mRNA levels of Dhcr24 (B), Fdft1 (C), and Sqle (D) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 10 to 18). (E) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle or 100 ng/ml KLA and labeled with [14C]acetate between 13 and 18 h after KLA addition. Neutral lipids were extracted, and HMG-CoA reductase-dependent synthesis was quantified as described in Materials and Methods. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 8). (F and G) C57BL/6J BMDM were treated with the vehicle or 100 ng/ml KLA for 8 h and then treated for an additional 8 h with 250 nM Torin1 or 10 μM PF-429242 in the continued presence of KLA. Following treatment, mRNA levels of Dhcr24 (F) and Sqle (G) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference between the vehicle and KLA within each pair; **, statistically significant difference among samples without KLA (white bars) determined by 2-way ANOVA (P ≤ 0.05; n = 8). (H and I) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were loaded with cholesterol, and efflux to APOA1 (H) or high-density lipoprotein (HDL) (I) was measured as described in Materials and Methods. *, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− mice determined by Student’s t test (P ≤ 0.05; n = 8). Data are means ± standard errors.
The induction of cholesterol biosynthesis detected at late stages of the TLR4 response (Fig. 7) is consistent with the work of Kusnadi et al., who observed an increase in cholesterol biosynthetic gene expression at similar late time points after treatment of macrophages with TNF (10). In contrast, several studies have suggested that sterol synthesis is decreased in TLR4-activated macrophages (6, 8, 9). Differences in the identity, strength, and duration of the inflammatory stimuli; types of macrophages used; as well as cell culture conditions may contribute to the somewhat contradictory results among experiments. Measurements of sterol synthesis in cells continuously cultured with labeled acetate for 24 h (6, 8) may also overlook acute increases in the synthesis rate, such as that measured between 13 and 18 h after KLA treatment (Fig. 7E). In support of a relatively late increase in sterol synthesis after TLR4 activation, time course analysis indicates that under our conditions, the expression of many cholesterol biosynthetic enzymes, including HMG-CoA reductase (Hmgcr) and lanosterol 14α-demethylase (Cyp51), is repressed 8 h after KLA treatment (Fig. 8). Araldi et al. (8) also described repression of Cyp51 expression at this time point. Finally, between 8 and 24 h, there is a gradual increase in cholesterol biosynthetic gene expression and sterol synthesis that is absent in LXR-deficient BMDM.
FIG 8.
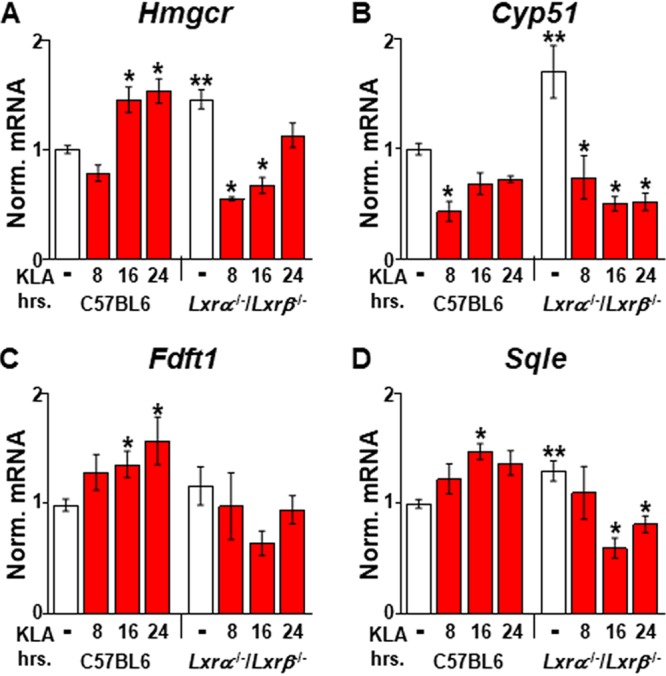
Effect of LXRs on cholesterol biosynthetic gene expression. C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle or 100 ng/ml KLA for the times shown. Following treatment, mRNA levels of Hmgcr (A), Cyp51a1 (B), Fdft (C), and Sqle (D) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same genotype; **, statistically significant difference between C57BL/6 and Lxrα−/−/Lxrβ−/− vehicle controls determined by 2-way ANOVA (P ≤ 0.05; n = 8).
Control of IFN-β signaling by LXRs.
We also examined the RNA-Seq data for mRNAs that demonstrate an elevated response to TLR4 activation in Lxrα−/−/Lxrβ−/− BMDM relative to controls. Gene ontology (GO) analysis of this set using Metascape identified the cellular response to interferon beta (q = 0.03) as the most enriched term. The cellular response to interferon beta GO term (GO 0035498) includes 40 mouse genes, 38 of which are detected in the RNA-Seq data set. Examination of the RNA-Seq data indicates that 31 out of the 40 genes (78%) exhibit increased expression in KLA-treated Lxrα−/−/Lxrβ−/− BMDM (Fig. 9A). Figure 9B to D show quantitative PCR (qPCR) validation demonstrating that 3 well-characterized IFN-β-regulated genes, the guanylate binding protein 9 gene (Gbp9), the interferon-regulated factor 1 gene (Irf1), and Stat1, are hyperinduced 16 h after KLA treatment in Lxrα−/−/Lxrβ−/− BMDM. The RNA-Seq data suggest that LXRs contribute to a negative-feedback loop that is required for the proper shutdown of IFN-β signaling.
FIG 9.
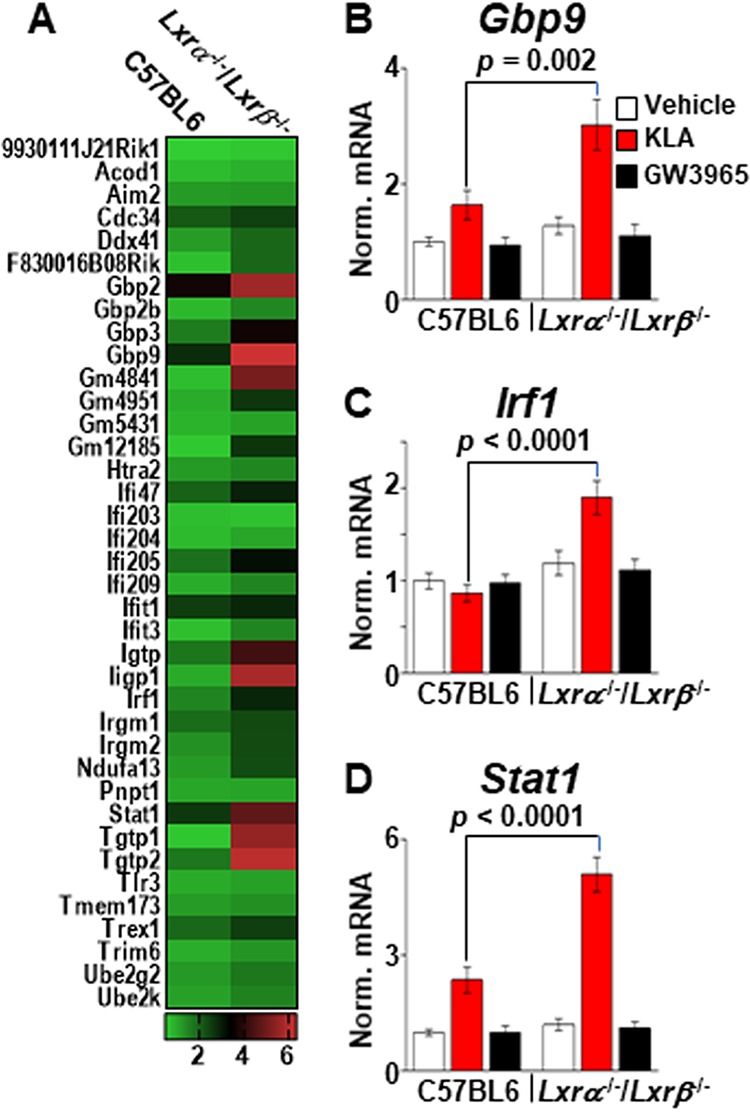
Increased ISG expression in LXR knockout macrophages. (A) Heat map of RNA-Seq data (n = 3/group) comparing 38 ISGs in KLA-treated C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM. (B to D) C57BL/6 and Lxrα−/−/Lxrβ−/− BMDM were treated with the vehicle, 100 ng/ml KLA, or 1.0 μM GW3965 for 16 h. Following treatment, mRNA levels of Gbp9 (B), Irf1 (C), and Stat1 (D) were quantified by real-time PCR and normalized to the value for cyclophilin. Statistically significant differences between C57BL/6 and Lxrα−/−/Lxrβ−/− mice determined by 2-way ANOVA (P ≤ 0.05; n = 10) are noted. Data are means ± standard errors.
Long-chain unsaturated fatty acids have been shown to exhibit anti-inflammatory properties and thus have been suggested to contribute to the resolution of inflammatory signaling (3, 7, 46). To determine if the potentiation of IFN-β signaling observed in KLA-treated Lxrα−/−/Lxrβ−/− BMDM results from a failure to induce fatty acid synthesis, IFN-β target gene expression was examined in BMDM treated with 5-(tetradecyloxy)-2-furoic acid (TOFA), an inhibitor of acetyl-CoA carboxylases alpha and beta (47), the rate-limiting enzymes in fatty acid synthesis. TOFA treatment of BMDM inhibits the incorporation of radiolabeled acetate into fatty acid (Fig. 10A). Nevertheless, the addition of TOFA to cells beginning 8 h after KLA treatment does not lead to increased expression of ISGs (Fig. 10B to D). Thus, the regulation of IFN-β signaling by LXRs appears to be independent of the control of fatty acid synthesis. Examination of published LXR ChIP-Seq data (7) suggests that LXR binds within 20 kb of the transcription start sites of 22 of the 40 genes associated with the cellular response to the interferon beta GO term. In 15 of the genes, LXR binding overlaps the binding sites for STAT1 detected in IFN-β-treated BMDM (7, 34). Consistent with the possibility that cross talk between STAT1 and LXR contributes to the regulation of type I interferon signaling, increased binding of LXRα to regulatory regions near the Gbp9 and Stat1 genes is detected 16 h after KLA treatment (Fig. 11).
FIG 10.
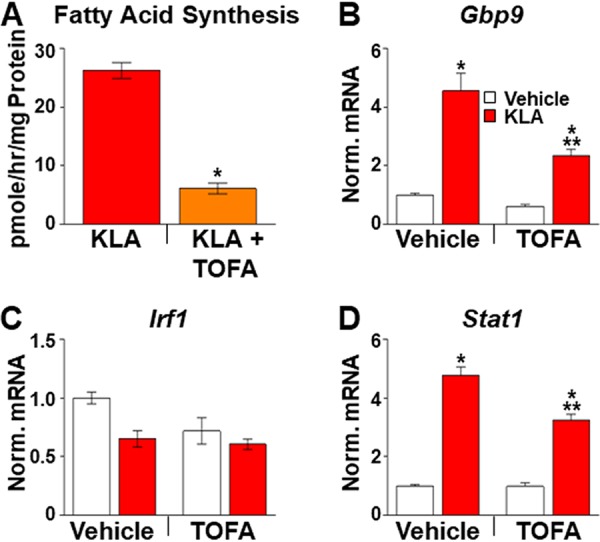
Inhibition of fatty acid synthesis does not increase ISG expression. (A) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for a total of 18 h. At 12.5 h, half of the wells were treated with 10 μM TOFA to inhibit fatty acid synthesis. Finally, cells were labeled with [14C]acetate between 13 and 18 h after KLA addition in the continued presence of KLA and/or TOFA. Upon completion of the experiment, fatty acids were extracted and quantified as described in Materials and Methods. *, statistically significant difference from the vehicle control determined by Student’s t test (P ≤ 0.05; n = 8). (B to D) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 8 h and then treated for an additional 8 h with 10 μM TOFA in the continued presence of KLA. Following treatment, mRNA levels of Gbp9 (B), Irf1 (C), and Stat1 (D) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference between vehicle- and KLA-treated cells with and without TOFA; **, statistically significant difference between KLA-treated and KLA- and TOFA-treated cells determined by 2-way ANOVA (P ≤ 0.05; n = 8).
FIG 11.
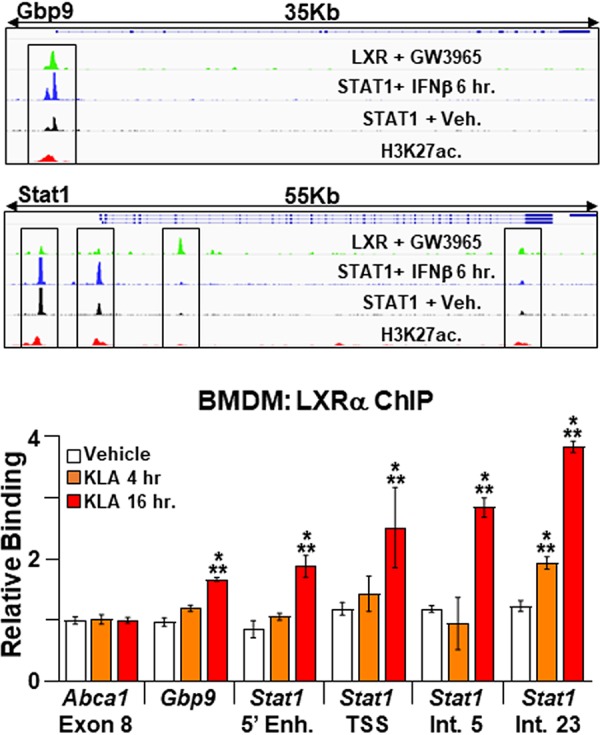
Binding of LXRα to ISG regulatory elements. (Top) IGV screenshots demonstrating binding of STAT1, LXRs, and histone H3 acetylated at lysine K27 (H3K27ac) to regulatory elements at the Gbp9 and Stat1 loci in BMDM. Boxed regions were examined by ChIP-PCR. STAT1 ChIP-Seq data are derived from data reported under GEO accession number GSE33913 (34), LXR ChIP-Seq data are derived from data reported under GEO accession number GSE79424 (7), and H3K27ac ChIP-Seq data are derived from the Encyclopedia of DNA Elements (ENCODE). (Bottom) C57BL/6 BMDM were treated with the vehicle or 100 ng/ml KLA for 4 or 16 h. ChIP assays were performed with an LXRα antibody, and real-time PCR was used to measure binding at the Gbp9 and Stat1 regulatory elements boxed in the top panel. A nonspecific site in exon 8 of the Abca1 gene was used as a control. Data are expressed relative to the value for the nonspecific site. *, statistically significant difference from the vehicle control within each group; **, statistically significant difference between specific and nonspecific binding within each treatment group determined by 1-way ANOVA (P ≤ 0.05; n = 6).
TLR4 activation leads to an IFN-β-dependent increase in 25-hydroxycholesterol, a cholesterol derivative with anti-inflammatory activity (1, 4, 5). The ability of 25-hydroxycholesterol to directly bind LXRs and function as an agonist (48, 49) raised the possibility that this ligand-receptor pair works together to induce lipogenic gene expression and/or to mediate feedback repression of IFN-β signaling. Deletion of the gene encoding cholesterol-25-hydroxylase (Ch25h), the enzyme that generates 25-hydroxycholesterol from cholesterol, however, has no effect on the KLA-dependent induction of lipogenic gene expression or on ISG regulation (data not shown). To explore a role for other endogenous cholesterol-derived LXR ligands in the TLR4 response, we took advantage of a point mutation described in the human LXRα ligand binding domain, tryptophan 443 to phenylalanine (W443F), that disrupts binding of cholesterol-derived ligands while still allowing binding of the potent synthetic agonist T0901317 (GW3965 was not examined) (50). Transient-transfection experiments in 293T cells using a luciferase reporter indicate that the transcriptional activity of W443F is reduced in vehicle-treated cells compared to wild-type LXRα (Fig. 12A, white bars), consistent with a reduced activity of the W443F mutant in response to endogenous ligands. The W443F mutant is expressed at slightly higher levels than wild-type LXRα (Fig. 12B), indicating that the reduced transcriptional activity measured in vehicle-treated cells does not result from lower protein levels. On the other hand, W443F and wild-type LXRα respond equivalently to T0901317 (Fig. 12A, black bars). To examine the activity of W443F in macrophages, Lxrα−/−/Lxrβ−/− BMDM were infected with lentiviruses expressing LXRα, LXRα W443F, or an empty vector expressing only green fluorescent protein (GFP) using viral titers that produce approximately equal expression levels of wild-type and mutant receptors (Fig. 12C). Expression of wild-type LXRα leads to a significant induction of Scd2 mRNA in response to KLA, indicating that ectopic expression can at least partially rescue LXR activity in BMDM. Expression of W443F, however, does not rescue the KLA-dependent induction of Scd2 (Fig. 12D, red bars), suggesting that a cholesterol-derived LXR ligand is required for the positive induction of lipogenesis in response to TLR activation. Consistent with data from the reporter gene assay, both receptors increase Scd2 expression in response to T0901317 (Fig. 12D, black bars). In contrast to the KLA-dependent induction of Scd2, W443F represses Gbp9 and Stat1 as well as wild-type LXRα (Fig. 12E and F), indicating that LXR-dependent repression of ISG expression occurs via a mechanism independent of cholesterol-derived ligand binding.
FIG 12.
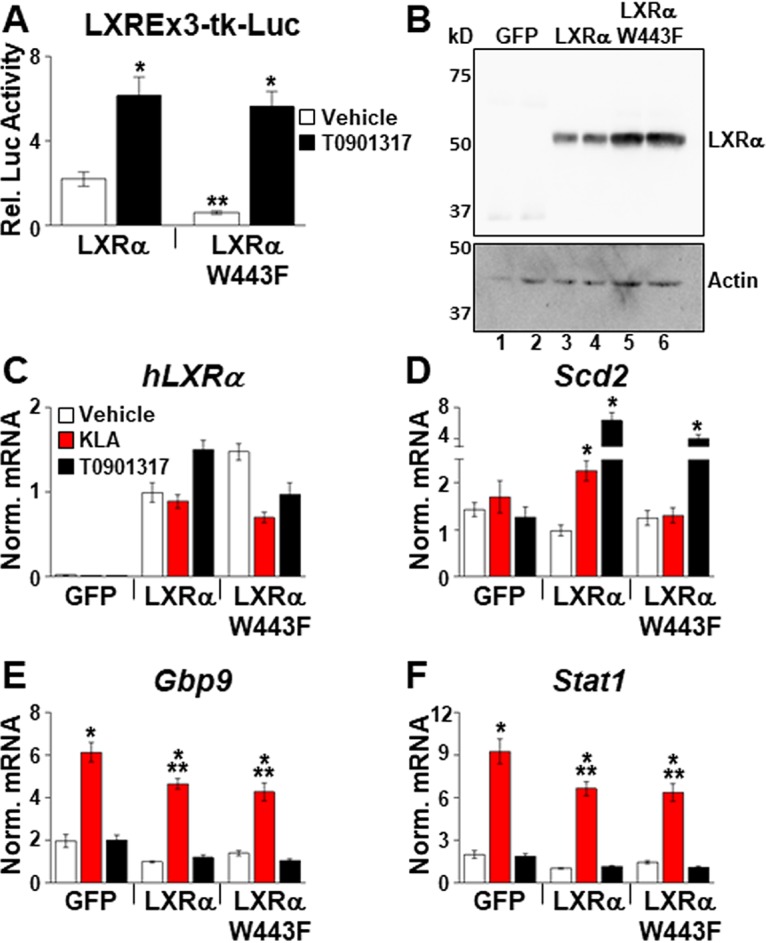
Endogenous LXR ligands are required for LXR-dependent regulation of Scd2 in response to TLR activation. (A) HEK293 cells were transfected with plasmids expressing hLXRα or hLXRα W443F or an empty expression vector, a luciferase reporter with LXR binding sites, and a β-galactosidase expression plasmid as described in Materials and Methods. After transfection, cells were treated with the vehicle or 1.0 μM T0901317 for 36 h, and luciferase activity was measured and normalized to β-galactosidase activity. *, statistically significant difference between the vehicle and T0901317; **, statistically significant difference between hLXRα and hLXRα W443F determined by 1-way ANOVA (P ≤ 0.05; n = 8). (B) HEK293 cells were transfected with plasmids expressing hLXRα, hLXRα W443F, or GFP alone, and nuclear extracts were prepared as described in Materials and Methods. LXR levels were examined by Western blotting using an antibody that detects the Xpress epitope tag at the amino terminus. (C to F) Lxrα−/−/Lxrβ−/− BMDM were infected with lentivirus expressing GFP alone, hLXRα, or hLXRα W443F as described in Materials and Methods. Following infection, cells were treated with the vehicle, 200 ng/ml KLA, or 1.0 μM T091317 for 17 h, and the mRNA levels of hLXRα (C), Scd2 (D), Gbp9 (E), and Stat1 (F) were quantified by real-time PCR and normalized to the value for cyclophilin. *, statistically significant difference from the vehicle control within the same virus infection; **, statistically significant difference among KLA-treated cells determined by 2-way ANOVA (P ≤ 0.05; n = 8). Data are means ± standard errors.
DISCUSSION
Recent studies examining the relationship between lipid metabolism and immune cell function have provided new insights into mechanisms regulating inflammation (1–10). Our studies indicate that TLR activation induces LXRα expression in a type I interferon-dependent manner and that LXR activity is necessary for the subsequent reinitiation of lipid synthesis at late stages of an inflammatory response. Long-chain unsaturated fatty acids have been associated with anti-inflammatory activity (46, 51), and Oishi et al. suggested that the reprogramming of fatty acid synthesis that occurs during the latter part of the TLR4 response contributes to the resolution of inflammation (7). Our results are consistent with the work of Oishi et al., demonstrating that SREBP1c expression and fatty acid synthesis are induced relatively late after TLR4 activation. Oishi et al., however, suggest that the induction of SREBP1 is LXR independent. In our BMDM experiments, genetic deletion of LXRs strongly derepresses fatty acid synthetic gene expression such that other signaling pathways impinging on Srebf1 regulation may not be detectable. LXR-dependent repression can be gene and tissue specific. For instance, we have previously shown that LXR knockout derepresses Srebf1c expression in the intestine but not in the liver or in thioglycolate-elicited peritoneal macrophages (45). Importantly, many of the experiments described by Oishi et al. use thioglycolate-elicited peritoneal macrophages. Srebf1 expression can also be influenced by sterol levels and by insulin signaling (15), so it is possible that variations in cell culture conditions or within the respective mouse colonies contribute to the observed differences in the requirement for LXR activity. Analysis of data from the Immunological Genome Project (52) indicates that inflammatory stimuli induce LXRα expression in alveolar and liver macrophages. Furthermore, a recent study of peripheral blood mononuclear cells from human patients demonstrates that respiratory infection increases LXR signaling (53). Together, these studies support the relevance of the TLR-IFN-LXR pathway uncovered by our experiments and suggest an important role of interferon-stimulated LXR expression in the immune response to pathogens.
Our observation that sterol synthesis increases at relatively late times after TLR4 stimulation is consistent with the work of Kusnadi et al., who detected an increase in sterol synthesis with similar kinetics after treatment with TNF (10). In contrast, York et al. and Blanc et al. propose that proinflammatory signals decrease macrophage sterol synthesis (6, 9). Variations in the identity, strength, and duration of the inflammatory stimuli; sources of macrophages; and cell culture conditions likely contribute to the disparate results. Our time course experiments may help resolve the inconsistencies among studies by further detailing the kinetics of these processes. In TLR4-activated macrophages, cholesterol biosynthetic gene expression is initially repressed at 8 h and then gradually reactivated at later stages of an inflammatory response. Therefore, measurements of sterol synthesis in cells continuously labeled with a precursor for 24 h (6, 8) may not have the sensitivity to detect more acute changes in synthetic rates. We suggest that the reactivation of both sterol and fatty acid synthesis may contribute to a switch from proinflammatory to proresolving phenotypes. Analysis of the LXR-dependent TLR response also highlights the remarkable signal-dependent plasticity in LXR-dependent regulation of lipid metabolism. Synthetic LXR agonists such as T0901317 and GW3965 induce gene networks promoting fatty acid synthesis and cholesterol efflux/excretion (12). Endogenous LXR ligands such as desmosterol have been reported to selectively induce cholesterol efflux genes and not lipogenesis, at least in part due to the ability of these molecules to interfere with SREBP processing (54, 55). Finally, TLR activation leads to LXR-dependent increases in fatty acid and sterol synthesis in a manner that requires mTOR and SREBP activity without inducing genes involved in cholesterol efflux.
Genetic deletion of LXR activity in vehicle-treated cells mimics the TLR4-dependent increase in cholesterol biosynthesis measured in LXR-positive cells. Thus, LXRs appear to function in a repressive pathway that keeps cholesterol biosynthesis low in the absence of inflammatory signals. In support of the direct regulation of cholesterol biosynthesis by LXRs, Wang et al. identified LXR binding sites in the promoters of human genes encoding several cholesterol biosynthetic enzymes and suggested that these sites function as negative regulatory elements (56). Published ChIP-Seq studies, however, have not demonstrated LXR binding in the vicinity of cholesterol biosynthetic genes in mouse BMDM (7, 57). In BMDM, our data and previous studies indicate that cholesterol efflux is elevated in LXR knockout cells due to a loss of direct repression of ABC transporter expression mediated by LXR-corepressor complexes (45). Increased cholesterol biosynthesis in LXR knockout cells may therefore represent an indirect response required to maintain normal intracellular cholesterol levels in the face of elevated cholesterol efflux. Future studies using CRISPR to delete potential LXREs will help to distinguish between direct and indirect mechanisms of LXR activity.
TLR activation promotes the expression of IFN-β, which can function in an autocrine or paracrine manner to activate ISGs. ISGs constitute a second wave of inflammatory gene expression that can act to either enhance or decrease inflammation depending on the context (33, 58). Our work defines LXRα as an ISG that is induced by IFN-β in a STAT1-dependent manner. Furthermore, in LXR knockout BMDM, a subset of ISGs is hyperinduced by KLA treatment, suggesting that LXRs contribute to a negative-feedback loop that shuts down IFN-β signaling during the later stages of inflammation. Elevated expression of ISGs at late time points is not observed in BMDM treated with inhibitors of fatty acid synthesis, indicating that the LXR-dependent regulation of ISG expression proceeds through a pathway that is independent of the control of fat synthesis. ISG expression is not influenced by synthetic LXR agonists or antagonists and thus differs from agonist-dependent inhibition of proinflammatory genes such as Tnf described in other studies (22, 59–61), which requires treatment with LXR ligands prior to inflammatory challenges. The ability to partially rescue ISG repression with the W443F mutant also suggests that ligand binding may not be necessary for this activity. Interestingly, Lee et al. have shown that LXRs can directly interact with STAT1 and inhibit STAT1 DNA binding (62). Analysis of published ChIP-Seq data (7, 34) further indicates that both proteins can bind to overlapping regulatory regions near several ISGs. Finally, at late stages of the inflammatory response, increased binding of LXRα to regulatory regions near the Gbp9 and Stat1 genes can be detected. Based on these studies, we favor a model where increasing LXR levels contribute to decreasing STAT1 activity via competition for overlapping binding sites and/or by direct interactions (Fig. 13). Nevertheless, we cannot rule out the possibility that the genetic deletion of LXR modulates ISG expression indirectly.
FIG 13.
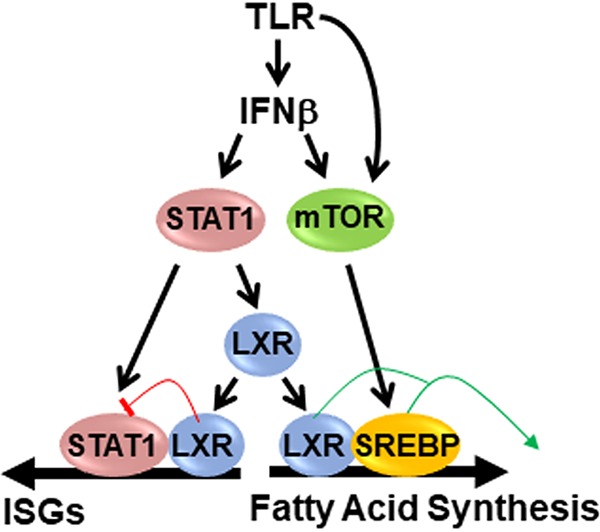
Model of TLR-dependent LXR activity. Type I interferons activate STAT1, which increases LXR expression. TLR signaling also increases mTOR activity via interferon-dependent and -independent mechanisms. LXRs cooperate with mTOR-activated SREBP1 to increase fatty acid synthesis. LXRs also inhibit STAT1 activity, leading to decreased ISG expression.
LXRs are ligand-dependent transcription factors; however, the ligand and/or signal mediating LXR activity during the TLR response remains to be determined. TLR4 activation increases the levels of 25-hydroxycholesterol, a putative LXR agonist with known anti-inflammatory properties (1, 4, 5, 48, 49), raising the possibility that this ligand-receptor pair functions to put a brake on inflammation. Genetic deletion of cholesterol-25-hydroxylase, the enzyme that synthesizes 25-hydroxycholesterol, however, has no effect on lipogenic gene expression or on LXR-regulated ISG expression in BMDM. Nevertheless, rescue experiments exploiting the W443F mutant suggest that ligand binding is necessary for the LXR-dependent lipogenic response to TLR activation. We propose that the combination of elevated LXR levels detected late in the inflammatory response, relatively weak endogenous LXR ligands that preexist in cells, and mTOR-dependent SREBP activity generates a unique cellular environment that facilitates the selective regulation of lipogenesis (Fig. 13). Future studies that define the nature of the inflammation-dependent signals that regulate LXR activity should provide new insight into the regulation and resolution of inflammation.
MATERIALS AND METHODS
Reagents.
GW3965, T0901317, 5-(tetradecyloxy)-2-furoic acid (TOFA), torin1, and PF-429242 were purchased from Cayman Chemicals. Compactin and carrier-free mouse IFN-β were purchased from R&D Systems. Kdo2-lipid A (KLA) was purchased from Avanti Polar Lipids, and stock solutions were prepared as described by the Lipid Maps Consortium (74). AcLDL and disuccinimidyl glutarate (DSG) were purchased from Alfa Aesar. Apolipoprotein A1 (APOA1) was purchased from MilliporeSigma. [2-14C]acetate and [1,2-3H]cholesterol were purchased from Perkin Elmer. GSK2033 was provided by GlaxoSmithKline.
Animals.
All animal experiments were approved by the Institutional Animal Care and Research Advisory Committee of the University of Virginia. Lxrα−/−/Lxrβ−/− mice were described previously (63). STAT1 knockout mice (129S6/SvEv-Stat1tm1Rds; model number 2045) and 129S6 (model number 129SVE) controls were purchased from Taconic Biosciences. C57BL/6J mice (stock number 000664) and cholesterol-25-hydroxylase knockout mice (64) (B6.129S6 Ch25htm1Rus/J; stock number 016263) were purchased from Jackson Laboratories. SREBP1a- and SREBP1c-deficient mice were previously described (65, 66). To examine the effect of IFN-β on LXR expression in mice, 10,000 U of carrier-free IFN-β in 100 μl of phosphate-buffered saline (PBS) was injected into the peritoneal space. After 8 h, mice were euthanized, and the peritoneal space was flushed with 10 ml of PBS. The recovered cells were pelleted, and RNA was isolated as described below.
Bone marrow-derived macrophage.
BMDM were isolated as previously described (45, 63) and plated at a concentration of 2 × 106 cells/ml in 12-well plates (1 ml/well) for RNA analysis or in 24-well plates (0.5 ml/well) for lipid synthesis assays. After 4 days at 37°C with 5% CO2, the medium was removed, cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and fresh BMDM differentiation medium was added. After an additional 24 h at 37°C with 5% CO2, cells were treated as described in the figure legends. For experiments using BMDM, bone marrow was isolated from 6 to 8 mice, and cells from each mouse were cultured individually. Gene expression analyses and measurements of fatty acid synthesis were then carried out using 4 replicas for each mouse.
Lentivirus construction and infection of BMDM.
Human LXRα cDNA was cloned into pcDNA3.1HisA, and mutagenesis was carried out using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). Sequence-verified mutant cDNAs, including an amino-terminal 6-histidine tag and an Xpress epitope tag, were subcloned into the pCDH-EF1-MCS-IRES-copGFP lentivirus vector (System Biosciences), and high-titer virus stocks were prepared by the Duke University Viral Vector Core. To rescue LXR activity, bone marrow cells from Lxrα−/−/Lxrβ−/− mice were isolated as described above, resuspended in BMDM differentiation medium at a concentration of 1.5 × 106 cells/ml, and plated in 24-well plates (0.5 ml/well). After 4 days at 37°C with 5% CO2, the medium was removed, cells were washed twice with DPBS, and fresh BMDM differentiation medium containing 8.0 μg/ml Polybrene and approximately 5 × 106 viral genomes/ml of the appropriate virus was added. Infection was allowed to proceed for 48 h at 37°C with 5% CO2, the medium was removed, cells were washed twice with DPBS, and fresh BMDM differentiation medium was added. Cells were cultured for an additional 24 h before ligands were added.
Transient transfection.
HEK293 cells cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin, and 1 mM sodium pyruvate at 37°C with 5% CO2 were transferred to 96-well plates (1 × 104 cells/well; 100 μl/well) and transfected using FuGENE6 (Promega). Each reaction mixture contained 50 ng of pCMX-β-galactosidase, 30 ng of the reporter plasmid LXREx3-TK-Luc (67), 30 ng of the appropriate pcDNA3.1HisA-hLXRα expression vector, and 0.28 μl of FuGENE6 in a volume of 10 μl. After transfection, cells were cultured for an additional 36 h in the absence or presence of T0901317. After ligand treatment, 100 μl/well lysis buffer (25 mM Tricine [pH 7.8], 10% glycerol, 1.0% Triton X-100, 5 mM dithiothreitol) was added, and luciferase activity was normalized to β-galactosidase activity.
THP-1 cell culture and IFN-β neutralization.
THP-1 human monocyte cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin, 1 mM sodium pyruvate, and 0.05 mM beta-mercaptoethanol at 37°C with 5% CO2. Differentiation of THP-1 cells into macrophages was performed by treatment with phorbol 12-myristate 13-acetate (PMA) at a final concentration of 60 ng/ml overnight. Following PMA treatment, cells were washed twice with DPBS, and fresh medium without PMA was added. After an additional 24 h, cells were treated with KLA, LXR ligands, or ruxolitinib as described in the figure legends. To block IFN-β signaling, cells were treated with 2 μg/ml anti-human IFN-β (catalog number 500-P32B; PeproTech).
RNA extraction, reverse transcription, and real-time quantitative RT-PCR.
Following treatment with LXR ligands, KLA, or enzyme inhibitors, total RNA was isolated using RNeasy minikits (Qiagen), treated with 2 U of DNase, and reverse transcribed using the Applied Biosystems high-capacity cDNA reverse transcription (RT) kit (Thermo Fisher). Real-time quantitative PCR assay mixtures contained 20 ng of cDNA, 360 nM gene-specific forward and reverse primers, and 6.25 μl of iQ SYBR green supermix (Bio-Rad). Amplification was carried out using a MyIQ single-color real-time PCR detection system (Bio-Rad). For each mRNA, individual cDNA samples were assayed in duplicate and normalized to cyclophilin mRNA levels. Primers are listed in Table 1.
TABLE 1.
Oligonucleotides for qPCR
| Species | Gene or genetic elementa | Sequences (forward, reverse) |
|---|---|---|
| Mouse | Abca1 | 5′-GCTCTCAGGTGGGATGCAG-3′, 5′-GGCTCGTCCAGAATGACAAC-3′ |
| Mouse | Cyp51a1 | 5′-AACAACACAGCAGAGGCTGA-3′, 5′-ACAGTTCAGTCGGGAAAAGG-3′ |
| Mouse | Ch25h | 5′-GGACACCATAAGGACAAGGGA-3′, 5′-CACGAACACCAGGTGCTG-3′ |
| Mouse | Dhcr24 | 5′-ACCACTTCGTGGAAGGGTTG-3′, 5′-ACTGCCAATGCTATTCAGCTT-3′ |
| Mouse | Fads2 | 5′-AAGGGAGGTAACCAGGGAGAG-3′, 5′-CCGGTGGGACCATTTGGTAA-3′ |
| Mouse | Fdft1 | 5′-ATGGAGTTCGTCAAGTGTCTAGG-3′, 5′-CGTGCCGTATGTCCCCATC-3′ |
| Mouse | Gbp9 | 5′-GGTCACCGGGAATAGACTGG-3′, 5′-GGGCCACACTTGTCATAGCA-3′ |
| Mouse | Hmgcr | 5′-CTTGTGGAATGCCTTGTGATTG-3′, 5′-AGCCGAAGCAGCACATGAT-3′ |
| Mouse | Ifnb | 5′-CAGCTCCAAGAAAGGACGAAC-3′, 5′-GGCAGTGTAACTCTTCTGCAT-3′ |
| Mouse | Irf1 | 5′-GGCCGATACAAAGCAGGAGAA-3′, 5′-GGAGTTCATGGCACAACGGA-3′ |
| Mouse | Nr1h2 (Lxrβ) | 5′-CTCCCACCCACGCTTACAC-3′, 5′-GCCCTAACCTCTCTCCACTCA-3′ |
| Mouse | Nr1h3 (Lxrα) | 5′-AGGAGTGTCGACTTCGCAAA-3′, 5′-CTCTTCTTGCCGCTTCAGTTT-3′ |
| Mouse | Ppia (cyclophilin gene) | 5′-CGATGACGAGCCCTTGG-3′, 5′-TCTGCTGTCTTTGGAACTTTGTC-3′ |
| Mouse | Scd1 | 5′-CCGGAGACCCCTTAGATCGA-3′, 5′-TAGCCTGTAAAAGATTTCTGCAAACC-3′ |
| Mouse | Scd2 | 5′-GCATTTGGGAGCCTTGTACG-3′, 5′-AGCCGTGCCTTGTATGTTCTG-3′ |
| Mouse | Sqle | 5′-GCTGGGCCTTGGAGATACAG-3′, 5′-CAGTGGGTACGGAATTTGAAC-3′ |
| Mouse | Srebf1a | 5′-GATGTGCGAACTGGACACAGC-3′, 5′-GAGAAGCTCTCAGGAGAGTTGG-3′ |
| Mouse | Srebf1c | 5′-CGTCTGCACGCCCTAGG-3′, 5′-CTGGAGCATGTCTTCAAATGTG-3′ |
| Mouse | Stat1 | 5′-TCACAGTGGTTCGAGCTTCAG-3′, 5′-CGAGACATCATAGGCAGCGTG-3′ |
| Mouse | Tnf | 5′-CTGAGGTCAATCTGCCCAAGTAC-3′, 5′-CTTCACAGAGCAATGACTCCAAAG-3′ |
| Human | NR1H2 (LXRβ) | 5′-CAGATGGACGCTTTCATGCG-3′, 5′-CTGCTGTTTCCGAATCTTCTTCT-3′ |
| Human | NR1H3 (LXRα) | 5′-CTTGCTCATTGCTATCAGCATCTT-3′, 5′-ACATATGTGTGCTGCAGCCTCT-3′ |
| Human | PPIA (cyclophilin gene) | 5′-ACGGCGAGCCCTTGG-3′, 5′-TTTCTGCTGTCTTTGGGACCT-3′ |
| Mouse | Abca1 LXRE (ChIP) | 5′-GCTTTCTGCTGAGTGACTGAACTAC-3′, 5′-GAATTACTGCTTTTTGCCGCG-3′ |
| Mouse | Abca1 exon 8 (ChIP) | 5′-CTGCATTTGAGCTCATACACTAAG-3′, 5′-AACACTGTGGTGGCTTCA-3′ |
| Mouse | Gbp9 TSS (ChIP) | 5′-CTGGAGATGGGTTCCAAAGAA-3′, 5′-GTCAGAGGATGCTTAGTGTCAG-3′ |
| Mouse | Srebf1c LXRE (ChIP) | 5′-GAACCAGCGGTGGGAACACAGAGC-3′, 5′-GACGGCGGCAGCTCGGGTTTCTC-3′ |
| Mouse | Stat1 enhancer (ChIP) | 5′-CGGCATTGCTTTCGCTTT-3′, 5′-ACGTGCTTTCTGGGAACTC-3′ |
| Mouse | Stat1 intron 5 (ChIP) | 5′-CCTCGTTCTTTAGGTCAAGGTTAG-3′, 5′-TGCCATGAACTCTGAGCATAC-3′ |
| Mouse | Stat1 intron 23 (ChIP) | 5′-AGGAACAGAGCCACCTCTAA-3′, 5′-CTGGATACAGGAGTCAGCAAAG-3′ |
| Mouse | Stat1 TSS (ChIP) | 5′-TGTCATCCCGCAGAGAGAA-3′, 5′-GAAGTGAGAACGGCAGGATAAG-3′ |
An alternate gene name or the oligonucleotide use is in parentheses. TSS, transcription start site.
Quantification of cholesterol and fatty acid synthesis.
BMDM cultured in 24-well plates were treated with or without 100 ng/ml KLA. At 12.5 h after KLA addition, compactin (50 μM) or TOFA (10 μM) was added to replica wells with and without KLA to inhibit cholesterol synthesis or fatty acid synthesis, respectively. After 30 min, cells were labeled for 5 h (13 to 18 h after the initial KLA treatment) with 8 μCi/ml [2-14C]acetate, and lipids were extracted as described previously by Hawkins et al. (42). Briefly, cells were lysed in 0.5 ml of 0.2 N KOH, and a 50-μl aliquot was removed for total protein determination by a Bio-Rad assay. The remaining cell suspension was combined with the medium in 15-ml polypropylene tubes, 0.3 ml of 50% KOH and 2.0 ml of 100% ethanol were added, and the samples were saponified at 75°C for 2 h and then incubated overnight at room temperature. The saponified samples were extracted 3 times with 4.5 ml hexane, and the organic fractions containing cholesterol and other neutral lipids were pooled in 50-ml polypropylene tubes, dried under nitrogen, and resuspended in 200 μl of toluene. The remaining aqueous layers containing fatty acid sodium salts were acidified by the addition of 0.5 ml 12 M HCl and extracted twice with 4.5 ml hexane. The organic fractions containing fatty acids were pooled in 15-ml glass tubes, dried under nitrogen, and resuspended in 200 μl of toluene. Radioactivity was measured by scintillation counting. Counts per minute were converted to picomoles of cholesterol or fatty acids by scintillation counting of a known quantity of [2-14C]acetate. For calculations, fatty acids were assumed to be C14. To determine the synthesis of neutral lipids dependent on HMG-CoA reductase activity, the average counts per minute in neutral lipid extracts from the wells treated with compactin were subtracted from those of the wells without compactin. Similarly, the average counts per minute in acidified lipid extracts from the wells treated with TOFA were subtracted from those of the wells without TOFA to determine incorporation dependent on fatty acid synthesis.
Extract preparation and Western blot analysis.
Nuclear and cytoplasmic extracts were prepared as described previously by Osburn et al. (68). Extracts were resolved on 10% or 12% SDS-PAGE gels, transferred onto polyvinylidene difluoride (PVDF) membranes, blocked in Tris-buffered saline–0.05% Tween 20 (TBST) containing 5% nonfat milk, and incubated overnight at room temperature in in TBST plus 5% nonfat milk with antibodies against LXRα (catalog number PP-PPZ0412-00; R&D Systems) (1:3,000 dilution), the Xpress epitope (catalog number 46-0528; Thermo Fisher) (1:3,000 dilution), SREBP1 (catalog number 557036; BD Pharmingen) (1:500 dilution), SCD2 (catalog number sc-518034; Santa Cruz Biotechnology) (1:500), or actin (catalog number 4907S; Cell Signaling) (1:1,000). Following primary antibody incubation, blots were washed and incubated for 2 h at room temperature with alkaline phosphatase-conjugated secondary antibodies (catalog number T2191 or T2192; Thermo Fisher) diluted 1:5,000 in TBST plus 5% nonfat milk. To visualize the signal, blots were washed with TBST, treated with CDP-Star reagent (catalog number T2305; Thermo Fisher), and imaged using a Fujifilm LAS-3000 imager.
RNA sequencing.
Bone marrow cells were isolated from 10- to 12-week-old male C57BL/6J or Lxrα−/−/Lxrβ−/− mice (n = 3/genotype) as described above. Cells were pelleted; resuspended in Dulbecco’s modified Eagle medium containing 20% heat-inactivated fetal bovine serum, 20 ng/ml macrophage colony-stimulating factor, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin, and 1 mM sodium pyruvate at a concentration of 2 × 106 cells/ml; and plated in 6-well plates (2 ml/well). Cells from individual mice were plated separately. After 4 days at 37°C with 5% CO2, the medium was removed, cells were washed twice with DPBS, and fresh medium was added. After an additional 24 h at 37°C with 5% CO2, cells were treated with the vehicle, 100 ng/ml KLA, 1.0 μM GW3965, or 50 μg/ml AcLDL (catalog number J65029; Alfa Aesar) for 17 h. After drug treatment, RNA was isolated, and RNA-Seq libraries from individual mice were produced using 500 ng of RNA and the SMARTer stranded total RNA sample prep kit-hi mammalian (Clontech). Libraries were sequenced at the HudsonAlpha Genomics Services Laboratory. Transcript abundances were summed to the gene level (Ensembl) and length scaled to produce counts appropriate for statistical analysis using the tximport package (69) in the R statistical computing environment. The DESeq2 Bioconductor package (70, 71) was then used to normalize count data, estimate dispersion, and fit a negative binomial model for each gene. The Benjamini-Hochberg false discovery rate procedure was used to reestimate the adjusted P values for Ensembl gene identifications (72). Finally, a negative binomial linear modeling framework implemented in DESeq2 was used to test the effect of LXR knockout, KLA, GW3965, and AcLDL individually and to determine the nonlinear interaction effect of KLA, GW3965, or AcLDL in combination with LXR knockout.
Cholesterol efflux.
BMDM isolated from C57BL/6J or Lxrα−/−/Lxrβ−/− mice, as described above, were plated into 96-well plates (1 × 105 cells/well) and loaded with 0.03 μCi/ml of [1,2-3H]cholesterol with 1.0% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin, and 1 mM sodium pyruvate for 24 h at 37°C with 5% CO2. The labeling medium was removed, cells were washed twice with DBPS, and serum-free DMEM containing 20 μg/ml bovine serum albumin (BSA), 10 μg/ml APOA1, or 20 μg/ml high-density lipoprotein particles was added. After 20 h at 37°C with 5% CO2, the medium was removed and pelleted at 1,000 × g for 5 min at room temperature, and the amount of radioactivity in the supernatant was measured by scintillation counting. Cells in the plate were washed once with DPBS and extracted with 150 μl of isopropanol/well overnight at room temperature. Radioactivity in the cell extracts was determined by scintillation counting. Cholesterol efflux was expressed as the percentage of counts per minute in the medium divided by the total counts per minute (medium plus cells). Acceptor-dependent efflux was determined by subtracting the efflux measured in the presence of BSA.
ChIP assays.
Approximately 3 × 107 bone marrow cells isolated from C57BL/6J mice were plated in 15-cm plates and differentiated into macrophages as described above. Macrophages were treated with the vehicle or 100 ng/ml KLA for 17 h and cross-linked at room temperature for 30 min with 2.0 mM DSG in PBS as described previously by Daniel et al. (73). Following DSG treatment, cells were additionally cross-linked with 1.0% formaldehyde at room temperature for 10 min. Cross-linking was terminated by the addition of 2.0 M glycine to a final concentration of 0.15 M, and cells were incubated for an additional 5 min at room temperature. After termination of the cross-linking reaction, medium was removed, cells were washed twice with DPBS, and 3 ml of cell lysis/wash buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 1.0% SDS) was added. The cross-linked cells were removed by scraping and transferred to 15-ml polypropylene tubes. The scraping was repeated, and the pooled cells from each plate were pelleted at 3,000 rpm for 10 min at 4°C. The cell pellet was resuspended in 1.0 ml of cell lysis/wash buffer, transferred to 1.0-ml polystyrene tubes, passed through a 1-ml insulin syringe, and pelleted at 13,000 rpm for 1 min at 4°C. The cell pellet was resuspended in 1.0 ml of cell lysis/wash buffer, passed through a 1-ml insulin syringe a second time, and pelleted at 13,000 rpm for 1 min at 4°C. The final pellet was resuspended in 300 μl of lysis buffer (50 mM Tris [pH 8.0], 1% SDS, 10 mM EDTA). Chromatin was sheared by sonication using a Misonix S-4000 ultrasonic processor with a cup horn sonicator for 5 min (10 s on/30 s off) at an amplitude of 75. Following sonication, chromatin was pelleted at 13,000 rpm for 10 min at 4°C, and supernatants were transferred to new tubes. The amount of chromatin in the soluble supernatant was estimated by measuring the optical density at 260 nm, and 5 μg of the sample was removed to serve as the input. For LXRα ChIP, 150 μg of sheared chromatin was diluted to 3 ml with ChIP dilution buffer (16.7 mM Tris [pH 8.0], 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethanesulfonyl fluoride), 7.5 μg of anti-LXRα antibody was added (catalog number PP-PPZ0412-00; R&D Systems), and the samples were rocked overnight at 4°C. Protein A-Dynabeads (30 μl) (catalog number 1002D; Thermo Fisher) were washed 3 times in DPBS plus 0.5% bovine serum albumin and incubated overnight in 0.5 ml DPBS plus 0.5% bovine serum albumin (BSA) at 4°C. The following morning, beads were captured on a magnet, resuspended in 200 μl DPBS plus 0.5% BSA, and added to the chromatin-antibody samples. The antibody was allowed to bind to beads for 4 h at 4°C, the beads were captured on a magnet, unbound supernatants were removed, and the bound chromatin-antibody conjugates were washed 6 times with 3 ml of cell lysis/wash buffer for 3 min with inversion at 4°C. After the final wash, the beads were resuspended in 1.0 ml of Tris-EDTA (TE) (pH 8.0) and transferred to 1.5-ml DNA Lobind tubes (catalog number 022431021; Eppendorf). Beads were captured on a magnet, supernatants were removed, and chromatin was eluted by resuspending the beads in 100 μl of a freshly prepared solution containing 100 mM NaHCO3 and 1.0% SDS by shaking at 1,000 rpm for 15 min at room temperature. Beads were captured on a magnet, supernatants containing the eluted chromatin were transferred to 1.5-ml DNA Lobind tubes, and the elution step was repeated. The eluted fractions were pooled (200-μl total volume), 8 μl of 5.0 M NaCl was added, and the samples were incubated overnight at 65°C to reverse cross-links. After reversing cross-links, 8 μl of 1.0 M Tris, 4 μl of 500 mM EDTA, and 1 μl of 10 mg/ml RNase A were added, and samples were incubated at 37°C for 30 min. After RNase treatment, 1 μl of 20 mg/ml protease K was added, and samples were incubated at 45°C for 2 h. Input samples were diluted to 200 μl with ChIP dilution buffer, 8 μl of 5.0 M NaCl was added, and input samples were then treated identically as described above for ChIP samples. Following proteinase K treatment, samples were extracted once with phenol and once with phenol-chloroform, and glycogen was added to a final concentration of 0.5 mg/ml. DNA was precipitated by the addition of 23 μl of 3.0 M sodium acetate (pH 5.5) and 2 volumes of ethanol. DNA was pelleted by centrifugation at 13,000 rpm at 4°C for 30 min, washed once with 70% ethanol, dried, and resuspended in 80 μl of H2O. Real-time quantitative PCR assay mixtures contained 5 μl of ChIP samples or 20 ng of the input, 360 nM gene-specific forward and reverse primers, and 6.25 μl of Sso Advanced universal SYBR green supermix (Bio-Rad). Amplification was carried out using a MyIQ single-color real-time PCR detection system (Bio-Rad). Samples were assayed in duplicate, and data are expressed relative to a nonspecific control. Primers are listed in Table 1.
Statistical analysis.
All analysis was carried out using GraphPad Prism. Data sets were first tested for a normal distribution using the D’Agostino-Pearson normality test. Experiments with normal distributions were then examined using parametric tests (Student’s t test or 1-way or 2-way analysis of variance [ANOVA]), depending on the experiment, as described in the figure legends. Mann-Whitney tests were used to examine experiments that did not meet a normal distribution because of too few samples.
Data availability.
RNA-Seq data are available at the Gene Expression Omnibus (GEO) under accession number GSE118656.
ACKNOWLEDGMENTS
This work was supported by a grant from the NIH/NHLBI (HL48044) to T.F.O. and by grants from the NIH/NIDDK (1R01DK119182-01A) and the American Heart Association (15GRNT25560038) to I.G.S.
We thank Norbert Leitinger and Thurl Harris for comments on the manuscript.
REFERENCES
- 1.Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, Craigon M, Ruzsics Z, Haas J, Angulo A, Griffiths WJ, Glass CK, Wang Y, Ghazal P. 2013. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38:106–118. doi: 10.1016/j.immuni.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ. 2014. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li P, Spann NJ, Kaikkonen MU, Lu M, Oh DA, Fox JN, Bandyopadhyay G, Talukdar S, Xu J, Lagakos WS, Patsouris D, Armando A, Quehenberger O, Dennis EA, Watkins SM, Auwerx J, Glass CK, Olefsky JM. 2013. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell 155:200–214. doi: 10.1016/j.cell.2013.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. 2013. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38:92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. 2014. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345:679–684. doi: 10.1126/science.1254790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, Cunningham CR, Tarling EJ, Wilks MQ, Casero D, Gray DH, Yu AK, Wang ES, Brooks DG, Sun R, Kitchen SG, Wu TT, Reue K, Stetson DB, Bensinger SJ. 2015. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, Kolar MJ, Matsuzaka T, Hayakawa S, Tao J, Kaikkonen MU, Carlin AF, Lam MT, Manabe I, Shimano H, Saghatelian A, Glass CK. 2017. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab 25:412–427. doi: 10.1016/j.cmet.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araldi E, Fernández-Fuertes M, Canfrán-Duque A, Tang W, Cline GW, Madrigal-Matute J, Pober JS, Lasunción MA, Wu D, Fernández-Hernando C, Suárez Y. 2017. Lanosterol modulates TLR4-mediated innate immune responses in macrophages. Cell Rep 19:2743–2755. doi: 10.1016/j.celrep.2017.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, Khondoker M, Dickinson P, Sing G, Rodriguez-Martin S, Phelan P, Forster T, Strobl B, Muller M, Riemersma R, Osborne T, Wenk MR, Angulo A, Ghazal P. 2011. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol 9:e1000598. doi: 10.1371/journal.pbio.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kusnadi A, Park SH, Yuan R, Pannellini T, Giannopoulou E, Oliver D, Lu T, Park-Min KH, Ivashkiv LB. 2019. The cytokine TNF promotes transcription factor SREBP activity and binding to inflammatory genes to activate macrophages and limit tissue repair. Immunity 51:241–257. doi: 10.1016/j.immuni.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SD, Tontonoz P. 2015. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 242:29–36. doi: 10.1016/j.atherosclerosis.2015.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulman IG. 2017. Liver X receptors link lipid metabolism and inflammation. FEBS Lett 591:2978–2991. doi: 10.1002/1873-3468.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong C, Tontonoz P. 2014. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 14.Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 15.Jeon TI, Osborne TF. 2012. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, Fernando G, Grimm D, Shipkova P, Zhang R, Garcia R, Zhu J, He A, Malone H, Martin R, Behnia K, Wang Z, Barrett YC, Garmise RJ, Yuan L, Zhang J, Gandhi MD, Wastall P, Li T, Du S, Salvador L, Mohan R, Cantor GH, Kick E, Lee J, Frost RJ. 2016. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab 24:223–233. doi: 10.1016/j.cmet.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. 2000. Role of LXRs in control of lipogenesis. Genes Dev 14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishibashi M, Varin A, Filomenko R, Lopez T, Athias A, Gambert P, Blache D, Thomas C, Gautier T, Lagrost L, Masson D. 2013. Liver X receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: a key role for lysophosphatidylcholine acyltransferase 3. Arterioscler Thromb Vasc Biol 33:1171–1179. doi: 10.1161/ATVBAHA.112.300812. [DOI] [PubMed] [Google Scholar]
- 20.Qin Y, Dalen KT, Gustafsson JA, Nebb HI. 2009. Regulation of hepatic fatty acid elongase 5 by LXRalpha-SREBP-1c. Biochim Biophys Acta 1791:140–147. doi: 10.1016/j.bbalip.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Varin A, Thomas C, Ishibashi M, Menegaut L, Gautier T, Trousson A, Bergas V, de Barros JP, Narce M, Lobaccaro JM, Lagrost L, Masson D. 2015. Liver X receptor activation promotes polyunsaturated fatty acid synthesis in macrophages: relevance in the context of atherosclerosis. Arterioscler Thromb Vasc Biol 35:1357–1365. doi: 10.1161/ATVBAHA.115.305539. [DOI] [PubMed] [Google Scholar]
- 22.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. 2003. Crosstalk between LXR and Toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell 12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 23.Fowler AJ, Sheu MY, Schmuth M, Kao J, Fluhr JW, Rhein L, Collins JL, Willson TM, Mangelsdorf DJ, Elias PM, Feingold KR. 2003. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J Invest Dermatol 120:246–255. doi: 10.1046/j.1523-1747.2003.12033.x. [DOI] [PubMed] [Google Scholar]
- 24.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. 2003. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 25.Ignatova ID, Angdisen J, Moran E, Schulman IG. 2013. Differential regulation of gene expression by LXRs in response to macrophage cholesterol loading. Mol Endocrinol 27:1036–1047. doi: 10.1210/me.2013-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson JK, Koenis DS, Scheij S, Cook EC, Moeton M, Santos A, Lobaccaro JA, Baron S, Zelcer N. 2017. EEPD1 is a novel LXR target gene in macrophages which regulates ABCA1 abundance and cholesterol efflux. Arterioscler Thromb Vasc Biol 37:423–432. doi: 10.1161/ATVBAHA.116.308434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Çimen I, Kocatürk B, Koyuncu S, Tufanlı Ö, Onat UI, Yıldırım AD, Apaydın O, Demirsoy Ş, Aykut ZG, Nguyen UT, Watkins SM, Hotamışlıgil GS, Erbay E. 2016. Prevention of atherosclerosis by bioactive palmitoleate through suppression of organelle stress and inflammasome activation. Sci Transl Med 8:358ra126. doi: 10.1126/scitranslmed.aaf9087. [DOI] [PubMed] [Google Scholar]
- 28.D’Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. 2006. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol 176:3087–3097. doi: 10.4049/jimmunol.176.5.3087. [DOI] [PubMed] [Google Scholar]
- 29.Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, Silva AR, Bozza MT, Castro-Faria-Neto HC, Bandeira-Melo C, Bozza PT. 2007. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol 179:8500–8508. doi: 10.4049/jimmunol.179.12.8500. [DOI] [PubMed] [Google Scholar]
- 30.Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, Jung ME, Ford DA, Tontonoz P. 2013. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab 18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, Osborne TF. 2011. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab 13:540–549. doi: 10.1016/j.cmet.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Li S, Wu J, Xia C, Lala DS. 2003. Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol 17:1019–1026. doi: 10.1210/me.2002-0399. [DOI] [PubMed] [Google Scholar]
- 33.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T. 2011. IkappaB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci U S A 108:21170–21175. doi: 10.1073/pnas.1119137109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LCF, Yángüez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, König R, Stertz S, García-Sastre A, Chanda SK. 2015. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bakan I, Laplante M. 2012. Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol 23:226–234. doi: 10.1097/MOL.0b013e328352dd03. [DOI] [PubMed] [Google Scholar]
- 37.Howell JJ, Manning BD. 2011. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 22:94–102. doi: 10.1016/j.tem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bodur C, Kazyken D, Huang K, Ekim Ustunel B, Siroky KA, Tooley AS, Gonzalez IE, Foley DH, Acosta-Jaquez HA, Barnes TM, Steinl GK, Cho KW, Lumeng CN, Riddle SM, Myers MG Jr, Fingar DC. 2018. The IKK-related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists. EMBO J 37:19–38. doi: 10.15252/embj.201696164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur S, Lal L, Sassano A, Majchrzak-Kita B, Srikanth M, Baker DP, Petroulakis E, Hay N, Sonenberg N, Fish EN, Platanias LC. 2007. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J Biol Chem 282:1757–1768. doi: 10.1074/jbc.M607365200. [DOI] [PubMed] [Google Scholar]
- 40.Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN, Platanias LC. 2003. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J Biol Chem 278:27772–27780. doi: 10.1074/jbc.M301364200. [DOI] [PubMed] [Google Scholar]
- 41.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hawkins JL, Robbins MD, Warren LC, Xia D, Petras SF, Valentine JJ, Varghese AH, Wang IK, Subashi TA, Shelly LD, Hay BA, Landschulz KT, Geoghegan KF, Harwood HJ Jr.. 2008. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther 326:801–808. doi: 10.1124/jpet.108.139626. [DOI] [PubMed] [Google Scholar]
- 43.Chu K, Miyazaki M, Man WC, Ntambi JM. 2006. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol 26:6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P. 2002. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem 277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 45.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK. 2003. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol 23:5780–5789. doi: 10.1128/mcb.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calder PC. 2017. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans 45:1105–1115. doi: 10.1042/BST20160474. [DOI] [PubMed] [Google Scholar]
- 47.Halvorson DL, McCune SA. 1984. Inhibition of fatty acid synthesis in isolated adipocytes by 5-(tetradecyloxy)-2-furoic acid. Lipids 19:851–856. doi: 10.1007/bf02534514. [DOI] [PubMed] [Google Scholar]
- 48.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. 2007. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab 5:73–79. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. 1999. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A 96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. 2003. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J 22:4625–4633. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tabas I, Glass CK. 2013. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heng TS, Painter MW, Immunological Genome Project Consortium . 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 53.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, Johnson J, Lee-McMullen B, Chen S, Metwally AA, Tran TDB, Nguyen H, Zhou X, Albright B, Hong BY, Petersen L, Bautista E, Hanson B, Chen L, Spakowicz D, Bahmani A, Salins D, Leopold B, Ashland M, Dagan-Rosenfeld O, Rego S, Limcaoco P, Colbert E, Allister C, Perelman D, Craig C, Wei E, Chaib H, Hornburg D, Dunn J, Liang L, Rose SMS, Kukurba K, Piening B, Rost H, Tse D, McLaughlin T, Sodergren E, Weinstock GM, Snyder M. 2019. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature 569:663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, Seidman JS, Henke A, Roland JT, Ozeki KA, Thompson BM, McDonald JG, Bahadorani J, Tsimikas S, Grossman TR, Tremblay MS, Glass CK. 2018. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc Natl Acad Sci U S A 115:E4680–E4689. doi: 10.1073/pnas.1714518115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH Jr, Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW, Glass CK. 2012. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151:138–152. doi: 10.1016/j.cell.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. 2008. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem 283:26332–26339. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramon-Vazquez A, de la Rosa JV, Tabraue C, Lopez F, Diaz-Chico BN, Bosca L, Tontonoz P, Alemany S, Castrillo A. 2019. Common and differential transcriptional actions of nuclear receptors liver X receptors alpha and beta in macrophages. Mol Cell Biol 39:e00376-18. doi: 10.1128/MCB.00376-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sheikh F, Dickensheets H, Gamero AM, Vogel SN, Donnelly RP. 2014. An essential role for IFN-beta in the induction of IFN-stimulated gene expression by LPS in macrophages. J Leukoc Biol 96:591–600. doi: 10.1189/jlb.2A0414-191R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. 2009. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. 2007. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. 2015. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JH, Park SM, Kim OS, Lee CS, Woo JH, Park SJ, Joe EH, Jou I. 2009. Differential SUMOylation of LXRalpha and LXRbeta mediates transrepression of STAT1 inflammatory signaling in IFN-gamma-stimulated brain astrocytes. Mol Cell 35:806–817. doi: 10.1016/j.molcel.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 63.Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, Li AC, Schulman IG. 2010. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res 51:900–906. doi: 10.1194/jlr.M900096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 2009. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A 106:16764–16769. doi: 10.1073/pnas.0909142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Im SS, Hammond LE, Yousef L, Nugas-Selby C, Shin DJ, Seo YK, Fong LG, Young SG, Osborne TF. 2009. Sterol regulatory element binding protein 1a regulates hepatic fatty acid partitioning by activating acetyl coenzyme A carboxylase 2. Mol Cell Biol 29:4864–4872. doi: 10.1128/MCB.00553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. 2002. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 67.Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. 1995. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 68.Osburn DL, Shao G, Seidel HM, Schulman IG. 2001. Ligand-dependent degradation of retinoid X receptors does not require transcriptional activity or coactivator interactions. Mol Cell Biol 21:4909–4918. doi: 10.1128/MCB.21.15.4909-4918.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soneson C, Love MI, Robinson MD. 2015. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. [Google Scholar]
- 73.Daniel B, Balint BL, Nagy ZS, Nagy L. 2014. Mapping the genomic binding sites of the activated retinoid X receptor in murine bone marrow-derived macrophages using chromatin immunoprecipitation sequencing. Methods Mol Biol 1204:15–24. doi: 10.1007/978-1-4939-1346-6_2. [DOI] [PubMed] [Google Scholar]
- 74.Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, Myers DS, Glass CK, Hardiman G, Reichart D, Merrill AH Jr, Sullards MC, Wang E, Murphy RC, Raetz CR, Garrett TA, Guan Z, Ryan AC, Russell DW, McDonald JG, Thompson BM, Shaw WA, Sud M, Zhao Y, Gupta S, Maurya MR, Fahy E, Subramaniam S. 2010. A mouse macrophage lipidome. J Biol Chem 285:39976–39985. doi: 10.1074/jbc.M110.182915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA-Seq data are available at the Gene Expression Omnibus (GEO) under accession number GSE118656.