

Abstract
Objective
To examine whether genetic variation affecting the expression or function of lipid‐lowering drug targets is associated with Alzheimer disease (AD) risk, to evaluate the potential impact of long‐term exposure to corresponding therapeutics.
Methods
We conducted Mendelian randomization analyses using variants in genes that encode the protein targets of several approved lipid‐lowering drug classes: HMGCR (encoding the target for statins), PCSK9 (encoding the target for PCSK9 inhibitors, eg, evolocumab and alirocumab), NPC1L1 (encoding the target for ezetimibe), and APOB (encoding the target of mipomersen). Variants were weighted by associations with low‐density lipoprotein cholesterol (LDL‐C) using data from lipid genetics consortia (n up to 295,826). We meta‐analyzed Mendelian randomization estimates for regional variants weighted by LDL‐C on AD risk from 2 large samples (total n = 24,718 cases, 56,685 controls).
Results
Models for HMGCR, APOB, and NPC1L1 did not suggest that the use of related lipid‐lowering drug classes would affect AD risk. In contrast, genetically instrumented exposure to PCSK9 inhibitors was predicted to increase AD risk in both of the AD samples (combined odds ratio per standard deviation lower LDL‐C inducible by the drug target = 1.45, 95% confidence interval = 1.23–1.69). This risk increase was opposite to, although more modest than, the degree of protection from coronary artery disease predicted by these same methods for PCSK9 inhibition.
Interpretation
We did not identify genetic support for the repurposing of statins, ezetimibe, or mipomersen for AD prevention. Notwithstanding caveats to this genetic evidence, pharmacovigilance for AD risk among users of PCSK9 inhibitors may be warranted. ANN NEUROL 2020;87:30–39
There are no preventive or disease‐modifying treatments for Alzheimer disease (AD). Expanding the indications of drugs of proven efficacy into other indications might be an effective strategy to provide new clinical treatments and preventative medicines for AD.1 Opportunities for indication expansion should be widespread, considering arguments based on first principles,2 and empirical evidence from genome‐wide association studies (GWASs) showing that the same gene can influence risk of more than one disease (pleiotropy).3
Medications that decrease circulating low‐density lipoprotein cholesterol (LDL‐C), such as statins, have been proposed as candidate therapies for AD. Hyperlipidemia in midlife is a risk factor for late onset AD in prospective epidemiological studies,4 and associations of higher LDL‐C with increased cerebral β‐amyloid load have also been observed in autopsy and in vivo imaging studies.5, 6 Similarly, AD risk is lower among statin users, and this association appears to be more pronounced with longer treatment exposure and the use of more potent drugs.7 In contrast, corresponding observational data on other lipid‐lowering drug classes are scant and inconclusive.7 Large randomized controlled trials (RCTs) may help to clarify the effects of dyslipidemia treatments on AD incidence without confounding, but such evidence is limited,8 and the slowly evolving pathogenesis of AD (over at least 1 decade)9, 10 means it is ill‐suited as an endpoint in trials of lipid‐lowering drugs with relatively short periods of intervention and follow‐up (typically 2–5 years).
Genetic epidemiology provides another means to address these questions. The expression or function of protein drug targets can be influenced by variants within or near the genes that encode them, and the genetic effects can be used to anticipate the effects of drug action.11 Because genotypes are inherited randomly at conception in an analogous manner to treatment allocation in clinical trials, associations of variants with biomarkers and disease outcomes are not expected to be subject to biases from confounding and reverse causation seen in other types of observational epidemiology—a principle leveraged in an approach known as “Mendelian randomization” (MR).12 Moreover, genotypes are mostly anticipated to confer lifelong differences in traits. Hence, MR studies can help to guide drug target validation by predicting the consequences of long‐term therapeutic exposure.13 In this study, we examined whether AD risk is influenced by variation in the genes encoding the targets of a range of medications that are currently licensed and recommended for the treatment of primary or familial hypercholesterolemia to prevent coronary heart disease.
Subjects and Methods
Study Design
We used a 2‐sample MR study design for these analyses,14 based mainly on genetic variants located in or near genes that encode the relevant drug targets (cis‐MR).13 We first identified differences in circulating LDL‐C that are associated with single nucleotide polymorphisms (SNPs) at the genomic regions of interest in sets of publicly available genetic association summary statistics, because the lowering of LDL‐C in circulation is a major, established physiological response produced by the use of the lipid‐lowering therapeutics. We then combined the LDL‐C association statistics for variant sets with corresponding genome‐wide association statistics for AD risk from 2 large, independent case–control datasets, to predict the effects of drug use on AD development.
As depicted in the study overview (Fig 1), we performed 5 main MR analyses: one assessing the effect of a general reduction of LDL‐C on AD risk (ie, achievable by any means), and the remainder for specific effects of variation at 4 gene regions (HMGCR, PCSK9, APOB, and NPC1L1) that are indicative of the long‐term use of different lipid‐lowering drug classes that are currently licensed and guideline‐supported for lowering LDL‐C in the primary prevention of coronary heart disease, familial hypercholesterolemia, or both. These genes encode the targets of statins (HMG‐CoA reductase [HMGCR]), proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (eg, evolocumab and alirocumab), apolipoprotein B‐100 (ApoB) antisense classes (eg, mipomersen), and NPC1‐like intracellular cholesterol transporter 1 (NPC1L1) inhibitors (eg, ezetimibe), respectively.
Figure 1.
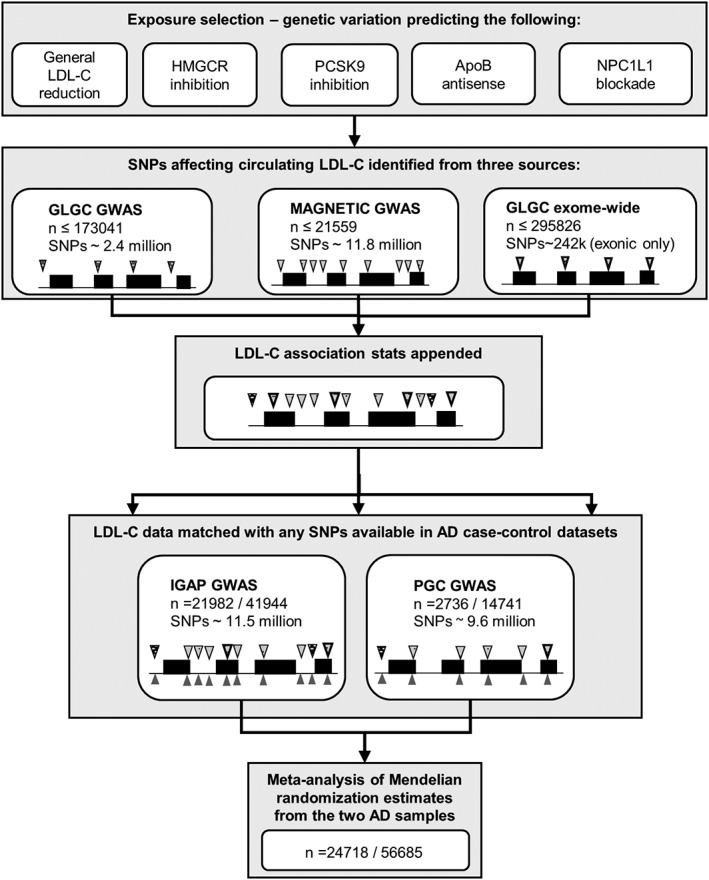
Study overview. Black lines depict an illustrative gene region, with raised boxes showing exons. Wedges represent the presence of specific genetic variants (single nucleotide polymorphisms [SNPs]) measured throughout the gene. Different patterns of the wedges illustrate the combination of sources of low‐density lipoprotein cholesterol (LDL‐C) association statistics across variants from 1 of 3 LDL‐C genome‐wide association studies (GWASs; second and third panels). The pairing of wedges above and below the gene outline in panel 4 depicts the harmonization of appended LDL‐C statistics with corresponding estimates of Alzheimer disease (AD) risk for each variant within the 2 AD datasets separately. The varying number of wedges present across each dataset represents the differences in densities of SNPs that were genotyped or imputed in the data. GLGC = Global Lipid Genetics Consortium; IGAP = International Genomics of Alzheimer's Project; PGC = Psychiatric Genomics Consortium.
In post hoc analyses to follow up on genetic analyses modeling PCSK9 inhibition in relation to AD risk that were weighted by LDL‐C, we also examined whether genetically indexed variation in circulating PCSK9 concentration is associated with AD risk.
Deriving SNP Associations with LDL‐C
SNP–LDL‐C association statistics were extracted from 3 GWASs of circulating LDL‐C concentrations—2 from the Global Lipid Genetics Consortium15, 16 and 1 from the MAGNETIC consortium.17 These contained different sets of genotyped and imputed SNP data and varying sample sizes, and hence estimated any genetic effect with varying precision. Due to partial overlap between the GWAS samples, the 3 sets of estimates were appended together, rather than combined in a meta‐analysis to prevent double counting (see Fig 1). Where the same SNP–LDL‐C associations were present in more than one GWAS dataset, we retained the estimate from the largest of the samples. In combining these sets of GWAS summary statistics, we aimed to leverage the maximum degree of genetic variation measured at each region (optimizing statistical power to detect associations of regional variation with AD risk). The sample sizes for SNP–LDL‐C association statistics ranged from 14,004 to 295,826. Each of the LDL‐C GWASs had homogeneous samples (all of European ancestry) and was modeled with consistent statistical procedures and transformations of the LDL‐C distributions.
For general LDL‐C models, we used independent SNPs throughout the genome that had replicated evidence for association with LDL‐C in any of the 3 GWAS datasets. We excluded a SNP in APOE on chromosome 19 due to a strong, established pleiotropic effect of this locus on AD risk, and another variant on chromosome 19 that exhibited linkage disequilibrium (LD) with the SNPs that constitute the ε2/3/4 genotypes in APOE. A total of 59 SNPs remained after LD clumping (steps described further in the Statistical Analysis section) and were taken forward to harmonize with AD data. In primary gene region models, we extracted data on all SNPs within 1 kilobase in either direction of the start and stop coordinates for the genes, according to Human Genome reference release GRCh37 positions. In secondary models for gene regions, we expanded the selection to all SNPs within ±100 kilobases of the genes’ coordinates. Data on the beta coefficients, standard errors, coded effect, and alternate alleles and allele frequencies per SNP were extracted to combine with summary statistics from the AD datasets.
Analyses to assess the effect of modifying circulating PCSK9 on AD risk were based on SNP–PCSK9 association statistics for 6 variants identified in a GWAS of circulating PCSK9 in suspected or confirmed coronary artery disease (CAD) patients of European ancestry.18 We used SNP–PCSK9 association statistics from a subgroup analysis of statin‐naive participants in the GWAS (n = 2,022).
AD Data
We assessed whether LDL‐associated variants also associate with late onset AD risk (age of onset ≥65 years) using data from 2 large genome‐wide analyses from the International Genomics of Alzheimer's Project (IGAP) and the Psychiatric Genomics Consortium (PGC).19, 20 The total sample size was 24,718 cases and 56,685 controls, all of whom had European ancestry (summary details are given in Table 1). Full information on genetic quality control procedures and association modeling are provided in the study references.19, 20
Table 1.
Summary Information on AD Datasets
| Study | ||
|---|---|---|
| IGAP19 | PGC20 | |
| Cases, n | 21,982 | 2,736 |
| Controls, n | 41,944 | 14,741 |
| Sample sources | Case–control studies, longitudinal cohorts | Register‐based follow‐up of twin cohorts/case–control DemGene study |
| Country of origin | Europe, Canada, USA | Sweden/Norway |
| Case mean AAO, yra | 71.1–82.6 | 77.0–80.5 |
| Control mean AAE, yra | 51.0–78.9 | 58.8–75.5 |
| % female, cases/controls | 59.3–67.3/51.8–60.6 | 52.5–66.4/48.0–51.1 |
| AD ascertainment | Clinical assessment, MRI‐ or autopsy‐confirmed, and/or diagnoses from health care records | Clinical assessment and/or diagnoses from health care records |
| Genetic data | Genome‐wide genotyping, imputation using 1000 Genomes project, phase 2 release | Genome‐wide genotyping, imputation using 1000 Genomes project, phase 3 release |
Ranges of mean or percentage values are presented for IGAP stage 1 and PGC data, which combined 4 consortia/studies and 3 samples in these genome‐wide association studies, respectively.
AAE = age at examination or last follow‐up; AAO = age at onset; AD = Alzheimer disease; IGAP = International Genomics of Alzheimer's Project; MRI = magnetic resonance imaging; PGC = Psychiatric Genomics Consortium.
Positive Control Analyses
To help validate the chosen modeling strategy, we also merged the same sets of LDL‐C summary statistics with genetic data on the risk of CAD and type 2 diabetes mellitus (T2D), for which we may expect to see effects of LDL‐C modification (ie, positive control analyses). These models used summary GWAS data on CAD risk from the CARDIoGRAM consortium (n = 22,233 cases, 64,762 controls)21, 22 and T2D risk from the DIAGRAM consortium (n = 12,171 cases, 56,862 controls).22 Samples included participants of European ancestry only.
Statistical Analysis
Data for SNP associations with LDL‐C or PCSK9 and with risk of AD or cardiometabolic outcomes were harmonized to match coded effect alleles consistently; we excluded ambiguous SNPs from analyses (those with palindromic genotypes and minor allele frequencies between 0.4 and 0.5).
To assess a potential effect of LDL‐C lowering on AD, regardless of drug target, we used the fixed‐effects inverse variance weighted (IVW) meta‐analysis method, which combines ratios of coefficients (Wald estimators) for the association estimates of each eligible SNP with LDL‐C and AD.23 We repeated analyses using alternative methods—weighted median and MR Egger estimators—which provide different degrees of robustness to bias from genetic pleiotropy.24, 25
For the primary gene (drug target)‐specific models, data on all successfully harmonized SNPs within the gene regions (±1 kilobase flank) were eligible for inclusion. We then used LD clumping to remove the excess of most highly correlated variants at each locus (with pairwise r 2 values >0.6), retaining the member of each correlated pair with the lowest p value for association with LDL‐C. Remaining SNPs were modeled together using a principal components (PC)‐based approach to handle estimates from correlated variants.26 This method relies on the use of reference data to estimate the correlations between variants in summary GWAS datasets, for which we used correlation matrices derived from 503 participants of European ancestry in the 1000 Genomes project, phase 3.27 We tested the robustness of using these reference data by inputting several matrix types from different subpopulations of the 1000 Genomes project sample, including the actual correlations from the PGC data. We also tested the consistency of PC estimates by specifying 3 values of κ (0.9, 0.99, and 0.999), which would be expected to model between 90 and 99.9% of variance in LDL‐C attributable to genetic variants in each gene region (results from the 99% models are presented in each figure).26 For comparison, we also conducted analyses for gene region models with a wider range of SNPs for inclusion (within ±100 kilobases of the genes), and with MR estimates provided by the IVW method to enroll only independent SNPs with GWAS significant associations with LDL‐C (SNP–LDL association p values <5 × 10−8). Independence between SNPs was defined by pairwise r 2 values <0.2 in one set of models, and then repeated with SNPs clumped using a more conservative threshold (r 2 < 0.01). Funnel plots were produced to test whether overall MR estimates were consistent with the estimates provided by the most precise individual SNP estimators, and leave‐one‐out plots were produced to examine whether models were influenced by any individual outlying SNP estimates.28 Using LD‐clumped variants, we also conducted MR Egger and weighted median models for any gene region–AD variant set that included 3 or more SNPs. We also conducted both PC and LD‐clumped gene region models for analyses of cardiometabolic outcomes.
For both general LDL‐C and gene (drug target)‐specific models, we opted to analyze effects on AD risk separately in the 2 AD GWAS datasets, due to these having varying SNP data available and different sample structures (a mix of matched case–control studies, samples from longitudinal cohorts, and health care register–derived samples). Finally, we combined the 2 results for each trait–AD association into overall MR estimates using fixed‐effects IVW meta‐analyses. Results were scaled to express odds ratios (ORs) for AD and cardiometabolic outcomes per 1 standard deviation (SD) lower LDL‐C, to indicate the expected directions of effect that would follow from the use of lipid‐lowering drugs.
Post hoc analyses for circulating PCSK9 involved the same steps and MR methods used to model the effect of a general LDL‐C lowering on AD risk. Again, we also estimated the effects of lowering PCSK9 on CAD for comparison. Because SNP–PCSK9 associations were expressed in logn‐transformed PCSK9 units, results from these MR models were scaled to show CAD and AD risk differences per halving of circulating PCSK9.
Analyses were undertaken in Stata version 15 (StataCorp, College Station, TX), PLINK 1.9 (http://www.cog-genomics.org/plink/1.9/), and R version 3.4.1, with the aid of the packages MR Base and MendelianRandomization.29, 30
This research involved the reuse of existing anonymized, study‐level summary data only. All of the original studies had received informed consent from participants and ethical approvals.
Results
After harmonizing exposure (LDL‐C or PCSK9) and disease outcome genetic associations, and subsequent LD clumping, main MR models included between 2 and 55 variants (Supplementary Tables 1–5).
A general, long‐term reduction in circulating LDL‐C, indexed using eligible variants throughout the genome, was not estimated to affect AD risk (Fig 2). Findings were also null in alternate MR analyses using weighted median and MR Egger methods (Table 2), and these findings did not suggest any notable bias from directional pleiotropy or weak instruments in these models; for example, in analyses of the IGAP dataset, the MR Egger intercept test was 1.00 (p = 0.39), with I2 gx of 98.7% in both datasets.
Figure 2.
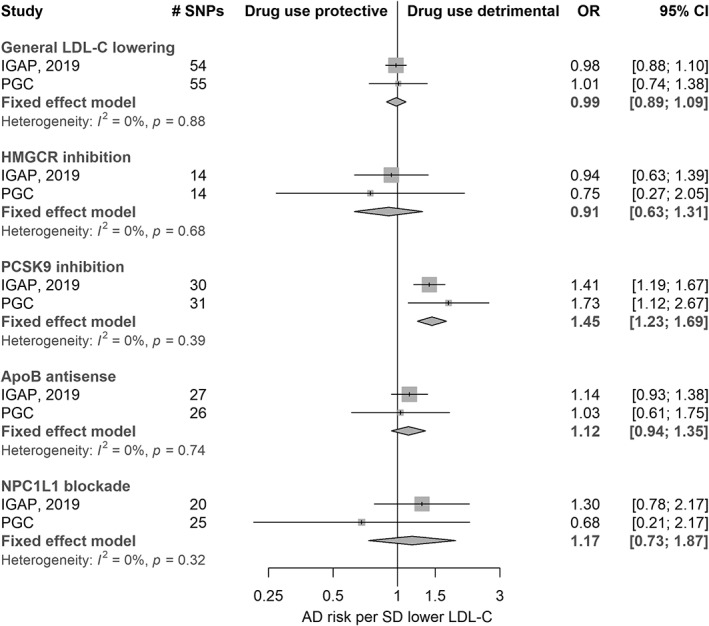
Meta‐analysis of Mendelian randomization estimates for Alzheimer disease (AD) risk according to a lifelong reduction in circulating low‐density lipoprotein cholesterol (LDL‐C) and exposure to the modulation of several related drug targets (n = 24,718 cases, 56,685 controls). The first group of results shows estimates for the effect of a general, long‐term reduction of LDL‐C (achievable by any means) on AD risk. The second to fifth group labels are representative of genetic variation at gene regions (HMGCR, PCSK9, APOB, and NPC1L1) that predict the effects of specific therapeutic target modulation, followed by example drug classes that affect these targets. CI = confidence interval; IGAP = International Genomics of Alzheimer's Project; OR = odds ratio; PGC = Psychiatric Genomics Consortium; SD = standard deviation; SNP = single nucleotide polymorphism.
Table 2.
MR Estimates for the Effect of LDL‐C Lowering on AD Risk in IGAP and PGC Datasets
| Study | Method | Odds Ratio | 95% CI | p |
|---|---|---|---|---|
| IGAP | IVW | 0.98 | 0.88–1.10 | 0.75 |
| MR Egger | 1.04 | 0.88–1.22 | 0.67 | |
| MR Egger intercept test | 1.00 | 0.99–1.00 | 0.39 | |
| Wtd median | 0.95 | 0.82–1.11 | 0.53 | |
| PGC | IVW | 1.01 | 0.74–1.38 | 0.96 |
| MR Egger | 1.41 | 0.89–2.25 | 0.15 | |
| MR Egger intercept test | 0.98 | 0.96–1.00 | 0.06 | |
| Wtd median | 1.19 | 0.81–1.75 | 0.38 |
IVW results are illustrated in the top section of Figure 2. Odds ratios and 95% CIs are per 1 standard deviation lower LDL‐C. IGAP and PGC models included 54 and 55 single nucleotide polymorphisms, respectively.
AD = Alzheimer disease; CI = confidence interval; IGAP = International Genomics of Alzheimer's Project; IVW = inverse variance weighted; LDL‐C = low‐density lipoprotein cholesterol; MR = Mendelian randomization; PGC = Psychiatric Genomics Consortium; Wtd = weighted.
Gene region models using the PC‐based approach for HMGCR, APOB, and NPC1L1 did not provide clear evidence to suggest that the use of the corresponding lipid‐lowering drug classes would affect AD risk (see Fig 2). Results for these regions were largely consistent when repeated with IVW models; all point estimates for HMGCR were on the side of neuroprotection and all estimates for APOB and NPC1L1 were on the side of risk, but confidence intervals (CIs) in all instances were wide and included the null. In contrast, variants in the vicinity of PCSK9 implied that exposure to PCSK9 inhibitors could increase the risk of AD (OR = 1.45, 95% CI = 1.23–1.69, p = 4.4 × 10−6). Estimates were similar in meta‐analyses of IVW models with liberal clumping (OR = 1.37, 95% CI = 1.12–1.66) and conservative clumping (OR = 1.44, 95% CI = 0.94–2.20). There was no evidence of heterogeneity between estimates from the 2 AD samples (p values for heterogeneity tests in all models ≥0.11). Funnel plots did not indicate asymmetry in SNP estimates across the scale of precision in IVW models, and leave‐one‐out plots did not suggest that any of the IVW findings were greatly influenced by single outlying Wald estimators (plots available on request). MR Egger and weighted median results for LD‐clumped variant sets are shown in Supplementary Table 6. In general, these did not deviate greatly from IVW findings, but most estimates were very imprecise (particularly those from MR Egger). Point estimates varied widely between IVW, weighted median, and MR Egger methods for PCSK9 inhibition and AD risk in IGAP data, but were more consistent in the PGC dataset.
Sensitivity analyses indicated that the choice of reference data used to model variant correlations would not be expected to influence findings from PC models notably. ORs for the effects of PCSK9 inhibition on AD risk in the PGC sample ranged from 1.64 (95% CI = 1.17–2.31) per SD lower LDL‐C using allele frequencies from a sample with Finnish ancestry to 1.73 (95% CI = 1.12–2.66) using allele frequencies of samples with ancestries from across Europe. In comparison, the corresponding estimate using the sample's actual allele frequencies was 1.69 (95% CI = 1.14–2.52). Changing input parameters for PC models introduced some slight variation in the magnitudes of estimates. For example, using κ = 0.90 or κ = 0.999 instead of 0.99 in PC models (ie, enrolling either 90% or 99.9% of variance in LDL‐C concentrations attributable to genetic variation in the gene region) yielded meta‐analysis effect estimates for PCSK9 inhibition on AD risk of 1.35 (95% CI = 1.09–1.67) and 1.62 (95% CI = 1.41–1.86), respectively. Despite these varying magnitudes of estimates, the use of different PC model permutations would not change the overall interpretation of these findings.
Using the same approach to predict the effects of long‐term modulation of these targets on cardiometabolic outcomes (positive control models), genetically predicted exposures to all lipid‐lowering drug classes were associated with a lower risk of CAD (Fig 3). There were trends for increased risk of T2D with predicted exposure to statins and inhibitors of PCSK9 and NPC1L1 (as has been observed previously31, 32), although results from NPC1L1 variants were estimated with limited precision.
Figure 3.
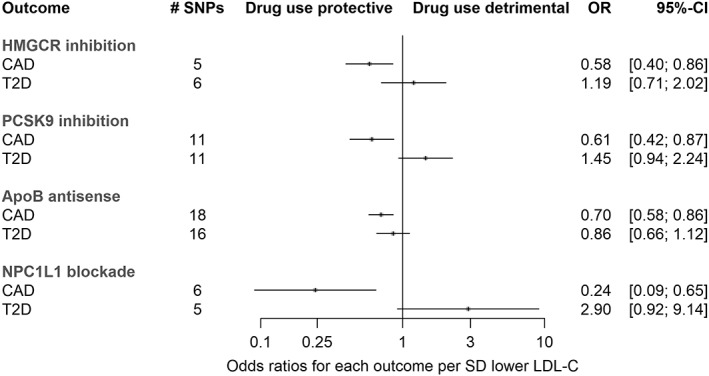
Mendelian randomization estimates for the effects of exposure to drug target modulation on coronary artery disease (CAD) and type 2 diabetes mellitus (T2D). Results were produced using variants within gene regions (±1 kilobase flanks) and the principal components–based method, as per analyses for the Alzheimer disease results presented in Figure 1. Sample sizes: CAD, n = 22,233 cases, 64,762 controls; T2D, n = 12,171 cases, 56,862 controls. CI = confidence interval; LDL‐C = low‐density lipoprotein cholesterol; OR = odds ratio; SD = standard deviation; SNP = single nucleotide polymorphism.
Given the finding that genetic indexing of PCSK9 inhibition predicted higher AD risk in our main models, we further examined whether differences in exposure to circulating PCSK9 may be related to AD risk using MR, and compared these results to the predicted effect of circulating PCSK9 variation on CAD (Table 3). As expected, lowering PCSK9 concentration (which correlates modestly with lower circulating LDL‐C) was predicted to reduce CAD risk. In IVW models for AD, point estimates from IGAP and PGC were both on the side of risk per halving of circulating PCSK9, but the IGAP sample result and the meta‐analysis finding both included the null. Point estimates from MR Egger and weighted median methods were very similar to IVW estimates from both AD samples, but the findings had very little precision due to the use of few available SNPs to index PCSK9 differences.
Table 3.
MR Estimates for the Effect of Lowering PCSK9 on CAD and AD Risk
| Study | SNPs, n | Method | Odds Ratio | 95% CI | p |
|---|---|---|---|---|---|
| CAD | 2 | IVW | 0.57 | 0.36–0.89 | 0.01 |
| IGAP | 3 | IVW | 1.10 | 0.85–1.43 | 0.47 |
| MR Egger | 1.10 | 0.67–1.81 | 0.78 | ||
| MR Egger intercept test | 1.00 | 0.93–1.07 | 0.99 | ||
| Wtd median | 1.09 | 0.81–1.47 | 0.56 | ||
| PGC | 3 | IVW | 1.98 | 1.02–3.81 | 0.04 |
| MR Egger | 1.95 | 0.58–6.54 | 0.48 | ||
| MR Egger intercept test | 1.00 | 0.84–1.19 | 0.98 | ||
| Wtd median | 1.72 | 0.81–3.65 | 0.16 | ||
| AD meta‐analysis | 3 | IVW | 1.19 | 0.94–1.51 | 0.16 |
Odds ratios and 95% CIs are per halving of circulating PCSK9 concentrations.
AD = Alzheimer disease; CAD = coronary artery disease; CI = confidence interval; IGAP = International Genomics of Alzheimer's Project; IVW = inverse variance weighted; MR = Mendelian randomization; PGC = Psychiatric Genomics Consortium; SNP = single nucleotide polymorphism; Wtd = weighted.
Discussion
This MR study did not identify support for the repurposing of statins, or medications that inhibit NPC1L1 or block ApoB production, to delay or prevent AD onset. Our results also raise the possibility (but in no way confirm) that exposure to PCSK9 inhibitors might predispose individuals to AD, although with the magnitude of relative risk observed being smaller than for the estimated degree of protection from CAD.
If the use of lipid‐lowering medications in general does not affect AD risk, this may signify the absence of a substantial role of primary hypercholesterolemia in AD etiology. The only major RCT to have addressed the effect of therapeutic LDL‐C reduction on dementia risk to date found equal incidence (0.3%) in both simvastatin and placebo‐allocated arms after 5 years of follow‐up (albeit in a sample aged 40–80 years at baseline, thus including many individuals too young to be at risk of late onset AD).33 Taking this finding alongside no prominent genetic associations observed for general LDL‐C lowering (as others have observed previously),34, 35 and unclear or null results for HMGCR and 2 other drug targets, suggests that previous prospective observational associations of hyperlipidemia with AD, and of higher LDL‐C with AD‐related neuropathology, may have been overstated due to residual confounding. Associations of lipid‐lowering drug use with AD risk would also be prone to bias via confounding by indication.36
A previous analysis has also addressed whether variation at HMGCR and PCSK9 is associated with AD risk using MR.37 After excluding pleiotropic variation at APOE, the authors found no association of a genetically predicted reduction in LDL‐C exposure with AD in a sample that overlaps with one of those in the current analyses (a previous IGAP GWAS from 2013).38 Their result using HMGCR and PCSK9 variation (combined in a single IVW model) from IGAP data had a consistent direction and similar magnitude of association as in our findings, but conflated the potentially independent associations of the different gene regions with AD, and may have lacked precision due to the use of fewer variants and a smaller sample.37 Using a more comprehensive survey of genetic variation at PCSK9 separately in 2 large AD datasets, our analyses yielded consistent estimates of higher AD risk from lower PCSK9 function, regardless of the MR method used. We also report tentative MR evidence to suggest that lower circulating PCSK9 might reduce AD risk, although these findings were inconclusive, because the precision of the models was limited by the use of few variants with which to proxy exposure to PCSK9 concentrations.
Considering the full range of our findings together, if PCSK9 function has a role in determining AD risk, this is likely to be through pathways other than the regulation of peripheral LDL‐C. Experimental evidence has linked PCSK9 dysfunction to cerebral β‐amyloid production and neuronal cell death—both hallmarks of AD.39 PCSK9 may also promote neurogenesis during development,40 so any impact on AD risk could potentially be mediated by establishing “brain reserve” in early life,41 as opposed to a hastening of AD pathogenesis in adulthood. However, concerns of adverse neurocognitive effects of PCSK9 inhibition have been raised from a network meta‐analysis of RCTs of PCSK9 inhibitors in middle‐aged and older individuals.42 Another MR analysis has also suggested potentially detrimental effects of lower PCSK9 function on cognitive ability in a cohort of participants aged 38 to 73 years.43 A further consideration is whether PCSK9 inhibition might harm the cerebral vasculature (independently of effects on atherosclerosis). Cerebrovascular damage might influence AD neurodegeneration directly,44 or affect risk estimates if AD case samples include misclassified vascular (or mixed) dementia patients. However, other MR studies and RCTs have found conflicting evidence of the relationship of PCSK9 inhibition with stroke risk,26, 45, 46 and we have no knowledge of evidence implicating PCSK9 function in cerebral small vessel disease or vascular dementia.
Given that PCSK9 inhibitors are an efficacious second‐line drug class for the treatment of primary hypercholesterolemia and mixed dyslipidemia, we present these suggestive findings cautiously. We note that the point estimates from the much larger sample (IGAP) were less pronounced than those from the smaller sample across the various types of models, although several estimates from IGAP were still of a concerning magnitude individually. Moreover, when considering SNPs with the greatest evidence of functional relevance in the PCSK9 region, such as the rare missense variant rs11591147, genotype–AD associations were on the side of higher risk per LDL‐lowering allele but were not statistically significant individually in either dataset or when meta‐analyzed. Further genetic studies can help to confirm or refute these results in several ways, including (1) repeating MR models of PCSK9 function in larger GWASs of AD risk, which would ideally include direct genotyping of functionally relevant SNPs in the PCSK9 gene region; (2) assessing the associations of PCSK9 variants with AD endophenotypes, for example, from large‐scale GWASs of fluid and/or imaging biomarkers of β‐amyloid load; and (3) recall‐by‐genotype studies to examine the presence of subclinical AD pathologies and symptoms in older individuals carrying loss‐ or gain‐of‐function PCSK9 mutations. Because PCSK9 inhibitors have been licensed since 2015 in several territories, pharmacoepidemiologic studies of the consequences of PCSK9 inhibitor use versus other lipid‐lowering agents for AD risk could also be conducted with large samples of individuals in national or insurance‐based prescription registers, as could the follow‐up of AD risk differences among past participants in PCSK9 inhibitor trials.
The principal strength of this study was the use of 2 large AD samples in which to test for the consistency, and improve the precision, of MR estimates. The merging of LDL‐C summary statistics and use of methodology to combine correlated variation in MR models allowed us to predict the effects of manipulating drug targets more precisely, even when using variants solely within gene regions. Using cis‐acting variants alone should be less prone to bias from horizontal pleiotropy than the use of variation across wide flanking regions, because there is a higher probability of SNPs incidentally tagging AD risk variants elsewhere in the genome with increasing physical distance from the gene encoding a target of interest.
Limitations include the prediction solely of on‐target effects of drug use by our models; they do not encapsulate off‐target consequences of using the related therapeutic classes. Second, genetics may not be informative about particular pharmacological aspects of drug exposure. For instance, monoclonal antibodies, such as evolocumab and alirocumab, are not expected to cross the blood–brain barrier (at least while the barrier's impermeability to large molecules is intact).47 Hence, peripherally administered PCSK9 inhibitors may not lead to cerebral exposure, which means the genetically‐instrumented risk might not be relevant in therapeutic practice. Analogous circumstances have been examined for T2D, where evidence from human genetics suggests reduced PCSK9 activity leads to greater diabetes risk, but there is no clear evidence to show that this risk increase has borne out among RCT participants exposed to PCSK9 inhibitors.31, 48 The disparities may be reconciled if localized PCSK9 function in pancreatic tissue were to affect glycemic control independently of any inhibition of PCSK9 in circulation, as has been suggested by data from tissue‐selective Pcsk9 knockout mice.49 If data from larger GWASs of circulating PCSK9 become available, improved MR analyses of circulating PCSK9 concentrations and AD risk (ie, using multiple genome‐wide SNPs to index differences in exposure) would help to evaluate the role of peripheral PCSK9 inhibition specifically. Third, given that genetic effects are often lifelong, we cannot distinguish critical periods of exposure to these targets in which disease risk might be specifically affected, such as neurodevelopment. Fourth, differences between the IGAP and PGC samples are also worth noting; in particular, a modest proportion of AD cases in the PGC sample were diagnosed from linkage to in‐ and outpatient health care records after the latest clinical assessments of these individuals were conducted (up to 741 cases, 27% of the PGC case sample). Individuals with other forms of dementia may have been misclassified as AD cases, or AD cases as controls, at higher rates with this type of ascertainment than in case–control or cohort studies with direct clinical assessments and/or pathological confirmation of AD.50 This could bias exposure–outcome association estimates if the exposures under study relate to the chance of misclassification differentially, for example, if an exposure increases the risk of hospitalization and, therefore, raises the likelihood of AD detection. However, this would not be expected to explain an inverse association of a trait affecting CAD risk with lower AD risk, as we observe for PCSK9 variation. Furthermore, a large majority of the cases in the PGC sample, as well as IGAP, were diagnosed reliably with AD at memory clinics according to standard criteria.
Conclusions
Evidence from this study does not support RCTs to investigate the repurposing of statins, or inhibitors of NPC1L1‐ or ApoB‐blocking therapeutics, for AD prevention. Notwithstanding the several caveats discussed here, pharmacovigilance for neurocognitive effects and the scope for increased AD burden in users of PCSK9 inhibitors may be warranted.
Author Contributions
All authors contributed to the conception and design of the study; D.M.W. conducted the analysis of data, and contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary table 1 information on variant sets used to assess the effect of a general LDL‐C reduction on Alzheimer's disease (AD) risk
Supplementary table 2: information on gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on AD risk in principal components MR models
Supplementary table 3: information on alternate gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on AD risk in IVW MR models with uncorrelated variants
Supplementary table 4: information on gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on cardiometabolic outcomes
Supplementary table 5: information on genome‐wide variants used to assess the effects of lowering circulating PCSK9 on AD and CAD
Supplementary table 6: Alternate MR methods for examining gene region variants in relation to AD risk, using two LD‐clumping strategies instead of principal component methodology
Acknowledgment
D.M.W. received support for this work from the Foundation for Geriatric Diseases at Karolinska Institute (2018‐01252). A.F.S. is supported by BHF grant PG/18/5033837.
We thank IGAP for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in the analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. We also thank the participants, researchers, and staff associated with the many other studies from which we used data for this report; and Drs I. Jansen, J. Savage, and D. Posthuma of the PGC for sharing additional data on the PGC GWAS sample to use in these analyses.
References
- 1. Corbett A, Pickett J, Burns A, et al. Drug repositioning for Alzheimer's disease. Nat Rev Drug Discov 2012;11:833–846. [DOI] [PubMed] [Google Scholar]
- 2. Hingorani AD, Kuan V, Finan C, et al. Improving the odds of drug development success through human genomics: modelling study (Preprint). [DOI] [PMC free article] [PubMed]
- 3. Pickrell JK, Berisa T, Liu JZ, et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 2016;48:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meng X‐F, Yu J‐T, Wang H‐F, et al. Midlife vascular risk factors and the risk of Alzheimer's disease: a systematic review and meta‐analysis. J Alzheimers Dis 2014;42:1295–1310. [DOI] [PubMed] [Google Scholar]
- 5. Matsuzaki T, Sasaki K, Hata J, et al. Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama study. Neurology 2011;77:1068–1075. [DOI] [PubMed] [Google Scholar]
- 6. Reed B, Villeneuve S, Mack W, et al. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 2014;71:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsson SC, Markus HS. Does treating vascular risk factors prevent dementia and Alzheimer's disease? A systematic review and meta‐analysis. J Alzheimers Dis 2018;64:657–668. [DOI] [PubMed] [Google Scholar]
- 8. McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev 2016;(1):CD003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70:960–969. [DOI] [PubMed] [Google Scholar]
- 10. Jack CR Jr, Wiste HJ, Weigand SD, et al. Age‐specific and sex‐specific prevalence of cerebral β‐amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross‐sectional study. Lancet Neurol 2017;16:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hingorani A, Humphries S. Nature's randomised trials. Lancet 2005;366:1906–1908. [DOI] [PubMed] [Google Scholar]
- 12. Lawlor DA, Harbord RM, Sterne JAC, et al. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–1163. [DOI] [PubMed] [Google Scholar]
- 13. Swerdlow DI, Kuchenbaecker KB, Shah S, et al. Selecting instruments for Mendelian randomization in the wake of genome‐wide association studies. Int J Epidemiol 2016;45:1600–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burgess S, Scott RA, Timpson NJ, et al. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015;30:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willer CJ, Schmidt EM, Sengupta S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu DJ, Peloso GM, Yu H, et al. Exome‐wide association study of plasma lipids in >300,000 individuals. Nat Genet 2017;49:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kettunen J, Demirkan A, Würtz P, et al. Genome‐wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun 2016;7:11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pott J, Schlegel V, Teren A, et al. Genetic regulation of PCSK9 (proprotein convertase subtilisin/kexin type 9) plasma levels and its impact on atherosclerotic vascular disease phenotypes. Circ Genom Precis Med 2018;11:e001992. [DOI] [PubMed] [Google Scholar]
- 19. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 2019;51:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 2019;51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schunkert H, König IR, Kathiresan S, et al. Large‐scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011;43:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morris AP, Voight BF, Teslovich TM, et al. Large‐scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgess S, Zuber V, Valdes‐Marquez E, et al. Mendelian randomization with fine‐mapped genetic data: choosing from large numbers of correlated instrumental variables. Genet Epidemiol 2017;41:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. 1000 Genomes Project Consortium , Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burgess S, Bowden J, Fall T, et al. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 2017;28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hemani G, Zheng J, Elsworth B, et al. The MR‐Base platform supports systematic causal inference across the human phenome. eLife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46:1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol 2017;5:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lotta LA, Sharp SJ, Burgess S, et al. Association between low‐density lipoprotein cholesterol–lowering genetic variants and risk of type 2 diabetes: a meta‐analysis. JAMA 2016;316:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heart Protection Study Collaborative Group . Effects of cholesterol‐lowering with simvastatin on stroke and other major vascular events in 20 536 people with cerebrovascular disease or other high‐risk conditions. Lancet 2004;363:757–767.15016485 [Google Scholar]
- 34. Østergaard SD, Mukherjee S, Sharp SJ, et al. Associations between potentially modifiable risk factors and Alzheimer disease: a Mendelian randomization study. PLoS Med 2015;12:e1001841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larsson SC, Traylor M, Malik R, et al. Modifiable pathways in Alzheimer's disease: Mendelian randomisation analysis. BMJ 2017;359:j5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev 2016;(1):CD003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benn M, Nordestgaard BG, Frikke‐Schmidt R, Tybjærg‐Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer's disease and Parkinson's disease: Mendelian randomisation study. BMJ 2017;357:j1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benn M, Nordestgaard BG, Frikke‐Schmidt R, Tybjærg‐Hansen A. Re: Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer's disease and Parkinson's disease: Mendelian randomisation study. BMJ 2017;357:j1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non‐acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep 2008;9:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poirier S, Prat A, Marcinkiewicz E, et al. Implication of the proprotein convertase NARC‐1/PCSK9 in the development of the nervous system. J Neurochem 2006;98:838–850. [DOI] [PubMed] [Google Scholar]
- 41. Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012;11:1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lipinski MJ, Benedetto U, Escarcega RO, et al. The impact of proprotein convertase subtilisin‐kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta‐analysis. Eur Heart J 2015;37:536–545. [DOI] [PubMed] [Google Scholar]
- 43. Lyall D, Ward J, Banach M, et al. PCSK9 genetic variants, life‐long lowering of LDL‐cholesterol and cognition: a large‐scale Mendelian randomization study. bioRxiv 2018;335877. [Google Scholar]
- 44. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain 2013;136:2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao AS, Lindholm D, Rivas MA, et al. Large‐scale phenome‐wide association study of PCSK9 variants demonstrates protection against ischemic stroke. Circ Genom Precis Med 2018;11:e002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milionis H, Barkas F, Ntaios G, et al. Proprotein convertase subtilisin kexin 9 (PCSK9) inhibitors to treat hypercholesterolemia: effect on stroke risk. Eur J Intern Med 2016;34:54–57. [DOI] [PubMed] [Google Scholar]
- 47. Pardridge WM. Drug transport across the blood–brain barrier. J Cereb Blood Flow Metab 2012;32:1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Carvalho LSF, Campos AM, Sposito AC. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and incident type 2 diabetes: a systematic review and meta‐analysis with over 96,000 patient‐years. Diabetes Care 2018;41:364–367. [DOI] [PubMed] [Google Scholar]
- 49. Da Dalt L, Ruscica M, Bonacina F, et al. PCSK9 deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low‐density lipoprotein receptor. Eur Heart J 2018;40:357–368. [DOI] [PubMed] [Google Scholar]
- 50. Rizzuto D, Feldman AL, Karlsson IK, et al. Detection of dementia cases in two Swedish health registers: a validation study. J Alzheimers Dis 2018;61:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1 information on variant sets used to assess the effect of a general LDL‐C reduction on Alzheimer's disease (AD) risk
Supplementary table 2: information on gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on AD risk in principal components MR models
Supplementary table 3: information on alternate gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on AD risk in IVW MR models with uncorrelated variants
Supplementary table 4: information on gene‐specific variant sets used to assess the effects of lipid‐lowering drug targets on cardiometabolic outcomes
Supplementary table 5: information on genome‐wide variants used to assess the effects of lowering circulating PCSK9 on AD and CAD
Supplementary table 6: Alternate MR methods for examining gene region variants in relation to AD risk, using two LD‐clumping strategies instead of principal component methodology