

Abstract
In humans, certain mutations in the gene encoding aldehyde dehydrogenase 7A1 are associated with pyridoxine-dependent epilepsy (PDE). Understanding the impact of PDE-causing mutations on the structure and activity of ALDH7A1 could allow for the prediction of symptom-severity and aid the development of patient-specific medical treatments. Herein, we investigate the biochemical and structural consequences of PDE missense mutations targeting residues in the aldehyde substrate binding site: N167S, P169S, A171V, G174V, and W175G. All but G174V could be purified for biochemical and X-ray crystallographic analysis. W175G has a relatively mild kinetic defect, exhibiting a 5-fold decrease in kcat with no change in Km. P169S and N167S have moderate defects, characterized by catalytic efficiencies of 20- and 100-times lower than wild-type, respectively. A171V has a profound functional defect, with catalytic efficiency 2000-times lower than wild-type. The crystal structures of the variants are the first for any PDE-associated mutant of ALDH7A1. The structures show that missense mutations that decrease the steric bulk of the side chain tend to create a cavity in the active site. The protein responds by relaxing into the vacant space, and this structural perturbation appears to cause misalignment of the aldehyde substrate in W175G and N167S. The P169S structure is nearly identical to that of the wild-type enzyme; however, analysis of B-factors suggests the catalytic defect may result from altered protein dynamics. The A171V structure suggests that the potential for steric clash with Val171 prevents Glu121 from ion pairing with the amino group of the aldehyde substrate.
Keywords: X-ray crystallography, enzyme kinetics, aldehyde dehydrogenase 7A1, pyridoxine-dependent epilepsy, missense mutation
Introduction
The enzyme aldehyde dehydrogenase 7A1 (ALDH7A1) catalyzes the final step in the lysine catabolic pathway – the NAD+-dependent oxidation of α-aminoadipate semialdehyde (AASAL) to α-aminoadipate (AA) (Fig. 1A). ALDH7A1 is a member of the ALDH superfamily, which is a large group of enzymes that catalyze the oxidation of various aldehydes [1–3]. ALDHs share a conserved catalytic mechanism that begins with nucleophilic attack by the catalytic cysteine (Cys302 in ALDH7A1) on the aldehyde to produce a hemithioacetal intermediate [4]. Hydride transfer to NAD+ generates NADH and the acyl-enzyme intermediate. Finally, hydrolysis of the acyl-enzyme intermediate yields the carboxylic acid product. ALDHs are known to form dimers, tetramers, and hexamers in solution [5, 6]. ALDH7A1 forms a concentration-dependent dimer-tetramer equilibrium in solution [7, 8], and the binding of NAD+ to the enzyme dramatically enhances formation of the active tetramer [9].
Fig. 1.
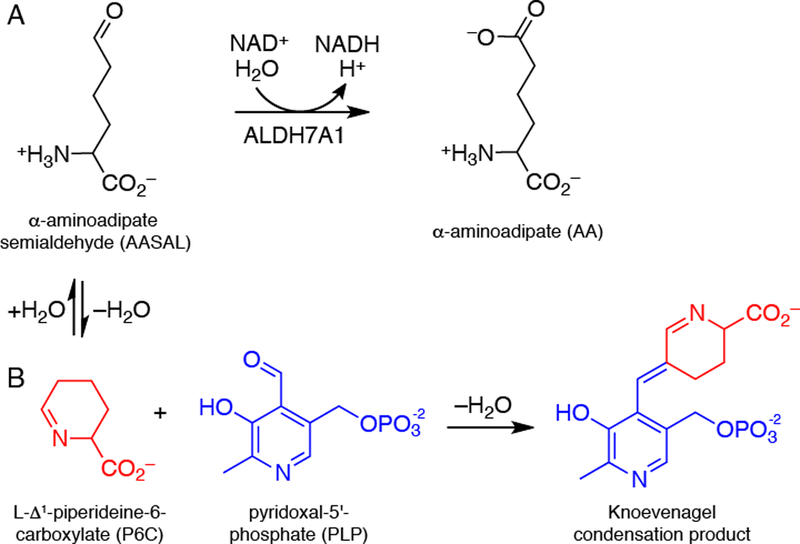
Chemical reactions relevant to ALDH7A1 and PDE. (A) The reaction catalyzed by ALDH7A1. (B) The reaction between P6C and PLP, which results in covalent inactivation of PLP. P6C forms a nonenzymatic equilibrium with AASAL.
Certain mutations in the ALDH7A1 gene cause pyridoxine-dependent epilepsy (PDE), an autosomal recessive metabolic disease characterized by seizures and in some cases intellectual disability [10, 11]. The estimated carrier frequency of ALDH7A1 mutations is 1:127, and the estimated incidence of ALDH7A1 deficiency is 1:64,352 births [12]. The molecular basis of PDE is a decrease of the ubiquitous enzyme cofactor pyridoxal 5´-phosphate (PLP) caused by Knoevenagel condensation of PLP with Δ1-piperideine-6-carboxylic acid (P6C), the cyclized form of AASAL (Fig. 1B) [13]. For this reason, patients with PDE are often treated with vitamin B6. Despite adequate seizure control, 75% of patients suffer intellectual developmental disability [10].
The mutational spectrum of PDE is vast, including more that 113 different mutations within the 18 exons of the ALDH7A1 gene. Approximately 60 of the mutations result in a single amino acid residue change in the protein (missense mutations) [10, 11]. Understanding the impact of PDE-causing mutations on the structure and activity of ALDH7A1 could allow for the prediction of symptom-severity and aid the development of patient-specific medical treatments. However, detailed biochemical and structural characterizations are lacking for the majority of the clinical missense mutations, with the exception of a group of six mutations targeting oligomer interfaces that were recently shown to abolish catalytic activity by inhibiting formation of the active tetrameric form of ALDH7A1 [7].
Herein, we investigate the biochemical and structural consequences of PDE-associated mutations that affect residues in the substrate AASAL binding site: N167S [14–21], P169S [22, 23], A171V [13, 24, 25], G174V [24, 26, 27], and W175G [22, 28, 29] (Fig. 2). Approximately 16 cases of patients harboring one of these mutations have been reported in the literature [13–29]. All of these patients have biallelic mutations consistent with autosomal recessive inheritance, and the majority are compound heterozygous for one of the mutations studied here. Asn167 is part of the oxyanion hole, which provides electrostatic stabilization of the anionic hemithioacetal intermediate [4]. Trp175 is part of an aromatic box (with Phe168 and Phe468), which contacts the aliphatic part of AASAL. The other residues occupy the second interaction sphere around AASAL. All of these residues are highly conserved among human ALDHs, except for Ala171 and Gly174 (Fig. 2C). All mutant variants except G174V could be produced as purified, stable recombinant proteins for biochemical and structural analysis. Surprisingly, the analyzed mutant proteins exhibited catalytic activity, albeit lower than that of wild-type ALDH7A1. Crystal structures of the mutant variants provide insight into the basis for the compromised enzymatic activity. Altogether, our results provide new molecular level insight into the pathology of PDE, which may be useful for therapeutic design.
Fig. 2.
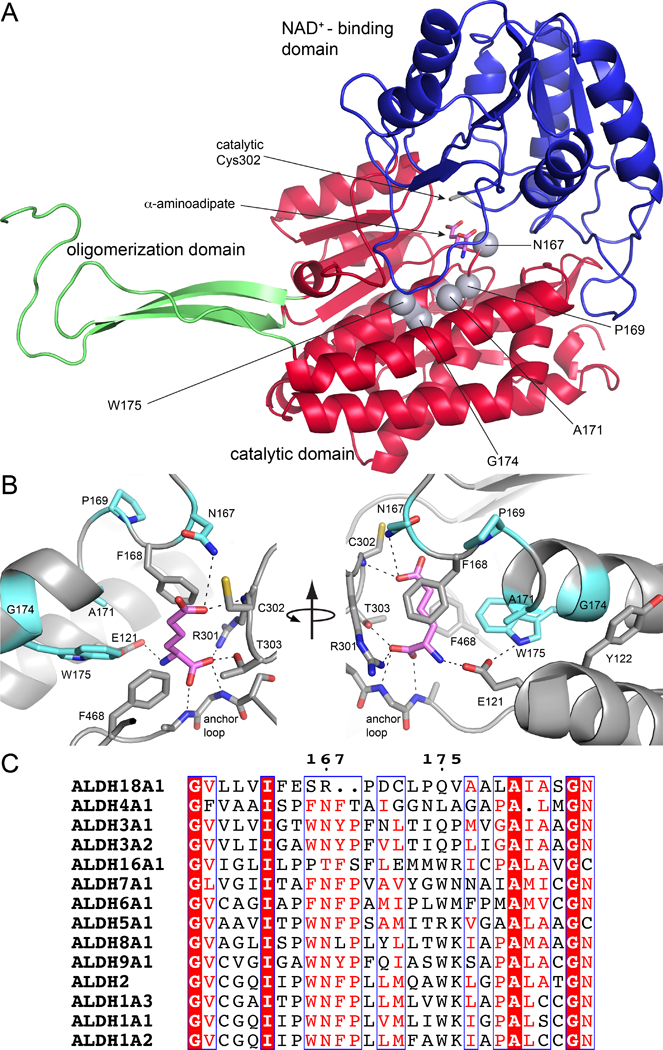
Structural context and conservation of the mutations studied. (A) The protomer of wild-type ALDH7A1 complexed with the product AA (PDB ID: 4ZUL). AA is in pink. Gray spheres indicate the mutated residues. (B) Close-up view of the AASAL-binding pocket highlighting the mutated residues in light blue. (C) A portion of a global sequence alignment of human ALDHs. The numbers refer to ALDH7A1.
Results
Steady-State Kinetic Analysis
Wild-type and mutant variant ALDH7A1 enzymes were analyzed using steady-state kinetics with AASAL as the variable substrate and NAD+ fixed at a saturating concentration. The wild-type enzyme displayed Michaelis-Menten behavior with kinetic constants comparable to those reported previously by us and others (Fig. 3A, Table 1) [8, 30, 31]. Enzymatic activity was observed for all the mutant variants, except for G174V, which was poorly soluble and could not be analyzed.
Fig. 3.
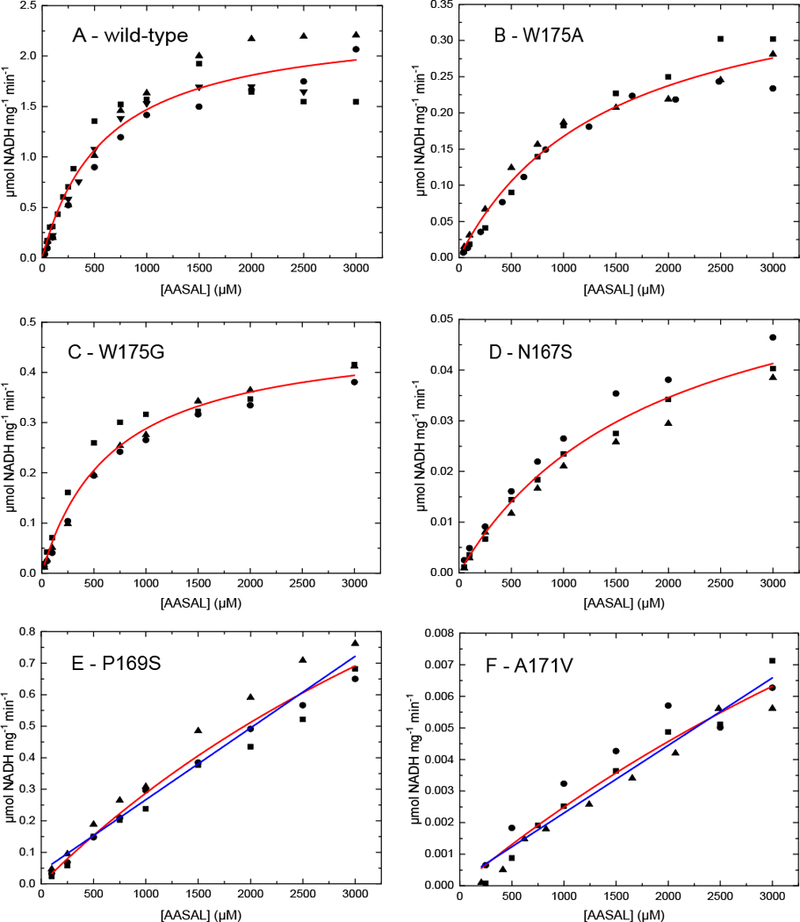
Steady-state kinetics of (A) wild-type and (B-F) mutant ALDH7A1 using AASAL as the variable substrate at a fixed NAD+ concentration of 2.5 mM (20°C, pH 8.0). The circles, squares, and triangles represent different replicate experiments performed at the same substrate concentration. Four trials were performed for the wild-type enzyme, and three trials were performed for each variant. The red curves are fits of all the replicates to the Michaelis-Menten equation. The blue lines in panels E and F are linear regression fits performed to estimate catalytic efficiency. Note the scales of the vertical axes differ.
Table 1.
Kinetic Parameters for Wild-Type and Mutant ALDH7A1 Enzymesa
|
Enzyme |
kcat (s−1) | Factor decrease in kcat |
Km (μM) | Factor increase in Km |
kcat/Km (M−1s−1) | Factor decrease in kcat/Km |
|---|---|---|---|---|---|---|
| ALDH7A1 | 2.18 ± 0.095 | 1 | 600 ± 76 | 1 | 3600 ± 4802 | 1 |
| W175A | 0.38 ± 0.025 | 6 | 1430 ± 204 | 2 | 266 ± 42 | 14 |
| W175G | 0.45 ± 0.018 | 5 | 680 ± 74 | 1 | 660 ± 77 | 5 |
| N167S | 0.063 ± 0.006 | 35 | 1900 ± 320 | 3 | 33 ± 6.4 | 110 |
| P169S | 2.14 ± 0.580 | 1 | 7000 ± 2500 | 12 | 300 ± 140 | 12 |
| A171V | 0.025 ± 0.012 | 87 | 9920 ± 5760 | 17 | 2.5 ± 1.9 | 1400 |
| P169Sb | - | - | 230 ± 6.1 | 16 | ||
| A171Vb | - | - | 2.1 ± 0.06 | 1700 | ||
The assays were performed at pH 8.0 with AASAL as the variable substrate at a fixed concentration of NAD+ of 2.5 mM. The parameters kcat and Km and the estimated uncertainties were obtained by fitting all the replicate trials for a given enzyme to the Michaelis-Menten equation using Origin software (4 replicates per AASAL concentration for wild-type; 3 replicates per AASAL concentration for each variant).
The catalytic efficiency was estimated from linear regression.
The W175G and N167S mutants clearly displayed Michaelis-Menten behavior (Fig. 3C,D); however, the catalytic efficiency was five-fold lower than wild-type for W175G and 110-fold lower for N167S (Table 1). To test the effect of having a minimal side chain at residue 175, we generated the mutant variant W175A. W175A had a catalytic efficiency approximately three-fold lower than W175G and 14-fold lower than wild-type (Table 1).
For the A171V and P169S variants, the dependence of the rate on aldehyde concentration was approximately linear, and it was not possible to saturate these enzymes with AASAL (Fig. 3E,F). The catalytic efficiency therefore was estimated from linear regression, revealing kcat/Km values of 2.1 M−1s−1 for A171V and 230 M−1s−1 for P169S (Table 1). These values are 1700-fold and 16-fold lower than wild-type, respectively.
Crystal Structures of W175G and W175A
The structures of W175G and W175A complexed with NAD+ were determined to understand the effects of the mutations on cofactor recognition. Strong electron density allowed modeling of the entire cofactor in both structures (Fig. 4A). NAD+ binds in the expected site in the Rossmann fold domain and adopts the canonical conformation observed in ALDH7A1 (Fig. 4B; e.g., PDB ID 2J6L [32] and 4ZUK [33]) and other ALDH superfamily enzymes. In this conformation, a hydrogen bond with the carbonyl of Leu269 establishes the orientation of the nicotinamide carboxamide group, the conserved Glu399 forms two hydrogen bonds with the nicotinamide ribose, Ser247 stabilizes the pyrophosphate, and Lys190 and Thr164 form hydrogen bonds with the adenine ribose. These results suggest that the mutations do not disrupt the cofactor binding site.
Fig. 4.
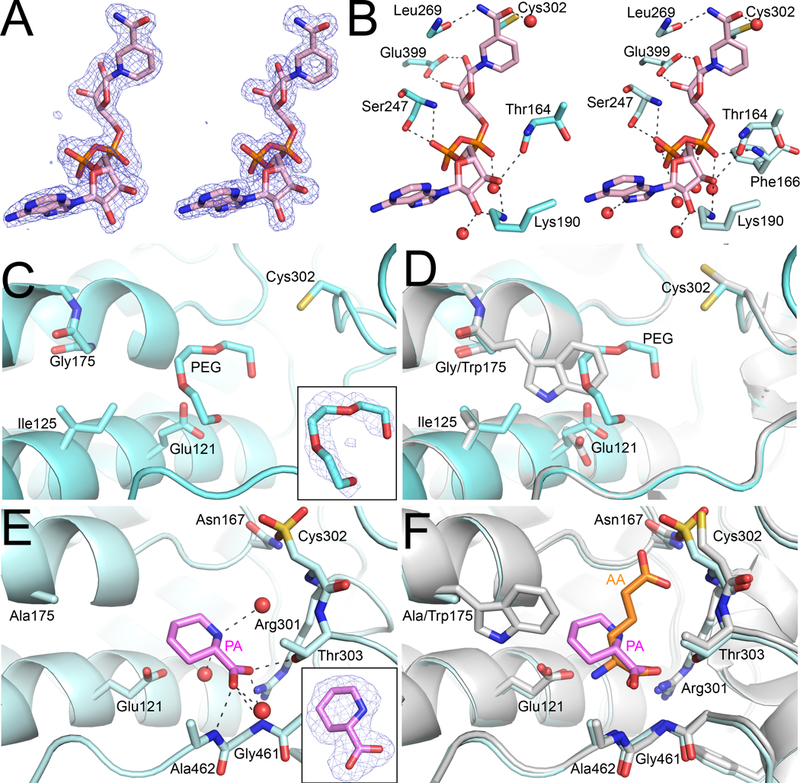
Structural impacts of ALDH7A1 W175G and W175A mutations. (A) Polder maps (4σ) showing electron density for NAD+ in both the W175G (left) and W175A (right) mutant structures. (B) The structural context of NAD+ in W175G and W175A mutant structures. (C) The active site of W175G showing a PEG molecule occupying the missing Trp side chain of W175G. (D) An overlay with the wild-type product complex (PDB ID: 4ZUK; white) for comparison. (E) The active site of W175A with PA bound. The inset shows electron density for PA from a polder map contoured to 4σ. Note that Cys302 shows apparent oxidation to sulfonic acid in this subunit. (F) An overlay with the wild-type product complex (PDB ID: 4ZUL; white) for comparison.
Removal of the bulky indole group of Trp175 creates a cavity in the AASAL pocket, and the protein and solvent respond to partially fill this cavity in W175G and W175A. In particular, Glu121 and Ile125 adopt new rotamers that move their side chains into the space occupied by Trp175 in the wild-type enzyme (Fig. 4C,D). Also, electron density consistent with a PEG molecule was observed in all eight subunits of both W175G-NAD+ and W175A-NAD+ (Fig. 4C). PEG overlaps with the indole of Trp175 of the wild-type enzyme, highlighting the extra space in the active sites of the mutant enzymes (Fig. 4D).
Attempts to determine structures of W175G and W175A complexed with AA were unsuccessful; however, we obtained a structure of W175A complexed with the P6C analog L-pipecolic acid (PA). Electron density was sufficient to model PA in all eight subunits (Fig. 4E). PA binds in the AASAL site with its carboxylate interacting with the anchor loop, similar to AA in the wild-type enzyme (Fig. 4E,F). Notably, the binding of PA induces the same cascade of conformational changes observed with the binding of AA to wild-type ALDH7A1: rearrangement of the anchor loop, rotations of Arg301 and Phe292 toward the AASAL site, and the 16 Å closure of the C-terminal arm [33]. Despite these similarities with the wild-type enzyme, PA is rotated by 180° relative to AA such that the amino group faces away from Glu121 (Fig. 4E). In this pose, the ring of PA occupies part of the Trp175 indole space (Fig. 4F). These results suggest that one function of Trp175 in the wild-type enzyme may be to correctly orient the distal end of AASAL so that it ion pairs with Glu121.
Crystal Structures of N167S
The structures of N167S complexed with NAD+ or the product AA were determined at 1.75 Å resolution and 1.9 Å resolution, respectively. The electron density maps clearly indicate that NAD+ is bound to N167S in the canonical conformation (Fig. 5A,B). Thus, the mutation does not disrupt the cofactor binding site. In both co-crystal structures, Ser167 occupies the highest frequency rotamer for serine (χ1=64°), which positions the hydroxyl group in the location of the Cγ atom of the Asn167 of the wild-type enzyme. Replacement of the Asn side chain with the shorter Ser side chain affects the local hydrogen bonding pattern. In the wild-type enzyme, the side chain of Asn167 forms a hydrogen bond with the backbone carbonyl of Gln300, and this interaction is lost in N167S.
Fig. 5.
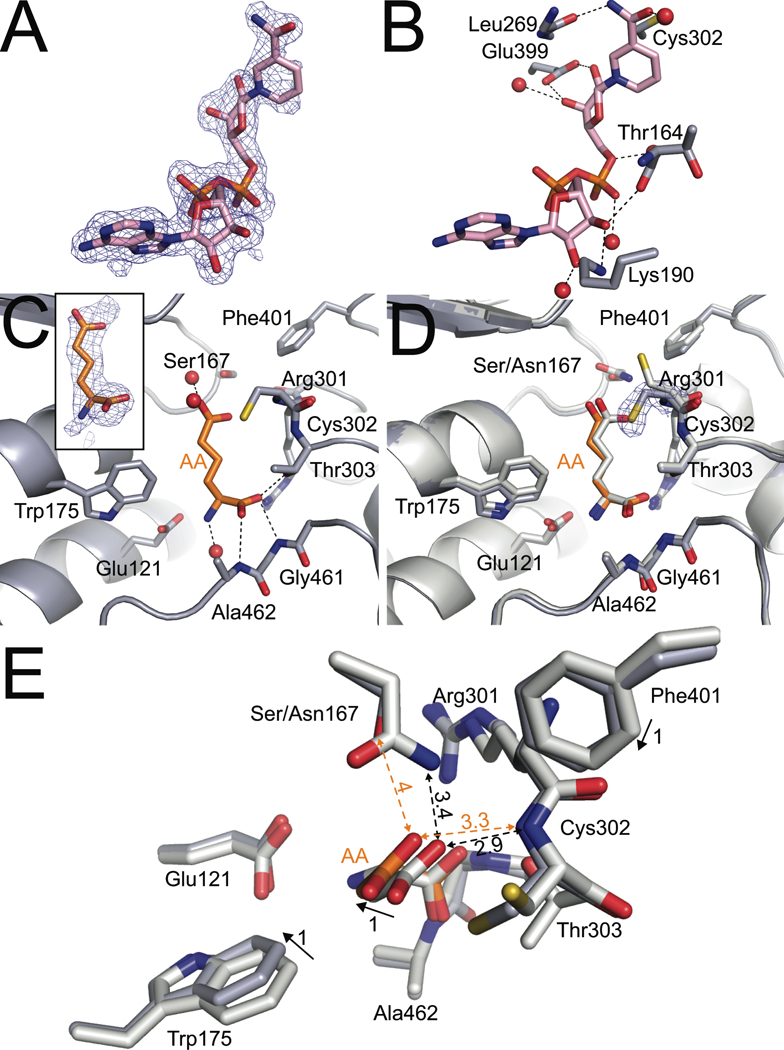
Structural impacts of ALDH7A1 N167S mutation. (A) Polder maps (4σ) showing electron density for NAD+ in the structure of N167S complexed with NAD+. (B) The structural context of NAD+ in the N167S structure. (C) The active site of N167S complexed with the product AA showing the hydrogen bonding pattern of AA. The inset shows electron density for AA (polder map, 4σ). (D) An overlay with the wild-type product complex (PDB ID: 4ZUL; white) for comparison. Electron density around Cys302 of N167S is a 2Fo-Fc map contoured at 1σ. (E) A simplified top view of the N167S active site showing the effects of the N167S mutation on the structures of the mutant (gray protein, orange AA) and wild-type (white protein and AA) active sites. The product AA has been colored white in the wild-type structure for clarity. Solid arrows depict the movement of a side chain between structures. Dashed, double-headed arrows depict the distance between atoms. The numbers next to the arrows denote distances in Å.
To understand how the mutation affects the conformation of the substrate AASAL, the structure of N167S-AA was determined at 1.9 Å resolution. Electron density was sufficient to model AA in three chains (B, E, F), with the strongest density observed in chain F (Fig. 5C). The amino acid backbone portion of the AA molecule, which is distal to the site of the mutation, hydrogen bonds to residues Gly461 and Ala462 similarly as to what is observed the wild-type enzyme (Fig. 5C,D). The mutation of Asn167 to Ser, however, causes the ε-carboxylate of AA to tilt away from Cys302 by 1 Å toward the space occupied by the side chain of Asn167 in the wild-type enzyme (Fig. 5D,E). This shift occurs in all three AA molecules. The mutation may perturb the strength of the hydrogen bond interaction between the side chain of residue 167 and the substrate. In the wild-type enzyme, Asn167 forms a weak hydrogen bond with the ε-carboxylate of AA, with an interaction distance ranging from 3.4 Å to 4.0 Å, depending on the subunit. In N167S, the corresponding interaction distance with Ser167 is 3.8 – 4.1 A.
The 1 Å movement of ε-carboxylate of AA is accompanied by several subtle structural changes in the protein. Phe401 moves by 1 Å toward the space occupied by the carboxamide group of Asn167 in the wild-type enzyme, while the side chain of Trp175 shifts 1 Å away from AA (Fig. 5C,D). We note that the overall Cα RMSD between the variant and wild-type structure is only 0.30 Å, implying that the observed displacements of 1Å are meaningful. The extra room created by these shifts allows Cys302 to adopt rotamer 3 in subunit F of N167S-AA, whereas Cys302 exclusively occupies rotamer 1 in the wild-type structure complexed with AA (Fig. 5D). Thus, as observed in the Trp175 variants, the protein relaxes into the cavity created by a mutation that reduces the steric bulk of the side chain. In the case of the N167S mutant variant, however, the creation of a cavity appears to induce a cascade of concerted movements involving both the protein and the ligand.
Crystal Structure of P169S–AA
The P169S mutation is located in the second sphere of interaction of the aldehyde substrate, yet this mutation caused a significant decrease in catalytic efficiency (12-fold). The structure of P169S complexed with AA was determined at 2.05 Å resolution. Electron density was sufficient to model AA in four chains (B, E, G, and H) (Fig. 6A). Despite the observed defect in enzymatic activity, the hydrogen-bonding pattern of the product AA in the active site (Fig. 6A) resembles that observed in the wild-type enzyme (Fig. 6B). Additionally, the overall active site architecture and side chain positions do not appear perturbed as in the Trp175 and Asn167 mutant variants.
Fig. 6.
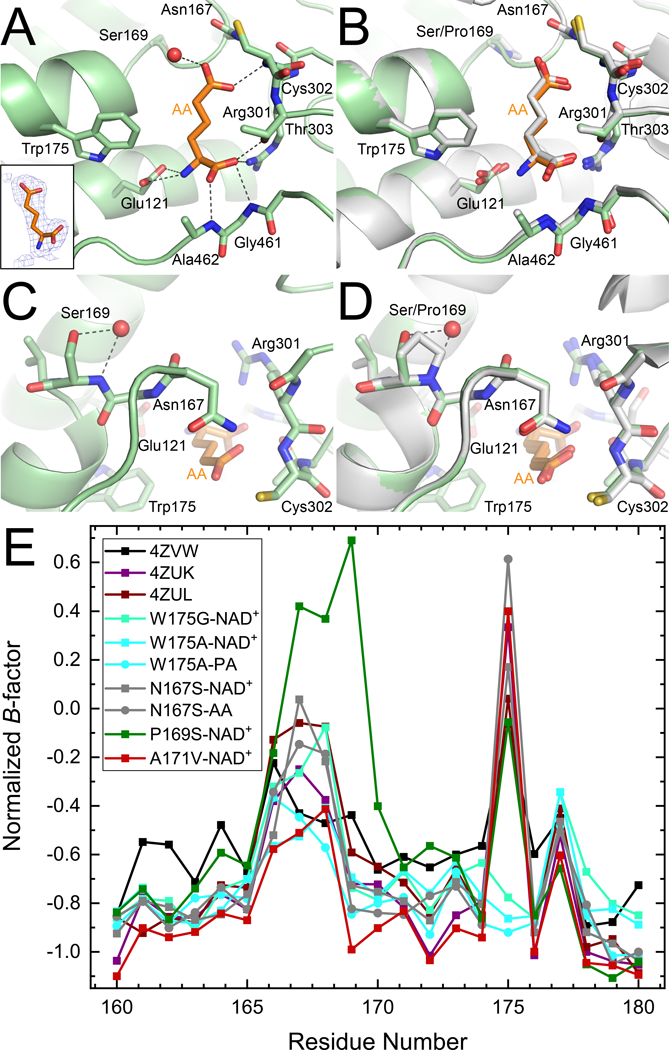
Structural impacts of ALDH7A1 P169S mutation. (A) The active site of P169S complexed with the product AA showing the hydrogen bonding pattern of AA. The inset (left) shows electron density for AA from a polder map contoured to 4σ. (B) An overlay with the wild-type structure (PDB ID: 4ZUL) for comparison (right). (C) The structural context of the P169S mutation. (D) An overlay with the wild-type structure (PDB ID: 4ZUL; right, white) for comparison. (E) Normalized B-factors for wild-type ALDH7A1 and mutant variant structures in the region of the mutations (residues 160 – 180). The wild-type structures included in this analysis are the apo enzyme (4ZVW), NAD+ complex (4ZUK), and AA complex (4ZUL). We note that all the structures have the same C2 lattice.
At the site of the mutation, the hydroxyl of Ser169 occupies the Cγ atom position of Pro169 in the wild-type enzyme (Fig. 6C). The cavity left by the mutation is filled by a water molecule, which forms hydrogen bonds with Ser169 (Fig. 6C). Ser169 and the new water molecule together mimic the 5-membered ring of Pro169 (Fig. 6D).
Because there are no obvious structural perturbations, the decreased activity of P169S may be due to non-structural factors, such as protein dynamics. To investigate this idea, we examined the crystallographic B-factors of the wild-type and mutant variant structures. This analysis was performed on normalized B-factors, which have been shown to be useful for comparing protein dynamics across multiple structures (see [34] and references therein). P169S displays conspicuously higher normalized B-factors than the other structures in residues 167 – 170 (Fig. 6E). This result is consistent with the P169S mutation increasing the flexibility of the active site.
Crystal Structure of A171V–NAD+
The A171V mutation occurs in the second sphere of interaction of the aldehyde substrate and produces a profound catalytic defect (1700-times lower catalytic efficiency). Unlike the other mutations described so far, A171V increases the steric bulk of the side chain. We determined the structure of A171V bound to NAD+ to 2.05 Å resolution. As in the other mutant structures presented in the current work, there is strong electron density for the entire NAD+ molecule (Fig. 7A), and the cofactor conformation and hydrogen-bonding pattern in the cofactor-binding site are not altered (Fig. 7B).
Fig. 7.
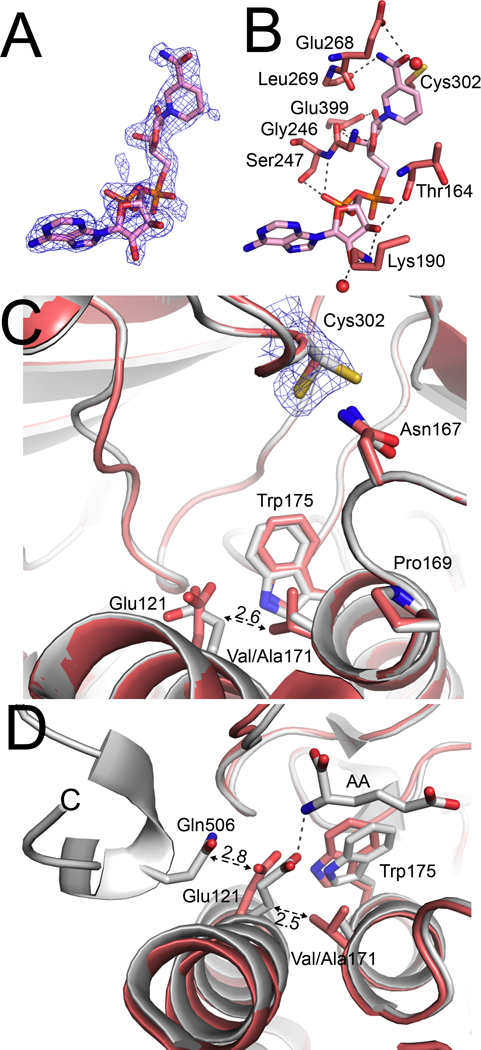
Structural impacts of ALDH7A1 A171V mutation. (A) A polder map contoured to 4σ showing electron density for NAD+ in the structure of A171V complexed with NAD+. (B) The structural context of NAD+ in A171V mutant structure. (C) The active site of A171V-NAD+ (red) and an overlay with the wild-type ALDH7A1-NAD+ structure (PDB ID: 4ZUK; white) for comparison. The dashed double-headed arrow indicates the clash distance of 2.6 Å that would have occurred if Glu121 had not changed conformation in A171. Electron density around Cys302 of A171V is a 2Fo-Fc map contoured at 1σ. (D) Overlay of A171V-NAD+ (red) and wild-type ALDH7A1-AA (PDB ID: 4ZUL; white) suggesting how steric clash with Val171 prevents Glu121 from ion pairing to AASAL and inhibits closure of the active site. The dashed, double-headed arrows depict the potential clashes that prevent active site closure. The numbers next to the arrows denote distances in Å. The dashed line denotes the ion pair formed between Glu121 and AA in the closed state of the wild-type enzyme.
The introduction of the valine side chain perturbs the local structure around position 171. Val171 adopts rotamer 3, which positions one of its Cγ atoms near the side chain of Glu121. The potential for steric clash forces a conformational change in Glu121 characterized by a displacement of its Cγ atom by 1.0 – 1.9 Å, depending on the chain considered (Fig. 7C). We note the overall RMSD of the A171V structure from the wild-type structure is only 0.25 Å, suggesting that the observed conformational change of Glu121 is meaningful. As previously described, Glu121 of the wild-type enzyme forms an ion pair with the amino group of the substrate AASAL (Fig. 2B). However, in A171V, the potential for steric clash with Val171 likely prevents Glu121 from ion pairing with AASAL (Fig. 7D). It is also possible that A171V is defective in active site closure, which is due to the aforementioned steric clash forcing Glu121 into the space occupied by Gln506 in the closed state (see the Discussion).
Discussion
In this study, we showed that several PDE-related mutations targeting residues in the aldehyde binding site of ALDH7A1 decrease catalytic activity. Three kinetic phenotypes were observed: mild, moderate, and severe. W175G has a relatively mild kinetic defect, exhibiting a 5-fold decrease in kcat with no change in Km. P169S and N167S have moderate defects, characterized by large increases in Km and catalytic efficiencies of 20–100 times lower than wild-type. A171V has a severe functional defect, characterized by catalytic efficiency almost 2000 times lower than wild-type. Interestingly, the magnitude of the kinetic defect correlated neither with the physicochemical nature of the mutation, the proximity of the mutated residue to the AASAL substrate, nor sequence conservation. For example, the W175G mutation resulted in a mild defect, although W175G is a highly non-conservative mutation, and Trp175 makes direct van der Waals contact with the substrate (3.6 Å). In contrast, A171V has a severe defect, yet Ala-to-Val is a more conservative substitution than Trp-to-Gly, and Ala171 does not directly contact the substrate (> 6 Å). Furthermore, Trp175 is highly conserved in human ALDHs, while significant sequence variation is allowed at Ala171, including Val in ALDH1A1 and ALDH3A2 (Fig. 2C). These results highlight the challenge of predicting the functional consequences of disease-related mutations, even when the structure of the wild-type protein is known.
Our result that W175G shows significant activity appears to be consistent with the relatively late onset of seizures in patients carrying the W175G mutation [35]. We note that our results differ somewhat from a previous study that found no measurable activity for recombinant W175G using assays of crude E. coli cell extracts [36]. This result shows the benefit of using purified enzyme for evaluating the kinetic properties of PDE variants of ALDH7A1.
The relatively modest impact of removing the side chain of Trp175 on catalytic function may reflect a redundancy of aromatic box interactions in the aldehyde binding site. The aromatic box is a conserved feature of aldehyde recognition by ALDHs and consists of three to four aromatic residues that contact the substrate [37]. ALDH7A1 has a 3-residue box consisting of Trp175, Phe168 and Phe468, and these three residues form a cage around the aliphatic part of AASAL (Fig. 2B). Apparently, the removal of one wall of the box is not especially detrimental to function, implying that redundancy of interaction is built into the aromatic box.
Our structural results provide new insight into the role of the oxyanion hole residue Asn167. It has traditionally been thought that the role of the conserved Asn is to provide a hydrogen bond donor to the anionic hemithioacetal intermediate, which appears in the reaction mechanism immediately after nucleophilic attack by Cys302 [38]. Our results suggest an additional role in helping align AASAL for nucleophilic attack by Cys302. The mutation of Asn167 to Ser causes the ε-carboxylate of AA to tilt away from catalytic Cys302 by 1 Å into vacant space normally occupied by the side chain carboxamide of Asn167 in the wild-type enzyme. This result implies that the aldehyde group of AASAL may not be positioned optimally for nucleophilic attack in N167S. Thus, we suggest that Asn167 plays a dual role in catalysis: its steric bulk is important for aligning the aldehyde for nucleophilic attack, and its hydrogen bonding capacity helps stabilize the hemithioacetal intermediate.
The crystal structure of P169S provided no obvious explanation for the kinetic defect of this enzyme. The mutation of proline to serine is expected to increase the flexibility of the active site. Indeed, the normalized B-factors of residues 167 – 170 are elevated in P169S, implying increased conformational flexibility in the second sphere around AASAL. It is well known that changes in active site and second sphere flexibility and dynamics are linked to changes in catalysis in a number of enzymes [39, 40], including cyclophilin A [41, 42], dihydrofolate reductase [43, 44], pentaerythritol tetranitrate reductase [45], and liver alcohol dehydrogenase [46, 47]. The latter three of these four examples all catalyze a hydride transfer step, which is highlighted as the aspect of the catalytic mechanism that is affected by changes in protein dynamics and flexibility. This is of note because the catalytic mechanism of ALDH7A1 relies on a hydride transfer. We note that enzyme thermodynamics and dynamics cannot be assessed using protein crystallography. Therefore, in the absence of any obvious structural changes, and given that Pro-to-Ser likely increases flexibility, we suggest that dynamics could potentially underlie the catalytic defect of P169S.
A171V has a severe catalytic defect, although the affected residue is in the second sphere around AASAL. The structure suggested a few possible explanations. The most obvious one is that steric clash with Val171 prevents Glu121 from making an ion pair with the amino group of AASAL. The negligible activity of A171V attests to the importance of this enzyme-substrate interaction. It is also possible that steric clash involving Val171 may prevent closure of the active site. As described previously, the active site of ALDH7A1 exhibits well-defined open and closed conformational states, with the binding of AA (and by inference, AASAL) stabilizing the closed conformation [33]. The C-terminus (residues 501 – 511) plays a central role in active site closure. During closure, the C-terminus executes a large swinging motion that translates Gln506 by 16 Å, from the surface of the enzyme into van der Waals contact with Glu121. The proximity of Gln506 to Glu121 helps position the latter to ion pair with AASAL (Fig. 7D). As mentioned above, steric clash with Val171 displaces Glu121 by > 1.0 Å. This movement forces Glu121 to occupy the space reserved for Gln506 in the closed state (Fig. 7D). Thus, the mutation of Ala171 to Val not only prevents Glu121 from interacting with AASAL, but may also prevent proper closure of the active site.
Finally, G174V is only mutation in this local cluster of PDE-related active site mutations that was not included in this study. This mutation is clearly the most detrimental as evidenced by our inability to recover adequate amounts of recombinant soluble enzyme to perform characterization. Inspection of position 174 in the wild-type structure reveals the Cα of Gly174 is in van der Waals contact with the aromatic ring of Tyr122 (3.4 Å, Fig. 2B, right panel). Likewise, Tyr122 is in close contact with eight other side chains. Thus, this region of the protein structure is very crowded and apparently cannot accommodate the bulky side chain of Val174 within the native fold of the enzyme.
Concluding Remarks
Herein, we examined the kinetics and three-dimensional structures of five PDE missense mutants of ALDH7A1. The crystal structures are the first of any ALDH7A1 PDE variant. Four of the five variants exhibited catalytic activity and could be organized into three kinetic defect phenotypes: mild (W175G), moderate (P169S, N167S), and severe (A171V). In general, mutations that increase the steric bulk of the side chain have a greater negative impact on function than those that reduce steric bulk. For mutations that decrease steric bulk, the enzyme tends to relax into the extra space created by the mutation, and this structural perturbation likely results in misalignment of the aldehyde substrate, leading to decreased catalytic activity (e.g., W175G, N167S). The mutation of proline to serine in P169S had little impact on the three-dimensional structure, suggesting this mutation may affect enzyme dynamics. The profound deleterious effects observed with two mutations that increase bulk (A171V, G174V) likely indicate that this region of the enzyme is tightly packed, and the tolerance for additional molecular volume is extremely low.
Materials and Methods
Site-Directed Mutagenesis
Site-directed mutagenesis was performed using the QuikChange Lightning site-directed mutagenesis kit (Agilent). The mutations were verified by DNA sequencing, protein mass spectrometry ( Table S1), and X-ray crystallography.
Protein Expression and Purification
Wild-type and mutant ALDH7A1 enzymes were expressed in Escherichia coli and purified with immobilized nickel affinity chromatography and size exclusion chromatography using protocols similar to those we described previously [7]. Briefly, cells were broken using sonication in a lysis buffer of 50 mM HEPES pH 8.0, 300 mM NaCl, 20 mM imidazole, 5% (v/v) glycerol, and 1% (v/v) Triton-X-100. After loading the sample on the Ni-NTA column, the resin was washed with 20 bed volumes of 50 mM HEPES pH 8.0, 300 mM NaCl, 50 mM imidazole, and 5% (v/v) glycerol, and the target protein was eluted with 50 mM HEPES pH 8.0, 300 mM NaCl, 300 mM imidazole, and 5% (v/v) glycerol. Following cleavage of the His-tag with tobacco etch virus protease, the sample was dialyzed overnight at 4°C against 50 mM HEPES pH 8.0, 300 mM NaCl, and 5% (v/v) glycerol. The sample was passed over Ni-NTA resin, and the flow-through was concentrated and dialyzed overnight at 4°C against 50 mM HEPES, pH 8.0, 100 mM NaCl, 1 mM Tris(hydroxypropyl) phosphine, and 5% (v/v) glycerol. The protein was further purified by size-exclusion chromatography on a Superdex 200 10–30 column with a mobile phase of 50 mM HEPES pH 8.0, 100 mM NaCl, and 5% (v/v) glycerol. The purified protein was concentrated to over 10 mg mL−1, aliquoted, flash cooled in liquid nitrogen, and stored at −80°C.
All mutant variant proteins were analyzed by mass spectrometry, as described previously [8], to confirm the presence of the desired mutation. For each sample, the experimental molecular masses were within 1.35 Da of the expected molecular masses of each variant (Supplementary Table S1).
Steady-State Kinetics Assays
ALDH7A1 enzymatic activity was measured by monitoring NADH production at a wavelength of 340 nm using an Agilent 8453 Hewlett-Packard G1103A spectrophotometer. Each assay was performed at 20°C for 300 seconds with measurements taken at 3-second intervals. The reaction assay buffer was 50 mM pyrophosphate buffer at pH 8.0. Enzyme stock solutions were prepared by diluting to the desired concentration with 50 mM pyrophosphate buffer at pH 8.0 supplemented with 5 mM NAD+. AASAL was used as the variable substrate (10 μM – 3000 μM) with NAD+ at a fixed saturating concentration (2.5 mM). AASAL was synthesized and quantitated as previously reported [8]. Buffer, NAD+, and AASAL were mixed by inversion in a 1 cm 284 QS quartz cuvette (Fisher Scientific) and blanked before addition of enzyme. The final enzyme concentrations in the assays were as follows: 0.07 μM wild-type, 2.2 μM N167S, 0.36 μM P169S, 3.3 μM A171V, 1.1 μM W175A, and 0.36 μM W175G. Triplicate data sets were collected, and kinetic constants were estimated by globally fitting the initial rate data to the Michaelis-Menten equation using Origin 2017. It was not possible to saturate A171V and P169S with AASAL; in these cases, linear regression was applied, and the slope of the best-fit line was interpreted as an approximation of kcat/Km.
Protein Crystallization
Crystallization trials were performed at 20°C using sitting drop vapor diffusion. Wild-type ALDH7A1 crystals were grown in space group C2 as described previously using a reservoir containing 0.2 M MgCl2, 0.1 M Bis-Tris pH 4.2 – 6.7, and 25% (w/v) PEG 3350 [33]. These crystals were used to make microseed stocks to aid crystallization of the mutant variants.
Crystals of the mutant enzymes complexed with NAD+ were grown by co-crystallization using 5 – 6 mg mL−1 enzyme and 15 mM NAD+. Except for A171V, the reservoir was 0.1 M MgCl2, 0.1 M sodium acetate trihydrate, 20% (w/v) PEG 4000, and 0.1 M Tris, with varying pH (N167S, pH 6.7; W175A, pH 7.5; W175G, pH 6.8). Crystallization trials were set up with an Oryx8 robot (Douglas Instruments) with microseeding from wild-type seeds. The crystals were prepared for low temperature data collection by soaking in a cryobuffer consisting of the reservoir supplemented with 15% (v/v) PEG 200 and 5 mM NAD+, followed by flash-cooling in liquid nitrogen. A171V complexed with NAD+ was grown in 0.2 M MgCl2, 25% (w/v) PEG 3350, and 0.1 M Bis-Tris pH 5.8, and 18% (v/v) ethylene glycol was used as the cryoprotectant.
The W175A and N167S mutants complexed with L-pipecolic acid (PA) were grown in 0.1 M MgCl2, 0.1 M Tris pH 7.2, 0.1 M sodium acetate trihydrate, and 20% (w/v) PEG 4000. The crystals were grown by co-crystallization using 5 – 6 mg mL−1 enzyme and with PA concentration of 175 mM for W175A and 210 mM for N167S. Trials were set up with an Oryx8 robot with microseeding from wild-type seeds. Crystals were cryoprotected with the reservoir solution supplemented with 15% (v/v) PEG 200 and 30 mM PA.
Crystals of mutant enzymes complexed with AA were grown in 0.2 M MgCl2, 25% (w/v) PEG 3350, and 0.1 M Bis-Tris (pH 5.3 for N167S and pH 6.0 for P169S). Crystals were grown by co-crystallization using 5 mg mL−1 enzyme and 65 – 70 mM AA. The Oryx8 robot was used to set up trials with microseeding from wild-type seeds. The cryoprotectant used was the reservoir solution supplemented with 18% (v/v) ethylene glycol and 10 mM AA.
X-ray Diffraction Data Collection and Structure Determination
X-ray diffraction data sets were collected at ALS beamline 4.2.2 using a Taurus-1 CMOS detector in shutterless mode. Each data sets consisted of 900 images covering a rotation range of 180° with a total exposure time of 360 sec. The data sets were integrated and scaled with XDS [48]. Datasets for W175G-NAD+, W175A-NAD+, N167S-NAD+, P169S-AA, and N167S-AA consisted of high-resolution and low-resolution scans; merging of scans was performed with XSCALE [49]. Data processing statistics are listed in Tables 2 and 3.
Table 2.
Data Collection and Refinement Statistics for W175G and W175A
| W175G -NAD+ | W175A- NAD+ | W175A-PA | |
|---|---|---|---|
| Space group | C2 | C2 | C2 |
| Beamline | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 |
| Unit cell parameters (Å,°) | a = 155.2, b = 161.5,c = 158.6, β = 94.7 | a = 156.1, b = 162.3, c = 159.6, β = 94.4 | a = 155.3, b = 160.0,c = 158.3, β = 95.1 |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 |
| Resolution (Å) | 62.78 – 1.85 (1.88–1.85) | 53.91 – 1.70 (1.73 – 1.70) | 62.90 – 1.88 (1.91 – 1.88) |
| Observationsa | 1,073,897 | 1,994,816 | 1,137,039 |
| Unique reflections | 312,381 | 429,401 | 316,551 |
| Rmerge(I)a | 0.046 (0.324) | 0.072 (0.270) | 0.052 (0.520) |
| Rmeas(I)a | 0.062 (0.451) | 0.087 (0.366) | 0.071 (0.701) |
| Rpim(I)a | 0.042 (0.312) | 0.047 (0.246) | 0.048 (0.467) |
| Mean I/σa | 14.6 (2.9) | 13.4 (3.1) | 14.0 (1.8) |
| Completeness (%)a | 94.6 (92.7) | 99.2 (89.4) | 99.6 (99.8) |
| Multiplicitya | 3.4 (3.0) | 4.6 (2.8) | 3.6 (3.2) |
| No. of protein residues | 4072 | 4072 | 4070 |
| No. of protein atoms | 30854 | 31223 | 30780 |
| No. of NAD+ atoms | 352 | 352 | N/A |
| No. of PA atoms | N/A | N/A | 72 |
| No. of waters | 1592 | 3323 | 2057 |
| Rcrysta | 0.169 (0.258) | 0.147 (0.205) | 0.172 (0.290) |
| Rfreea,b | 0.212 (0.310) | 0.175 (0.234) | 0.215 (0.331) |
| rmsd bonds (Å) | 0.006 | 0.006 | 0.006 |
| rmsd angles (°) | 0.770 | 0.791 | 0.767 |
| Ramachandran plotc | |||
| Favored (%) | 96.79 | 96.84 | 96.81 |
| Outliers (%) | 0.22 | 0.20 | 0.20 |
| Clashscore (PR)c | 2.75 (99) | 1.54 (99) | 2.75 (99) |
| MolProbity score (PR)c | 1.32 (98) | 1.08 (100) | 1.26 (99) |
| Average B (Å2) | |||
| Protein | 26.5 | 15.0 | 28.6 |
| NAD+ | 27.6 | 14.0 | N/A |
| PA | N/A | N/A | 39.9 |
| Water | 27.7 | 23.0 | 30.9 |
| Coord. error (Å)d | 0.21 | 0.15 | 0.22 |
| PDB code | 6O4B | 6O4C | 6O4D |
Values for the outer resolution shell of data are given in parenthesis.
5% test set.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate from PHENIX.
Table 3.
Data Collection and Refinement Statistics for N167S, P169S, and A171V
| N167S-NAD+ | N167S-AA | P169S-AA | A171V-NAD+ | |
|---|---|---|---|---|
| Space group | C2 | C2 | C2 | C2 |
| Beamline | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 |
| Unit cell parameters (Å,°) | a = 154.9, b = 161.7,c = 158.4, β= 94.7 | a = 155.2, b = 161.5,c = 158.6, β = 94.7 | a = 155.3, b = 160.0,c = 158.3, β = 95.1 | a = 156.0, b = 161.4, c = 159.2, β = 95.0 |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 | 1.000 |
| Resolution (Å) | 56.49 – 1.75 (1.78 – 1.75) | 48.79 – 1.90 (1.93 – 1.90) | 49.07 – 2.05 (2.09 – 2.05) | 62.86 – 2.05 (2.09 – 2.05) |
| Observationsa | 1,352,436 | 2,575,937 | 1,321,614 | 876,881 |
| Unique reflections | 366,672 | 284,125 | 238,744 | 244,055 |
| Rmerge(I)a | 0.042 (0.399) | 0.116 (0.831) | 0.101 (0.829) | 0.060 (0.536) |
| Rmeas(I)a | 0.058 (0.561) | 0.130 (0.982) | 0.122 (1.129) | 0.083 (0.722) |
| Rpim(I)a | 0.039 (0.393) | 0.057 (0.510) | 0.067 (0.761) | 0.046 (0.410) |
| Mean I/σa | 14.4 (2.3) | 15.3 (1.9) | 12.1 (1.1) | 12.5 (1.8) |
| Completeness (%)a | 94.2 (94.8) | 96.5 (74.3) | 99.3 (92.4) | 99.7 (97.5) |
| Multiplicitya | 3.7 (3.4) | 9.1 (5.8) | 5.5 (3.1) | 3.6 (3.0) |
| No. of protein residues | 4067 | 4068 | 4066 | 3977 |
| No. of protein atoms | 30877 | 31179 | 30767 | 29946 |
| No. of NAD+ atoms | 352 | N/A | N/A | 352 |
| No. of AA atoms | N/A | 33 | 44 | N/A |
| No. of waters | 2050 | 1758 | 952 | 1343 |
| Rcrysta | 0.168 (0.269) | 0.173 (0.275) | 0.1922 (0.339) | 0.1743 (0.288) |
| Rfreea,b | 0.202 (0.303) | 0.2178 (0.299) | 0.2503 (0.381) | 0.2189 (0.332) |
| rmsd bonds (Å) | 0.006 | 0.006 | 0.007 | 0.007 |
| rmsd angles (°) | 0.791 | 0.793 | 0.807 | 0.816 |
| Ramachandran plotc | ||||
| Favored (%) | 96.99 | 96.72 | 96.32 | 96.92 |
| Outliers (%) | 0.20 | 0.20 | 0.22 | 0.20 |
| Clashscore (PR)c | 3.15 (98) | 2.83 (99) | 3.55 (99) | 2.28 (99) |
| MolProbity score (PR)c | 1.28 (98) | 1.28 (99) | 1.52 (97) | 1.26 (99) |
| Average B (Å2) | ||||
| Protein | 29.8 | 24.0 | 38.0 | 28.2 |
| NAD+ | 33.7 | N/A | N/A | 33.9 |
| AA | N/A | 33.0 | 51.6 | N/A |
| Water | 32.7 | 26.5 | 34.6 | 29.2 |
| Coord. error (Å)d | 0.21 | 0.22 | 0.31 | 0.25 |
| PDB code | 6O4E | 6O4F | 6O4G | 6O4H |
Values for the outer resolution shell of data are given in parenthesis.
5% test set.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate from PHENIX.
In all cases, the space group is C2 with the following unit cell dimensions: a = 155–156 Å, b = 160–162 Å, c = 158.5–159.5 Å, and β = 94–95º; there are eight molecules in the asymmetric unit arranged as two tetramers. Models for refinement were prepared by molecular replacement in CCP4i MolRep [50]. For structures co-crystallized with NAD+, Protein Data Bank (PDB) entry 4ZUK was used as the search model, and for structures co-crystallized with AA or PA, PDB entry 4ZUL was used as the search model. Each model was modified such that Cys302 and corresponding mutated residue were truncated to an alanine, and residues 500 – 511, which correspond to the mobile C-terminus [33], were deleted. PHENIX [51] was used for refinement, and COOT [52] was used for model building. The structures were validated using the PDB validation server and MolProbity [53].
Structural and Amino Acid Sequence Analyses
The root mean square deviation (RMSD) of the variant structure from the wild-type structure was calculated using CNS version 1.3 [54]. For each of the eight chains in the asymmetric unit, the variant structure was fitted to the appropriate wild-type structure using the Cα atom positions, and the RMSD was averaged over the chains. These calculations yielded average Cα RMSDs in the range of 0.14 – 0.33 Å.
CNS was also used to calculate normalized B-factors for each structure. The normalized B-factor is defined as Bnorm = (B - <B>)/σB, where B is the isotropic atomic B-factor from crystallographic refinement, <B> is the mean of B for the structure, and σB is the standard deviation of B [55]. Normalized B-factors have been shown to be useful for comparing motions across multiple structures (see [34] and references therein).
Amino acid sequence alignments were created with Clustal Omega [56] and visualized with ESPript [57].
Supplementary Material
Acknowledgements
We thank Beverly DaGue of the University of Missouri Charles W. Gehrke Proteomics Center for performing mass spectrometry analysis. Research reported in this publication was supported by the NIGMS of the National Institutes of Health under award number R01GM093123 (to J.J.T.). We thank Jay Nix for his assistance with data collection at Advanced Light Source beamline 4.2.2. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract No. DE-AC02–05CH11231.
Abbreviations:
- AA
α-aminoadipate
- AASAL
α-aminoadipate semialdehyde
- ALDH
aldehyde dehydrogenase
- ALDH7A1
aldehyde dehydrogenase 7A1
- P6C,
Δ1-piperideine-6-carboxylic acid
- PA
L-pipecolic acid
- PDE
pyridoxine-dependent epilepsy
- PLP
pyridoxal 5′-phosphate
Footnotes
Enzymes: Aldehyde dehydrogenase 7A1 (1.2.1.31)
Databases: Coordinates have been deposited in the Protein Data Bank under the following accession codes: 6O4B, 6O4C, 6O4D, 6O4E, 6O4F, 6O4G, 6O4H.
Conflict of Interest: The authors declare no competing financial interest.
References
- 1.Sophos NA & Vasiliou V (2003) Aldehyde dehydrogenase gene superfamily: the 2002 update, Chem Biol Interact. 143–144, 5–22. [DOI] [PubMed] [Google Scholar]
- 2.Vasiliou V & Nebert DW (2005) Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family, Hum Genomics. 2, 138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasiliou V, Thompson DC, Smith C, Fujita M & Chen Y (2013) Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells, Chem Biol Interact. 202, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD & Vasiliou V (2012) Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application, Pharmacol Rev. 64, 520–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo M, Singh RK & Tanner JJ (2013) Structural determinants of oligomerization of delta(1)-pyrroline-5-carboxylate dehydrogenase: identification of a hexamerization hot spot, J Mol Biol. 425, 3106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner JJ (2015) SAXS fingerprints of aldehyde dehydrogenase oligomers, Data in Brief. 5, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korasick DA, Tanner JJ & Henzl MT (2017) Impact of disease-Linked mutations targeting the oligomerization interfaces of aldehyde dehydrogenase 7A1, Chem Biol Interact. 276, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korasick DA, Wyatt JW, Luo M, Laciak AR, Ruddraraju K, Gates KS, Henzl MT & Tanner JJ (2017) Importance of the C-Terminus of Aldehyde Dehydrogenase 7A1 for Oligomerization and Catalytic Activity, Biochemistry. 56, 5910–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korasick DA, White TA, Chakravarthy S & Tanner JJ (2018) NAD(+) promotes assembly of the active tetramer of aldehyde dehydrogenase 7A1, FEBS Lett. 592, 3229–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Karnebeek CD, Tiebout SA, Niermeijer J, Poll-The BT, Ghani A, Coughlin CR 2nd, Van Hove JL., Richter JW, Christen HJ, Gallagher R, Hartmann H & Stockler-Ipsiroglu S. (2016) Pyridoxine-dependent epilepsy: An expanding clinical spectrum, Pediatr Neurol. 59, 6–12. [DOI] [PubMed] [Google Scholar]
- 11.Stockler S, Plecko B, Gospe SM Jr., Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, Tein I, Jakobs C, Clayton P & Van Hove JL (2011) Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up, Molecular genetics and metabolism. 104, 48–60. [DOI] [PubMed] [Google Scholar]
- 12.Coughlin CR 2nd, Swanson MA, Spector E, Meeks NJL, Kronquist KE, Aslamy M, Wempe MF, van Karnebeek CDM, Gospe SM, Aziz VG Jr, Tsai BP, Gao H, Nagy PL, Hyland K, van Dooren SJM, Salomons GS. & Van Hove JLK. (2019) The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy, Journal of inherited metabolic disease. 42, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B & Clayton PT (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures, Nature medicine. 12, 307–9. [DOI] [PubMed] [Google Scholar]
- 14.Tincheva S, Todorov T, Todorova A, Georgieva R, Stamatov D, Yordanova I, Kadiyska T, Georgieva B, Bojidarova M, Tacheva G, Litvinenko I & Mitev V (2015) First cases of pyridoxine-dependent epilepsy in Bulgaria: novel mutation in the ALDH7A1 gene, Neurological Sciences. 36, 2209–2212. [DOI] [PubMed] [Google Scholar]
- 15.Mercimek-Mahmutoglu S, Donner EJ & Siriwardena K (2013) Normal plasma pipecolic acid level in pyridoxine dependent epilepsy due to ALDH7A1 mutations, Molecular Genetics and Metabolism. 110, 197. [DOI] [PubMed] [Google Scholar]
- 16.Mercimek-Mahmutoglu S, Cordeiro D, Cruz V, Hyland K, Struys EA, Kyriakopoulou L & Mamak E (2014) Novel therapy for pyridoxine dependent epilepsy due to ALDH7A1 genetic defect: l-arginine supplementation alternative to lysine-restricted diet, European Journal of Paediatric Neurology. 18, 741–746. [DOI] [PubMed] [Google Scholar]
- 17.Nasr E, Eva M, Anette F, Elizabeth JD & Saadet M-M (2014) Long-Term Treatment Outcome of two Patients With Pyridoxine-Dependent Epilepsy Caused byALDH7A1 Mutations: Normal Neurocognitive Outcome, Journal of Child Neurology. 30, 648–653. [DOI] [PubMed] [Google Scholar]
- 18.Perez B, Gutierrez-Solana LG, Verdu A, Merinero B, Yuste-Checa P, Ruiz-Sala P, Calvo R, Jalan A, Marin LL, Campos O, Ruiz MA, San Miguel M, Vazquez M, Castro M, Ferrer I, Navarrete R, Desviat LR, Lapunzina P, Ugarte M & Perez-Cerda C (2013) Clinical, biochemical, and molecular studies in pyridoxine-dependent epilepsy. Antisense therapy as possible new therapeutic option, Epilepsia. 54, 239–48. [DOI] [PubMed] [Google Scholar]
- 19.Bennett CL, Chen Y, Hahn S, Glass IA & Gospe SM Jr. (2009) Prevalence of ALDH7A1 mutations in 18 North American pyridoxine-dependent seizure (PDS) patients, Epilepsia. 50, 1167–75. [DOI] [PubMed] [Google Scholar]
- 20.Friedman SD, Ishak GE, Poliachik SL, Poliakov AV, Otto RK, Shaw DW, Willemsen MA, Bok LA & Gospe SM Jr. (2014) Callosal alterations in pyridoxine-dependent epilepsy, Developmental medicine and child neurology. 56, 1106–10. [DOI] [PubMed] [Google Scholar]
- 21.Jung S, Tran NT, Gospe SM Jr. & Hahn SH (2013) Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS, Mol Genet Metab. 110, 237–40. [DOI] [PubMed] [Google Scholar]
- 22.Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, Hemingway C, Marlow N, Rennie J, Baxter P, Dulac O, Nabbout R, Craigen WJ, Schmitt B, Feillet F, Christensen E, De Lonlay P, Pike MG, Hughes MI, Struys EA, Jakobs C, Zuberi SM & Clayton PT (2010) Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency), Brain. 133, 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliveira R, Pereira C, Rodrigues F, Alfaite C, Garcia P, Robalo C, Fineza I, Gonçalves O, Struys E, Salomons G, Jakobs C & Diogo L (2013) Pyridoxine-dependent epilepsy due to antiquitin deficiency: achieving a favourable outcome, Epileptic Disorders. 15, 400–406. [DOI] [PubMed] [Google Scholar]
- 24.Plecko B, Hikel C, Korenke GC, Schmitt B, Baumgartner M, Baumeister F, Jakobs C, Struys E, Erwa W & Stöckler-Ipsiroglu S (2005) Pipecolic Acid as a Diagnostic Marker of Pyridoxine-Dependent Epilepsy, Neuropediatrics. 36, 200–205. [DOI] [PubMed] [Google Scholar]
- 25.Schmitt B, Baumgartner M, Mills PB, Clayton PT, Jakobs C, Keller E & Wohlrab G (2010) Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency, Developmental medicine and child neurology. 52, e133–42. [DOI] [PubMed] [Google Scholar]
- 26.Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E, Jakobs C, Hartmann H, Luecke T, di Capua M, Korenke C, Hikel C, Reutershahn E, Freilinger M, Baumeister F, Bosch F & Erwa W (2007) Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene, Human mutation. 28, 19–26. [DOI] [PubMed] [Google Scholar]
- 27.Kanno J, Kure S, Narisawa A, Kamada F, Takayanagi M, Yamamoto K, Hoshino H, Goto T, Takahashi T, Haginoya K, Tsuchiya S, Baumeister FAM, Hasegawa Y, Aoki Y, Yamaguchi S & Matsubara Y (2007) Allelic and non-allelic heterogeneities in pyridoxine dependent seizures revealed by ALDH7A1 mutational analysis, Molecular Genetics and Metabolism. 91, 384–389. [DOI] [PubMed] [Google Scholar]
- 28.Mefford HC, Zemel M, Geraghty E, Cook J, Clayton PT, Paul K, Plecko B, Mills PB, Nordli DR & Gospe SM (2015) Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination, Neurology. 85, 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baxter P, Griffiths P, Kelly T & Gardner-Medwin D (1996) Pyridoxine-Dependent Seizures: Demographic, Clinical, MRI and Psychometric Features, and Effect of Dose on Intelligence Quotient, Developmental Medicine & Child Neurology. 38, 998–1006. [DOI] [PubMed] [Google Scholar]
- 30.Brocker C, Lassen N, Estey T, Pappa A, Cantore M, Orlova VV, Chavakis T, Kavanagh KL, Oppermann U & Vasiliou V (2010) Aldehyde Dehydrogenase 7A1 (ALDH7A1) Is a Novel Enzyme Involved in Cellular Defense against Hyperosmotic Stress, Journal of Biological Chemistry. 285, 18452–18463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koncitikova R, Vigouroux A, Kopecna M, Andree T, Bartos J, Sebela M, Morera S & Kopecny D (2015) Role and structural characterization of plant aldehyde dehydrogenases from family 2 and family 7, Biochemical Journal. 468, 109–23. [DOI] [PubMed] [Google Scholar]
- 32.Brocker C, Lassen N, Estey T, Pappa A, Cantore M, Orlova VV, Chavakis T, Kavanagh KL, Oppermann U & Vasiliou V (2010) Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress, J Biol Chem. 285, 18452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo M & Tanner JJ (2015) Structural basis of substrate recognition by aldehyde dehydrogenase 7A1, Biochemistry. 54, 5513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson TW, Gallego RA, Brooun A, Gehlhaar D & McTigue M (2018) Reviving B-Factors: Retrospective Normalized B-Factor Analysis of c-ros Oncogene 1 Receptor Tyrosine Kinase and Anaplastic Lymphoma Kinase L1196M with Crizotinib and Lorlatinib, ACS Med Chem Lett. 9, 878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, Hemingway C, Marlow N, Rennie J, Baxter P, Dulac O, Nabbout R, Craigen WJ, Schmitt B, Feillet F, Christensen E, De Lonlay P, Pike MG, Hughes MI, Struys EA, Jakobs C, Zuberi SM & Clayton PT (2010) Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency), Brain : a journal of neurology. 133, 2148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coulter-Mackie MB, Li A, Lian Q, Struys E, Stockler S & Waters PJ (2012) Overexpression of human antiquitin in E. coli: enzymatic characterization of twelve ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy, Molecular genetics and metabolism. 106, 478–81. [DOI] [PubMed] [Google Scholar]
- 37.Riveros-Rosas H, Gonzalez-Segura L, Julian-Sanchez A, Diaz-Sanchez AG & Munoz-Clares RA (2013) Structural determinants of substrate specificity in aldehyde dehydrogenases, Chem Biol Interact. 202, 51–61. [DOI] [PubMed] [Google Scholar]
- 38.Luo M, Gates KS, Henzl MT & Tanner JJ (2015) Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1, ACS Chem Biol. 10, 693–7. [DOI] [PubMed] [Google Scholar]
- 39.Agarwal PK (2006) Enzymes: An integrated view of structure, dynamics and function, Microb Cell Fact. 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pabis A, Risso VA, Sanchez-Ruiz JM & Kamerlin SC (2018) Cooperativity and flexibility in enzyme evolution, Curr Opin Struct Biol. 48, 83–92. [DOI] [PubMed] [Google Scholar]
- 41.Henzler-Wildman K & Kern D (2007) Dynamic personalities of proteins, Nature. 450, 964–72. [DOI] [PubMed] [Google Scholar]
- 42.Eisenmesser EZ, Bosco DA, Akke M & Kern D (2002) Enzyme dynamics during catalysis, Science. 295, 1520–3. [DOI] [PubMed] [Google Scholar]
- 43.Mhashal AR, Pshetitsky Y, Eitan R, Cheatum CM, Kohen A & Major DT (2018) Effect of Asp122 Mutation on the Hydride Transfer in E. coli DHFR Demonstrates the Goldilocks of Enzyme Flexibility, J Phys Chem B. 122, 8006–8017. [DOI] [PubMed] [Google Scholar]
- 44.Klinman JP & Kohen A (2014) Evolutionary aspects of enzyme dynamics, J Biol Chem. 289, 30205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iorgu AI, Baxter NJ, Cliff MJ, Levy C, Waltho JP, Hay S & Scrutton NS (2018) Nonequivalence of Second Sphere “Noncatalytic” Residues in Pentaerythritol Tetranitrate Reductase in Relation to Local Dynamics Linked to H-Transfer in Reactions with NADH and NADPH Coenzymes, Acs Catal. 8, 11589–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colby TD, Bahnson BJ, Chin JK, Klinman JP & Goldstein BM (1998) Active site modifications in a double mutant of liver alcohol dehydrogenase: structural studies of two enzyme-ligand complexes, Biochemistry. 37, 9295–304. [DOI] [PubMed] [Google Scholar]
- 47.Billeter SR, Webb SP, Agarwal PK, Iordanov T & Hammes-Schiffer S (2001) Hydride transfer in liver alcohol dehydrogenase: quantum dynamics, kinetic isotope effects, and role of enzyme motion, J Am Chem Soc. 123, 11262–72. [DOI] [PubMed] [Google Scholar]
- 48.Kabsch W (2010) XDS, Acta Crystallographica Section D: Biological Crystallography. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kabsch W (2010) Integration, scaling, space-group assignment and post-refinement, Acta Crystallographica Section D. 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lebedev AA, Vagin AA & Murshudov GN (2008) Model preparation in MOLREP and examples of model improvement using X-ray data, Acta Crystallographica Section D: Biological Crystallography. 64, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC & Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallographica Section D. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG & Cowtan K (2010) Features and development of Coot, Acta Crystallographica Section D: Biological Crystallography. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS & Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids, Nucleic Acids Research. 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brunger AT (2007) Version 1.2 of the Crystallography and NMR system, Nat Protoc. 2, 2728–33. [DOI] [PubMed] [Google Scholar]
- 55.Parthasarathy S & Murthy MR (1997) Analysis of temperature factor distribution in high-resolution protein structures, Protein Sci. 6, 2561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD & Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, Mol Syst Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robert X & Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server, Nucleic Acids Res. 42, W320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.