

Abstract
Transcription factors critical for the transition of normal breast epithelium to ductal carcinoma in situ (DCIS) and invasive breast cancer are not clearly defined. Here, we report that the expression of a subset of YAP‐activated and YAP‐repressed genes in normal mammary and early‐stage breast cancer cells is dependent on the nuclear co‐activator AIB1. Gene expression, sequential ChIP, and ChIP‐seq analyses show that AIB1 and YAP converge upon TEAD for transcriptional activation and repression. We find that AIB1‐YAP repression of genes at the 1q21.3 locus is mediated by AIB1‐dependent recruitment of ANCO1, a tumor suppressor whose expression is progressively lost during breast cancer progression. Reducing ANCO1 reverts AIB1‐YAP‐dependent repression, increases cell size, and enhances YAP‐driven aberrant 3D growth. Loss of endogenous ANCO1 occurs during DCIS xenograft progression, a pattern associated with poor prognosis in human breast cancer. We conclude that increased expression of AIB1‐YAP co‐activated targets coupled with a loss of normal ANCO1 repression is critical to patterns of gene expression that mediate malignant progression of early‐stage breast cancer.
Keywords: 1q21.3, AIB1, ANCO1, ductal carcinoma in situ, YAP
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Signal Transduction
The nuclear co‐activator AIB1 recruits the tumor suppressor ANCO1 to the YAP‐TEAD transcription factor complex to repress breast cancer genes like S100A and SPRR. ANCO1 loss leads to their reactivation, promoting oncogenic YAP‐TEAD activity and disease progression.
Introduction
AIB1/SRC‐3/NCOA3 is an oncogene that is amplified and overexpressed in breast cancer 1, 2. We and others have shown in transgenic models that AIB1 expression is critical to the transition of normal tissue to early‐stage breast cancer 3, 4, 5. Reducing AIB1 levels delays progression of ductal carcinoma in situ (DCIS) to invasive ductal carcinoma, suggesting that it plays a pivotal role in early‐stage triple‐negative breast cancer (TNBC) 6. In normal human breast tissue, AIB1 levels are low, but significantly increase at the DCIS stage of breast cancer 6. AIB1 functions as a transcription co‐activator and has been shown to interact with many transcription factors, such as nuclear receptors, NFκB, and E2F, to promote transcription in physiology and disease contexts 7. Further, we and others have shown that AIB1 can also recruit transcription repressor proteins, such as the putative tumor suppressor ANCO1/ANKRD11, to suppress transcription at select sites 8, 9, 10, 11, which may also contribute to malignant progression. Thus, AIB1 appears capable of both activating and repressing distinct sets of genes and thus orchestrate the development of early‐stage breast cancer.
AIB1 has been shown to interact with the TEAD family of transcription factors 12, though the impact of this interaction is unclear. TEADs, and their well‐studied binding partners, the orthologue co‐activators YAP and TAZ, control transcription of many oncogenic target genes in a majority of solid tumor types 13, 14, 15. In TNBC, YAP is important for disease progression and promotes the transcription of target genes that are correlated with increased metastasis and decreased survival 16, 17, 18. The extent to which the oncogenic activity of AIB1 can be attributed to interaction with TEADs and YAP/TAZ is not known. Previous studies have indicated that AIB1 is involved in the transcription of YAP‐TEAD target genes 16, 17, although some reports have suggested that AIB1 can transactivate TEADs independently of YAP or TAZ 12, 18.
Here, we report that through distinct mechanisms, AIB1 functions as a cofactor to activate and repress subsets of YAP‐TEAD target genes during TNBC progression. We found that AIB1‐mediated repression is dependent on recruitment of the tumor suppressor, ANCO1/ANKRD11, to TEAD to suppress transcription of the S100A and SPRR gene families at the 1q21.3 cytoband. Functionally, we show that both AIB1‐YAP‐activated and ANCO1‐AIB1‐YAP‐repressed gene expression are required for distinct and different YAP conferred aberrant growth phenotypes in vitro and the transition to invasive cancer in vivo.
Results and Discussion
AIB1 regulates induction and repression of subsets of YAP‐TEAD target genes
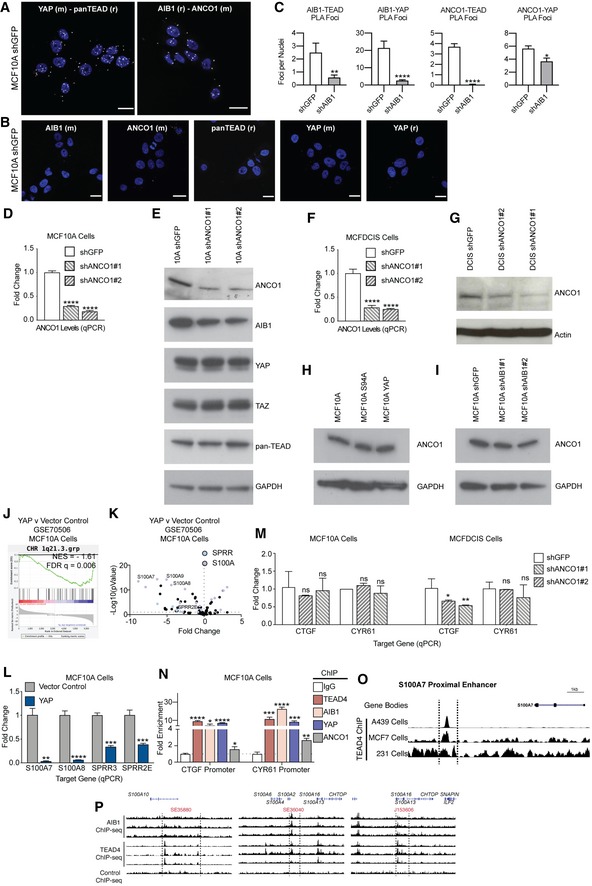
The co‐activators YAP/TAZ and their target genes are elevated in triple‐negative breast cancer 19, 20, 21, 22, 23. Further, synergistic interactions of YAP/TAZ and AIB1 at TEAD‐binding sites have been described in select cancers 16 but whether interactions of these co‐activators with the transcription factor TEAD play a role in the development and progression of TNBC is not known. To investigate this, we first asked whether AIB1, YAP/TAZ, and TEAD complexes exist in normal mammary epithelial as well as early‐stage breast cancer cells. For this, we performed a series of co‐immunoprecipitations (Co‐IP) in extracts from the immortalized normal mammary epithelial MCF10A cell line 24 as well as from the MCFDCIS cell line, which is capable of forming DCIS lesions that progress to invasive breast cancer in vivo 6, 25, 26. Immunoprecipitation with a pan‐TEAD, AIB1, or a YAP/TAZ antibody of endogenous proteins confirmed that robust complexes involving AIB1, YAP/TAZ, and TEAD can be detected in extracts from MCF10A or MCFDCIS cells (Fig EV1A).
Figure EV1. AIB1 interacts with YAP and TEAD in normal mammary epithelial and breast cancer cell lines.
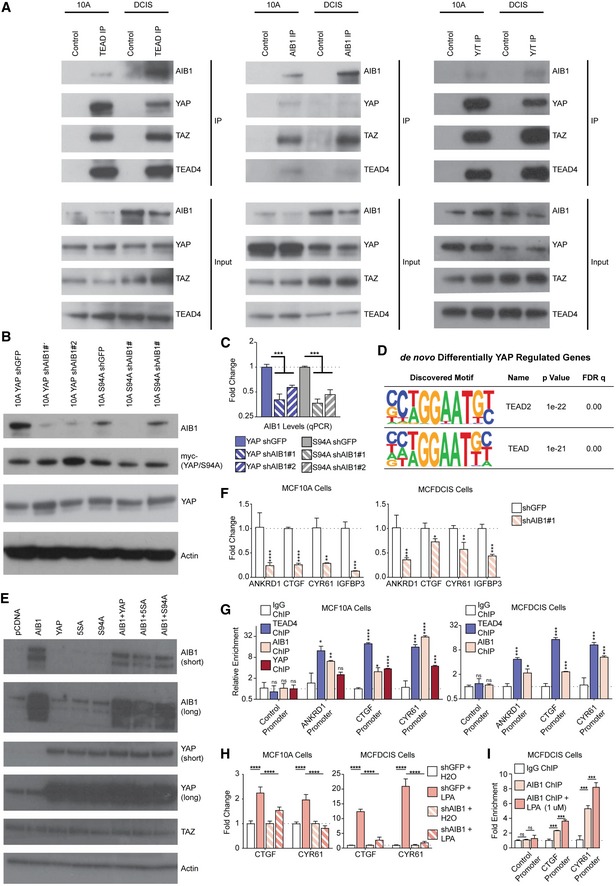
- AIB1 interacts with YAP/TAZ and TEAD proteins, as part of a larger complex in normal mammary epithelial (MCF10A) and DCIS (MCFDCIS) cells. Co‐immunoprecipitations performed with antibodies as indicated in lanes. Immunoblots show co‐IP and input lysate; membranes were probed with antibodies as indicated.
- Western blot analysis of myc‐tagged YAP or YAP S94A levels after AIB1 knockdown in the MCF10A cell lines shown in Fig 1A–C.
- Validation of AIB1 knockdown in the different MCF10A cell lines shown in Fig 1A–C, assessed by RT–qPCR.
- TEAD motifs are significantly enriched upstream of the de novo YAP signature genes in MCF10A cells. HOMER de novo motif analysis in promoters and proximal enhancers (−5,000 to +500 of TSS; Hg38) of genes described in Fig 1A–C.
- Transfection of indicated proteins (Fig 1D) in HEK293T does not increase endogenous expression of additional AIB1, YAP, or TAZ. Western blot analysis of reporter assay lysate expressing ectopically expressed proteins.
- AIB1 controls endogenous TEAD target levels in MCF10A and MCFDCIS cells. Levels of endogenous TEAD targets were assessed by RT–qPCR.
- AIB1 is recruited to promoters of TEAD target genes. Chromatin immunoprecipitation (ChIP) performed with indicated antibodies in MCF10A and MCFDCIS cells. Levels of precipitated chromatin assessed by qPCR.
- AIB1 is required for lysophosphatidic acid (LPA)‐induced YAP activation. Levels of endogenous TEAD targets were assessed by RT–qPCR following 1 μM LPA treatment for 1 h.
- AIB1 is increasingly recruited to promoters of TEAD target genes following LPA induction of YAP activity. ChIP performed with indicated antibodies in MCFDCIS cells following 1 μM LPA treatment for 1 h.
To examine the effect of crosstalk of AIB1 and YAP in the context of TEAD‐binding sites in non‐transformed cells, we experimentally modulated the proteins' levels and assessed gene expression changes in normal MCF10A cells. We created cell lines overexpressing either wild‐type YAP, or the TEAD‐binding‐deficient mutant YAP S94A 27, and subsequently infected these derivative lines with two separate shRNA constructs targeting AIB1 or GFP as a control (Fig EV1B and C). We analyzed the RNA‐seq data from these cell lines first by stratifying genes based on the effect of YAP overexpression or AIB1 knockdown (cutoffs of ±2 fold change relative to YAP S94A) and then subsequently subgrouped YAP‐modulated genes based on the effects of AIB1 knockdown (expression/repression reversal greater than 25% with both shAIB1 constructs; Dataset EV1). As illustrated in the Venn diagram (Fig 1A), we observed 272 genes that were activated by YAP overexpression, of which 102 genes were reduced by AIB1 knockdown. Conversely, YAP overexpression repressed 611 genes and AIB1 reduction reversed the YAP‐dependent repression of 292 of these genes (Fig 1A). AIB1 knockdown alone downregulated 246 genes, and de‐repressed expression of 315 genes independent of YAP overexpression, potentially through effects on endogenous YAP or TAZ in MCF10A cells (Fig 1B). We thus have defined four subgroups of genes in MCF10A cells: YAP‐activated, AIB1‐YAP co‐activated, YAP‐repressed, and AIB1‐YAP co‐repressed genes (Fig 1B and C).
Figure 1. AIB1 and YAP coregulate a subset of TEAD target genes.
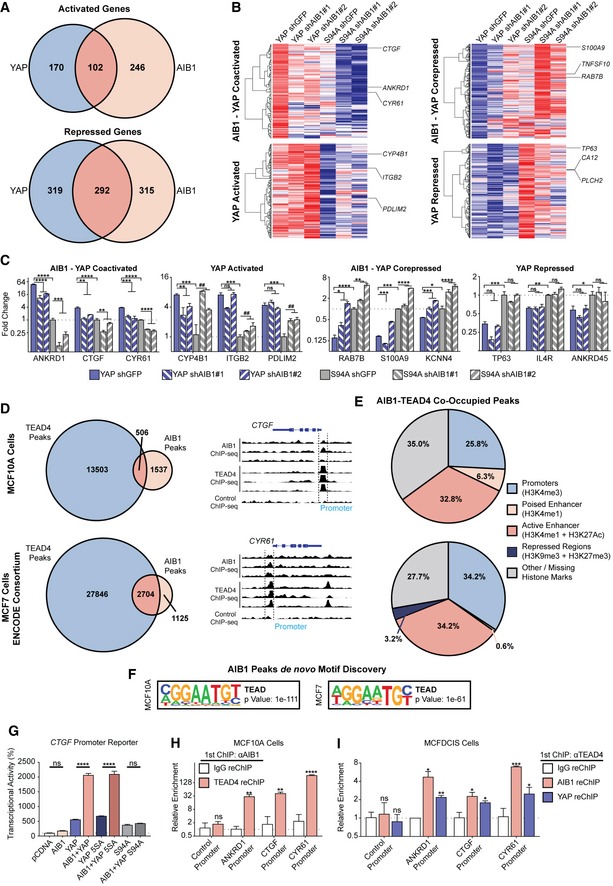
- AIB1 and YAP co‐activate and co‐repress a subset of TEAD target genes. Venn diagrams showing differentially regulated genes in MCF10A cell lines that overexpress YAP or YAP S94A in the presence of control shGFP or shAIB1 as indicated. RNA‐sequencing performed in triplicate.
- Heatmaps of AIB1‐YAP co‐activated, YAP‐activated, AIB1‐YAP co‐repressed, and YAP‐repressed genes shown in panel (A). Red and blue indicate high and low levels, respectively.
- Select genes are regulated by YAP and AIB1, and others are YAP‐regulated independently of AIB1. Endogenous gene expression changes assessed by RT–qPCR.
- Significant genomic co‐occupancy between AIB1 and TEAD in normal breast and breast cancer cells. Overlap of AIB1 and TEAD4 ChIP‐seq peaks in MCF10A and MCF7 cell lines. Select peaks from MCF10A ChIP‐seq shown. ChIP‐sequencing performed in triplicate.
- AIB1 and TEAD are localized at promoters and enhancers globally. Feature distribution based on overlapping AIB1‐TEAD4 peaks with respective MCF10A and MCF7 histone ChIP‐seq peaks.
- TEAD‐binding motifs are enriched within AIB1 occupied sites across the genome. HOMER de novo motif analysis of AIB1 ChIP‐seq peaks in MCF10A and MCF7 cells.
- AIB1 modulates TEAD transcriptional activity in a YAP‐dependent manner. HEK293T cells were transfected with CTGF promoter reporter and expression vectors of indicated proteins.
- AIB1 is engaged with TEAD on co‐activated target promoters in normal cells. Sequential ChIP‐reChIP performed with indicated antibodies in MCF10A cells. Levels of precipitated chromatin assessed by qPCR.
- AIB1 is engaged with TEAD on co‐activated target promoters in early‐stage breast cancer cells. Sequential ChIP‐reChIP performed with indicated antibodies in MCFDCIS cells. Levels of precipitated chromatin assessed by qPCR.
Interestingly, well‐defined direct YAP‐TEAD target genes are split between our YAP‐ and AIB1‐YAP‐regulated groups 28, 29. For example, the expression of ANKRD1, CTGF, and CYR61 are dependent on AIB1, whereas ITGB2, PDLIM2, and CYP4B1 are driven by YAP independent of AIB1. Similarly, RAB7B, TNFSF10, and S100A9 repression requires AIB1, whereas CA12, TP63, and IL4R can be suppressed by YAP alone (Fig 1B and C). These data suggest that AIB1 impacts transcription and repression of nearly half of YAP‐TEAD targets in normal epithelial cells indicating it is a critical coregulator of a distinct subset of YAP‐TEAD activated and repressed target genes.
To determine whether there is direct cooperation of these two co‐activators at the promoters and enhancer of the co‐regulated genes, we performed AIB1 and TEAD4 ChIP‐seq data in MCF10A; we also analyzed data from MCF7 breast cancer cells produced by the ENCODE consortium 30. We observed significant genomic co‐occupancy between TEAD and AIB1 in both MCF10A and MCF7 cells (P < 0.001, both groups), suggesting their interaction may promote cooperative transcription activation and repression when engaged on the chromatin (Fig 1D). AIB1 and TEAD co‐engagement was predominantly localized to promoters and active enhancers (Fig 1E), corroborating previous reports 28, 31, 32. Interesting, we also noted their engagement on transcriptionally repressed regions and loci lacking activating histone marks, which may explain the large number of AIB1‐YAP co‐repressed genes (Fig 1A, B and E). De novo motif discovery identified significant enrichment of TEAD‐binding motifs within AIB1 ChIP‐seq peaks, thereby independently validating the global coincidence of AIB1 and TEAD localization (Fig 1F). We subsequently performed motif discovery of YAP‐regulated and AIB1‐YAP co‐regulated genes to ensure a significant portion of differentially expressed genes were a direct effect of AIB1‐YAP convergent activity on TEAD. In line with our ChIP‐seq data, we observed a significant enrichment of TEAD motifs in the promoters and proximal enhancer of regulated genes in our YAP‐activated and YAP‐repressed genes (Fig EV1D). To validate the transcriptional effects of AIB1 on TEAD‐dependent transcription, we employed a luciferase reporter driven by the endogenous CTGF promoter, which we had observed to be an AIB1‐YAP co‐activated gene (Fig 1B and C). While the overexpression of AIB1 alone did not significantly increase the luciferase signal compared to the baseline, AIB1 synergized with wild‐type YAP and significantly increased signal relative to controls and YAP alone (Fig 1G). This effect was further enhanced with the expression of the hyperactive mutant YAP 5SA, whereas the synergistic effect was lost with the expression of TEAD‐binding‐deficient YAP S94A (Fig 1G). Western blot analysis of cellular lysates showed that endogenous protein levels of YAP and TAZ were unchanged by the overexpression of AIB1, likewise AIB1 or TAZ expression was unchanged by either the wild‐type or mutant YAP co‐activator expression (Fig EV1E). Thus, taken together, these data indicate that the effects of AIB1 following interaction with TEAD at gene promoters are dependent on the presence of the YAP co‐activator and that AIB1 modulates the magnitude of transcription activation of YAP‐TEAD targets.
We next asked whether similar AIB1‐YAP co‐activation also occurred in early‐stage TNBC cells. In agreement with our findings in MCF10A cells, we found that knockdown of AIB1 by shRNA in MCFDCIS breast cancer cells significantly reduced the endogenous mRNA expression levels of TEAD target genes ANKRD1, CTGF, CYR61, and IGFBP3 (Fig EV1F). To validate that these gene expression changes were due to interactions of AIB1 with complexes on chromatin, we performed chromatin immunoprecipitation (ChIP) and found that TEAD, YAP, and AIB1 were significantly enriched at TEAD sites within the promoters of AIB1‐YAP co‐activated target genes in MCF10A and MCFDCIS cells (Fig EV1G). Further, sequential ChIP‐reChIP indicated that AIB1 and TEAD proteins were complexed at their target genes (Fig 1H and I). Thus, the interaction between AIB1‐TEAD appears to be conserved during malignant progression and AIB1 and YAP synergize to co‐activate baseline target gene expression in this context.
To determine whether induced transcription also requires AIB1, we examined the well‐described induction of YAP activity by lysophosphatidic acid (LPA) through G‐protein‐coupled receptor (GPCR) signaling 33. LPA significantly upregulated YAP activity as measured by CTGF and CYR61 mRNA levels in the MCF10A and MCFDCIS cells (Fig EV1H). Conversely, knockdown of AIB1 significantly reduced YAP responsiveness to LPA, suggesting AIB1 is required for propagating intracellular GPCR effects in MCF10A and MCFDCIS cells. Consistent with this finding, ChIP‐qPCR validated increased recruitment of AIB1 to TEAD target promoters following LPA treatment (Fig EV1I). Thus, AIB1 is also critical for maintenance and propagation of GPCR‐induced AIB1‐YAP co‐activated transcription in TNBC cells.
Finally, to ensure that the YAP S94A was an appropriate control for TEAD‐mediated effects in our experiments, we compared the effects of YAP and YAP S94A compared to parental and vector controls. The overexpression of YAP or S94A did not affect the levels of AIB1 or TEAD in MCF10A cells (Fig EV2A). Further, we verified by co‐IP that the mutant YAP cannot interact with TEAD (Fig EV2B). We also wanted to verify that the expression differences we described (Fig 1) were a product of YAP activity through TEAD, rather than off‐target effects of the S94A point mutant protein. We therefore examined our RNA‐seq data from YAP S94A overexpressing MCF10A cells compared to RNA‐seq expression data from the vector control (GSE70506). We analyzed these data using the same cutoffs for our RNA‐seq analysis, and we did not observe a significant difference in gene regulation when comparing this independent data set to our YAP/S94A comparison and saw significant correlation between both groups (Fig EV2C; Dataset EV2). Further, we repeated this analysis after removing our YAP signature genes (Fig 1A) to ensure that well‐described YAP target genes were not masking a significant off‐target effect of the S94A mutant. In doing so, we effectively compared the YAP S94A gene expression to the vector control, and we did not observe a significant difference in gene regulation, suggesting the YAP S94A mutant is functionally similar to a vector control (Fig EV2C). Finally, genes within the YAP signature (Fig 1A) did not behave different in the YAP/Vector data set (Fig EV2D) suggesting that almost all of the YAP effect is mediated by TEAD in these cells. AIB1 manipulation did not affect the levels of YAP, TAZ, or TEAD in MCF10A cells (Fig EV2E). Thus, we conclude that the YAP S94A is an appropriate control for the read out of TEAD‐binding effects of YAP in the MCF10A cells.
Figure EV2. YAP S94A‐expressing cells are similar to vector control.
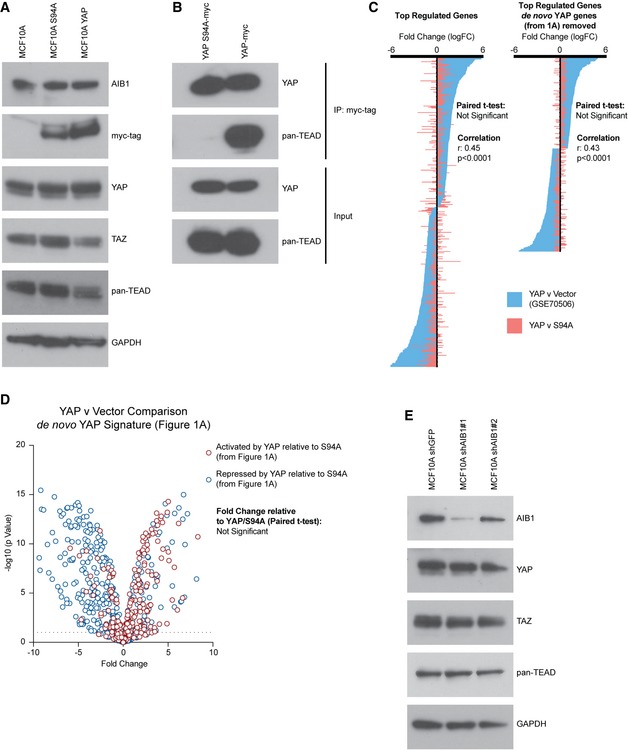
- Overexpression of YAP or YAP S94A does not significantly alter the expression of endogenous AIB1, YAP, TAZ, or TEADs. Western blot analysis of indicated proteins following YAP S94A or YAP overexpression compared to parental baseline. Asterisk indicates non‐specific band.
- YAP S94A does not interact with TEAD. Co‐immunoprecipitation of myc‐tagged YAP S94A or YAP performed with myc antibody. Immunoblots show co‐IP and input lysate; membranes were probed with antibodies as indicated.
- Gene expression changes between YAP and YAP S94A are not significantly different from YAP relative to vector control (GSE70506) and are significantly correlated. Waterfall plot comparing significantly regulated genes (> 2FC an P < 0.05), to significantly regulated genes following removal of de novo YAP signature genes (Fig 1) in MCF10A YAP vs. vector control to YAP vs. YAP S94A.
- De novo YAP signature expression changes between YAP and vector control are not significantly different than YAP relative to YAP S94A. Volcano plot showing change of YAP‐activated and YAP‐repressed genes (described in Fig 1A and B) in YAP vs. vector control.
- Knockdown of AIB1 does not affect stabilization of YAP, TAZ, or TEADs. Western blot analysis of indicated proteins following AIB1 knockdown.
Source data are available online for this figure.
AIB1 recruits the tumor suppressor ANCO1/ANKRD11 for YAP‐TEAD‐dependent repression at the 1q21.3 locus
An unexpected finding of our RNA‐seq data was that a large fraction of repressed gene expression by YAP was dependent on AIB1. There have been some reports on the direct repressive activities by YAP 29, 34; yet, the majority of analyses have focused on transcriptional activation. We and others have previously demonstrated that AIB1 can recruit the tumor suppressor ANCO1/ANKRD11 to repress transcription of some nuclear receptor‐regulated genes 8, 9. We therefore hypothesized that ANCO1 could be involved in AIB1‐YAP‐driven transcription repression. To first determine whether ANCO1 protein complexed with AIB1, YAP, and TEAD could exist in the nuclei of normal breast epithelium, we utilized proximity ligation assays (PLA) to determine whether close interactions of these factors takes place in situ. We used ANCO1‐AIB1 and YAP‐TEAD interaction as robust positive controls for the assay (Fig EV3A). We found that ANCO1‐TEAD and ANCO1‐YAP form a complex within the nucleus of MCF10A cells (Fig 2A). Furthermore, the PLA signal was significantly reduced upon knockdown of AIB1, and single antibody controls confirmed PLA specificity, therefore suggesting that ANCO1 recruitment to TEAD is AIB1‐dependent (Figs 2A, and EV3B and C). Though we detect all members in complex, it is still unclear whether AIB1 interacts with YAP directly or indirectly convergently through TEADs. However, taken together, these data indicate that ANCO1 forms a complex with AIB1, YAP, and TEAD in the nuclei of cells and that AIB1 is a critical mediator of TEAD‐ANCO1 interactions. It should be noted that although Drosophila homologues of YAP and AIB1 (Yki and Tai, respectively) have been shown to directly interact through WW‐PPxY interaction, this domain has been lost in NCOA/SRC/p160 family during evolution 35, 36, 37, 38 and cannot explain AIB1‐YAP interactions in human cells.
Figure EV3. ANCO1 knockdown in normal mammary epithelial and early‐stage breast cancer cell lines.
- Positive control PLA, showing foci (white foci, falsely colored 594 nm signal) of interaction between YAP‐TEAD and AIB1‐ANCO1. Representative image; scale bars shown indicate 25 μm.
- Single antibody negative PLA controls showing baseline signal in MCF10A shGFP (control) cells. Scale bars shown indicate 25 μm.
- PLA foci are significantly reduced between ANCO1‐TEAD, ANCO1‐YAP, AIB1‐TEAD, and AIB1‐YAP following AIB1 knockdown. Quantification of proximity ligation assays shown in Fig 2A.
- Levels of endogenous ANCO1 in MCF10A cells following knockdown of ANCO1; levels were assessed by RT–qPCR.
- Levels of AIB1, YAP, TAZ, and TEADs are unaffected by ANCO1 depletion. Immunoblot of MCF10A cells following knockdown of ANCO1.
- Levels of endogenous ANCO1 in MCFDCIS cells following infection of shANCO1; levels were assessed by RT–qPCR.
- Immunoblot of MCFDCIS cells following knockdown of ANCO1.
- Levels of ANCO1 are not changed by overexpression of YAP or YAP S94A. Immunoblot of ANCO1 in MCF10A cells following overexpression.
- Levels of ANCO1 are not changed by AIB1 knockdown. Immunoblot of ANCO1 in MCF10A cells following AIB1 knockdown.
- 1q21.3 genes are suppressed by YAP overexpression relative to vector control. Gene set enrichment analysis of RNA‐seq in MCF10A cells overexpressing YAP or empty vector (GSE70506).
- Genes regulated by YAP overexpression on the 1q21.3 locus. Volcano plot of 1q21.3 genes with indicated S100A and SPRR family genes from GSE70506.
- YAP represses 1q21.3 localized genes. Levels of endogenous S100 and SPPR genes in MCF10A cell lines following overexpression of YAP or vector control; levels were assessed by RT–qPCR.
- Levels of endogenous TEAD targets in MCF10A and MCFDCIS cell lines following infection of shANCO1; levels were assessed by RT–qPCR.
- ANCO1 is moderately enriched at CTGF and CYR61 promoters; in contrast, AIB1, TEAD4, and YAP are robustly recruited to these promoters. Chromatin immunoprecipitation (ChIP) performed in MCF10A cells. Levels of precipitated chromatin assessed by qPCR.
- AIB1 and TEAD are detected at superenhancer sites in normal mammary cells that were identified in other cancer cell lines. Analysis of AIB1 and TEAD4 ChIP‐seq peaks at 1q21.3 in MCF10A cells.
Figure 2. AIB1 recruits ANCO1 to YAP‐TEAD to repress transcription at 1q21.3.
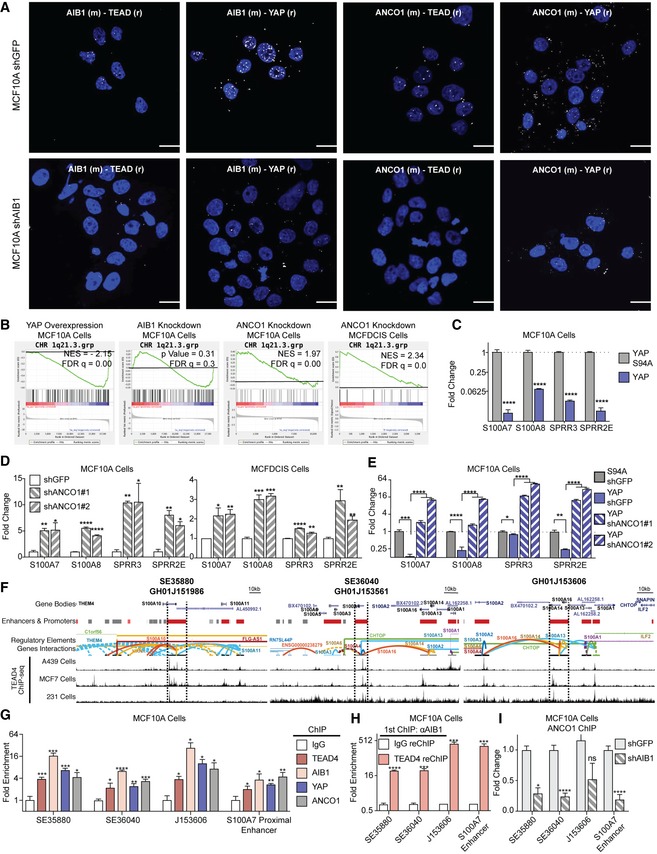
- ANCO1 is recruited to YAP‐TEAD by AIB1. Positive proximity ligation assay (PLA) signal (white foci, falsely colored 594 nm signal) shown between ANCO1‐TEAD, ANCO1‐YAP, AIB1‐TEAD, and AIB1‐YAP in control cells is lost in AIB1 knockdown. Representative images of proximity ligation assay (PLA) between indicated proteins in MCF10A shGFP (control) and shAIB1 cell lines using antibodies (species) as indicated; scale bars shown indicate 25 μm.
- YAP overexpression represses, AIB1 knockdown dysregulates, and ANCO1 knockdown amplifies genes at the 1q21.3 locus in MCF10A and MCFDCIS cells. Gene set enrichment analysis of RNA‐seq data and cDNA array data generated from MCF10A cell lines as indicated.
- YAP represses 1q21.3 localized genes. Levels of endogenous S100 and SPPR genes in MCF10A cell lines following overexpression of YAP/S94A; levels were assessed by RT–qPCR.
- ANCO1 knockdown increases expression of S100 and SPPR genes in MCF10A and MCFDCIS cells; levels were assessed by RT–qPCR.
- YAP‐driven suppression of 1q21 genes is reverted upon ANCO1 knockdown. Levels of endogenous 1q21.3 genes were assessed in YAP or S94A‐expressing, control or shANCO1 MCF10A cells by RT–qPCR.
- TEAD, YAP, AIB1, and ANCO1 are engaged at annotated enhancer regions within 1q21.3. Chromatin immunoprecipitation (ChIP) performed with indicated antibodies in MCF10A cells.
- AIB1 is engaged with TEAD on superenhancer sites at 1q21.3 in normal cells. Sequential ChIP‐reChIP performed with indicated antibodies in MCF10A cells. Levels of precipitated chromatin assessed by RT‐qPCR.
- ANCO1 is recruited to superenhancers at 1q21.3 by AIB1 and is significantly reduced at 1q21.3 following AIB1 knockdown. ChIP performed with ANCO1 antibodies in control or shAIB1 cells. Levels of precipitated chromatin assessed by RT‐qPCR.
To describe gene expression changes resulting from ANCO1‐AIB1‐YAP activity, we compared gene expression profiles of our MCF10A and MCFDCIS cells following ANCO1 knockdown (Fig EV3D–G). We observed that the S100A and SPRR gene families at the 1q21.3 locus were strongly repressed by YAP overexpression, while ANCO1 knockdown dramatically upregulated this same signature in MCF10A and MCFDCIS cells (Fig 2B; Dataset EV3). This was particularly noteworthy because the 1q21.3 locus has been associated with basal breast cancer aggressiveness 39, and S100A7 has been shown to be significantly enriched in more aggressive, high‐grade triple‐negative DCIS lesions 40, 41, 42, 43, 44, 45. We validated that YAP repressed 1q21.3 genes in MCF10A and MCFDCIS cells by RT–qPCR (Fig 2C) and conversely that knockdown of the repressor ANCO1 by shRNA caused significant upregulation of these gene families in both cell lines (Fig 2D). Knockdown of ANCO1 in MCF10A‐YAP‐shGFP cells reverted YAP‐driven repression of S100A7, S100A8, SPRR3, and SPRR2E (Fig 2E). Modulation of ANCO1 did not affect the levels of YAP, TAZ, or TEAD, and had only modest effects on AIB1 (Fig EV3E); neither changes in levels of YAP or YAP S94A or AIB1 affected the stability of ANCO1 (Fig EV3H and I). Similar repressive effects of YAP on 1q21.3 genes were observed in comparison to either a vector control or to YAP S94A (Fig EV3J–L). Finally, ANCO1 knockdown did not affect the expression levels of AIB1‐YAP co‐activated genes in MCF10A or MCFDCIS cells (compared to the magnitude of the reversion of YAP‐repression effect), and there was minimal ANCO1 chromatin engagement on these activated genes compared to YAP, TEAD4, and AIB1 (Fig EV3M and N). Thus, much like our previous paradigm involving AIB1‐estrogen receptor repression 9, we saw that ANCO1 mediated repression was limited to only a subset of AIB1‐YAP‐repressed TEAD target genes. Taken together, these data suggest that ANCO1 is not recruited or recruited inefficiently to AIB1‐YAP co‐activated targets, but it is critical for AIB1‐YAP co‐repression.
To confirm that ANCO1/AIB1/YAP/TEAD complexes are engaged at 1q21.3, we utilized publicly available ChIP‐seq and Hi‐C data 28, 46, 47, 48, 49 to identify loci of TEAD enrichment at enhancer regions annotated to interact with and modulate expression of S100A and SPPR genes (Figs 2F and EV3O). We validated the presence of peaks at these sites in our ChIP‐seq data and subsequently noted a significant enrichment of AIB1, YAP, TEAD, and ANCO1 at these loci by ChIP in parental MCF10A cells (Figs 2G and EV3P). Sequential reChIP of AIB1 and TEAD showed the proteins were indeed co‐engaged at these 1q21.3 superenhancers (Fig 2H). Finally, ANCO1 recruitment to these sites is significantly reduced upon AIB1 knockdown, highlighting the critical role of AIB1 recruitment of ANCO1 to TEAD (Fig 2I). Overall the data suggest that YAP repression of genes at the 1q21.3 locus is mediated by recruitment of AIB1 and ANCO1.
YAP‐driven invasion requires AIB1 and is enhanced by ANCO1 loss
Our data demonstrate that a large portion of both YAP‐activated and YAP‐repressed gene expression is regulated by AIB1 and ANCO1. To determine how these regulatory events contribute to malignant progression, we examined the 3D growth of MCF10A mammospheres in Matrigel 50 of cell lines overexpressing wild‐type YAP or YAP S94A ± shRNA constructs with or without knockdown of AIB1 or ANCO1. Similar to previous reports 51, MCF10A‐YAP‐shGFP cells formed aberrant spheres lacking circularity and defined borders that invaded the surrounding matrix, whereas normal spheres circular and well polarized were formed by control MCF10A‐S94A‐shGFP cells (Fig 3A). Knockdown of AIB1 completely attenuated the YAP‐driven invasive phenotype (Fig 3A and B). However, knockdown of ANCO1 sustained the invasive phenotype and increased the size of the abnormal spheres (Fig 3A–C). Interestingly, we found that ANCO1 reduction alone increased 3D sphere size of MCF10A‐shANCO1 cells in the Matrigel assay (Fig EV4A and B). Endogenous ANCO1 levels were decreased in our MCF10A‐YAP‐shGFP spheres, suggesting that the YAP conferred aberrant growth and increased sphere size is a product of both AIB1‐YAP co‐activated gene expression and endogenous decrease of ANCO1 repression (Fig EV4C). The increase in sphere size promoted by ANCO1 loss may be attributed to an increase in individual cell size of shANCO1 cells (Fig EV4D and E). YAP has been previously implicated in increasing cell size through MTOR signaling [52, 53, preprint: 54], and we observed increased MTOR signaling from AIB1 and ANCO1 knockdown in our model system (Fig EV4F). Interestingly, the S100A proteins that are regulated by the ANCO1/AIB1/YAP complex have previously been implicated in upregulation of MTOR signaling 55, 56, 57. These findings suggest cell size control by MTOR may be downstream of YAP, AIB1, and ANCO1. Genes downstream of AIB1‐YAP co‐activation appear to contribute to malignant progression and proliferation, whereas the genes downstream of ANCO1‐AIB1‐YAP repression impact overall sphere size while maintaining features of malignancy. Thus, the loss of AIB1‐YAP repression by decreased ANCO1 coupled with AIB1‐YAP co‐activation seems to contribute independently to the invasive phenotype.
Figure 3. AIB1 and ANCO1 regulate YAP‐induced invasion in vivo .
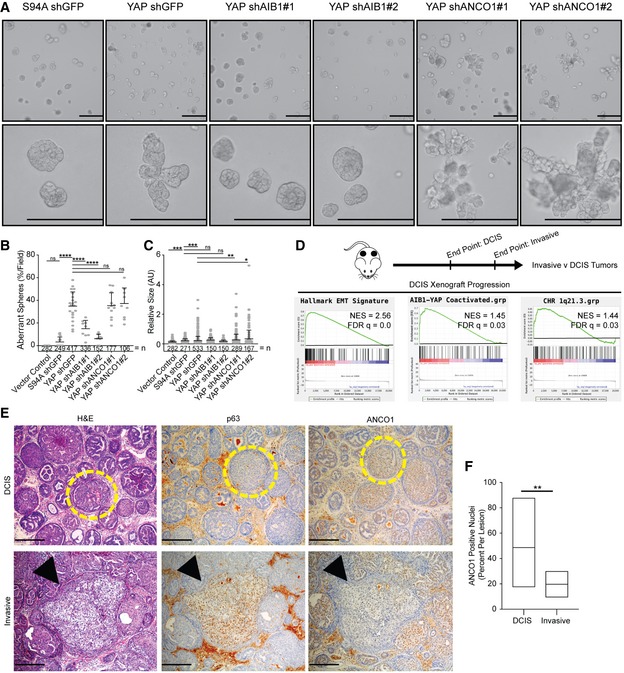
- YAP‐driven aberrant sphere formation of MCF10A cell lines is ablated by AIB1 knockdown, but further enhanced by ANCO1 knockdown. Indicated cell lines were plated on Matrigel and allowed to form mammospheres for 7 days. 10× and 40× magnifications shown, scale bar = 200 μm.
- Quantification of aberrant spheres per field displayed in panel (A), n = number of spheres.
- Quantification of size (arbitrary units) of aberrant spheres per field displayed in panel (A), relative to control YAP S94A shGFP spheres. n = number of spheres.
- MCFDCIS xenograft tumors have increased expression of AIB1‐YAP, EMT, and 1q21.3 genes during the invasive transition. Schematic of experiment shown. Gene set enrichment analysis of RNA‐seq data generated from MCFDCIS xenografts at indicated time points.
- Invasive lesions have significantly less ANCO1 than encapsulated DCIS lesions. Representative images of H&E, p63, and ANCO1‐stained MCFDCIS xenografts as DCIS and invasive lesions. Yellow circles indicate DCIS lesions, and black arrows indicate invasive lesions. 20× magnification; scale bar = 100 μm.
- Quantification of ANCO1 positivity in DCIS and invasive lesions shown in panel (E).
Figure EV4. ANCO1 reduction results in larger single cells and spheres in 3D culture.
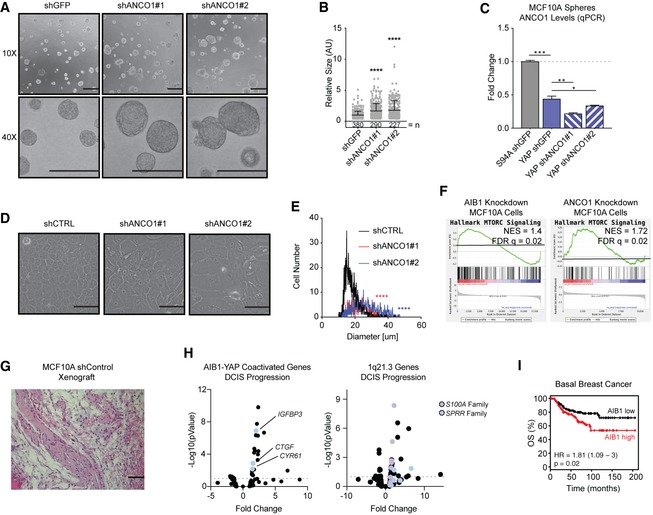
- ANCO1 knockdown increases the size of MCF10A spheres. Indicated cells were plated on Matrigel and allowed to form spheres for 7 days. 10× and 40× magnifications shown, scale bar = 100 μm.
- Quantification of size (arbitrary units) of aberrant spheres per field displayed in panel (A), relative to control 10A shGFP spheres. n = number of spheres.
- ANCO1 levels decrease with YAP overexpression in spheres. ANCO1 mRNA levels were assessed by RT–qPCR.
- ANCO1 loss increases cell size. Representative brightfield images of MCF10A cells with control shRNA or shANCO1. Scale bar = 100 μm.
- Quantification of cell size following ANCO1 knockdown in panel (C).
- AIB1 and ANCO1 knockdown amplifies MTORC1 downstream genes. Gene set enrichment analysis of RNA‐seq data and cDNA array data generated from MCF10A cell lines as indicated.
- MCF10A cells are non‐tumorigenic in vivo. Representative H&E staining of MCF10A xenograft in athymic nude mice at day 39 after cell implantation. Fibrotic, benign lesions shown, 20× magnification; scale bar = 100 μm.
- AIB1‐YAP co‐activated genes and genes at the 1q21.3 locus increase during DCIS progression. Volcano plot of RNA‐seq data from MCFDCIS xenografts during invasive progression. Select upregulated genes indicated.
- High AIB1 levels are a poor prognostic indicator in basal breast cancer in patients. KM plot showing overall survival in patients with stratified ANCO1 expression. n = 360 patients.
AIB1‐YAP co‐activation and loss of ANCO1 repression promote DCIS progression to invasive cancer
We next addressed the role of ANCO1/AIB1/YAP/TEAD crosstalk in early breast cancer progression in vivo. The MCF10A cell line is non‐tumorigenic when implanted in mice (Fig EV4G); however, the MCFDCIS cell line forms DCIS lesions that progress to locally invasive triple‐negative breast cancer in athymic nude mice over a defined timeline 6, 26, 58. Furthermore, we have shown that progression in this model can be inhibited by reducing AIB1 levels 6. Using RNA‐sequencing (GSE130903; Dataset EV4), we analyzed gene expression in xenografts that underwent local invasion (5.5 weeks post‐implantation) compared to early‐stage 0 DCIS xenografts (4 weeks post‐implantation) 58 and observed a significant enrichment in AIB1‐YAP target genes, EMT hallmark genes, and 1q21.3 locus gene signatures (Fig 3D). Specifically, we observed significant increases in the levels of AIB1‐YAP co‐activated genes (de novo signature from Figs 1B and EV4H). The increase in the level of S100A and SPRR genes during the invasive transition (Fig EV4H) indicated that like our in vitro data, these changes may be associated with decreased expression of ANCO1 and loss of the associated repression phenotype. In order to specifically categorize the novel role of ANCO1 during progression, we stained MCFDCIS xenografts by IHC. Lesions that stained positive for p63, a marker of local invasion in this DCIS model 26, showed significantly less nuclear ANCO1 staining than encapsulated DCIS lesion, suggesting invasion is inversely correlated with levels of nuclear ANCO1 (Fig 3E and F). Therefore, we propose that the demarcation of high‐grade DCIS lesions by S100A7 suggested by previous reports 41, 42, 43, 59 is a product of loss of ANCO1. These findings are consistent with our in vitro and 3D spheroid findings, suggesting that AIB1‐YAP activity coupled with ANCO1 loss could play a role in the progression of TNBC.
Loss of ANCO1 is a poor prognostic marker
We next wanted to determine the relevance of our findings to the clinical presentation of breast cancer. AIB1 and YAP have previously been shown to be oncogenes, as have their co‐activated target genes. Using the GTEX and TCGA RNA‐seq gene expression profiling, we found that ANCO1 mRNA levels are significantly decreased in breast cancer relative to normal tissue (Fig 4A). Consistent with this, we find in normal human breast tissue that ANCO1 levels are high and localized to the basal, myoepithelial cells surrounding the ductal cells (Fig 4B). During the transformation to stage 0 ductal carcinoma in situ (DCIS) lesions, some patients maintain expression and nuclear localization of ANCO1, while some lose ANCO1 expression entirely (Fig 4B). Further analysis showed that low levels of ANCO1 are a poor prognostic marker in basal breast cancer, whereas high AIB1 is a poor prognostic marker (Figs 4C and EV4I) 60. Finally, we analyzed patient tumor data housed within TCGA to validate our proposed ANCO1/AIB1/YAP/TEAD paradigm. We stratified patients by median expression of ANCO1 into high or low expression groups. AIB1‐YAP co‐activated targets CTGF and CYR61 showed significant positive correlation in patients as expected, and there is no correlation with ANCO1 expression level (Fig 4D). Similarly, as previously reported 61 CTGF and ACTB showed little correlation in expression, there was also no significant difference in expression associated with ANCO1 level (Fig 4D). We next assessed the levels of 1q21.3 genes in patients and observed that mRNA levels of S100A7, S100A8, SPRR3, and SPRR2E are all significantly inversely correlated with AIB1‐YAP co‐activated genes in the presence of high ANCO1 levels (Fig 4D). This inverse correlation is significantly decreased in low ANCO1, indicating that AIB1‐YAP co‐activation and ANCO1‐AIB1‐YAP co‐repression complexes modulate transcription of distinct target genes, but their activities are coordinated.
Figure 4. Loss of ANCO1 in breast cancer perturbs ANCO1‐dependent YAP repression and is a marker for poor prognosis.
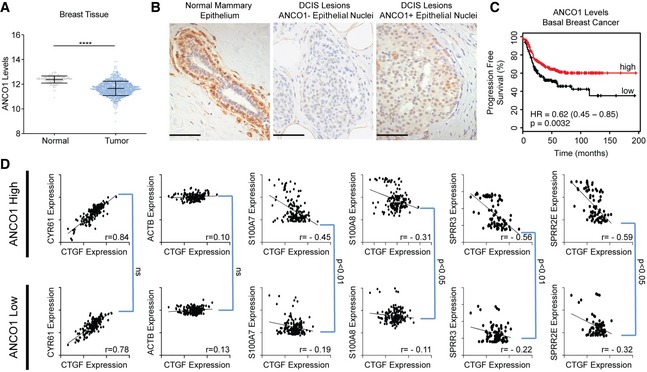
- ANCO1 levels decrease in breast cancer. ANCO1 mRNA expression from normal tissue (GTEX) and tumor (TCGA) for breast samples.
- Representative IHC images of ANCO1 staining in normal human breast and DCIS lesions. DCIS lesions can be stratified to either ANCO1‐negative (center panel) or ANCO1‐positive (right panel). Scale bar = 50 μm.
- High ANCO1 levels are a good prognostic indicator in basal breast cancer in patients. KM plot showing progression free survival in patients with stratified ANCO1 expression. n = 360 patients.
- Patient breast tumor samples with reduced ANCO1 levels have high expression of 1q21.3 genes, without altered AIB1‐YAP co‐activated gene expression. Dot plots showing TCGA Pan‐Cancer data accessed via Xenabroswer. Patients were stratified to high or low expression of ANCO1 by median expression.
Conclusion
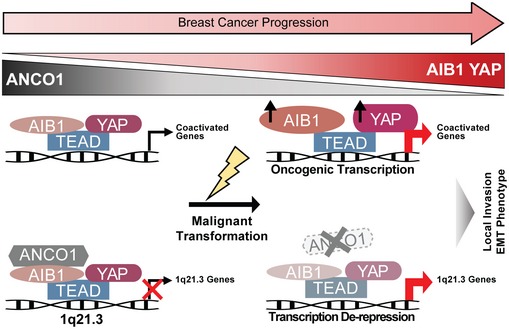
Here, we demonstrate that convergent activities of ANCO1 and AIB1 upon TEAD govern the activation and repression of a subset of YAP target genes during basal breast cancer progression. Our findings suggest a model (Fig 5) whereby in normal tissues, low levels of AIB1 result in low levels of YAP‐activated transcription, but in parallel, these low AIB1 levels are sufficient to maintain ANCO1 repressive transcription complexes on distinct subsets of YAP‐TEAD target genes. The loss of ANCO1 and higher expression of AIB1‐YAP in breast cancer lead to both de‐repression of transcription at 1q21.3 and activation of a genome‐wide oncogenic transcription program. Given the increasingly appreciated role of 1q21.3 genes in breast cancer development and progression 39, it may be that the loss of ANCO1 repression of S100A and SPRR proteins plays a critical role during the early stages of breast cancer initiation and this is consistent with lower ANCO1 levels in patient cancer sample being associated with a worse prognosis. Since the wider transcriptome of human breast cancer is not radically changed between DCIS and invasive cancer 62, it may well be that it is the temporal pattern of select gene expression changes effected by AIB1/YAP/ANCO1 that is critical to the development and progression of DCIS.
Figure 5. Both de‐repression and activation of YAP gene expression play distinct roles in early progression of breast cancer, mediated through changes in AIB1 and ANCO1 levels.
Materials and Methods
Cell lines and cell culture
HEK293T cells were maintained in DMEM (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS). MCF10A and MCFDCIS 6 cell lines were maintained in DMEM:F12 (Invitrogen, Carlsbad, CA) with 5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml insulin. All cell lines were grown at 37°C with 5% CO2. Cells were counted and size assessed with the Beckman Coulter Multisizer 3 Coulter Counter.
Transfections and infections
Cells were transfected using FuGENE6 (Promega, Madison, WI). Plasmids used are described in Key Resource Table. Oligonucleotide sequences for shRNA knockdown were cloned in to pLKO.1 construct (see Appendix Table S3).
Virus was made by transfecting HEK293T cells with packaging and envelope plasmids (see Appendix Table S3), and lentiviral or retroviral element containing plasmids. Media containing virus was collected 48 and 72 h post‐transfection and used for infection.
Western blot
Whole cell lysate was isolated from cells using either NP‐40 lysis buffer [50 mM Tris–HCl (pH 8.0), 170 mM NaCl, 0.5% NP‐40, 50 mM NaF and 1 mM PMSF] or RIPA lysis buffer [50 mM Tris–HCl (pH 8.0), 1% Triton X, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl] and allowed to lyse on ice for 15 min. Samples were centrifuged at 15,000 × g for 10 min, and supernatant was collected. Protein was quantified by Bradford assay. Samples were prepared using NuPAGE reagents according to the manufacturer's protocol and transferred to PVDF membranes using the iBlot dry blotting system according to the manufacturer's protocol using P0 preset template, or a 20 V, 10‐min transfer for large proteins (Thermo Fisher, Waltham, MA). Membranes were probed with indicated antibodies, with concentrations described in Appendix Table S1.
Luciferase reporter assays
Cells were transfected with indicated reporter and TK‐promoter‐driven Renilla luciferase control and indicated overexpression constructs for 24–48 h. Luciferase activity was quantified using the Dual‐Luciferase System (Promega, Madison, WI) according to the manufacturer's protocol. All luciferase reporter assays were done in triplicate.
RT–qPCR
RNA was isolated from cultured cells, spheres, or tissues using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol and quantified by NanoDrop (Thermo Fisher, Waltham, MA). For tumor samples, RNA was extracted from tissue samples using the RNeasy Kit following homogenization using the QIAshredder (Qiagen, Hilden, Germany). Reverse transcription was done using iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA) according to the manufacturer's protocol. Real‐time quantitative RT–PCR was performed with iQ SYBR Green Supermix (Bio‐Rad, Hercules, CA). Primers used for RT–qPCR were purchased from Integrated DNA Technologies, and their sequences are described in Appendix Table S2. Fold change was calculated by 2−ΔΔCT normalized to human actin or human B2M, as housekeeping genes. All RT–qPCR assays were done in triplicate.
Immunoprecipitation
Whole cell lysate was isolated from cells using either NP‐40 lysis buffer or RIPA lysis buffer. A portion of the lysate was reserved as input and prepared according to the Western blot methodologies (above). The lysates were precleared with unblocked Sepharose antibody purification resin (Amersham Pharmacia Biotech, Uppsala, Sweden). Immunoprecipitation was performed overnight at 4°C while rocking with indicated antibodies, at concentrations described in Appendix Table S1. Blocked GammaBind Plus Sepharose antibody purification resin (GE Life Sciences, Pittsburgh, PA) was added to the immunoprecipitation solution and rocked for 2–4 h. Samples were then washed repeatedly with lysis buffer, and then denatured using NuPAGE reagents, and assessed by Western blot.
For overexpression–immunoprecipitation, cells were transfected 24–48 h before lysis and immunoprecipitation.
Chromatin immunoprecipitation
Cells were crosslinked with 1.2% formaldehyde at room temperature for 15 min and then stopped with 125 mM glycine. Cells were rinsed twice with cold PBS and collected in PBS with EDTA‐free protease and phosphatase inhibitors (Roche, Indianapolis, IN). Cells were resuspended in nuclei isolation buffer (50 mM Tris–HCl, 60 mM KCl, 0.5% NP‐40, EDTA‐free protease and phosphatase inhibitors) and centrifuged to isolate the nuclei. Nuclei were lysed in Nuclei Lysis Buffer (50 mM Tris–HCl, 1% SDS, 10 mM EDTA, pH 8.1 with protease and phosphatase inhibitors). Chromatin was sonicated using a Diagenode Bioruptor (Denville, NJ) on “High” for 15–25 cycles (30 s on/30 s off). Sonicated samples were centrifuged at 4°C for 10 min at full speed, and supernatant was diluted 10× with Dilution Buffer [0.01% SDS, 1.1% Triton X‐100, 1.2 mM EDTA, 16.7 mM Tris–HCl (pH 8.1), 167 mM NaCl, EDTA‐free protease and phosphatase inhibitors]. Samples were precleared with unblocked Sepharose antibody purification resin (Amersham Pharmacia Biotech, Uppsala, Sweden) Immunoprecipitation was performed overnight at 4°C while rocking with indicated antibodies, at concentrations described in Appendix Table S1. Blocked GammaBind Plus Sepharose antibody purification resin (GE Life Sciences, Pittsburgh, PA) was added to the immunoprecipitation solution and rocked for 2–4 h. The beads bound to immunocomplexes were sequentially washed with low salt (20 mM Tris–HCl, 2 mM EDTA, 0.1% SDS, 1% Triton X‐100, and 150 mM NaCl), high salt (20 mM Tris–HCl, 2 mM EDTA, 0.1% SDS, 1% Triton X‐100, and 500 mM NaCl), and LiCl Buffer [0.25 M LiCl, 1% NP‐40, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM Tris–HCl (pH 8.1)]. Precipitates were eluted with freshly made 1% SDS and 0.1 M NaHCO3. Elutes were de‐crosslinked using 200 μM of NaCl at 65°C overnight while shaking. DNA was then purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol, and relative amount of chromatin was assessed by ChIP‐qPCR. All ChIP‐qPCR assays were done in triplicate. Primers used for ChIP‐qPCR were purchased from Integrated DNA Technologies, and their sequences are described in Appendix Table S2. All ChIP reactions were done in triplicate.
For sequential reChIP, chromatin was eluted in 10 mM DTT in fresh elution buffer to prevent leak from the first antibody. Eluted chromatin was diluted 20× in Dilution Buffer, and the immunoprecipitation, wash, and elution steps were repeated before de‐crosslinking.
Proximity ligation assay
Cells were plated on coverslips and allowed to attach overnight. Cells were crosslinked with 4% formaldehyde at room temperature for 15 min. Cells were rinsed with PBS and permeabilized with 0.1% Triton X in PBS for 15 min. Proximity ligation assay was then done with Duolink In Situ PLA Kit (Sigma, St. Louis, MO) according to the manufacturer's protocol. Antibody concentrations used are described in Appendix Table S1. Images were acquired with Leica SP8 confocal microscope.
Sphere assay and quantification
MCF10A cells were trypsinized and counted. 40 μl of Matrigel Basement Matrix, Reduced Growth Factor (Corning, Big Flats, NY), was plated in 8‐well chamber CultureSlides (Corning, Big Flats, NY) and allowed to solidify. 5,000 cells were resuspended in a 2% Matrigel DMEM:F12 Assay Media solution, containing 2% horse serum, 5 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml insulin, and plated atop of the solidified Matrigel. Media was refreshed every 48–72 h. Single cell suspension was verified by brightfield microscopy on day 0, and cells were allowed to form mammospheres for 7 days.
Images were acquired with Olympus IX‐71 Inverted Epifluorescence Microscope. Quantification was done using ImageJ. At least 5 fields of 10× photographs were randomly selected per condition. For aberrant sphere quantification, spheres were counted based on abnormal circulatory. For sphere size quantification, spheres were all counted using “Analyze Particles” function if the particle was larger than 50 pixels. Sphere size quantification was carried out by ImageJ, with a particle size threshold (0.05—inf. inches, 10× images). IHC nuclei quantification was carried out by ImageJ, with a particle size threshold of (50—inf. pixels, 20× images).
In order to collect the spheres for RNA isolation, media was removed from the chamber slides and the Matrigel layer with adhered spheres was incubated with 5 U/ml dispase (Stem Cell Technologies, Vancouver, Canada) for 30 min at 37°C. Solution was collected, centrifuged to pellet spheres, and washed with PBS before RNA isolation with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Reverse transcription–qPCR (RT–qPCR) was then carried out as previously described.
cDNA array, RNA‐seq, ChIP‐seq, and Hi‐C data analysis
ChIP‐seq
Samples were prepared with the Active Motif's Low Cell ChIP‐seq Kit according to the manufacturer's protocol. Libraries were sent to Novogene for sequencing, and 45 million paired reads per sample were acquired on an Illumina HiSeq 2500 Sequencer. Raw FASTQ files were aligned to Hg38 with BWA‐MEM, and peaks were called with MACS2. ChIP‐seq tracks made using bedtools and visualized on IGV. Publicly available ChIP‐seq files were downloaded from Cistrome DB, ENCODE, or EMBO, and processed with ChIPseeker in R. Additionally, tracks were downloaded from the Cistrome platform and visualized on the UCSC Genome Browser.
RNA‐seq
RNA was extracted from cell lines in triplicate using RNeasy Mini Kit with on‐column DNAse digestion, and quality was assessed by Bioanalyzer. All samples submitted for sequencing had an RNA Integrity Value > 9.0. RNA‐sequencing libraries were prepared using TruSeq Total RNA with RiboZero Gold library (Illumina, San Diego, CA, USA) at the UCLA Neuroscience Genomics Core, and 50 million paired reads per sample were acquired on an Illumina HiSeq 2500 Sequencer. A STAR index was generated using GTF annotation from GENCODEv28. Raw FASTQ files were aligned to GRCh38 with STAR and normalized and background‐corrected using EdgeR. Derived counts per million and P value were used to create a rank ordered list, which was then analyzed by GSEA. For gene groupings, genes were selected if YAP overexpression yielded +2‐fold change in gene expression levels compared to S94A, and then sequentially grouped based on effect of AIB1 knockdown. Heatmaps were generated by heatmap function in R.
Illumina cDNA array
RNA was extracted from cell lines in duplicate using RNeasy Mini Kit (Qiagen, Hilden, Germany), and quality was assessed by Bioanalyzer. All samples submitted for hybridization had an RNA integrity value > 9.0. RNA was amplified in a single round with the Illumina TotalPrep RNA Amplification Kit (Ambion). Amplified RNA was hybridized to Illumina HumanHT‐12 v4 Expression BeadChip according to the manufacturer's instructions. Illumina files were background‐corrected and quantile‐normalized using the limma in R. Derived fold change and P value were used to create a rank ordered list, which was subsequently analyzed by GSEA.
Hi‐C
Hi‐C and gene‐enhancer interaction tracks were downloaded from the GeneHancer platform and visualized on the UCSC Genome Browser.
Xenograft studies
MCF10A and MCFDCIS transduced with control or ANCO1 shRNAs were resuspended at a density of 7.5 × 105 cells in 1:1 culture medium/Matrigel™ (Corning) and were injected subcutaneously into 6‐ to 8‐week‐old female nude mice. MCF10A and MCFDCIS xenografted mice were euthanized by CO2 inhalation 39 and 48 days after cell implantation, respectively. Xenograft tissues were collected, fixed in 10% formalin for histopathology, or were snap‐frozen for gene expression analyses.
IHC/IF
Paraffin‐embedded sections (5 μm) obtained from human reduction mammoplasty, breast cancer biopsies, and MCF10A and MCFDCIS xenograft tissues were hematoxylin and eosin (H&E) and immunohistochemistry (IHC)‐stained. H&E and IHC staining were carried out by the Georgetown University Histopathology & Tissue Shared Resource using standard methods. For IHC analysis, paraffin‐embedded sections were deparaffinized, rehydrated, antigens exposed with citrate buffer pH 6.0 (00‐5000, Thermo Fisher Scientific) at 100°C, and were quenched with 3% hydrogen peroxide (H325‐500, Fisher Scientific). To block non‐specific binding, tissue sections were incubated with 10% normal goat serum. The detection of p63 and ANCO1 protein expression was accomplished by incubating tissue sections overnight at 4°C with described antibody dilution of the primary monoclonal antibody according to the manufacturers’ directions (see Appendix Table S1 for antibody concentrations). The detection of the primary antibody was performed using DAKO Envision Plus HRP Kit (Dako Corporation). The slides were counterstained with hematoxylin solution (MHS16, Sigma‐Aldrich). Stained tissues were photographed using an Olympus BX40 microscope. Quantification of the histological areas and the percentage of cells with ANCO1 nuclear staining were carried out using Fiji software.
Animal models
Female athymic nude mice were obtained from Envigo (Huntingdon, UK) and were maintained in the Georgetown University Division of Comparative Medicine. Animal experiments were conducted in compliance with protocols approved by the Georgetown University Institutional Animal Care and Use Committee.
Human samples
Human formalin‐fixed, paraffin‐embedded tissue sections of normal breast reduction mammoplasty and DCIS were de‐identified and obtained from Georgetown University tissue bank shared resource. The samples were processed and stained according to histochemical protocols described.
Patient gene expression analysis
Normalized patient data were downloaded either from the GEPIA database (for Breast GTEX and TCGA data), or Xenabrowser (Pan‐Can TCGA data). TCGA data were stratified at the median by ANCO1 expression and subsequently analyzed.
Quantification and statistical analysis
All statistical tests were carried out using GraphPad Prism 8.0, Microsoft Excel, GSEA, or ChIPseeker and EdgeR package in R. Statistical tests used described in figure legends.
Kaplan–Meier plots were generated and analyzed at http://kmplot.com/analysis.
Author contributions
MHK, VO, GMS, AW, and ATR designed the experiments and wrote the manuscript. MHK, VO, ST, and MY performed the biochemical and cell biological experiments. MHK, GTG, WBK, and MOS performed the bioinformatics analysis. All authors discussed the results and contributed to the manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Drs. Elena Tassi, Gray Pearson, Rebecca Riggins, Surojeet Sangupta, and Chunling Yi for their thoughtful input and discussions. We thank Dr. Kun‐Liang Guan for reagents. We thank Alqassem Abuarqoub and Chan Nguyen for their help with the image quantitation. This work was funded by T32 Training Grant in Tumor Biology CA009686 (PI: ATR), F31CA232664 (PI: MHK), R01CA231291 (PI: AW), and R01CA205632 (PI: ATR) from the National Cancer Institute and also by the Mistletoe Foundation (Research Fellowship; PI: MHK). The project described used the Tissue Culture & Biobanking, Flow Cytometry & Cell Sorting, Microscopy & Imaging, Animal Model, Histopathology & Tissue, and Genomics & Epigenomics Shared Resources which are partially supported by Award Number P30CA051008 (PI: Weiner) from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
EMBO Reports (2020) 21: e48741
Data availability
The RNA‐sequencing data have been deposited to the Gene Expression Omnibus under record GSE132475; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132475. The ChIP‐sequencing data have been deposited to the Gene Expression Omnibus under record GSE137284; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137284.
References
- 1. Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968 [DOI] [PubMed] [Google Scholar]
- 2. List HJ, Reiter R, Singh B, Wellstein A, Riegel AT (2001) Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res Treat 68: 21–28 [DOI] [PubMed] [Google Scholar]
- 3. Tilli MT, Reiter R, Oh AS, Henke RT, McDonnell K, Gallicano GI, Furth PA, Riegel AT (2005) Overexpression of an N‐terminally truncated isoform of the nuclear receptor coactivator amplified in breast cancer 1 leads to altered proliferation of mammary epithelial cells in transgenic mice. Mol Endocrinol 19: 644–656 [DOI] [PubMed] [Google Scholar]
- 4. Fereshteh MP, Tilli MT, Kim SE, Xu J, O'Malley BW, Wellstein A, Furth PA, Riegel AT (2008) The nuclear receptor coactivator amplified in breast cancer‐1 is required for neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Can Res 68: 3697–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Torres‐Arzayus MI, de Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M (2004) High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 6: 263–274 [DOI] [PubMed] [Google Scholar]
- 6. Ory V, Tassi E, Cavalli LR, Sharif GM, Saenz F, Baker T, Schmidt MO, Mueller SC, Furth PA, Wellstein A et al (2014) The nuclear coactivator amplified in breast cancer 1 maintains tumor‐initiating cells during development of ductal carcinoma in situ . Oncogene 33: 3033–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kushner MH, Riegel AT, Sharif GM (2018) The AIB1/NCOA3/SRC‐3 oncogene. In Oncogenes and Carcinogenesis, pp 1–23. London: IntechOpen. [Google Scholar]
- 8. Zhang A, Yeung PL, Li C‐W, Tsai S‐C, Dinh GK, Wu X, Li H, Chen JD (2004) Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J Biol Chem 279: 33799–33805 [DOI] [PubMed] [Google Scholar]
- 9. Garee JP, Chien CD, Li JV, Wellstein A, Riegel AT (2014) Regulation of HER2 oncogene transcription by a multifunctional coactivator/corepressor complex. Mol Endocrinol 28: 846–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noll JE, Jeffery J, Al‐Ejeh F, Kumar R, Khanna KK, Callen DF, Neilsen PM (2011) Mutant p53 drives multinucleation and invasion through a process that is suppressed by ANKRD11. Oncogene 31: 2836–2848 [DOI] [PubMed] [Google Scholar]
- 11. Lim SP, Wong NC, Suetani RJ, Ho K, Ng JL, Neilsen PM, Gill PG, Kumar R, Callen DF (2012) Specific‐site methylation of tumour suppressor ANKRD11 in breast cancer. Eur J Cancer 48: 3300–3309 [DOI] [PubMed] [Google Scholar]
- 12. Belandia B, Parker MG (2000) Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem 275: 30801–30805 [DOI] [PubMed] [Google Scholar]
- 13. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes‐associated protein localized in the cytoplasm. Genes Dev 15: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahoney WM, Hong J‐H, Yaffe MB, Farrance IKG (2005) The transcriptional co‐activator TAZ interacts differentially with transcriptional enhancer factor‐1 (TEF‐1) family members. Biochem J 388: 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zanconato F, Cordenonsi M, Piccolo S (2016) YAP/TAZ at the roots of cancer. Cancer Cell 29: 783–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park J‐S et al (2016) Tead and AP1 coordinate transcription and motility. Cell Rep 14: 1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim T, Hwang D, Lee D, Kim JH, Kim S‐Y, Lim D‐S (2017) MRTF potentiates TEAD‐YAP transcriptional activity causing metastasis. EMBO J 36: 520–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Y, Huang T, Cheng A, Yu J, Kang W, To K (2016) The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci 17: 138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A et al (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- 20. Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, Pan D (2014) A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 28: 432–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S (2013) Inactivation of the Hippo tumour suppressor pathway by integrin‐linked kinase. Nat Commun 4: 591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding N, Huang T, Yuan J, Mao J, Duan Y, Liao W, Xiao Z (2019) Yes‐associated protein expression in paired primary and local recurrent breast cancer and its clinical significance. Curr Probl Cancer 43: 429–437 [DOI] [PubMed] [Google Scholar]
- 23. Wang X, Su L, Ou Q (2012) Yes‐associated protein promotes tumour development in luminal epithelial derived breast cancer. Eur J Cancer 48: 1227–1234 [DOI] [PubMed] [Google Scholar]
- 24. Soule HD, Maloney TM, Wolman SR, Peterson WD, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC (1990) Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF‐10. Can Res 50: 6075–6086 [PubMed] [Google Scholar]
- 25. Miller FR, Santner SJ, Tait L, Dawson PJ (2000) MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ . J Natl Cancer Inst 92: 1185–1186 [DOI] [PubMed] [Google Scholar]
- 26. Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y et al (2008) Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 13: 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC et al (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome‐wide association between YAP/TAZ/TEAD and†AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim M, Kim T, Johnson RL, Lim D‐S (2015) Transcriptional co‐repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep 11: 270–282 [DOI] [PubMed] [Google Scholar]
- 30. The ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D et al (2015) YAP1 exerts its transcriptional control via TEAD‐mediated activation of enhancers. PLoS Genet 11: e1005465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zwart W, Theodorou V, Kok M, Canisius S, Linn S, Carroll JS (2011) Oestrogen receptor‐co‐factor‐chromatin specificity in the transcriptional regulation of breast cancer. EMBO J 30: 4764–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu F‐X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H et al (2012) Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Estarás C, Hsu H‐T, Huang L, Jones KA (2017) YAP repression of the WNT3gene controls hESC differentiation along the cardiac mesoderm lineage. Genes Dev 31: 2250–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang C, Yin M‐X, Wu W, Dong L, Wang S, Lu Y, Xu J, Wu W, Li S, Zhao Y et al (2016) Taiman acts as a coactivator of Yorkie in the Hippo pathway to promote tissue growth and intestinal regeneration. Cell Discov 2: 16006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu J, Li Q (2003) Review of the in VivoFunctions of the p160 steroid receptor coactivator family. Mol Endocrinol 17: 1681–1692 [DOI] [PubMed] [Google Scholar]
- 37. Zhang C, Robinson BS, Xu W, Yang L, Yao B, Zhao H, Byun PK, Jin P, Veraksa A, Moberg KH (2015) The ecdysone receptor coactivator Taiman links Yorkie to transcriptional control of germline stem cell factors in somatic tissue. Dev Cell 34: 168–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oh H, Slattery M, Ma L, White KP, Mann RS, Irvine KD (2014) Yorkie promotes transcription by recruiting a histone methyltransferase complex. Cell Rep 8: 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goh JY, Feng M, Wang W, Oguz G, Yatim SMJM, Lee PL, Bao Y, Lim TH, Wang P, Tam WL et al (2017) Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med 23: 1319–1330 [DOI] [PubMed] [Google Scholar]
- 40. Enerbäck C, Porter DA, Seth P, Sgroi D, Gaudet J, Weremowicz S, Morton CC, Schnitt S, Pitts RL, Stampl J et al (2002) Psoriasin expression in mammary epithelial cells in vitro and in vivo . Can Res 62: 43–47 [PubMed] [Google Scholar]
- 41. Porter D, Lahti‐Domenici J, Keshaviah A, Bae YK, Argani P, Marks J, Richardson A, Cooper A, Strausberg R, Riggins GJ et al (2003) Molecular markers in ductal carcinoma in situ of the breast. Mol Cancer Res 1: 362–375 [PubMed] [Google Scholar]
- 42. Emberley ED, Niu Y, Njue C, Kliewer EV, Murphy LC, Watson PH (2003) Psoriasin (S100A7) expression is associated with poor outcome in estrogen receptor‐negative invasive breast cancer. Clin Cancer Res 9: 2627–2631 [PubMed] [Google Scholar]
- 43. Emberley ED, Alowami S, Snell L, Murphy LC, Watson PH (2004) S100A7 (psoriasin) expression is associated with aggressive features and alteration of Jab1 in ductal carcinoma in situof the breast. Breast Cancer Res 6: 4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krop I, März A, Carlsson H, Li X, Bloushtain‐Qimron N, Hu M, Gelman R, Sabel MS, Schnitt S, Ramaswamy S et al (2005) A putative role for psoriasin in breast tumor progression. Can Res 65: 11326–11334 [DOI] [PubMed] [Google Scholar]
- 45. Doebar SC, Sieuwerts AM, de Weerd V, Stoop H, Martens JWM, van Deurzen CHM (2017) Gene expression differences between ductal carcinoma in situ with and without progression to invasive breast cancer. Am J Pathol 187: 1648–1655 [DOI] [PubMed] [Google Scholar]
- 46. Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, Zhu M, Wu J, Shi X, Taing L et al (2017) Cistrome Data Browser: a data portal for ChIP‐Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 45: D658–D662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint‐André V, Sigova AA, Hoke HA, Young RA (2013) Super‐enhancers in the control of cell identity and disease. Cell 155: 934–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M et al (2017) GeneHancer: genome‐wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017: 1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, Cooper GM, Reddy TE, Crawford GE, Myers RM (2013) Distinct properties of cell‐type‐specific and shared transcription factor binding sites. Mol Cell 52: 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- 51. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng C‐X, Brugge JS, Haber DA (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA 103: 12405–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, Guan K‐L (2012) YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR‐29. Nat Cell Biol 14: 1322–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Csibi A, Blenis J (2012) Hippo–YAP and mTOR pathways collaborate to regulate organ size. Nat Cell Biol 14: 1244–1245 [DOI] [PubMed] [Google Scholar]
- 54. Mugahid D, Kalocsay M, Gruver S, Peshkin L, Kirschner MW (2018) YAP independently regulates cell size and population growth dynamics via non‐cell autonomous mediators. bioRxiv 10.1101/482836 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leclerc E, Vetter SW (2015) The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim Biophys Acta 1852: 2706–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brenner AK, Bruserud ÃY (2018) S100 proteins in acute myeloid leukemia. Neoplasia 20: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuberappa PH (2016) Certainty of S100 from physiology to pathology. J Clin Diagn Res 10: ZE10–ZE15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kietzman WB, Graham GT, Ory V, Sharif G, Kushner MH, Gallanis GT, Kallakury B, Wellstein A, Riegel AT (2019) Short and long‐term effects of CDK4/6 inhibition on early stage breast cancer. Mol Cancer Ther 10.1158/1535-7163.MCT-19-0231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P, Schwendener RA, Bai XF, Shilo K, Zou X et al (2012) S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Can Res 72: 604–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123: 725–731 [DOI] [PubMed] [Google Scholar]
- 61. Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu F‐X, Karin M, Pan D et al (2015) A YAP/TAZ‐induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 29: 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma X‐J, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM et al (2003) Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA 100: 5974–5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
The RNA‐sequencing data have been deposited to the Gene Expression Omnibus under record GSE132475; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132475. The ChIP‐sequencing data have been deposited to the Gene Expression Omnibus under record GSE137284; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137284.