

Abstract
Protease-activated receptor 2 (PAR-2) has been demonstrated to promote invasion and metastasis of certain cancer cells. This study aimed to investigate the mechanism by which PAR-2 regulated invasion and migration of esophageal cancer (EC) cells EC109. A successfully constructed PAR-2 shRNA lentiviral vector (Lenti-PAR-2 shRNA) was stably transfected into EC109 cells, and the expression of PAR-2 in infected cells was detected by quantitative real-time PCR (qRT-PCR) and western blotting. Specific inhibitors, PD98059 (for MEK/ERK) and LY294002 (for PI3K/Akt), were used to confirm the role of MEK/ERK and PI3K/Akt signaling pathways, respectively, in PAR-2-regulated invasion and migration of EC109 cells. A significant decrease in PAR-2 mRNA and protein expression was detected in EC109 cells stably transfected with Lenti-PAR-2 shRNA. The PAR-2 agonist could dramatically promote cell invasion and migration, up-regulate the expression of MMP-9 and TM4SF3, and activate MEK/ERK and PI3K/Akt signaling pathways. However, PAR-2 gene silencing attenuated PAR-2-mediated enhancement of invasion and migration of EC109 cells, significantly down-regulated the mRNA and protein expression of MMP-9 and TM4SF3, and inhibited ERK (Try202/204) and Akt (Ser473) phosphorylation. An effect similar to PAR-2 silencing could be achieved with the two specific MEK/ERK and PI3K/Akt pathway inhibitors. Consequently, these results demonstrated that PAR-2 promoted invasion and migration of esophageal cancer cells EC109 by activating MEK/ERK and PI3K/Akt signaling pathways, which was accompanied by up-regulation of MMP-9 and TM4SF3 expression. Hence, PAR-2 may be a potential candidate for anti-metastasis treatment of EC.
Keywords: Protease-activated receptor 2, esophageal cancer, invasion, migration, MEK/ERK, PI3K/Akt
Introduction
Esophageal cancer (EC) is one of the main public health concerns in China, where it is the eighth most common malignancy, It is the sixth leading cause of cancer-related death in the world [1]. Insidious onset, rapid progression and low long-term survival rate are the main features of EC [2]. The poor prognosis of EC is largely due to its high invasiveness and early extensive metastasis; thus, blocking or preventing the invasion and migration of EC is essential to the development of effective therapy and new drugs.
Protease-activated receptor 2 (PAR-2) belongs to the family of G-protein coupled receptors (GPCRs) expressed on the surface of cell membranes. GPCRs are irreversibly activated by serine-dependent proteolytic cleavage. This unique process exposes a N-terminal sequence called a tethered ligand which binds and activates the receptor trans-membrane domain, triggering a cellular cascade in response to multiple inflammatory and carcinogenic stimuli [3,4]. The association of PAR-2 expression and cancer has long been reported. Recent studies found that PAR-2 was highly expressed in various digestive system tumors such as EC, liver, colon, and pancreatic cancer, and was related to the high morbidity, rapid progression, and poor prognosis of these tumors [5-8]. Inci et al. found that PAR-2 receptor was distinctly stained in the luminal part of the esophageal epithelium, where trypsin coming from duodenogastric/esophageal refluxate could cleave and activate the receptor [9]. Duodenogastroesophageal reflux was considered to be the main factor of EC development [10], which suggested that PAR-2 was closely related to the development of EC.
Our previous study confirmed that endogenous trypsin and SLIGKV, an artificial PAR-2 agonist peptide, could activate PAR-2, leading to EC cell proliferation, invasion, and metastasis [11,12], and transfection of PAR-2 targeted shRNA plasmid into EC109 cells reversed this phenomenon [13]. However, the low transient transfection efficiency of common plasmids and the short duration of gene silencing greatly increase the difficulty of further investigating the underlying mechanisms. Lentivirus-mediated shRNA (Lenti-shRNA) can directly and efficiently infect cells, and compared with other vectors, Lenti-shRNA vector can be rapidly introduced into a specific cell line that can stably and continuously express the target gene for a long time [14]. In addition, PAR-2 regulated cell migration has been reported to involve several signaling pathways, including MEK/ERK and PI3K/Akt pathways [7,15,16].
In the present study, we used Lenti-shRNA to silence the expression of PAR-2 gene in EC109 cells, and explored whether it affected cell invasion and metastasis through the regulation of MEK/ERK and PI3K/Akt signaling pathways.
Material and methods
Lentiviral shRNA vectors and cell transfection
Three pairs of PAR-2 siRNA, targeting human PAR-2 gene (Gene ID: 2150) and a non-targeting siRNA, NC, were transfected respectively into conventional cultured EC109 cells (CCAS, Shanghai, China) with LipofetamineTM 2000 (Hanbio Biotechnology, Shanghai, China) according to the protocol. Western blotting was used to detect PAR-2 protein level after 72 h, and the most efficient silencing sequence was screened. To construct lentivirus, the PAR-2-gene cDNA of the above selected silencing sequence cloned by PCR was inserted into pHB-U6-MCS-CMV-ZsGreen-PGK-puromycin lentiviral vectors. The recombinant lentivirus, PAR-2 shRNA vector, was produced by co-transfection of 293T cells with PSPAX2 and PMD2G plasmids with LipofetamineTM 2000. Later, EC109 cells were transduced with lentiviral vector, and GFP expression was observed under a fluorescence microscopy 48 h after transfection. Puromycin was added to medium at the concentration of 1 μg/mL for stable cell line selection. The Lenti-NC vector was used as negative control. The stable PAR-2-gene-silencing cell line, EC109-PAR-2 shRNA, was obtained after antibiotic selection for 2 weeks. After cells were harvested, the expression level of PAR-2 gene was determined by western blotting and quantitative real-time PCR (qRT-PCR).
Cell treatment and experimental grouping
EC109 cells in the logarithmic growth phase were treated with various reagents for 24 hours and then grouped. PAR-2 agonist group (50 μmol/L SLIGKV-NH2, Abcam, Cambridge, MA, USA), PAR-2 anti-agonist group (50 μmol/L VKGILS-NH2, R&D Systems, Minneapolis, MN, USA), MEK/ERK inhibitor group [EC109 cells were pretreated with 10 μmol/L PD98059 (Abcam, Cambridge, MA, USA) for 1 h, then 50 μmol/L SLIGKV-NH2 was added], PI3K/Akt inhibitor group [EC109 cells were pre-treated with 20 μmol/L LY294002 (Abcam, Cambridge, MA, USA) for 1 h, then 50 μmol/L SLIGKV-NH2 was added]. The optimal concentrations of SLIGKV-NH2 and VKGILS-NH2 were chosen according to data obtained in preliminary experiments. The same batch of untreated EC109 cells and EC109-PAR-2 shRNA cells served as control group and PAR-2 shRNA group, respectively.
Cell viability assay
To determine the final concentration of both PD98059 and LY294002, EC109 cells (5 × 104/mL) were plated in 96-well plates and divided into a control group, a drug group, and a blank group not seeded with cells, and 5 replicate wells in each group were incubated overnight. Then, fresh medium (100 μL/well) was added into the blank group and the control group, and cells in the drug group were incubated with various concentrations of PD98059 (0.1, 1, 10, 20, 30 μmol/L) or LY294002 (10, 20, 30, 40, 50 μmol/L) for 24 h. Cell viability was then detected using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to the manufacturers’ instructions. The cell viability ratio was calculated by the following formula: cell viability (%) = average absorbance of (drug-blank)/average absorbance of (control-blank) × 100%, and the IC50 (half-inhibitory concentration of drug) value was calculated using GraphPad PRISM. 6.0 (GraphPad Software, CA, USA).
Cell growth curve
To assess cell proliferation, EC109 cells treated with different drugs were seeded in 96-well culture dishes (3000 to 5000 cells/well) and measured by recording time lapse images of the cells over 7 days. During this study, 5 fixed sites per well were monitored using a JuLITM Stage Real-Time cell recorder (NanoEnTek, Seoul, Korea) at 24 h intervals, and confluences were also measured by the system software to plot the cell survival curve. The experiments were carried out in triplicate.
Transwell invasion and migration assay
To detect the invasion ability of EC109 cells, transwell chambers with a pore size of 8.0 µm were coated evenly with 60 μl Matrigel, which was diluted 1:4 in serum-free RPMI-1640 medium and allowed to gel in an incubator for 1 h. 100 μl cell suspensions (5 × 104 cells/well) were seeded in the upper chambers, while 600 μl RPMI-1640 medium containing 15% FBS was added into the lower chambers, and then cultured for 24 h at 37°C in a CO2 incubator. After invasion, the upper chamber was removed with tweezers, and cells were fixed in 100% methanol for 30 min and stained with 0.1% crystal violet for 30 min. Cells that did not pass through the membrane were carefully wiped with a wet cotton swab. Under the microscope, we randomly selected five fields of view: upper, middle, lower, left and right, and the average of cells were calculated for statistical analyses.
In the migration experiments, all procedures followed the transwell invasion assay, except for the coating of the transwell chamber with Matrigel. All experiments in every group were performed in triplicate.
RNA extraction and qRT-PCR
Cells of each group were collected and the total RNA was extracted by Trizol reagent according to typical protocol. RNA concentration and quality were measured by a NanoPhotometer® Spectrophotometer (IMPLEN, CA, USA) [17]. Primers are listed in Table 1. cDNA synthesis and qRT-PCR used commercial kit according to manufacturer’s protocol, and 2 μl reverse transcription product was added as template into a 20 μl reaction. A PikoRealTM TCR0096 (Thermo Fisher Scientific, Waltham, MA, US) was used for the reaction, and the Ct values per well were obtained as raw data for analyses of results. The 2-ΔΔCt method was used to calculate the relative expression of the target gene in each group, and GAPDH or β-actin was selected as endogenous control gene to normalize samples. All reactions were performed in triplicate.
Table 1.
Sequences
| Gene | Sequence (5’-3’) |
|---|---|
| siRNA1 | GCAAAGAACGCUCUCCUUU |
| siRNA2 | CCAUGUACCUGAUCUGCUU |
| siRNA3 | GCACCAUCCAAGGAACCAA |
| NC | UUCUCCGAACGUGUCACGUAA |
| PAR-2 shRNA | FP: TTCTCCGAACGTGTCACGTAA |
| RP: TTACGTGACACGTTCGGAGAA | |
| NC shRNA | FP: GCACCATCCAAGGAACCAA |
| RP: TTGGTTCCTTGGATGGTGC | |
| PAR-2 | FP: AATATGGCTGCTGATTCTGCTGGTC |
| RP: ATAGGCAGAGGCTGTGAGGAAGG | |
| MMP-2 | FP: CACCTACACCAAGAACTTCCGTCTG |
| RP: GTGCCAAGGTCAATGTCAGGAGAG | |
| MMP-9 | FP: TCCTGGTGCTCCTGGTGCTG |
| RP: CTGCCTGTCGGTGAGATTGGTTC | |
| TM4SF3 | FP: GCTATAAAAGAAAGTCGCTGCA |
| RP: TTCATTCACAATGCGATCAGAC | |
| β-actin | FP: TGTTTGAGACCTTCAACACCC |
| RP: AGCACTGTGTTGGCGTACAGG |
Western blotting assay
Cells were washed three times with pre-cooled PBS and lysed in protein extraction reagent (RIPA: PMSF = 100:1) for 30 min. After centrifugation at 12,000 rpm for 10 min at 4°C, the supernatant was collected as total cell protein and stored at -80°C. The protein concentration was determined by the bicinchoninic acid method. Total proteins were boiled at 100°C for 10 min for denaturation, loaded onto a 10% polyacrylamide gel for electrophoresis, and then transferred to a PVDF membrane. Membranes were incubated in blocking buffer (5% non-fat milk diluted with TBST) at room temperature for 1 h, and washed with TBST (3 ×; 5 minutes each time). Later, membranes incubated with mouse anti-human β-actin and GAPDH monoclonal antibodies (Proteintech, Chicago, IL, USA), and rabbit anti-human PAR-2, ERK1, p-ERK (Try202/204), Akt, p-Akt (Ser473), MMP-9 and MMP-2 monoclonal antibodies, and mouse anti-human TM4SF3 polyclonal antibody (Abcam, Cambridge, MA, USA) (1:1,000) prepared with 5% non-fat milk co-incubated overnight at 4°C. After washing with TBST (3 ×; 5 min each time), membranes were incubated with the corresponding secondary antibody (1:5,000) diluted with TBST at room temperature for 1 h. Following incubation in ECL regent for 1 minute, membranes were exposed and scanned by a ChemiDocTM XRS + System with Image LabTM Software (Bio-RAD, Hercules, CA, USA). The gray value of western blotting signals was assessed by image analysis software. The protein levels were normalized using GAPDH or β-actin as the endogenous control, and the ratio of target protein signal values to those of the corresponding endogenous control signal value were used for statistical analysis.
Statistical analysis
All experimental data are presented as x̅ ± s and statistically analyzed using GraphPad PRISM software version 6.0 (GraphPad Software, CA, USA). Student’s t test was used to compare the differences between two groups. One-way ANOVA with Tukey’s post-hoc test was used to investigate the differences among multiple groups. In all analyses, P < 0.05 was considered statistically significant.
Results
Identification of PAR-2 gene silencing-stable cell lines
We designed three different PAR-2 specific targeting sequences to investigate the function of PAR-2 in the biological progression of EC. Fortunately, different concentrations of siRNA targeting human PAR-2 gene could significantly down-regulate the expression of PAR-2, especially siRNA3 at 100 nmol/L (Figure 1A, 1B); therefore, it was chosen for subsequent lentivirus vector construction and lentivirus packaging (Figure 1C). After transfection for 48 h, GFP expression was observed in EC109 cells transfected with PAR-2 shRNA or NC lentivirus under a fluorescence microscope (Figure 1D). The expression of PAR-2 protein was also significantly lower than that of NC group (P < 0.01, Figure 1E), and the relative expression level of PAR-2 mRNA in EC109-PAR-2 shRNA cell line was decreased by 0.211 time that of EC109-NC cells (P < 0.01, Figure 1F). These results showed that a PAR-2 gene silencing-stable cell line was successfully constructed.
Figure 1.
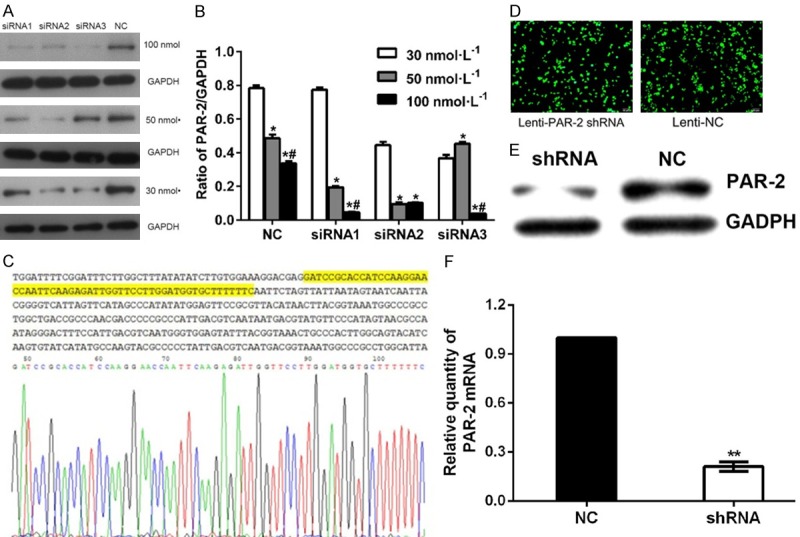
Construction of lentiviral vector and identification of Lentivirus-mediated PAR-2 gene silencing. (A, B) Western blot assay detected the expression level of PAR-2 protein in EC109 cells transfected with different concentrations of siRNA for 72 h. (C) PAR-2 shRNA plasmid sequencing peak. (D) Lenti-PAR-2 shRNA and Lenti-NC transfected EC109 cells show green fluorescence for 48 h (× 100). (E) Protein expression of PAR-2 in EC109 stable cell lines. **P < 0.01 vs NC group (F) The relative expression levels of PAR-2 mRNA in EC109 stable cell lines. *P < 0.05 vs 30 nmol/L group, #P < 0.05 vs 50 nmol/L group.
Effects of PAR-2 on invasion and migration of EC109 cells
The number of EC109 cells that migrated through the Matrigel barrier in the PAR-2 shRNA group was significantly lower than that in the blank group (40.8 ± 4.6 vs 184.5 ± 9.4, P < 0.01) and agonist group (40.8 ± 4.6 vs 255.3 ± 8.3, P < 0.01) (Figure 2B). Transwell cell migration assays showed the same pattern; the number of EC109 cells that migrated through the artificial basement membrane by deformation in the PAR-2 shRNA group was significantly decreased when compared with blank group (122 ± 9.6 vs 158.0 ± 6.1, P < 0.01) and agonist group (122 ± 9.6 vs 224.2 ± 8.8, P < 0.01). At the same time, the PAR-2 agonist dramatically enhanced the invasion and migration ability of EC109 cells, but had no significant effect on the ability of the PAR-2 shRNA group (Figure 2A). In summary, these results suggested that PAR-2 low expression could inhibit the capability of EC109 cell migration and invasion.
Figure 2.
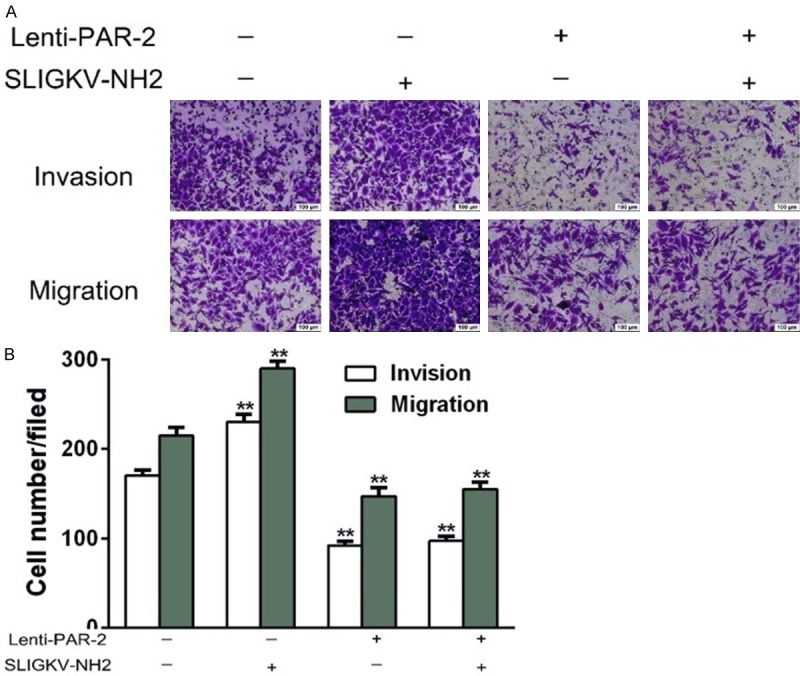
Effects of PAR-2 on invasion and migration of EC109 cells measured by transwell assay (× 200, x̅ ± s, n = 3). A. The effects of PAR-2 on invasion and migration of EC109 cells. EC109-PAR-2 shRNA and EC109 blank cells were treated with 50 μmol/L SLIGKV-NH2 for 24 h. The same batch of untreated PAR-2 shRNA and blank cells were also used for comparison analysis. B. Histogram of invasion and metastasis capacity of EC109 cells in different groups. **P < 0.01 vs blank group, ##P < 0.01 shRNA vs shRNA plus agonist.
Effects of PAR-2 on the mRNA and protein expression levels of cell invasion related molecules
In the blank group, agonist group, PAR-2 shRNA group, and PAR-2 shRNA plus agonist group, the relative expression levels of PAR-2 mRNA were 1.000 ± 0.000, 2.036 ± 0.095, 0.211 ± 0.017, and 0.305 ± 0.011; the relative expression levels of MMP-2 mRNA were 1.000 ± 0.000, 1.059 ± 0.059, 0.980 ± 0.020, and 0.989 ± 0.011; the relative expression levels of MMP-9 mRNA were 1.000 ± 0.000, 7.619 ± 0.136, 0.872 ± 0.77, and 0.873 ± 0.028; the relative expression levels of TM4SF3 mRNA were 1.000 ± 0.00, 0.809 ± 0.0139, 1.708 ± 0.014, and 1.760 ± 0.045 (Figure 3A).
Figure 3.
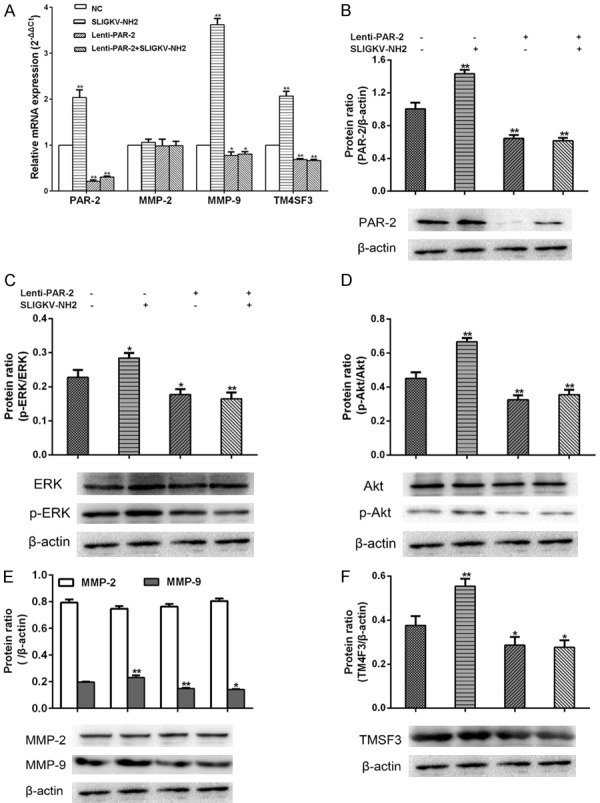
Effects of PAR-2 on the mRNA and protein expression levels of cell invasion related molecules (x̅ ± s, n = 3). A. The relative mRNA expression levels of PAR-2, MMP-2, MMP-9, and TM4SF3. B-F. Protein expressions of PAR-2, MMP-2, MMP-9, TM4SF3, ERK, p-ERK, Akt, and p-Akt. *P < 0.05 vs blank group, **P < 0.01 vs blank group.
The results of qRT-PCR and western blotting presented a parallel trend (Figure 3B-F). The protein expression of PAR-2, p-ERK (Try202/204), p-Akt (Ser473), MMP-9, and TM4SF3 in the PAR-2 shRNA group were significantly decreased (P < 0.05), but MMP-2 did not change obviously between blank and other treatment groups. However, PAR-2 activation significantly promoted the expression of the above-mentioned proteins (P < 0.05), whereas the PAR-2 agonist had no effect on the PAR-2 shRNA group.
MEK/ERK and PI3K/Akt signaling pathway inhibitors attenuated PAR-2-mediated enhancement of invasion and migration in EC109 cells
Two pharmacological inhibitors, PD98059 (for MEK/ERK pathway) and LY294002 (for PI3K/Akt pathway), were used to verify whether PAR-2-induced enhancement of invasion and metastasis was regulated by MEK/ERK and PI3K/Akt signaling pathways in EC109 cells. PD98059 and LY294002 significantly inhibited the proliferation of EC109 cells (P < 0.05), which was assessed by CCK8 assay after 24 h incubation (Figure 4A, 4B). The IC50 values of PD98059 and LY294002 were 10 μmol/L and 20 μmol/L, respectively. Subsequently, EC109 cells were exposed to different drugs for 7 days. The ability of cell proliferation was stronger in PAR-2 agonist group, when compared with blank control, while slower in PAR-2 shRNA group, the MAPK inhibitor group, and the PI3K/Akt inhibitor group (Figure 4C). Finally, PAR-2-induced cell invasion and metastasis were abolished by treatment with the two specific inhibitors (Figure 4D, 4E). As with the PAR-2 shRNA group, the ability of “three-dimensional” plane migration and erosion of Matrigel were significantly weaker after inhibitor treatment than those in the control group (P < 0.01), which suggested that the positive effect of PAR-2 on cell invasion and migration was regulated by MEK/ERK and/or PI3K/Akt signaling pathways in EC109 cells.
Figure 4.

MEK/ERK and PI3K/Akt signaling pathway inhibitors attenuated PAR-2-mediated enhancement of invasion and migration in EC109 cells (x̅ ± s, n = 3). CCK-8 assay is used to measure the effect of two pharmacologic inhibitors, PD98059 (for MEK/ERK pathway, A) and LY294002 (for PI3K/Akt pathway, B), on EC109 cells. (C) Cell growth curve in each group. (D) Cells passing through in every group in the transwell invasion and migration experiment. (E) Data summary of the invasion and migration capacity of EC109 cells in different groups. **P < 0.01 vs control group.
MEK/ERK and PI3K/Akt signaling pathway inhibitors attenuated PAR-2-mediated up-regulation of invasion related molecules in EC109 cells
The expression of PAR-2 mRNA in PAR-2 agonist group was increased, while decreased in PAR-2 shRNA group when compared with blank control (Figure 5A); The expression of MMP-9 (Figure 5C) and TM4SF3 (Figure 5D) was higher in PAR-2 agonist group but lower in PAR-2 shRNA group, MEK/ERK inhibitor group and PI3K/Akt inhibitor compared with blank control. However, the MMP-2 protein expression (Figure 5B) in each group did not change significantly.
Figure 5.
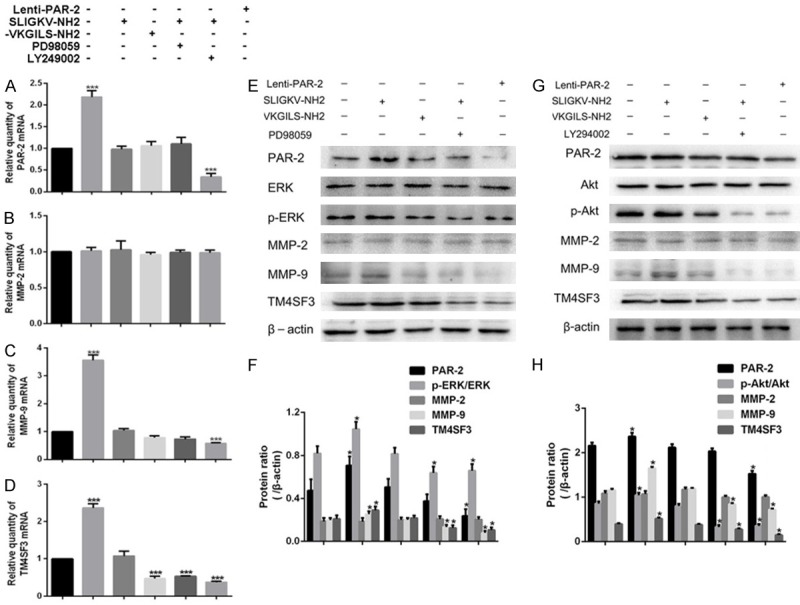
MEK/ERK and PI3K/Akt signaling pathway inhibitors attenuated PAR-2-mediated up-regulation of invasion related molecules in EC109 cells (x̅ ± s, n = 3). PAR-2 agonist group (50 μmol/L SLIGKV-NH2, Abcam, Cambridge, MA, USA), PAR-2 anti-agonist group (50 μmol/L VKGILS-NH2, R&D Systems, Minneapolis, MN, USA), MEK/ERK inhibitor group [EC109 cells were pretreated with 10 μmol/L PD98059 (Abcam, Cambridge, MA, USA) for 1 h, then 50 μmol/L SLIGKV-NH2 was added)], PI3K/Akt inhibitor group [EC109 cells were pre-treated with 20 μmol/L LY294002 (Abcam, Cambridge, MA, USA) for 1 h, then 50 μmol/L SLIGKV-NH2 was added)]. The optimal concentration of both SLIGKV-NH2 and VKGILS-NH2 was selected according to data obtained in preliminary experiments. The same batch of untreated EC109 cells and EC109-PAR-2 shRNA cells served as control group and PAR-2 shRNA group, respectively. A-D. The relative mRNA expression levels of PAR-2, MMP-2, MMP-9, and TM4SF3. E, F. Protein expressions of PAR-2, MMP-2, MMP-9, TM4SF3, ERK and p-ERK in blank control, PAR-2 agonist group, PAR-2 anti-agonist group, MEK/ERK inhibitor group, PAR-2 shRNA group. G, H. Protein expressions of PAR-2, MMP-2, MMP-9, TM4SF3, Akt and p-Akt in blank control, PAR-2 agonist group, PAR-2 anti-agonist group, PI3K/Akt inhibitor group, PAR-2 shRNA group. *P < 0.05 vs control group.
As shown in Figure 5E-H, we observed same tendencies of the above-mentioned protein expression in each group. In the PAR-2 agonist group, p-ERK (Try202/204) and p-Akt (Ser473) were significantly increased, and TM4SF3 and MMP-9 expressions were dramatically up-regulated, while they were decreased in PAR-2 shRNA group when compared with control group (P < 0.05). In addition, the expression of MMP-9 and TM4SF3 were inhibited by two pharmacologic inhibitors, accompanied by blockade of MEK/ERK and PI3K/Akt signaling pathways.
Discussion
RNA interference (RNAi) is a specific gene silencing phenomenon induced by dsRNA homologous to the target gene sequence [18]. The viral shRNA vector can also be used for RNA interference. Since lentivirus belongs to retrovirus group, it is feasible to inhibit continuous and stable expression of a target gene in transfected cells by using a Lenti-shRNA vector with antibiotic label [14]. In this experiment, we successfully established a cell line with PAR-2 gene silencing, and in this cell the mRNA and protein expression of PAR-2 were down-regulated to 21% and 30%, respectively, fully meeting the need for further study of PAR-2 function.
It has been reported that PAR-2 has the ability to promote cell invasion of colon cancer [7], breast cancer [15], and melanoma [19]. Knocking out PAR-2 in melanoma animal models limited the growth of primary tumors and reduces the amount of spontaneous metastases [19]. Wang et al. [5] revealed higher expression of PAR-2 in EC tissues than that in normal tissues. Meanwhile, our previous studies confirmed that high expression of PAR-2 was involved in EC invasion and metastatic phenotype [11]. In this study, we found that silencing PAR-2 inhibited the migration behavior of EC109 cells moving through transwell chambers, and arrested the invasion process of cells penetrating Matrigel, and these effects were enhanced in EC109 cells treated with PAR-2 agonist. Previous reports have shown that PAR-2 can inhibit MMP-9 activity in colon cancer cells [7]. TM4SF3 binds to prostaglandin F2a receptor regulatory protein (FPRP) in the presence of CD9 and CD81, thereby regulating the binding of GPCRs to their corresponding ligands [20]. There are also reports that MMP-2, MMP-9 [21,22], and TM4SF3 [23,24] play important roles in the invasion and migration of EC. Therefore, we proposed a hypothesis that MMP-2, MMP-9, or TM4SF3 may be involved in PAR-2-promoted cell invasion and metastasis. Consistent with phenotypic observations, we found that mRNA and protein expression of these metastasis-associated proteins, MMP9 and TM4SF3, were significantly reduced in EC109-PAR-2 shRNA cells, whereas increased in EC109 cells treated with PAR-2 agonist. Based on the above results, we initially considered that Lentivirus-mediated RNA knockdown of PAR-2 restricted EC cell invasion and migration, and the molecular mechanism may be associated with expression up-regulation of MMP-9 and TM4SF3.
Investigating which downstream signaling pathways PAR-2 regulates will help us better elucidate the molecular mechanisms of invasion and metastasis in EC cells. In this study, we found that the MEK/ERK and PI3K/Akt pathways were suppressed in PAR-2 gene silenced cells, but were enhanced in PAR-2 activated cells. Moreover, two pathway-specific inhibitors resulted in attenuation of the enhanced invasion and migration induced by PAR-2 activation in EC cells, and the increased expression levels of MMP-9 and TM4SF3. Our previous studies have revealed that PAR-2 promoted proliferation of EC109 cells by activating the MEK/ERK signaling pathway [12]. In addition, several studies in the past have shown that activation of MEK/ERK and PI3K/Akt signaling pathways could significantly promote malignant tumor invasion and metastasis, such as in esophageal [25], gastric [26], pancreatic [27], and breast cancers [28]. Li et al. found that activation of MEK/ERK and PI3K/Akt signaling pathways in breast cancer increased MMP-9 degradation of fibronectin and binding to β6 integrin, enhancing the aggressiveness of breast cancer cells [29]. Xia et al. found that miR302a was significantly down-regulated in EC cell lines, and up-regulation of miR302a markedly reduced phosphorylation of ERK1/2 and Akt and inhibited proliferation and invasion of EC cells [30]. These findings, together with results of the current study, indicated that PAR-2 may promote EC cell invasion and migration via MEK/ERK and PI3K/Akt pathway regulation.
In conclusion, the present study indicated that PAR-2 promoted invasion and migration of EC109 cells via MEK/ERK and PI3K/Akt pathway, and MMP-9 and TM4SF3 took part in this process.
Acknowledgements
This study was completed at the Institute of Rescue Medicine of the Affiliated Hospital of the Armed Police Logistics College. We would like to thank the laboratory teachers and classmates for their help. This work was supported by The National Natural Science Foundation of China [grant number 81273745]; the Seed Capital Fund of Affiliated Hospital of Logistic University of the Chinese People’s Armed Police Force (PAP) [grant number WHJ2016022]; the Major Scientific Research Projects of Tianjin Municipal Science and Technology Commission [grant number 15ZXLCSY00040]; and the Major Scientific Research Projects of Tianjin Municipal Science and Technology Commission [grant number 16ZXHLSY00120].
Disclosure of conflict of interest
None.
References
- 1.Testa U, Castelli G, Pelosi E. Esophageal cancer: genomic and molecular characterization, stem cell compartment and clonal evolution. Medicines (Basel) 2017;4:245–249. doi: 10.3390/medicines4030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 3.Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Dore AS, Geschwindner S, Grebner C, Hermansson NO, Jazayeri A, Johansson P, Leong L, Prihandoko R, Rappas M, Soutter H, Snijder A, Sundstrom L, Tehan B, Thornton P, Troast D, Wiggin G, Zhukov A, Marshall FH, Dekker N. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–115. doi: 10.1038/nature22309. [DOI] [PubMed] [Google Scholar]
- 4.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Liu HT, Li S, Li K, Lin N, Fan QX, Zheng YL. Prognostic value of protease-activated receptor 2 expression in oesophageal squamous cell carcinoma. J Int Med Res. 2010;38:1381–1388. doi: 10.1177/147323001003800420. [DOI] [PubMed] [Google Scholar]
- 6.Mussbach F, Ungefroren H, Gunther B, Katenkamp K, Henklein P, Westermann M, Settmacher U, Lenk L, Sebens S, Muller JP, Bohmer FD, Kaufmann R. Proteinase-activated receptor 2 (PAR2) in hepatic stellate cells - evidence for a role in hepatocellular carcinoma growth in vivo. Mol Cancer. 2016;15:54. doi: 10.1186/s12943-016-0538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Zhang X, Zhou H, Chen D, Xie H, Mu Y, Wu B, Yan J. Factor VIIa regulates the expression of caspase-3, MMP-9, and CD44 in SW620 colon cancer cells involving PAR2/MAPKs/NF-kappaB signaling pathways. Cancer Invest. 2013;31:7–16. doi: 10.3109/07357907.2012.743556. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda O, Egami H, Ishiko T, Ishikawa S, Kamohara H, Hidaka H, Mita S, Ogawa M. Expression of proteinase-activated receptor-2 in human pancreatic cancer: a possible relation to cancer invasion and induction of fibrosis. Int J Oncol. 2003;22:295–300. [PubMed] [Google Scholar]
- 9.Inci K, Edebo A, Olbe L, Casselbrant A. Expression of protease-activated-receptor 2 (PAR-2) in human esophageal mucosa. Scand J Gastroenterol. 2009;44:664–671. doi: 10.1080/00365520902783683. [DOI] [PubMed] [Google Scholar]
- 10.Olbe L. Strong activation of PAR-2 receptors: a common trigger for the development of gastrointestinal adenocarcinomas? Scand J Gastroenterol. 2007;42:1133–1137. doi: 10.1080/00365520601175983. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Xie LQ, Li X, Chen XY, Chen L, Zheng YM. Protease-activated receptor-2 agonists promote cell invasion and metastasis in human esophageal cancer cell line EC109. World Chinese Journal of Digestology. 2010;18:1313–1319. [Google Scholar]
- 12.Quanjun D, Qingyu Z, Qiliang Z, Liqun X, Jinmei C, Ziquan L, Shike H. Effect and mechanism of PAR-2 on the proliferation of esophageal cancer cells. Eur Rev Med Pharmacol Sci. 2016;20:4688–4696. [PubMed] [Google Scholar]
- 13.Chen J, Xie L, Zheng Y, Liu C. Effects of silenced PAR-2 on cell proliferation, invasion and metastasis of esophageal cancer. Oncol Lett. 2017;14:4115–4121. doi: 10.3892/ol.2017.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roy A, Ansari SA, Das K, Prasad R, Bhattacharya A, Mallik S, Mukherjee A, Sen P. Coagulation factor VIIa-mediated protease-activated receptor 2 activation leads to beta-catenin accumulation via the AKT/GSK3beta pathway and contributes to breast cancer progression. J Biol Chem. 2017;292:13688–13701. doi: 10.1074/jbc.M116.764670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka Y, Sekiguchi F, Hong H, Kawabata A. PAR2 triggers IL-8 release via MEK/ERK and PI3-kinase/Akt pathways in GI epithelial cells. Biochem Biophys Res Commun. 2008;377:622–626. doi: 10.1016/j.bbrc.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 17.Yang YH, Li MJ, Yi YJ, Li RF, Dong C, Zhang ZY. The root transcriptome of achyranthes bidentata and the identification of the genes involved in the replanting benefit. Plant Cell Rep. 2018;37:611–625. doi: 10.1007/s00299-018-2255-z. [DOI] [PubMed] [Google Scholar]
- 18.Rowley PT. RNA interference. N Engl J Med. 2003;348:564. doi: 10.1056/NEJM200302063480617. [DOI] [PubMed] [Google Scholar]
- 19.Olejar T, Vetvicka D, Zadinova M, Pouckova P, Kukal J, Jezek P, Matej R. Dual role of host par2 in a murine model of spontaneous metastatic B16 melanoma. Anticancer Res. 2014;34:3511–3515. [PubMed] [Google Scholar]
- 20.Little KD, Hemler ME, Stipp CS. Dynamic regulation of a GPCR-tetraspanin-G protein complex on intact cells: central role of CD81 in facilitating GPR56-Galpha q/11 association. Mol Biol Cell. 2004;15:2375–2387. doi: 10.1091/mbc.E03-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Back M, Ketelhuth DF, Agewall S. Matrix metalloproteinases in atherothrombosis. Prog Cardiovasc Dis. 2010;52:410–428. doi: 10.1016/j.pcad.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 22.González-Arriaga P, Pascual T, García-Alvarez A, Fernández-Somoano A, López-Cima MF, Tardón A. Genetic polymorphisms in MMP 2, 9 and 3 genes modify lung cancer risk and survival. BMC Cancer. 2012;12:121. doi: 10.1186/1471-2407-12-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Z, Ran YL, Hu H, Pan J, Li ZF, Chen LZ, Sun LC, Peng L, Zhao XL, Yu L, Sun LX, Yang ZH. TM4SF3 promotes esophageal carcinoma metastasis via upregulating ADAM12m expression. Clin Exp Metastasis. 2008;25:537–548. doi: 10.1007/s10585-008-9168-0. [DOI] [PubMed] [Google Scholar]
- 24.Kanetaka K, Sakamoto M, Yamamoto Y, Takamura M, Kanematsu T, Hirohashi S. Possible involvement of tetraspanin CO-029 in hematogenous intrahepatic metastasis of liver cancer cells. J Gastroenterol Hepatol. 2003;18:1309–1314. doi: 10.1046/j.1440-1746.2003.03182.x. [DOI] [PubMed] [Google Scholar]
- 25.Tian B, Chen X, Zhang H, Li X, Wang J, Han W, Zhang LY, Fu L, Li Y, Nie C, Zhao Y, Tan X, Wang H, Guan XY, Hong A. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget. 2017;8:42300–42313. doi: 10.18632/oncotarget.15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y, Ge Y, Jiang M, Zhou J, Luo D, Fan H, Shi L, Lin L, Yang L. MiR-592 promotes gastric cancer proliferation, migration, and invasion through the PI3K/AKT and MAPK/ERK signaling pathways by targeting spry2. Cell Physiol Biochem. 2018;47:1465–1481. doi: 10.1159/000490839. [DOI] [PubMed] [Google Scholar]
- 27.Amin H, Wani NA, Farooq S, Nayak D, Chakraborty S, Shankar S, Rasool RU, Koul S, Goswami A, Rai R. Inhibition of invasion in pancreatic cancer cells by conjugate of EPA with beta(3,3)-Pip-OH via PI3K/Akt/NF-kB pathway. ACS Med Chem Lett. 2015;6:1071–1074. doi: 10.1021/acsmedchemlett.5b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zibara K, Zeidan A, Mallah K, Kassem N, Awad A, Mazurier F, Badran B, El-Zein N. Signaling pathways activated by PACAP in MCF-7 breast cancer cells. Cell Signal. 2018;50:37–47. doi: 10.1016/j.cellsig.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Liu Z, Zhao C, Zhai L. Binding of MMP-9-degraded fibronectin to beta6 integrin promotes invasion via the FAK-Src-related Erk1/2 and PI3K/Akt/Smad-1/5/8 pathways in breast cancer. Oncol Rep. 2015;34:1345–1352. doi: 10.3892/or.2015.4103. [DOI] [PubMed] [Google Scholar]
- 30.Xia D, Tian S, Chen Z, Qin W, Liu Q. miR302a inhibits the proliferation of esophageal cancer cells through the MAPK and PI3K/Akt signaling pathways. Oncol Lett. 2018;15:3937–3943. doi: 10.3892/ol.2018.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]