

Abstract
Dysregulation of mitochondrial function often precedes malignant transformation of hematopoietic stem cells (HSCs). Mitochondria have a direct role in the maintenance of HSC functions. For example, D-2-hydroxyglutarate, generated due to the activity of mutated mitochondrial isocitrate dehydrogenase (IDH), has been implicated in the pathogenesis of leukemia. Furthermore, disturbances in the fatty acid breakdown and pyruvate oxidation are often seen in leukemic cells. These and other abnormalities expedite leukemogenesis and chemoresistance of leukemic cells. However, it needs to be elucidated whether these aberrations are the result or cause of leukemogenesis. Accordingly, for this review, a search was carried out in PubMed and Google Scholar databases until June 2019 to assess the relationship between metabolic pathways in altered mitochondria and leukemia development. In the present review, an overview of mitochondria-related mechanisms and their abnormalities in leukemia is presented, with mitochondrial pathways and factors, such as mitophagy, intermediary metabolism enzymes, oncometabolites and reactive oxygen species' generation, discussed as potential diagnostic and therapeutic targets in leukemia.
Keywords: Apoptosis, intermediary mitochondrial metabolism, leukemia, mitochondrial DNA, mitophagy, reactive oxygen species
INTRODUCTION
Mitochondria are involved in adenosine triphosphate (ATP) generation as well as in other essential cellular processes, such as apoptosis, metabolite synthesis, free radical generation and regulation of redox status.[1,2] The structural and biochemical features of mitochondria fluctuate in response to alterations in cellular physiology,[3,4] and any disturbances in mitochondrial processes may lead to dysfunctional and/or damaged mitochondria and disrupt normal cellular functions, which may initiate or aggravate pathophysiological conditions including cancer.
Leukemias are a group of highly heterogeneous cancers of the blood characterized by the excessive proliferation of immature or mature cells of lymphoid or myeloid origin in the peripheral blood and bone marrow [Table 1]. Leukemic cells develop from hematopoietic stem cells (HSCs), which are rare cells with self-renewal and multilineage differentiation capacity, through multiple and progressive genetic aberrations.[5,6]
Table 1.
Characteristics of leukemia subtypes
| Leukemia subtypes | Characteristics | ||||
|---|---|---|---|---|---|
| Clinical features | Laboratory findings | ||||
| Age | Onset | Symptoms | Peripheral blood | Bone marrow | |
| Acute lymphoblastic leukemia/ lymphoma | Most common hematological malignancy of children. Most common between 1 and 5 years of age and between 30 and 40 years | Abrupt | Bone marrow failure Extramedullary infiltration CNS involvement Testicular involvement |
Total WBC count: Markedly raised, ranging from 20×109/L to 200×109/L | Markedly hypercellular due to proliferation of blasts |
| Acute myelogenous leukemia | May develop at any age, but is more common in adults | Abrupt | Bone marrow failure Extramedullary infiltration |
Total WBC count: Markedly raised, ranging from 20×109/L to 100×109/L | Markedly hypercellular due to proliferation of blasts |
| Chronic lymphocytic leukemia | Between 50 and 60 years of age | Insidious | Fatigue, loss of weight and anorexia Generalized lymphadenopathy |
Total leukocyte count is increased (20-50×109/L) | Hypercellular marrow due to infiltration by mature lymphocytes |
| Chronic myelogenous leukemia | Usually occurs between 40 and 60 years of age | Insidious | Fatigue, weakness, weight loss and anorexia Splenomegaly Hepatomegaly |
Marked leukocytosis (12-600×109/L) Total leukocyte count usually exceeds 100×109/L Philadelphia chromosome |
Markedly hypercellular due to myeloid hyperplasia Philadelphia chromosome |
CNS – Central nervous system; WBC – White blood cell
Although several review articles have discussed individual pathways and/or mechanisms of mitochondrial function in HSCs and leukemia, a review article that considers mitochondria as the central focus is lacking. Therefore, the purpose of the present review is to present a holistic view of the significance of mitochondria in leukemia. PubMed and Google Scholar databases were searched until June 2019 according to the following strategy: mitochondrial pathways and factors, e.g., mitophagy, intermediary mitochondrial metabolism, oncometabolites, reactive oxygen species (ROS) generation and drugs that target the abnormal mitochondrial DNA (mtDNA) for treating leukemia. A total of 315 pertinent and contemporary studies were found, of which 192 met the initial criteria. Full-text articles were assessed if their titles had (1) leukemia in relation with mitochondrial genome; (2) mitophagy in leukemia; (3) apoptosis in leukemia; (4) intermediary mitochondrial metabolism in leukemia; (5) hypoxia and ROS in leukemia or (6) drugs targeting abnormal mtDNA for the treatment of leukemia. A total of 69 articles were identified. These articles were critiqued using critical reading and writing approaches. Author information, publication date, mitochondrial pathway and factor information were extracted from each relevant study and tabulated. After the initial data extraction, each category was grouped.
MITOCHONDRIAL GENOME AND LEUKEMIA
Mitochondria are unique cell organelles as they contain their own genome, also known as mitochondrial DNA (mtDNA). The replication and transcription of mtDNA are carried out in the displacement loop or D-loop region. Alterations of this region can have a significant impact on gene expression, with somatic mutations leading to a disruption in the electron transport chain. This increases the presence of ROS, leading to increased oxidative damage during cancer progression.[7,8]
The D-loop region is a regulatory region of mtDNA that is involved in replication and transcription and contains three hypervariable regions: HV-I, 16024-16383; HV-II, 57-372; and HV-III, 438-574. In the D-loop of pediatric acute myeloid leukemia (AML) patients, 1490 variations have been found at 237 positions.[8] Another study corroborated the association between the T16311C variation and poor prognosis in AML patients and found it to be the most frequently occurring (20%) single nucleotide polymorphism among these patients.[9] In the same study, increased nonsynonymous variations in cytochrome c oxidase-I and II (COX-I and COX-II) genes were also observed in AML subjects.[9] However, in a study by Zhou et al.,[10] no association was found between 2630 variations at 232 locations in the D-loop region and AML, except for T152C.
Mitochondria are essential for AML progression, a hypothesis supported by the higher mtDNA biosynthesis in AML blasts compared with normal hematopoietic cells.[11] Polymorphisms around H-strand replication origin (OH) and a conserved sequence block II were observed in acute lymphoblastic leukemia (ALL) and associated with disease burden and therapeutic response.[12]
Recently, Kodroń et al.[13] found a unique 3688G>A variation in ALL and its level varied in response to chemotherapy, making it a potential biomarker for monitoring the chemotherapeutic response in ALL [Figure 1].
Figure 1.
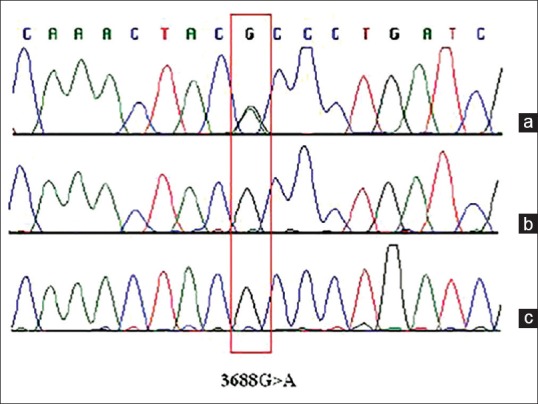
Molecular analysis of the ND1 gene in an acute lymphoblastic leukemia patient. This electropherograms of the ND1 gene show the 3688G>A transition only in a bone marrow sample collected at the time of leukemia diagnosis: (a) DNA isolated from bone marrow cells collected from the patient at the time of diagnosis. (b) DNA isolated from bone marrow cells collected from the patient at day 42 of treatment. (c) DNA isolated from the patient's blood. (Figure retrieved from the article of Kodroń et al.,[13] after permission for use)
In a study with a large cohort, it was seen that there was a strong positive association between a higher mtDNA copy number and chronic lymphocytic leukemia (CLL).[14] Recently, the same research group presented additional evidence and observed this association to be more pronounced among male smokers.[15] Finally, imatinib resistance in chronic myelogenous leukemia (CML) is associated with higher mtDNA instability and ROS levels.[16]
MITOPHAGY AND LEUKEMIA
Mitophagy refers to mitochondrial degradation, necessary for recycling of senescent or removal of damaged mitochondria.[17] Dysregulated mitophagy may result in hematological malignancies.[18] This is shown by haploinsufficiency of Beclin-1 leading to lymphoproliferative diseases, while invasive myeloproliferative diseases can be caused by defective autophagy-mediated through deletion of autophagy-related protein 7.[19]
Mutations in various autophagy-related genes have been associated with hematological malignancies. Aberrant expression of the BNIP3 and BNIP3 L genes has been observed in leukemic cell lines and AML.[20] WIPI-1 and WIPI-2 expression were found to be significantly reduced in AML and acute promyelocytic leukemia, respectively.[21] Mutations in FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD), common in AML cases, result in constitutive activation of FLT3 and promote leukemic cell proliferation and survival.[22] On drug-induced inhibition of FLT3-ITD signaling, ceramide synthesis gets activated and results in selective mitophagy and leukemic cell death. Mechanistically, dynamin-related protein 1 (Drp1) gets activated following protein kinase A (PKA) deactivation, and PKA is one of the downstream effectors of the FLT3-ITD-dependent oncogenesis.[22] Therefore, pharmacological targeting of FLT3-ITD might promote mitophagy. In contrast, Heydt et al.[23] reported increased basal autophagy in FLT3-ITD cells.
In addition, aberrant mitophagy (and/or autophagy) signaling pathways have also been seen in several hematological malignancies. The phosphatidylinositol-3-kinase/AKT (PI3K/Akt) and the mammalian target of rapamycin (mTOR) intracellular signaling pathways (PI3K/Akt/mTORC1) are important in regulating cell growth, as they repress mitophagy, which is abnormally activated in AML patients. However, the role of mitophagy in the pathogenesis of AML remains controversial.[24] Similarly, elevated notch signaling, which regulates autophagy and mitochondrial remodeling in murine HSCs, resulted in low autophagy and a preleukemic phenotype, and abrogation of elevated notch signaling restored the HSC function and multilineage differentiation potential.[25]
APOPTOSIS AND LEUKEMIA
Alteration in apoptosis may have a critical role in the pathogenesis of myelodysplastic syndrome (MDS) and MDS to AML transformation, as increased proliferation, but not reduced apoptosis, is characteristic of the transformation stage, whereas increased apoptosis is a predominant feature of early-stage MDS.[26] In addition, mutations in apoptotic proteins are associated with chemoresistance and increase in tumor aggressiveness.[27]
Double-stranded RNA-dependent protein kinase (PKR) is a mediator for the antiproliferative effects of cellular stresses in HSCs. When activated, it inhibits proliferation and initiates apoptosis by phosphorylating eukaryotic initiation factor 2α. A decrease in PKR has been observed in several malignancies including leukemia, resulting in decreased activity of protein phosphatase 2A (PP2A), a BCL2 phosphatase, and consequently, enhanced BCL2 phosphorylation and activity. Activated PKR is required for PP2A activation, which in turn regulates the intrinsic pathway of apoptosis through dephosphorylation of BCL2.[28] A G(-248) A polymorphism in BAX and a C(-717)A polymorphism in BCL2 have been observed in diffuse large B-cell lymphoma patients and are associated with reduced survival through increased chemoresistance.[29] Increase in BAX and BCL2 levels correlate with poor outcome in AML as well, although there is conflicting evidence, with some studies showing a positive relation, some showing a negative relation and some showing no relation.[30]
Polymorphisms in the BCL2L11 gene, coding for pro-apoptotic BIM (a proapoptotic member of BCL2 protein family) protein leads to decrease in the overall survival of children under chemotherapy for ALL.[31] BIM deletion/polymorphism promotes inferior response and intrinsic resistance to tyrosine kinase inhibitors in CML patients.[32,33]
Moreover, a minimal relationship was found between the induction of a therapy response in B-cell precursor ALL and deletion polymorphism of BIM mutation.[34] Low MCL-1 levels are required for complete remission in CLL, as high levels of this prosurvival protein are implicated in the development of CLL and in resistance to therapy. Hence, MCL-1 levels are directly related to the survival rates of patients.[35]
In addition, various studies have pointed out the significance of the antiapoptotic and proapoptotic protein ratio in predicting disease progression, therapy response, prognosis and overall survival in leukemic patients. Low Bax/Bcl-2 ratio is accompanied by a relapse of ALL among pediatric patients,[36] and a decrease in the Bcl-2/Bax ratio is indicative of the response to chemotherapy in CLL patients. A recent study reported that a Bax/Bcl-2 ratio of >1.5 is associated with a low-risk profile in CLL, and an elevated Bcl-2/Bax ratio negatively correlates with the lymphocyte doubling time in CLL.[37] A Bcl-2 promoter region polymorphism is more reliable than the BAX gene promoter polymorphism in any ALL scoring system.[38]
INTERMEDIARY MITOCHONDRIAL METABOLISM AND LEUKEMIA
A metabolic shift from tricarboxylic acid (TCA) cycle to fatty acid oxidation (FAO) has been observed in leukemic cells and is associated with increased proliferation, while amelioration of FAO resulted in apoptosis [Figure 2].[39,40] This corresponds to the upregulation of carnitine palmitoyltransferase 1A (CPT1A) that catalyzes the rate-limiting step of β-oxidation in mitochondria in AML cells.[41] Leukemic cells activate FAO not only by increasing the activity of CPT1 but also by downregulating its negative regulator malonyl CoA, which is synthesized by acetyl-CoA carboxylase.[41] Interestingly, hexokinase-II, glucose transporter-1, lactate dehydrogenase (LDH) and hypoxia-induced factor 1α (HIF-1α) have been found to be upregulated in AML patients with no remission compared with AML patients with complete remission.[42]
Figure 2.

Proposed paradigm for the reprograming of intermediary metabolism in leukemia cells. (a) In monoculture: leukemia cells maintain an intact Krebs cycle that help in oxidizing pyruvate-derived acetyl-CoA and fatty acid-derived acetyl-CoA which lead to induction of apoptosis. (b) In coculture: leukemia cells with bone marrow-derived stromal cells increase Krebs cycle activity which does not oxidize pyruvate-derived acetyl-CoA, but instead metabolizes most of fatty acid-derived acetyl-CoA that increasing resistance to apoptosis (Figure retrieved from the article of Vélez et al.,[39] after permission for use)
Several other enzymes of the TCA cycle also have been associated with leukemic progression. Fumarate hydratase 1, which converts fumarate into malate, is essential for the development, but not maintenance, of leukemia-initiating cells in MEIS1/Hoxa9-driven leukemia.[43] Pyruvate carboxylase catalyzes carboxylation of pyruvate to oxaloacetate; the first reaction of gluconeogenesis is highly activated in the AML K562 cell line.[44] Mutations in fumarate hydratase, IDH and succinate dehydrogenase result in accumulation of fumarate, 2-oxoglutarate and succinate, respectively. In turn, this inhibits ten-eleven translocase (TET) that converts 5-methylcytosine to 5-hydroxymethylcytosine, which epigenetically regulates the relevant genes in AML. The importance of balancing these cytosines is shown by how mutations in TET-2 are implicated in AML.[45]
Defects in the proteins of the electron transport chain are responsible for the dependency of leukemic cells on the glycolytic pathway for energy requirement and are associated with poor prognosis. For example, β-F1-ATPase (part of complex 5 of the respiratory chain) is downregulated in AML patients with no remission compared with patients with complete remission, indicating that low activity of ATP synthase in leukemia may be associated with poor prognosis.[42]
IDH is often mutated in AML and MDS.[46] Three isoforms of IDH are present inside the cell: nicotinamide adenine dinucleotide phosphate (NADPH)-dependent IDH1 and 2 and NADH-dependent IDH3. IDH1 is present in the cytoplasm and peroxisomes, while IDH2 and 3 reside in the mitochondria. IDH1 mutations are usually associated with solid malignancies, while IDH2 mutations are more common in hematological malignancies; associations of IDH3 mutations have not yet been reported.[47] In addition to acting as a TCA cycle enzyme, IDH has multiple functions such as HIFs degradation, regulation of cellular redox state, histone demethylation and DNA modification. Mutated IDH has neomorphic activity that converts 2-oxoglutarate (2-OG) into 2-hydroxyglutarate (2-HG).[48,49] Furthermore, demonstration of 2HG production is the common feature of IDH1 and IDH2 mutations, and the measurement of 2HG levels allows the identification of additional IDH mutations in AML patients.[50] These metabolites normally exist in cells in small quantities and increase by more than 100 folds in case of IDH mutation. Owing to structural similarity with 2-OG, R-2-HG competitively inhibits 2-OG-dependent enzymes, including the Jumonji domain-containing group of histone lysine demethylases and TET family of 5-methylcytosine hydroxylases. This results in genome-wide alterations in the DNA, and thus abnormal gene expression. R-2HG also stabilizes HIFs by inhibiting prolyl hydroxylases, indicating that, in hematological malignancies, enzymes participating in epigenetic regulation are also targets of R-2HG.[48,49]
HYPOXIA AND LEUKEMIA
The hypoxic environment of the bone marrow niche is essential for maintenance and quiescence of HSCs. HIFs play a major role in maintaining HSCs as deletions in these factors result in increased proliferation and decreased self-renewal capacity of HSCs. HIFs are activated during hypoxia.[51] In addition, oncometabolites such as fumarate and succinate and the glycolytic end products pyruvate and lactate stabilize HIF-1α by allosteric inhibition of prolyl hydroxylase. The activated HIF-1α translocates to the nucleus and heterodimerizes with aryl hydrocarbon receptor nuclear translocator (a subunit of HIF-1). This activates the transcription of the glucose transporter and glycolytic enzyme genes and represses proteins of mitochondrial respiration, thereby promoting preferential aerobic glycolysis.[51,52] The most prominent enzymes induced by HIF-1α are LDHA and pyruvate dehydrogenase kinase-1 (PDK1).[47] PDK1 plays an important role in regulating glucose and fatty acid metabolism and homeostasis through phosphorylation of the pyruvate dehydrogenase subunits 1 and 2, resulting in its inactivation. Inhibition of pyruvate dehydrogenase activity results in regulation of metabolite flux through the TCA cycle with downregulation of aerobic respiration and inhibition of acetyl CoA formation from pyruvate.
The deletion of LDHA leads to the inhibition of function of HSCs and hematopoietic progenitor cells. A decrease in PDK decreases suppression of oxidative phosphorylation. PKD1 is an important enzyme of the glycolytic pathway and regulates the flux of pyruvate from cytoplasm to mitochondria. It is overexpressed in cancer cells and induces apoptosis resistance. It has been found that 2,2-dichloroacetophenone (PKD1 inhibitor) increased apoptosis induction in AML cells and inhibited leukemic growth in a mouse AML model.[52] In addition, murine HSCs with higher ROS production have a decreased self-renewal capacity due to the inhibition of p38 mitogen-activated protein kinases (which is responsive to stress stimuli and involved in cell differentiation and apoptosis) and mTOR pathways. Increased ROS production has been described in AML, CML and MDS.[52]
HIFs have a controversial role in solid tumors, with some studies indicating that it has no role in solid tumors, while others supporting an oncogenic activity of HIFs.[53] Solid tumors offer hypoxic microenvironments, which upregulate HIFs that promote cancerous growth by increasing ATP generation through aerobic glycolysis, decreasing cellular oxygen requirement and increasing ROS generation. Therefore, high HIF levels in solid cancers are considered a marker for poor prognosis. Increased levels of HIFs have also been reported in almost all major types of leukemias and MDS.[54] However, the role of HIFs in leukemogenesis is debatable. A study showed a tumor-suppressive role of HIF-1α, as its loss resulted in disease aggravation in a murine AML model, while in a human study, the effects of HIF-1α deletion were found to have no effect on AML progression. Furthermore, conditional deletion of HIF-2α was shown to accelerate the development of leukemic stem cells and shorten the AML latency, and this effect was further strengthened by codeletion of HIF-1α.[54] The controversial role of HIFs in leukemogenesis is further highlighted by the contrasting results of two studies, wherein one study demonstrated the pro-leukemogenic role of HIFs in APL and another study revealed that suppression of HIF-1α delays leukemia progress.[55] In addition, ectopic expression of HIF-2α accelerates leukemic cell proliferation in mouse models, but higher expression of HIF-2α in human leukemia cells is not associated with poor prognosis.[56]
REACTIVE OXYGEN SPECIES AND LEUKEMIA
ROS are a heterogeneous group of molecules produced by the electron transport chain in the mitochondria or NADPH oxidases. They play an important role in various cellular processes, including an antimicrobial role during phagocytosis. However, their excessive production is implicated in pathological processes and leads to a state of oxidative stress. A state of chronic oxidative stress has been found in many hematopoietic malignancies, including AML, ALL, MDS and CML.[57]
Cells have an antioxidant system to control ROS levels in cell organelles, cytoplasm and even in the extracellular space. Superoxide dismutase reduces O2 •− to H2O2, which is further reduced to water by glutathione peroxidase in the mitochondria at the expense of reduced glutathione and NADPH.[58] High levels of H2O2 are often seen in cancer cells, where it may induce DNA damage and malignant cellular transformation by either activating an oncogene or deactivating a tumor-suppressor gene. In addition to genotoxicity, H2O2 also affects cellular proliferation and migration by mediating intracellular signaling. Consistently high ROS levels are detrimental to even cancer cells; therefore, ironically, cancer cells have better antioxidant defense mechanisms to counter the high ROS levels.[52] Several studies have implicated ROS in the initiation, progression and chemoresistance of leukemia. There are conflicting reports regarding the source of elevated ROS levels in leukemic cells and have been variously attributed to NADPH oxidases in CML and other mitochondrial enzymes in CLL,[59] whose levels are regulated by protein kinase C epsilon. Low ROS level is essential to keep HSCs in the quiescent state, and ROS generation in these cells is tightly regulated. Nuclear factor erythroid 2-related factor 2 (Nrf2), forkhead box O (FOXO) and ataxia telangiectasia mutated (ATM) are major factors that control the level of ROS by inducing antioxidant enzymes.[60]
Nrf2 is a global regulator of the oxidative stress response and is expressed constitutively in HSCs and regulated by Kelch-like ECH-associated protein 1 (KEAP1)-mediated degradation under physiological conditions.[61] Murakami et al.[61] observed that high expression of Nrf2 in KEAP1-deficient mice commits HSCs to the granulocyte-monocyte lineage at the expense of the erythroid-lymphoid lineage, although the underlying mechanisms are not known. Moreover, increased Nrf2-mediated signaling has been identified in AML and CLL cells. FOXO transcription factors, typically known as the longevity factors, also keep ROS levels in check in HSCs.[62] They activate antioxidant enzymes and DNA repair enzymes and/or repress mitochondrial respiration. Apart from regulating ROS levels, they have also been implicated in a variety of cellular processes such as apoptosis, cell cycle arrest, immune response, metabolism, stress resistance and longevity. Conditional deletion of FOXO in HSCs increased ROS generation and HSC depletion, indicating their essential role in HSC function.[62] Recently, Kazlov et al.[63] observed phosphorylation changes in 2716 proteins following oxidative damage and identified 11 candidate cytoplasmic proteins regulated by ATM. In addition, somatic as well as germline mutations were identified in the ATM gene in a significant number of CLL cases with the 11q deletion, along with mutated TP53 and 17p deletion. However, no strong role was seen between ATM mutation and function.[64]
Taken together, the hypoxic environment is important for maintaining HSCs quiescence and self-renewal, while ROS-mediated signaling promotes HSC differentiation. Oxidative stress leads to HSCs senescence and loss of self-renewal capacity and quiescence as well as leukemogenesis [Figure 3].[65]
Figure 3.

This is an overview of the main components of the HSC niche and their alterations in leukemia. Simplified schematic of the normal HSC niche (left) and leukemic niche (right). The diagram illustrates some of the best characterized candidate niche cells and factors that have been found altered in leukemias. The right panel summarizes niche abnormalities observed in various experimental models representing different leukemia types. (Figure retrieved from the article of Sánchez-Aguilera and Méndez-Ferrer[65] after permission for use). HSC – Hematopoietic stem cell; LSC – Leukemia stem cell
CLINICAL IMPLICATIONS
Recently, new drugs have been developed that target the abnormal mtDNA for treating different types of leukemia. One such drug is enasidenib (Idhifa), which targets IDH2, and was approved in 2017 by the US Food and Drug Administration (FDA) for the treatment of adult patients with relapsed or refractory AML.[66] Similarly, in April 2016, Venetoclax (ABT-199), an orally bioavailable inhibitor of Bcl-2 and a BH3-mimetic, was approved by the FDA for the treatment of CLL patients with 17p deletion,[67] and in November 2018, it was approved for the treatment of AML in patients aged ≥75 years.[68] In addition, Midostaurin, a small-molecule kinase inhibitor with activity against the receptor tyrosine kinase FLT3, targeted agents for the treatment of molecularly defined subtypes of AML.[68] Vitamin C has also used in the treatment of TET2-mutated patients. TET-mediated DNA oxidation induced by Vitamin C treatment in leukemia cells enhances their sensitivity to poly ADP ribose polymerase inhibition and could provide a safe and effective combination to block leukemia progression.[69]
CONCLUSION
Proper functioning of the mitochondria is crucial to HSC function, and any aberration may result in pathological conditions, including cancer. However, the metabolic cues or signals that initiate the abnormalities in HSC mitochondria are still obscure. Although there is ample evidence regarding the role of mitochondria in tumorigenesis, its relevance in hematological malignancies has only recently been elucidated. Despite the significant mitochondrial abnormalities seen in leukemic cells, its role in leukemogenesis and progression is relatively unknown. The increased risk of developing hematological malignancies relative to increased mtDNA copy number and nonsynonymous sequence variation of D-loop region of the mitochondria needs to be validated on large and multiethnic populations at various centers. Finally, mitochondrial pathways and factors, e.g., mitophagy, intermediary metabolism enzymes, oncometabolites and ROS generation, need to be studied further as potential diagnostic and therapeutic targets in leukemia.
Peer review
This article was peer reviewed by four independent and anonymous reviewers.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The author would like to thank Kodroń et al.,[13] Acta Biochemica; Vélez et al.,[39] Molecular and Cellular Oncology; and Sánchez-Aguileraand Méndez-Ferrer,[65] Cellular and Molecular Life Sciences; as three figures were retrieved from these articles. These figures helped in clarifying many points in this paper.
REFERENCES
- 1.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–7. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253–62. doi: 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK, et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature. 2015;525:124–8. doi: 10.1038/nature14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–17. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, et al. Stem cells. A symmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–3. doi: 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 7.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat Rev Genet. 2015;16:530–42. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi A, Pramanik R, Vishnubhatla S, Ali S, Bakhshi R, Chopra A, et al. Pattern of mitochondrial D-loop variations and their relation with mitochondrial encoded genes in pediatric acute myeloid leukemia. Mutat Res. 2018;810:13–8. doi: 10.1016/j.mrfmmm.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Silkjaer T, Nørgaard JM, Aggerholm A, Ebbesen LH, Kjeldsen E, Hokland P, et al. Characterization and prognostic significance of mitochondrial DNA variations in acute myeloid leukemia. Eur J Haematol. 2013;90:385–96. doi: 10.1111/ejh.12090. [DOI] [PubMed] [Google Scholar]
- 10.Zhou J, Gou H, Ye Y, Zhou Y, Lu X, Ying B. Sequence variations of mitochondrial DNA D-loop region in patients with acute myeloid leukemia. Oncol Lett. 2017;14:6269–76. doi: 10.3892/ol.2017.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liyanage SU, Hurren R, Voisin V, Bridon G, Wang X, Xu C, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129:2657–66. doi: 10.1182/blood-2016-10-741207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwok CS, Quah TC, Ariffin H, Tay SK, Yeoh AE. Mitochondrial D-loop polymorphisms and mitochondrial DNA content in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:e239–44. doi: 10.1097/MPH.0b013e31820a5ece. [DOI] [PubMed] [Google Scholar]
- 13.Kodroń A, Ghanim M, Krawczyk KK, Stelmaszczyk-Emmel A, Tońska K, Demkow U, et al. Mitochondrial DNA in pediatric leukemia patients. Acta Biochim Pol. 2017;64:183–7. doi: 10.18388/abp.2016_1444. [DOI] [PubMed] [Google Scholar]
- 14.Hosnijeh FS, Lan Q, Rothman N, San Liu C, Cheng WL, Nieters A, et al. Mitochondrial DNA copy number and future risk of B-cell lymphoma in a nested case-control study in the prospective EPIC cohort. Blood. 2014;124:530–5. doi: 10.1182/blood-2013-10-532085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim C, Bassig BA, Seow WJ, Hu W, Purdue MP, Huang WY, et al. Mitochondrial DNA copy number and chronic lymphocytic leukemia/small lymphocytic lymphoma risk in two prospective studies. Cancer Epidemiol Biomarkers Prev. 2015;24:148–53. doi: 10.1158/1055-9965.EPI-14-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blasiak J, Hoser G, Bialkowska-Warzecha J, Pawlowska E, Skorski T. Reactive oxygen species and mitochondrial dna damage and repair in BCR-ABL1 cells resistant to imatinib. Biores Open Access. 2015;4:334–42. doi: 10.1089/biores.2015.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–85. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018;23:86–100. doi: 10.1016/j.stem.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You L, Jin S, Zhu L, Qian W. Autophagy, autophagy-associated adaptive immune responses and its role in hematologic malignancies. Oncotarget. 2017;8:12374–88. doi: 10.18632/oncotarget.13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarini M, Machado-Neto JA, Duarte AD, Pericole FV, Vieira KP, Niemann FS, et al. BNIP3L in myelodysplastic syndromes and acute myeloid leukemia: Impact on disease outcome and cellular response to decitabine. Haematologica. 2016;101:e445–8. doi: 10.3324/haematol.2016.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brigger D, Proikas-Cezanne T, Tschan MP. WIPI-dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis. 2014;5:e1315. doi: 10.1038/cddis.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood. 2016;128:1944–58. doi: 10.1182/blood-2016-04-708750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heydt Q, Larrue C, Saland E, Bertoli S, Sarry JE, Besson A, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018;37:787–97. doi: 10.1038/onc.2017.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72:2337–47. doi: 10.1007/s00018-015-1867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellavia D, Palermo R, Felli MP, Screpanti I, Checquolo S. Notch signaling as a therapeutic target for acute lymphoblastic leukemia. Expert Opin Ther Targets. 2018;22:331–42. doi: 10.1080/14728222.2018.1451840. [DOI] [PubMed] [Google Scholar]
- 26.Langemeijer SM, Mariani N, Knops R, Gilissen C, Woestenenk R, de Witte T, et al. Apoptosis-related gene expression profiling in hematopoietic cell fractions of MDS patients. PLoS One. 2016;11:e0165582. doi: 10.1371/journal.pone.0165582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed JC. Targeted Therapy of Acute Myeloid Leukemia. New York, Heidelberg, Dordrecht, London: Springer; 2015. Roles of apoptosis-regulating Bcl-2 family genes in AML; pp. 47–65. [Google Scholar]
- 28.Cheng X, Bennett RL, Liu X, Byrne M, Stratford May W. PKR negatively regulates leukemia progression in association with PP2A activation, Bcl-2 inhibition and increased apoptosis. Blood Cancer J. 2013;3:e144. doi: 10.1038/bcj.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brito ABC, Delamain MT, de Oliveira C, Fanelli MF, Soares FA, de Souza CA, et al. Association of BAX G(-248) A and BCL2 C(-717) A polymorphisms with outcome in diffuse large B-cell lymphoma patients. Br J Haematol. 2017;177:650–4. doi: 10.1111/bjh.14089. [DOI] [PubMed] [Google Scholar]
- 30.Kulsoom B, Shamsi TS, Afsar NA, Memon Z, Ahmed N, Hasnain SN. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for Bcl-2-directed therapy? Cancer Manag Res. 2018;10:403–16. doi: 10.2147/CMAR.S154608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plesa M, Gagné V, Glisovic S, Younan M, Sharif-Askari B, Laverdière C, et al. Influence of BCL2L11 polymorphism on osteonecrosis during treatment of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2019;19:33–41. doi: 10.1038/s41397-017-0002-4. [DOI] [PubMed] [Google Scholar]
- 32.Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521–8. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 33.Ko TK, Chin HS, Chuah CT, Huang JW, Ng KP, Khaw SL, et al. The BIM deletion polymorphism: A paradigm of a permissive interaction between germline and acquired TKI resistance factors in chronic myeloid leukemia. Oncotarget. 2016;7:2721–33. doi: 10.18632/oncotarget.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang M, Miyake K, Kagami K, Abe M, Shinohara T, Watanabe A, et al. Lack of association between deletion polymorphism of BIM gene and in vitro drug sensitivity in B-cell precursor acute lymphoblastic leukemia. Leuk Res. 2017;60:24–30. doi: 10.1016/j.leukres.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Besbes S, Pocard M, Mirshahi M, Billard C. The first MCL-1-selective BH3 mimetics have therapeutic potential for chronic lymphocytic leukemia. Crit Rev Oncol Hematol. 2016;100:32–6. doi: 10.1016/j.critrevonc.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Stamati L, Avgeris M, Kosmidis H, Baka M, Anastasiou T, Piatopoulou D, et al. Overexpression of BCL2 and BAX following BFM induction therapy predicts ch-ALL patients' poor response to treatment and short-term relapse. J Cancer Res Clin Oncol. 2015;141:2023–36. doi: 10.1007/s00432-015-1982-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Principe MI, Dal Bo M, Bittolo T, Buccisano F, Rossi FM, Zucchetto A, et al. Clinical significance of bax/bcl-2 ratio in chronic lymphocytic leukemia. Haematologica. 2016;101:77–85. doi: 10.3324/haematol.2015.131854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moazami-Goudarzi M, Farshdousti-Hagh M, Hoseinpour-Feizi A, Talebi M, Movassaghpour-Akbari AA, Shams-Asanjan K, et al. The acute lymphoblastic leukemia prognostic scoring whether it is possible by BCL-2, BAX gene promoter genotyping. Caspian J Intern Med. 2016;7:105–13. [PMC free article] [PubMed] [Google Scholar]
- 39.Vélez J, Hail N, Jr, Konopleva M, Zeng Z, Kojima K, Samudio I, et al. Mitochondrial uncoupling and the reprograming of intermediary metabolism in leukemia cells. Front Oncol. 2013;3:67. doi: 10.3389/fonc.2013.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tcheng M, Samudio I, Lee EA, Minden MD, Spagnuolo PA. The mitochondria target drug avocatin B synergizes with induction chemotherapeutics to induce leukemia cell death. Leuk Lymphoma. 2017;58:986–8. doi: 10.1080/10428194.2016.1218005. [DOI] [PubMed] [Google Scholar]
- 41.Shi J, Fu H, Jia Z, He K, Fu L, Wang W. High expression of CPT1A predicts adverse outcomes: A potential therapeutic target for acute myeloid leukemia. EBioMedicine. 2016;14:55–64. doi: 10.1016/j.ebiom.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song K, Li M, Xu X, Xuan LI, Huang G, Liu Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett. 2016;12:334–42. doi: 10.3892/ol.2016.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guitart AV, Panagopoulou TI, Villacreces A, Vukovic M, Sepulveda C, Allen L, et al. Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J Exp Med. 2017;214:719–35. doi: 10.1084/jem.20161087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed MA, Ludwig C, Bunce CM, Khanim FL, Günther UL. Malonate as a ROS product is associated with pyruvate carboxylase activity in acute myeloid leukaemia cells. Cancer Metab. 2016;4:15. doi: 10.1186/s40170-016-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG, Jr, et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET Enzymes. J Biol Chem. 2016;291:4256–65. doi: 10.1074/jbc.M115.688762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178–84. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31:272–81. doi: 10.1038/leu.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bargiela D, Burr SP, Chinnery PF. Mitochondria and hypoxia: Metabolic crosstalk in cell-fate decisions. Trends Endocrinol Metab. 2018;29:249–59. doi: 10.1016/j.tem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Laukka T, Myllykoski M, Looper RE, Koivunen P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J Mol Biol. 2018;430:3081–92. doi: 10.1016/j.jmb.2018.06.048. [DOI] [PubMed] [Google Scholar]
- 50.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morikawa T, Takubo K. Hypoxia regulates the hematopoietic stem cell niche. Pflugers Arch. 2016;468:13–22. doi: 10.1007/s00424-015-1743-z. [DOI] [PubMed] [Google Scholar]
- 52.Testa U, Labbaye C, Castelli G, Pelosi E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp Hematol. 2016;44:540–60. doi: 10.1016/j.exphem.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 53.Velasco-Hernandez T, Cammenga J, Bryder D. Targeting hypoxia pathway in a model of acute myeloid leukemia. Exp Hematol. 2017;53:S61. [Google Scholar]
- 54.Vukovic M, Guitart AV, Sepulveda C, Villacreces A, O'Duibhir E, Panagopoulou TI, et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J Exp Med. 2015;212:2223–34. doi: 10.1084/jem.20150452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Migliavacca J, Percio S, Valsecchi R, Ferrero E, Spinelli A, Ponzoni M, et al. Hypoxia inducible factor-1α regulates a pro-invasive phenotype in acute monocytic leukemia. Oncotarget. 2016;7:53540–57. doi: 10.18632/oncotarget.10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forristal CE, Brown AL, Helwani FM, Winkler IG, Nowlan B, Barbier V, et al. Hypoxia inducible factor (HIF)-2α accelerates disease progression in mouse models of leukemia and lymphoma but is not a poor prognosis factor in human AML. Leukemia. 2015;29:2075–85. doi: 10.1038/leu.2015.102. [DOI] [PubMed] [Google Scholar]
- 57.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–26. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 58.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292:16804–9. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123:2663–72. doi: 10.1182/blood-2013-10-532200. [DOI] [PubMed] [Google Scholar]
- 60.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murakami S, Shimizu R, Romeo PH, Yamamoto M, Motohashi H. Keap1-Nrf2 system regulates cell fate determination of hematopoietic stem cells. Genes Cells. 2014;19:239–53. doi: 10.1111/gtc.12126. [DOI] [PubMed] [Google Scholar]
- 62.Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: Insights from the hematopoietic system. Cell Stem Cell. 2007;1:140–52. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 63.Kozlov SV, Waardenberg AJ, Engholm-Keller K, Arthur JW, Graham ME, Lavin M. Reactive Oxygen Species (ROS)-Activated ATM-dependent phosphorylation of cytoplasmic substrates identified by large-scale phosphoproteomics screen. Mol Cell Proteomics. 2016;15:1032–47. doi: 10.1074/mcp.M115.055723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sánchez-Aguilera A, Méndez-Ferrer S. The hematopoietic stem-cell niche in health and leukemia. Cell Mol Life Sci. 2017;74:579–90. doi: 10.1007/s00018-016-2306-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Myers RA, Wirth S, Williams S, Kiel PJ. Enasidenib: An Oral IDH2 Inhibitor for the treatment of acute myeloid leukemia. J Adv Pract Oncol. 2018;9:435–40. [PMC free article] [PubMed] [Google Scholar]
- 67.Gentile M, Petrungaro A, Uccello G, Vigna E, Recchia AG, Caruso N, et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017;26:1307–16. doi: 10.1080/13543784.2017.1386173. [DOI] [PubMed] [Google Scholar]
- 68.Knight T, Edwards H, Taub JW, Ge Y. Evaluating venetoclax and its potential in treatment-naïve acute myeloid leukemia. Cancer Manag Res. 2019;11:3197–213. doi: 10.2147/CMAR.S180724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170:1079–95.e23. doi: 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]