

SUMMARY
Drug-tolerant “persister” tumor cells underlie emergence of drug-resistant clones and contribute to relapse and disease progression. Here we report that resistance to the BCL-2 targeting drug ABT-199 in models of mantle cell lymphoma and double-hit lymphoma evolves from outgrowth of persister clones displaying loss of 18q21 amplicons that harbor BCL2. Further, persister status is generated via adaptive super-enhancer remodeling that reprograms transcription and offers opportunities for overcoming ABT-199 resistance. Notably, pharmacoproteomic and pharmacogenomic screens revealed that persisters are vulnerable to inhibition of the transcriptional machinery and especially to inhibition of cyclin-dependent kinase 7 (CDK7), which is essential for the transcriptional reprogramming that drives and sustains ABT-199 resistance. Thus, transcription-targeting agents offer new approaches to disable drug resistance in B-cell lymphomas.
In Brief
Zhao et al. show that resistance to the BCL-2 inhibitor ABT-199 is conferred by super enhancer reprogramming and loss of BCL2 amplicons in models of mantle cell lymphoma and double hit lymphoma. Combining ABT-199 with the CDK7 inhibitor THZ1 prevents or reverses BCL-2 inhibitor resistance.
Graphical abstract
INTRODUCTION
Acquired drug resistance limits effectiveness of targeted cancer therapies and occurs even following a robust response to treatment. While enormous effort has gone into understanding the molecular events manifest in acquired resistance, little attention has been given to what happens early during treatment when patients still respond to drug treatment. In addition to mutational mechanisms and tumor heterogeneity, an increasing body of evidence suggests that non-mutational mechanisms contribute to the emergence of resistance. In particular, tumor cell plasticity allows them to adapt to chemotherapy and targeted therapies, and this is often driven by epigenetic and transcriptional reprogramming (Hata et al., 2016; Knoechel et al., 2014; Koppikar et al., 2012; Ramirez et al., 2016; Sharma et al., 2010).
Emerging evidence suggests that, on drug treatment, small subpopulations of cancer cells evade drug pressure by entering a largely quiescent drug-tolerant “persister” (DTP) state. Further, some DTP cells can then expand in the presence of drug to become drug-tolerant expanded persisters (DTEP). Importantly, DTP/DTEP status is clinically relevant because: (1) DTP cells represent minimal residual disease (MRD), the small populations of cancer cells that survive therapy; (2) DTP/MRD serve as the reservoir for the expansion of subpopulations of cells that maintain resistance after therapy, and that then expand and lead to relapse; and (3) DTP/MRD and DTEP cells are barriers to successful therapy. Accordingly, finding new strategies that disable DTP and the emergence of DTEP would have a major impact in the clinic.
BCL-2 has major roles as an anti-apoptotic protein in hematological malignancies. In particular, B-cell lymphomas, such as mantle cell lymphoma (MCL) and double-hit lymphoma (DHL) often have dysregulated BCL-2 and are addicted to this oncoprotein to variable degrees (Ruefli-Brasse and Reed, 2017). Venetoclax (ABT-199), a novel, potent, and selective small-molecule BCL-2 inhibitor, is being clinically vetted and is an effective therapy for some B-cell lymphomas (Anderson et al., 2016; Leverson et al., 2017). Indeed, ABT-199 has the potential to be the standard of care for B-cell lymphomas, including MCL, yet many patients who initially respond to ABT-199 develop resistance (Choudhary et al., 2015; Esteve-Arenys et al., 2018; Fresquet et al., 2014; Thijssen et al., 2015). Thus, there is an urgent need to define mechanisms of ABT-199 resistance.
The bulk of tumor phenotypes, including clinical progression and therapeutic responses, are controlled by dysregulated transcriptional programs manifest in cancer cells. Several studies have shown DTP cells undergo transcriptional adaptation via epigenetic regulation and transcriptional reprograming during development of acquired drug resistance. Further, regulators of these transcriptional programs, for example BET bromodomain proteins that are required for transcriptional and enhancer activity, are emerging as attractive targets for new drugs that perturb their functions and the transcription programs they govern (Bradner et al., 2017; Nakagawa et al., 2018). Moreover, several studies have identified extremely large enhancer domains termed super-enhancers (SEs), which were identified based on histone H3 lysine 27 acetylation (H3K27ac) and span up to 50 kb (Hnisz et al., 2013; Whyte et al., 2013). Notably, SEs specifically regulate genes associated with cell identity and disease, including oncogenes (Ceribelli et al., 2016; Chapuy et al., 2013; Loven et al., 2013; Whyte et al., 2013). Accordingly, approaches that disable SEs have received attention as drug targets. Among these is RNA polymerase II (RNAPII) itself, which is regulated by a set of cyclin-dependent kinases (CDKs) having critical roles in transcription initiation and elongation (Larochelle et al., 2012). These transcriptional CDKs (e.g., CDK7 and CDK9) phosphorylate key serine residues of the C-terminal domain (CTD) of RNAPII that are necessary for transcription initiation and elongation (Larochelle et al., 2012), and these have emerged as attractive therapeutic targets. For example, THZ1, a selective covalent inhibitor of CDK7, has activity against several tumor types, including T-cell acute lymphoblastic leukemia (Kwiatkowski et al., 2014), MYCN-amplified neuroblastoma (Chipumuro et al., 2014), and small-cell lung cancer (Christensen et al., 2014).
Here we characterize MCL, a non-Hodgkin B-cell lymphoma that has very poor long-term survival (Schieber et al., 2018), and DHL, an extremely aggressive subtype of B-cell lymphoma that overexpresses both MYC and BCL-2 (Reagan and Davies, 2017), to define mechanisms that drive drug tolerance and resistance against ABT-199.
RESULTS
Evolution of Drug-Resistant Persisters in ABT-199-Treated MCL Models
To define the mechanisms that contribute to development of DTEP in MCL, we modeled the response to ABT-199 in an MCL-derived cell line (HBL-2) that has high sensitivity to ABT-199. Whereas the vast majority of pools of HBL-2 cells plated into 384-well plates were killed within a few days of exposure to a drug concentration at 50- to 100-fold the half maximal inhibitory concentration, we detected a small fraction of viable, largely quiescent cells (DTPs) in 12 wells 10–14 days following treatment with ABT-199 (Figure S1A). From these 12 DTPs, we were successful at generating three independent ABT-199 DTEPs (DTEP I10, J4, and L20) that were cloned and grew indefinitely in high doses of ABT-199 (Figures 1A, 1B, and S1B; Table S1). To test if resistance was a stable phenotype, DTEP cells were cultured without ABT-199 for 6–8 weeks and then retested for their sensitivity to the drug. The slope of the drug-sensitivity curves revealed that subpopulations of these “drug holiday” (dh) DTEP cells partially re-acquired sensitivity to ABT-199 (Figures 1C and S1B), consistent with epigenetic alterations observed in other models (Liau et al., 2017). To assess if resistance was associated with shifts in dependence on BCL-2 family members, BH3 peptide sensitivity screens and immunoblot analyses were performed. These studies revealed that ABT-199 DTEP clones are more dependent on MCL-1, and that they express increased levels of MCL-1 and much reduced levels of BCL-2 versus parental cells, which rather relied on BCL-2 given their differential sensitivity to BH3 peptides (Figures 1D and 1E). Consistent with these findings, DTEP cells were more sensitive to the MCL-1-specific inhibitor S63645 than parental HBL-2 cells (Figure S1C). Finally, activity-based proteomic profiling (ABPP) using an ATP-binding probe (Fang et al., 2015; Zhao et al.,2017) revealed that ABT-199 DTEP cells had selective and shared reprogramming of their kinome versus parental HBL-2 cells, in which at least half of the kinases altered were shared by all three independent DTEP lines (Figure S1D). Thus, rewired signaling programs that alter the expression of apoptotic regulators are associated with ABT-199 resistance in MCL models.
Figure 1. Phenotypes of ABT-199 (Venetoclax)-Resistant Mantle Cell Lymphoma.

(A) Top, experimental design for isolation of DTP cells and drug-tolerant expanded persisters (DTEP) of HBL-2 mantle cell lymphoma (MCL) cells. A total of 1,000 cells/well plated on 384-well collagen-coated plates were continuously treated with 20 nM ABT-199 (50–100× half maximal inhibitory concentration [IC50], IC50 was measured at 72 h after treatment). After 10–14 days, nearly all wells lacked viable cells as detected by live cell imaging, except for 12 wells, where drug-tolerant persisters (DTP) cells remained (bottom). Spatially separated colonies from these wells were then transferred to individual wells of a 96-well plate. Over the course of 3 months the selected colonies were expanded in medium containing 20 nM ABT-199 and three colonies survived and expanded to obtain the DTEP that are coined J4, L20, and I10.
(B) Dose-response curves to ABT-199, for parental HBL-2 cells and DTEP clones (L20, J4, I10) showing drug response and resistance to ABT-199 treatment (72 h, with technical replicates, n = 4) at each indicated dose.
(C) Drug-response curves of parental cells, DTEP cells, and DTEP cells after removing ABT-199 for a period of 6 weeks (“drug holiday” [dh]) to ABT-199 (100 nM) (technical replicates, n = 4).
(D) BH3 profiling of parental HBL-2 and DTEP cells, as measured by mitochondrial membrane depolarization. BAD is indicative of BCL-2 or BCL-XL dependency, HRK is specifically indicative of BCL-XL dependency, BAD and HRK (BAD-HRK) are indicative of BCL-2 dependency, and MS1 is indicative of an MCL-1 dependency.
(E) Levels of the indicated BCL-2 family proteins in parental HBL-2 cells versus ABT-199-resistant DTEP cells was determined by immunoblot analyses. Blots are representative of three independent experiments, which were quantified and the means are shown.
(B–E) Data are shown as mean ± SD. See also Figure S1.
Selective 18q21 Amplicon Loss and Acquired SE Remodeling of Transcriptional Programs Drive ABT-199 Drug Resistance in B-Cell Lymphoma Models
To gain insights into the mechanisms of ABT-199 resistance, RNA sequencing (RNA-seq) analyses were performed on parental HBL-2 and DTEP cells. These analyses revealed both shared and selective transcriptome reprogramming in the three DTEP cells, and confirmed that they represented independent resistant clones (Figure 2A; Table S2). To assess if these transcriptional changes were associated with genetic alterations, we performed copy-number variant (CNV) analysis and integrated the CNV and RNA-seq data. Strikingly, parental HBL-2 cells had an amplification of chromosome 18q21, and all three DTEP cells had distinct deletions of this amplicon, leaving one wild-type copy of chromosome 18 (Figure 2B; Table S3). Fluorescence in situ hybridization (FISH) analyses confirmed copy-number loss of chromosomal 18q21 in all DTEP cells (Figure 2C). Notably, RNA-seq analyses established that loss of the 18q21 amplicon in DTEP cells was associated with a marked downregulation of three apoptotic regulators located on chromosome 18q21, specifically of BCL2, the target of ABT-199, as well as of PMAIP1 (NOXA) and TCF4 (Figures 2A–2C).
Figure 2. 18q21 Amplicon Loss and Super-Enhancer Remodeling Drive ABT-199 Resistance in Mantle Cell Lymphoma.

(A) Unsupervised clustering of RNA-seq data from parental and DTEP cells in triplicate. Key genes involved in the evolution of DTEP are indicated.
(B) Copy number variant (CNV) analysis of DTEP and parental cells. Copy-number loss (deletion), red; copy-number gain (amplification), blue.
(C) Fluorescence in situ hybridization (FISH) analysis using a BCL2 probe in DTEP and parental cells. Cell nuclei are counterstained with DAPI in blue, the 5′ region of BCL2 gene was targeted with a red colored probe and the 3′ region of the BCL2 gene with a green colored probe. One isolated fusion signal represents one normal BCL2 gene, while five BCL2 fusion signals in cluster indicate BCL2 gene amplification (oval circle).
(D) Viability of BCL-2, NOXA, or TCF4 knockdown (KD) versus control (GFP-KD) parental HBL-2 cells following treatment with ABT-199 (left, 3.0 μM; right, 0.04 μM) was assessed by live cell imaging.
(E) Image-based cell-viability assays of DTEP control cells and cells overexpressing TCF4 and BCL2 in response to ABT-199 (upper left, 0.11 μM; upper right, 0.04 μM; middle/lower left, 1.1 μM; middle/lower right, 0.12 μM).
(F) Enhancers ranked by H3K27ac signals. Super-enhancer (SE)-associated genes were identified as H3K27ac signal densities that surpass the inflection point by ChIP-seq and are indicated in red font; typical enhancer (TE)-associated genes are noted in black font.
(G) Gene set enrichment analysis (GSEA). Top panels, gained (activated) SE-associated genes in DTEP cells. Bottom panels, SE-associated genes that are present in parental HBL-2 cells but that are suppressed (inactivated) in DTEP cells. Genes were ranked according to their expression fold change between DTEP and parental HBL-2 cells. NES, normalized enrichment score; FDR, false discovery rate.
(D and E) Experiments were performed in triplicate and results are representative of three independent experiments. See also Figures S2 and S3 and Tables S2, S3, and S4.
To establish the role of loss of the 18q21 amplicon and its associated apoptotic genes in ABT-199 resistance, gain- and loss-of-function studies were performed. Notably, silencing of BCL2, PMAIP1, or TCF4 genes in parental HBL-2 cells by CRISPR editing (Figure S2A, left) conferred partial (BCL2, TCF4) to nearly complete (PMAIP1) resistance to ABT-199 (Figure 2D). Further, the degree of ABT-199 resistance correlated with increased MCL-1 protein levels in NOXA or TCF4 knockdown cells, but not in BCL-2 knockdown cells that were more sensitive to ABT-199 (Figure S2A, left). Conversely, forced expression of BCL-2 in DTEP cells (Figure S2A, right) re-sensitized these otherwise resistant cells to ABT-199 treatment (Figure 2E).
Given the marked changes in the signaling programs and transcriptome of DTEPs versus parental MCL cells (Figures 2A and S1D), we assessed if reprogramming was associated with adaptive SE remodeling using chromatin immunoprecipitation sequencing (ChIP-seq) specific for acetylated lysine 27 of histone H3 (H3K27ac), a mark of SEs and actively transcribed genes (Brown et al., 2014; Hnisz et al., 2015; Rusan et al., 2018; Zawistowski et al., 2017). ChIP-seq and ranking enhancers by the amplitude and density of H3K27ac marks revealed distinct upregulated and reduced SEs, and SE-associated pathways, in parental versus DTEP cells (Figures 2F, S2B, and S2C; Table S4). ChIP-seq results confirmed loss of the 18q21 amplicon in all three DTEP cells (Figure S2D). Gene set enrichment analysis (GSEA) showed a strong correlation between activated (gained) or inactivated (lost or reduced) SEs and transcripts in both DTEP and parental cells (Figures 2G, S2E, and S2F). Concordant with RNA-seq and the 18q21 amplicon present in parental HBL-2 cells, there were marked increases in H3K27ac marks in the BCL2, TCF4, and PMAIP1 genes and their associated SEs in these cells, whereas H3K27ac marks at these genes and associated SEs were lost or reduced in DTEP cells (Figures 2F and S2F). Rather, resistance of all three DTEPs was associated with selective and marked increases in H3K27ac marks at SEs that drive elevated expression of the apoptotic and transcriptional regulators BCL2A1, FOXC1, IRF5, and IKZF1 (Figures 2A, 2F, and S2B). In accord with these findings, increased BCL2A1 expression has been associated with ABT-199 resistance in a DHL cell line (Esteve-Arenys et al., 2018). Thus, copy-number loss of BCL2 amplicons at 18q21 and adaptive SE-dependent transcriptional programming in this MCL model are associated with ABT-199 resistance.
To assess if DTEP clones I10, J4, and L20 were derived from selection and expansion of rare subpopulations of pre-existing persister cells in parental HBL-2 lymphoma that had various deletions of the 18q21 amplicon, or if amplicon loss was acquired along with phenotypic resistance mechanisms altering transcriptional landscapes, we derived single-cell clones using a limiting dilution approach from ABT-199-naive, parental HBL-2 cells (Figure S3A). From several hundred naive ABT-199-sensitive clones, we developed three “acquired” DTEP clones (P5F6-DTEP, P2D8-DTEP, and P1C10-DTEP; Figures S3A and S3B), and performed FISH and western blot analyses. Loss of the 18q21 amplicon was not observed in these DTEP clones, and BCL-2, NOXA, and TCF4 proteins were expressed at levels similar to parental HBL-2 cells (Figures S3C and S3D). Thus, the selection for ABT-199 resistance can be heterogeneous.
CDK7 Is a Vulnerability for ABT-199-Resistant MCL Models
To identify effectors that drive and sustain DTEP, a drug sensitivity screen was performed on HBL-2 cells versus DTEP I10, J4, and L20 cells using a panel of 60 small-molecule kinase inhibitors and epigenetic drugs. Parental HBL-2 cells were preferentially dependent on histone deacetylases (HDACs) and EZH2, while, strikingly, DTEP cells were highly sensitive to inhibitors of other epigenetic regulators (EZH1, CBP/EP300, G9a, EHMT1, and JMJD3), and of the PI3K-mTOR pathway, but were most vulnerable to THZ1, a CDK7 inhibitor that regulates RNAPII activity (Figures 3A and 3B) (Chipumuro et al., 2014; Christensen et al., 2014; Kwiatkowski et al., 2014; Wang et al., 2015). These findings agreed with Kyoto Encyclopedia of Genes and Genomes pathway analyses indicating an upregulation of transcriptional regulators in the SEs manifest in DTEP cells (Figure S2C), and with data showing that THZ1 suppresses transcriptional adaptive resistance mechanisms in solid tumors (Rusan et al., 2018). Notably, low doses of THZ1 rapidly compromised the survival of all three DTEP cells, yet had only modest activity against parental HBL-2 cells (Figure 3C). Similarly, low doses of THZ1 abolished the colony-forming capacity of DTEP cells in methylcellulose, but had no effect on parental HBL-2 cells (Figure 3D).
Figure 3. ABT-199-Resistant DTEP Cells Are Highly Sensitive to CDK7 Inhibition.

(A) Drug sensitivity shown as a heatmap of relative area under the curve differences between DTEP and parental cells for each indicated drug.
(B) Top drug sensitivity hits for parental HBL-2 cells (top) and DTEP cells (bottom) are listed.
(C) Image-based cell-viability assay of parental and DTEP cells treated with DMSO or THZ1 (100 nM) for the indicated time points.
(D) Clonogenic growth assay of parental and DTEP cells treated with DMSO or THZ1 (50 nM) for the indicated time points.
(E) Western blots of parental and DTEP cells treated with the indicated doses of THZ1 at different time points. CTD, C-terminal repeat domain of RNAPII; cPARP, cleaved PARP.
(A–E) Data are shown as mean ± SD and are representative of three independent experiments. See also Figure S4.
CDK7 affects transcription initiation via phosphorylation of serine 5 (Ser5) and Ser7 of the CTD of RNAPII, but also affects transcriptional elongation via its functions as a CDK-activating kinase for CDK9 (Larochelle et al., 2012; Wang et al., 2015; Zhou et al., 2012), which phosphorylates Ser2 of the CTD of RNAPII. As expected (Wang et al., 2015), THZ1 treatment provoked dose-dependent decreases in phosphorylation of all three of these serine residues of RNAPII but these effects were more profound in DTEP versus parental cells (Figure 3E). Further, THZ1 treatment also induced a more rapid and profound reduction in the levels of total RNAPII in DTEP cells versus parental cells (Figures 3E and S4A). THZ1 treatment also induced marked reductions in MYC levels, and increased cleavage of PARP, and these effects were again more pronounced in DTEP versus parental cells (Figures 3E and S4A). Finally, CDK7 inhibition had profound effects on the kinome of DTEP cells, where ABPP established that THZ1 treatment (50 nM, 6 h) attenuated activity of approximately 60% of the upregulated kinases manifest in all three DTEP clones, and especially pathways controlling transcription and apoptosis (Figures 3F and S4B–S4D). Thus, CDK7-directed responses are essential to sustain most of the acquired signaling circuits manifest in these DTEP cells.
CDK7 Is Necessary to Sustain SE-Driven Transcriptional Reprogramming in ABT-199-Resistant MCL Cells
The rapid and profound effects of THZ1 treatment on the kinome suggested that it would also have major effects on the transcriptome of DTEP cells. Indeed, RNA-seq analyses revealed that THZ1 treatment led to preferential and dose-dependent reductions in steady-state mRNA levels in DTEP cells (Figure 4A). Whereas high doses of THZ1 (250 nM) globally suppressed mRNA levels in both parental and DTEP cells, lower doses of THZ1 (50 nM) displayed selective effects on DTEP cells, in which CDK7 inhibition suppressed the expression of select mRNAs that were upregulated in DTEP cells but were downregulated by THZ1 treatment (DTEP-associated/THZ1-sensitive genes; Figures S5A and S5B). Similarly, a small cast of transcripts that are repressed in DTEP cells versus parental cells were upregulated by THZ1 treatment (Figure S5A).
Figure 4. CDK7 Inhibition Suppresses SE-Driven Transcriptional Reprogramming and Overcomes and Blocks the Evolution of ABT-199 Drug Resistance.

(A) Quartile boxplots of Log2 fold changes of global gene expression following THZ1 treatment (50 nM, 250 nM, 6 h) in parental HBL-2 cells and DTEP cells. Top, middle, and bottom lines of the boxplot represent third quantile, median and first quantile of all values, respectively. The whiskers, the two lines outside the box, represent the highest and lowest observations excluding the possible outliers (dots above or below the whisker lines are outliers).
(B) Global RNAPII ChIP-seq profiles showing the RNAPII-seq signal (y axis, RRPM/BP) across all RNAPII domains (>7 kb, x axis) in parental HBL-2 cells and DTEP cells (I10, J4) ± THZ1 treatment (50 nM, 6 h). The x axis represents size-scaled RNAPII domains, with or without flanking regions as indicated; TSS, transcription start site; PRPM/BP, Rx-normalized reads per million per base pair; BP, base pair.
(C) Quartile boxplots of Log2 fold changes in the expression of genes that are regulated by gained SEs (with increased RNAPII binding) following 6 h THZ1 treatment (left, 50 nM; right, 250 nM) in parental and DTEP cells. Top, middle, and bottom lines of the boxplot represent third quantile, median and first quantile of all values, respectively. The whiskers, the two lines outside the box, represent the highest and lowest observations excluding the possible outliers (dots above or below the whisker lines are outliers).
(D) GSEA of preferentially enriched for genes in DTEP cells sensitive to THZ1 treatment (50 nM) with NES at −2.06, −2.25, and −2.54, respectively.
(E) Venn diagram of shared SE-associated/THZ1-sensitive genes in three DTEP cells.
(F) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to identify the number of shared SE-associated/THZ1-sensitive genes among three DTEP cells.
(G) Principal-component analysis (PCA) of gene expression for parental and DTEP cells with and without THZ1 treatment.
See also Figures S5 and S6.
To assess if DTEP-associated/THZ1-sensitive transcripts were coupled with SE remodeling manifest in ABT-199-resistant MCL, ChIP-seq was performed with antibodies recognizing RNAPII and H3K27ac in control and THZ1-treated (50 nM, 6 h) parental HBL-2 and DTEP cells. These analyses revealed that the binding profiles of H3K27ac and RNAPII highly correlated and differed in DTEP versus parental cells (Figure S6A). Further, THZ1 treatment induced a more global reduction of RNAPII occupancy at transcriptional start sites in DTEP cells compared with parental cells (Figure 4B). Importantly, THZ1-induced more significant reductions in the expression of SE-associated transcripts in DTEP versus parental cells (Figures 4C and S6B), and the effects of THZ1 were more profound on SE-regulated genes than on transcripts driven by typical enhancers (Figure S6C). This was particularly evident in genes selectively upregulated in DTEP cells and especially those that control transcription (e.g., FOXC1, IRF5, and IKZF1), apoptosis (BCL2A1 and BCL11A1), and RNAPII activity (Figures 4D–4F and S6D). RNA-seq and western blot analyses confirmed the selective inhibitory effects of THZ1 treatment on these regulators in DTEP cells at both the RNA and protein level (Figures S6E and S6F). Strikingly, principal-component analysis revealed that THZ1 treatment in all three DTEP clones drives their transcriptional profile to shift toward that of parental MCL cells (Figure 4G). Thus, CDK7 activity is necessary to sustain expression of SE-driven transcriptional reprogramming in ABT-199-resistant MCL.
To confirm the effects of THZ1 were on target, CRISPR editing was used to knock down CDK7 in DTEP and parental MCL cells. As predicted, transient CDK7 knockdown attenuated the cytotoxic effects of THZ1 treatment on these cells (Figure 5A, upper). Further, as with THZ1 treatment, CDK7 knockdown led to reductions in expression of the SE-associated regulators BCL2A1, BCL11A1, FOXC1, IRF5 and IKZF1 upregulated in DTEP cells (Figure 5A, lower). To assess the contribution of these SE-associated genes and of MCL-1 to the maintenance of DTEP cells, BCL2A1, FOXC1, IRF5, MCL1, and IKZF1 were individually knocked down by CRISPR editing in all three DTEP cell lines (Figure 5B). Individual knockdown of IKZF1, IRF5, FOXC1, and MCL-1 had little-to-modest effects on DTEP growth or survival, whereas BCL2A1 knockdown effectively blocked DTEP cell proliferation (Figures 5C–5G). However, loss of these DTEP-associated genes was sufficient to at least partially restore the sensitivity of all three DTEP cells to ABT-199 (Figures 5C–5G). Thus, these SE-driven transcriptional targets converge and cooperatively contribute to ABT-199 resistance in this MCL model.
Figure 5. CDK7 and Super-Enhancer-Driven Genes Contribute to ABT-199 Resistance of DTEP Cells.

(A) Image-based cell-viability assays (upper panel) and western blots (lower panel) of control (GFP) or CDK7 knockdown derivatives of parental (left), I10 (middle), and L20 (right) cells treated with THZ1 (220 nM).
(B) Efficiency of CRISPR/Cas9-directed knockdown of BCL2A1, MCL-1, IKZF1, and IRF-5, and deletion of FOXC1, in DTEP cells, as determined by western blot.
(C–F) Viability of I10 (left), J4 (middle), and L20 (right) DTEP cells in paired control (GFP) and BCL2A1 (C), MCL-1 (D), IRF5 (E) knockdown, and FOXC1 (F) knock out cells ± ABT-199 treatment (C), 10 μM and (D–F), 3.3 μM.
(G) Image-based cell-viability assays of J4 in paired control (GFP) and IKZF1 knockdown cells ± ABT-199 treatment (3.3 μM).
(A–G) Data shown are representative of at least three independent experiments.
Dual CDK7/BCL-2 Inhibition Disables Emergence and Maintenance of ABT-199 Resistance
Given the essential role of BCL-2 in MCL, CDK7-dependent transcriptome reprogramming, and the low basal levels of BCL-2 still manifest in DTEP, we evaluated the effects of combined THZ1/ABT-199 treatment in DTEP lines and in primary MCL patient samples cultured on autologous bone marrow stroma. There were clear synergistic effects of combined ABT-199 + THZ1 treatment in all DTEP cell lines, and in primary MCL cells (Figures 6A, 6B, and S7A). Further, xenograft studies revealed robust responses of mice bearing parental HBL-2 lymphoma to ABT-199 treatment, whereas DTEP L20 lymphomas were resistant to ABT-199 but were highly sensitive to THZ1 treatment. Notably, the ABT-199 + THZ1 combination had superior efficacy in both parental and DTEP-L20-derived xenografts and provoked tumor regression (Figure 6C). No adverse effects from either THZ1 or combined ABT-199 + THZ1 treatments were noted. These observations agree with previous reports of no toxicity of single or combination THZ1 treatments (Cayrol et al., 2017; Chipumuro et al., 2014; Wang et al., 2015). Thus, CDK7 inhibitors represent an attractive therapeutic strategy for ABT-199-resistant MCL.
Figure 6. CDK7 Inhibition Synergizes with ABT-199, Overrides ABT-199 Resistance and Prevents Emergence of ABT-199 Resistance.

(A and B) Image-based cell-viability assays at different time points (upper) and doses (lower, at 48 h) of MCL cell lines (A) and primary MCL patient specimens (B). Combination index (CIs) for drug combinations were obtained with CalcuSyn software using percent inhibition (fraction affected [Fa]) resulting from combined action of the two drugs versus effects of either drug alone. CI values < 1.0 indicate synergism of the two agents. Viability of indicated cells was determined by cell-based image analysis. +, positive for 18q21 amplification; −, negative for 18q21 amplification (B).
(C) Upper, effect of combined ABT-199 + THZ1 treatment on tumor growth of parental and ABT-199-resistant DTEP mantle cell lymphoma xenografts. NOD-SCID mice bearing the indicated tumor xenografts were treated with THZ1 (5 mg/kg), ABT-199 (5 mg/kg), the combined ABT-199 + THZ1 (5 mg/kg each), or vehicle; n = 6 for all 8 cohorts. Lower, tumor volume of parental and ABT-199-resistant DTEP MCL xenografts. Mean volumes ± SD are presented. The red arrow indicates the treatment start time.
(D) Clonogenic potential of parental HBL-2 cells was assessed at the indicated intervals following treatment with vehicle, ABT-199 (20 nM), THZ1 (50 nM), or ABT-199 + THZ1 combination (20 and 50 nM, respectively). TTP, time to progression.
To assess if CDK7 inhibition would block the emergence of ABT-199 resistance, parental HBL-2 cells were cultured in the presence or absence of high doses of ABT-199 ± THZ1 and assessed for anchorage-independent colony formation. As expected, ABT-199 treatment triggered initial growth suppression, and this was then followed by the growth of drug-resistant DTEP colonies (Figure 6D). In contrast, THZ1 co-treatment abolished the emergence of ABT-199-resistant DTEP colonies (Figure 6D). Thus, CDK7 activity is also necessary for the emergence of ABT-199 resistance in this MCL model.
BCL2 Amplicon Loss and CDK7 Vulnerability Are Hallmarks of ABT-199 Resistance in Models of DHL, and in Primary MCL and DHL
We also tested if BCL2 amplicon loss and/or a reliance on CDK7 were involved in ABT-199 resistance of single-cell-derived DTEP clones of MCL, and in acquired DTEP that were derived from other validated models of MCL or DHL. Single-cell-derived DTEP clones of HBL-2 MCL cells that retain the BCL2 amplicon were also sensitive to THZ1 and were especially vulnerable to the ABT-199 + THZ1 combination (Figure S7A, left). To test this treatment strategy on DHL, we generated ABT-199-resistant DHL lymphoma cells, following culture of VAL DHL cells with a high dose of ABT-199 (VAL-DTEP; Figure 7A, upper). ABT-199-resistant VAL-DTEP cells were more sensitive to THZ1 treatment than parental VAL DHL cells (Figure 7A, lower), and VAL-DTEP cells were especially vulnerable to the ABT-199 + THZ1 combination (Figure S7A, right). Chou-Talalay analyses also revealed synergy for the ABT-199 + THZ1 combination in P2D8-DTEP, P5F6-DETP, P1C10-DTEP, and VAL-DTEP cells (Figures S7B–S7D). Similar synergistic or enhanced effects of ABT-199 and THZ1 were also observed in primary MCL patient samples (Figures S7C and S7D). Finally, even though ABT-199-resistant VAL-DTEP cells grew at accelerated rates versus parental DHL cells in vivo, VAL-DTEP-derived tumors were exquisitely sensitive to THZ1 treatment (Figure S7E), and increased sensitivity and synergistic effects of the ABT-199 + THZ1 combination were also manifest in primary DHL patient specimens cultured on autologous stroma (Figure 7B).
Figure 7. BCL2 Amplicon Loss and CDK7 Vulnerability Are Hallmarks of ABT-199-Resistant DHL Cells and Primary DHL Specimens.

(A) Image-based cell-viability assays of parental DHL VAL and VAL-DTEP cells treated with the indicated doses of ABT-199 (upper) or with 110 nM THZ1 (lower). Data shown are representative of three independent experiments.
(B) Left, image-based cell viability of primary double-hit lymphoma (DHL) patient samples cultured on lymphoma stromal cells following treatment with ABT-199 (20 nM), THZ1 (50 nM), or ABT-199 + THZ1 (20 and 50 nM, respectively). Right, synergistic effects for drug combinations were obtained through combination indices (CIs) with CalcuSyn software using percent inhibition (fraction affected [Fa]) resulting from combined action of the two drugs versus effects of either drug alone. CI values < 1.0 indicate synergism of the two agents.
(C) FISH analysis using a BCL2 probe in parental DHL VAL cells (VAL, left), and ABT-199-resistant VAL-DTEP cells (right). Cell nuclei are counterstained with DAPI in blue, the 5′ region of the BCL2 gene was targeted with a red colored probe and the 3′ region of BCL2 gene with a green colored probe.
(D) GSEA of DTEP-associated and THZ1-sensitive genes identified in DTEP MCL cell lines with THZ1-induced altered genes in primary ABT-199-resistant MCL patient samples. Genes were ranked according to their expression fold change between control and THZ1 treatment (50 or 250 nM, 6 h). NES, normalized enrichment score; FDR, false discovery rate.
(E) Western blot showing THZ1-induced degradation of the large subunit of RNAPII in parental HBL-2 and DTEP cells, and that pre-treatment with proteasome inhibitor MG132 impaired THZ1-induced degradation of RNAPII in all DTEP cells. Data shown are representative of three independent experiments.
See also Figures S7 and S8.
Strikingly, ABT-199 resistance in VAL-DTEP cells was also associated with selection for rare subpopulations having loss of a BCL2 amplicon that is manifest in parental cells (Figure 7C); thus, this mechanism can also contribute to the emergence of ABT-199 resistance in DHL cells. To verify this phenomenon in other models, we generated three independent DTEP derivatives of the Mino MCL line (DTEP2, 6, and 9), and also assessed the acute effects of ABT-199 treatment (48 h, 1 nM) on BCL2 copy number in primary lymphoma cells from an MCL patient (Pt61) that harbor the BCL2 18q21 amplification. FISH analyses of the BCL2 amplicon and qRT-PCR analysis of BCL2 mRNA levels revealed partial or complete loss of the 18q21 amplicon in the Mino DTEP cells and corresponding reductions in BCL2 mRNA levels versus those present in parental Mino cells (Figure S8A). Further, analyses of primary patient MCL cells surviving after a 2-day treatment with ABT-199 ex vivo showed significant reductions in the BCL2 amplicon and BCL2 mRNA levels (Figure S8B).
To further assess the dynamics of this selection response, we treated lymphoma-bearing recipient mice transplanted with HBL-2 MCL cells, or with a patient-derived xenograft (PDX) model of a DHL primary tumor that has the 18q21 amplicon, daily with ABT-199 (5 mg/kg, Ren et al., 2018) for 10 days, and then harvested tumors for FISH analyses. Both HBL-2 and DHL PDX tumors were highly sensitive to ABT-199, with dramatic reductions in tumor size after 10 days of ABT-199 treatment (not shown). Notably, ABT-199- but not vehicle-treated tumor cells displayed marked reductions in the BCL2 amplicon and BCL2 mRNA levels in these models (Figures S8C and S8D), emphasizing the dynamic and rapid nature of this selection process in vivo. Thus, 18q21 amplicon loss is a generalized mechanism for ABT-199 resistance of MCL and DHL harboring BCL2 locus amplification.
Akin to the findings observed in MCL models, treatment of VAL DHL-derived tumors with ABT-199 in vivo also selects for marked reductions in the expression of BCL2 and NOXA, and to increased expression of MCL1 and FOXC1 (Figure S8E). Further, qRT-PCR analyses of primary MCL patient specimens (n = 13) that had variable resistance to ABT-199 revealed BCL2 mRNA levels correlating with ABT-199 sensitivity, where high BCL2 levels connote ABT-199 sensitivity (Figure S8F). Finally, THZ1 treatment of primary MCL patient specimens led to significant effects on the expression of DTEP-specific/THZ1-sensitive genes, including the SE-driven genes BCL2A1, BCL11A, and FOXC1 (Figures 7D and S8G).
Conspicuous effects of THZ1 treatment, especially manifest in DTEP of both MCL and DHL, are profound and rapid reductions in the steady-state levels of the large subunit of RNAPII (Figure 7E), and of RNAPII phosphorylated on Ser2 and Ser7 (Figure S8H), which are required for transcriptional elongation (Wang et al., 2015). Interestingly, transcriptional elongation block of RNAPII provokes rapid ubiquitin-dependent proteasome degradation of the large subunit of RNAPII (Karakasili et al., 2014). Concordant with this control, pre-treatment with the proteasome inhibitor MG132 impaired THZ1-induced degradation of RNAPII in all DTEP cells (Figure 7E). Thus, THZ1 treatment disables ABT-199 resistance by promoting destruction of the large subunit of RNAPII, an essential component of the transcriptional apparatus.
DISCUSSION
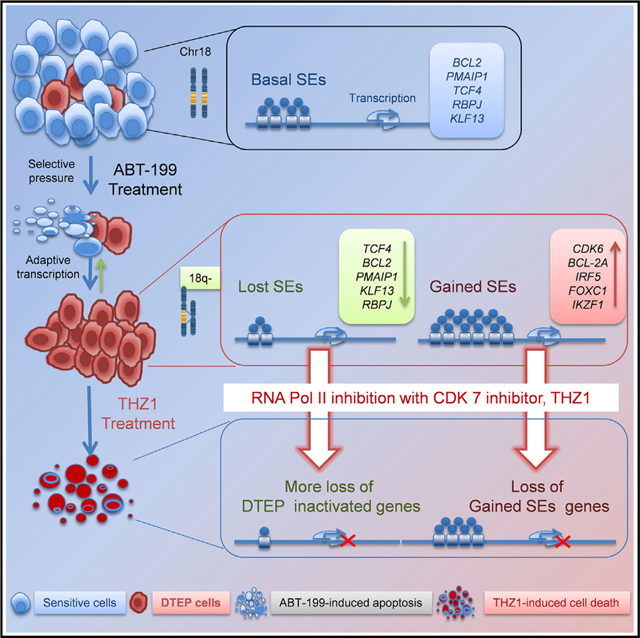
Historically, drivers of drug resistance have been associated with genetic alterations, which either pre-exist in a small fraction of cells in the population or arise de novo to mediate drug resistance (Hammerlindl and Schaider, 2018). However, emerging evidence has revealed that non-mutational adaptive responses that result in a reversible drug-tolerant phenotype precede emergence of permanent drug resistance following prolonged drug exposure (Hammerlindl and Schaider, 2018; Liau et al., 2017). Recently, it has been shown that adaptive SE-associated transcriptional programs promote resistance of solid tumors to targeted therapies, and that CDK7 inhibition disrupts these programs to provoke a therapeutic response (Rusan et al., 2018). While our studies revealed a generally similar adaptive CDK7-dependent, SE-associated transcriptional reprogramming as a mechanism of drug resistance to ABT-199 in MCL and DHL, our studies identified and established the roles of reprogrammed SE-associated genes needed to sustain ABT-199 resistance, and they revealed that resistance to this BCL-2 targeting drug is associated with upregulation of MCL-1. Furthermore, and importantly, in aggressive B cell lymphoma bearing 18q21 amplification, the selection for rare subpopulations having selective copy-number loss of BCL2 amplicons is revealed to contribute to the emergence and maintenance of ABT-199-resistant MCL and DHL (Figure 8). Excitingly, ABT-199 + THZ1 combination therapy is now shown to both prevent the onset of ABT-199 resistance in aggressive lymphoma models and to eradicate drug-resistant cells.
Figure 8. Model for the Evolution of ABT-199 Persistence/Resistance and CDK7 Vulnerability.

Model for the evolution of ABT-199 persistence/resistance in 18q21-amplified mantle cell lymphoma and double-hit lymphoma models, which occurs through the selection for rare clones having loss or reductions in copy number of 18q21 amplicons that harbor BCL2, and via adaptive super-enhancer (SE)-driven transcriptional reprogramming. The latter mechanism confers vulnerability to transcriptional inhibition by targeting CDK7.
Given the high levels of BCL-2 protein expressed from the 18q21 amplicon, the majority of MCL (HBL-2, Mino) and DHL (VAL) cells undergo apoptosis following ABT-199 treatment. However, small subsets of these DTP cells survive, and some then evolve and expand as ABT-199 DTEP cells. Analyses of ABT-199-resistant cells arising from single cells revealed loss of the 18q21 amplicon is not a requisite for resistance, as DTEP clones can be derived from cells that retain the amplicon. However, the fact that independent DTEP derivatives having distinct deletions or reductions in the copy number of the 18q21 amplicon arise from bulk MCL and DHL populations indicates that these rare clones could be the most fit in the population, due to ABT-199-driven selection for loss of apoptotic regulators, and that such clones would likely contribute to ABT-199 resistance in relapsed patients. In support of this notion, others have shown that early drug-tolerant clones are derived from rare pre-existing subpopulations (Hata et al., 2016; Knoechel et al., 2014; Ramirez et al., 2016). Further, our ex vivo analyses of primary MCL patient samples and in vivo xenograft and PDX models reveal that ABT-199-driven selection for clones having loss or reduced copy number of the 18q21 amplicon is a dynamic and rapid process, and suggest that this could lead to rapid treatment failure in patients having the 18q21 amplicon. Collectively, these results support the selection for 18q21 amplicon loss as a general mechanism of ABT-199 resistance for MCL and DHL harboring BCL2 locus amplification. Once ABT-199 achieves clinical approval for treating B-cell lymphomas, it will be important to assess if this mechanism is manifest in resistant or relapsed MCL or diffuse large B-cell lymphoma (DLBCL) having the 18q21 amplicon, and if 18q21 amplicon/copy-number gain is a predictive biomarker for applying ABT-199 therapy in B-cell malignancies.
Emphasizing the importance of our findings, 18q21 amplification occurs frequently in B-cell malignancies (Sanchez-Izquierdo et al., 2003), specifically in ~25% of primary MCL and in ~38% of aggressive DLBCL specimens (Bea et al., 2009). Notably, the 18q21 amplicon also drives expression of NOXA, a dedicated proapoptotic inhibitor of MCL-1 (Guikema et al., 2017), causing lymphoma to have the 18q21 amplicon more reliant on BCL-2 and thus ideal for ABT-199 therapy. Accordingly, loss of proapoptotic NOXA likely contributes to the resistance of DTEP cells to ABT-199. However, there are also clearly roles for loss or reductions in BCL2 copy number and expression in ABT-199 resistance, because forced overexpression of BCL-2 in otherwise ABT-199-resistant DTEP cells leads to a re-addiction to BCL-2. We propose that this likely occurs via the ability of overexpressed BCL-2 to sequester BAX, and that treatment with ABT-199 then releases BAX pools to provoke the formation of BAX:BAX homodimers and/or BAX:BAK heterodimers, which trigger apoptosis. However, this hypothesis merits further research.
SE and associated transcriptional reprogramming are also key contributors to ABT-199 resistance, and here the selection for increased transcription of compensatory genes when MCL or DHL cells are faced with this BCL-2-targeting agent is striking. In particular, gained SEs in DTEP cells include those that induce high levels of a cast of anti-apoptotic and cell fate regulators (BCL2A1, BCL11A1, FOXC1, IRF5, and IKZF1), and genetic studies have established that each contributes to sustaining ABT-199 resistance in lymphoma cells. It will therefore be interesting to test if each is also necessary for the emergence of DTEP and, if so, what alternatives, if any, the tumor cell can exploit to allow survival in the face of ABT-199. Again, in future studies it will be important to evaluate whether such compensatory SEs and targets are induced in MCL or DLBCL patients who have relapsed on ABT-199 treatment.
ChIP-seq and RNA-seq analyses, coupled with CDK7/transcription-targeting studies, clearly established SE remodeling drives transcriptome and kinome reprogramming in ABT-199 DTEP cells. Further, our functional drug screening and genetic studies revealed that the SE-driven reprograming of the transcriptome and kinome of lymphoma cells relies on CDK7, and that this dependency confers a therapeutic vulnerability that can be exploited to prevent emergence of resistance, and to kill ABT-199-resistant MCL and DHL cells. Notably, the resultant phenotype affords an evolutionary double bind, where combining potent agents that disable BCL-2 and CDK7 is an extremely potent combination strategy to treat ABT-199-resistant MCL and DHL that triggers rapid cell death of primary MCL and DHL patient specimens, and that provokes tumor regression of validated in vivo models of these B cell malignancies.
As selection for ABT-199 resistance involves increased expression of MCL-1- and SE-driven expression of the anti-apoptotic gene BCL2A1, small molecules targeting these BCL-2 family members should have activity in ABT-199-resistant patients, again particularly when combined with CDK7 inhibitors. Indeed, regardless of the evolutionary strategy applied to attain resistance, CDK7 is revealed as necessary to sustain SE-driven transcription and for the development and maintenance of DTEPs in MCL and DHL. Thus, disabling CDK7 in combination with ABT-199 is an attractive means to provoke tumor regression of otherwise refractory lymphoma, and such a combination strategy could be applied across a broad spectrum of hematological malignancies.
STAR★METHODS
CONTACTS FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jianguo Tao (Jianguo.Tao@moffitt.org).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Patients and Tumor Specimens
The primary samples from MCL, DHL and diffuse large B cell lymphoma (DLBCL) patients were obtained from fresh biopsy-derived lymphoma tissues (lymph nodes) and from peripheral blood following informed consent from patients and approval by the Moffitt Cancer Center/University of South Florida Institutional Review Board.
For preparation of viable, sterile, single cell suspensions, the lymph node tissue was diced and forced through a cell strainer into RPMI-1640 tissue culture medium. Cells, obtained after low-speed centrifugation, were re-suspended in media. Lymphoma cells from peripheral blood were isolated by Ficoll-Paque purification, and only lymphoma samples that had greater than 80% tumor cells were used for experiments.
The human specimen studies presented were approved by the Moffitt/University of South Florida Institutional Review Board and patients provided signed informed consent forms.
Mice
All animal studies were conducted in accordance with the NIH guidelines for animal care. All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Moffitt Cancer Center and the University of South Florida.
Cell Lines
Mantle cell lymphoma cell lines (MCL) HBL-2 and Mino were purchased from ATCC. The double hit lymphoma (DHL) cell line VAL (Ren et al., 2018) was purchased from DSMZ (ACC586, German Collection of Microorganisms and Cell Cultures, Braunschweig. Germany). These cells and their ABT-199-resistant derivatives were cultured in RPMI-1640 (Gibco-Invitrogen) with penicillin (100 U/mL) and streptomycin (100 μg/mL) and maintained at 37 °C in 5% CO2. Cell lines were routinely tested for mycoplasma using the Universal Mycoplasma Detection Kit from ATCC. Cell lines were authenticated using short tandem repeat (STR) DNA typing according to ATCC’s “Authentication of Human Cell Lines: Standardization of STR Profiling” ((ANSI), 2012). Genomic DNA [HBL-2-Parental, HBL-2-I10, HBL-2-J4, HBL-2-L20,] were submitted to Genetica Cell Line Testing (a LabCorp brand; Burlington, NC). Amplification and amplicon analyses were performed using PowerPlex 16 HS (Promega Corp), generating STR DNA profiles for 15 independent genetic sites and amelogenin (the sex identity locus) as well as a single mouse marker for the detection of mouse DNA contamination. Cell lines were considered and confirmed authenticated when the number of shared alleles across the eight core loci (THO1, D5S818, D13S317, D7S820, D16S539, CSF1PO, vWA, TPOX) and Amelogenin is ≥80%, as described by the ATCC ((ANSI), 2012). Results of cell lines authentication are provided in Table S1.
METHOD DETAILS
Mice
Six-to eight-week-old male NOD/SCID mice were purchased from the Jackson Laboratory and used for xenograft experiments as described (Zhao et al., 2017). One million HBL-2 parental or L20 DTEP cells were injected into the lower flank of NOD/SCID mice in a volume of 0.1 mL PBS. Mice were then randomized into control and treatment groups when tumor volume reached 0.2 cm3 (4 mice per group). Tumors were measured with calipers and when tumor volume reached 100–200 mm3 mice were randomized for treatment with ABT-199, THZ1 or vehicle. ABT-199 was dissolved in DMSO and then diluted at the final ratio: 5% DMSO+40% PEG 400 (or 300) +5% Tween 80+50% 0.9% NaCl. Drugs were given i.p. and the dose for combined ABT-199 + THZ1 was 5 mg/kg each. Mice were humanely sacrificed when the control tumor reached ~2,000 mm3 or after the loss of more than 10% of body weight.
Generation of ABT-199 Drug-Tolerant Persister (DTP) and DTP Expanded Persister (DTEP) Cell Lines
HBL-2 parental cells were seeded into 384-well collagen-coated plates at a density of 1,000 cells per well for 24 hr, and then cultured in the medium containing 20 nM ABT-199 for 10–14 days. Most cells died, leaving a few, isolated, drug-tolerant and slow-growing cells (DTP cells) in a few (12) wells, and only 1–2 colonies were observed in a few of these wells. Cells from clear single-colony containing wells were transferred to 96-well plates around day 14. Colonies were expanded from 96-well plates to 24-well plates to 6-well plates and finally to cell culture flasks (DTEP cells, J4, I10 and L20) in the presence of ABT-199 (20 nM, up to 6–8 weeks). To obtain sufficient cells for our large-scale experiments (drug screening, RNA-seq and ABPP assays), we expanded the DTEP and parental cells an additional 6–10 further passages (4–6 weeks). Cell lines were periodically examined and were negative for mycoplasma contamination during the course of this work. Cell culture media contained 20 nM ABT-199 for the entire duration of the DTEP generation time (~3 months) and was changed regularly (~every 3 days). To assess reversibility of these DTEPs, J4, I10 and L20 DTEPs were cultured with regular medium that did not contain ABT-199 for three months (drug holiday) and used for subsequent experiments.
Generation of Single-Cell Clones and ABT-199 DTEP Derivates
HBL-2 parental cells were seeded into 96-well plates at a density of 1 cell per well. After 2 weeks, approximately 30% of wells contained cells. These cells were expanded from 96-well plates to 24-well plates to 6-well plates, and finally to cell culture flasks (single-cell subclones). These subclones were further treated with ABT-199 (20 nM) and cell viability assay was performed. Three ABT-199-sensitive subclones (IC50 < 1 nM, P5F6, P2D8 and P1C10) were selected and 105 cells were plated in five 10 cm dishes and were then treated with 20 nM ABT-199. Fresh media-containing 20 nM ABT-199 was replaced every 3 days until clones of drug-resistant cells appeared, expanded in drug-containing media (P5F6-DTEP, P2D8-DTEP and P1C10-DTEP), and “stock cultures” of these DTEPs were maintained in 20 nM ABT-199 for use in experiments.
BCL-2 Overexpression and CRISPR/Cas9 Editing of SE Target Genes
Exogenous BCL-2 was overexpressed in HBL-2 cells and ABT-199 DTEP cells by retroviral transduction. Retroviral particles were generated by transient transfection of GP2–293T cells with pMIP-BCL2 and the packaging vector pCLampho. 48 hr after transfection, culture supernatants were harvested and filtered. Cells were infected with the virus-containing supernatant and 8 μg/mL polybrene. Three days after infection, cells were selected with 0.5 μg/mL puromycin.
For CRISPR/Cas9 editing of BCL2, TCF4, NOXA, BCL2A1, MCL1, FOXC1, IKZF1, and IRF5, gRNAs targeting these genes or GFP were cloned into a vector encoding espCas9. gRNA sequences are listed in KRT. Briefly, lentiviral particles were generated by transfection of HEK-293T cells with pLentiCRISPR V2 constructs and the packaging vectors pVSVg (AddGene, 8454) and psPAX2 (AddGene, 12260). 48 hr after transfection, culture supernatants containing virus were harvested and filtered. The filtrate was concentrated by ultra-centrifuging for 2 hr at 23,000 RPM, 4°C. Cells were infected with the concentrated viral supernatants and 8 μg/mL polybrene. For stable knockdown, 3 days after viral infection, cells were selected in 0.5 μg/mL puromycin. Puromycin-resistant cells were seeded in methocult™ medium (Stem Cell, 4034). Cell clones were picked up and were characterized by western blots to determine if the gene targeted protein was significantly reduced (knockdown) or completely depleted (knock out) after gRNA transduction and selection.
High-Throughput Small-Molecule Drug Screens
Using a semi-automated platform, we tested the potency of a 60 small molecule annotated library in HBL-2 parental and three DTEP cell lines (I10, J4 and L20). Cell viability was estimated by using Resazurin (R&D Systems, AR002). In brief, cells were seeded in 384-well plates with 2,000 cells per well in 25 μL medium. Cells were cultured in the presence of different compounds at serial threefold diluted concentrations. After 3 (kinase inhibitors) or 6 days (epigenetic inhibitors) of treatment, 6 μL of Resazurin reagent was added into each well and incubated for 2 hr. Plates were read at 560/590 nm wavelength to estimate cell proliferation. All kinase inhibitors and epigenetic drugs used in drug screening and cell-based drug screening assays were purchased from Selleckchem, except NVP2 which was from MedChemExpress (HY-12214A) and INCB054329, INCB053914, INCB059872, INCB050465, which were kindly provided by Incyte Corporation (Wilmington, DE). THZ1 was synthesized as described (Kwiatkowski et al., 2014).
Cell Viability Assays
5000 cells were seeded in 96-well plates in 50 μl medium, 50 μl drug medium at five or nine serial diluted concentrations were added to the cell suspension. 3000 cells were seeded in 384-well plates in 25 μl medium, 5 μl drug medium at five serial diluted concentrations were added to the cell suspension. After 72 hr incubation, 20 or 6 ul of Resazurin reagent were added into the 96-well or 384-well plates. The plates were read after two hour incubation using 544 nm excitation and 590 nm emission wavelengths. Relative cell viability was normalized to DMSO treated wells.
Image-Based Cell Viability Analysis
Cells were seeded in a 384-well plates of a reconstructed lymphoma TME, including high physiological densities (1–10 × 106 cells/mL), extracellular matrix (collagen, Advanced BioMatrix, #5005-B), and lymphoma stromal cells (HK cells or autologous stromal cells). A panel of drugs at five serial diluted concentrations were added to the media, and plates were continuously imaged every 30 min for 96 hr or 10 days. All images were analyzed using a digital image analysis algorithm to detect cell viability based on membrane motion (pseudo-colored in green), and changes in viability were quantified by area under curve (AUC) as described (Silva et al., 2015, 2017; Zhao et al., 2017).
To assess synergistic versus additive effects of combined drug treatment, the combination index (CI) was used. CI for each drug combination was calculated by median dose-effect analyses using the commercially available software CalcuSyn (Biosoft, Great Shelford, Cambridge, UK). CI values <1.0 represent synergism of the two drugs in the combination.
BH3 Profiling of Parental MCL and DTEP Cells
Parental HBL-2 MCL and DTEP cells were seeded at a density of 4 × 105 cells/mL in 10% FBS RPMI media 24 hr before BH3 profiling. Two million parental and DTEP cells were pelleted at 400 × g for 5 min at room temperature and resuspended in 2 mL DTEB (135 mmol/L Trehalose, 10 mmol/L HEPES-KOH, 0.1% w/v BSA, 20 μmol/L EDTA, 20 μmol/L EGTA, 50 mM KCl, 5 mM succinate, pH 7.4). A plate-based JC-1 BH3 profiling assay was performed as described (Vo et al., 2012). Cells were permeabilized with digitonin, exposed to BH3 peptides, and mitochondrial transmembrane potential loss was monitored using the ratiometric dye JC-1.
Fluorescence In Situ Hybridization (FISH)
Cells were washed with PBS once after harvest, and treated with 0.075 M KCl for 15 min, then cell pellet was fixed with fixative solution (3:1 methanol to acidic acid). Cell suspension was dropped to a slide and dry in room temperature. Slide was dehydrated by incubating in 70%, 85%, and 100% of ethanol for 2 min each. Then slide were probed with BCL2 dual color break apart probe (CytoCell, Cambridge, UK), and follow with 75°C and 37°C of denaturation and incubation procedure overnight in Thermobrite. Next day, slide was washed with 0.4 X SSC at 72°C. Cell was counterstained with DAPI, and analyzed under DM5500 Leica fluorescence microscope, and images were recorded in CytoVision analysis system.
RNA-Sequencing with ERCC RNA Spike-In Mix
All samples were prepared in biological triplicates. 1×106 cells were treated for 6 hr with either 50 or 250 nM THZ1 or DMSO (vehicle control). Total RNA was isolated using RNA isolation kit, RNeasy Plus Mini (Qiagen Cat# 74134) and a set of external RNA controls using ERCC RNA Spike-In Mix (Life Technologies, Cat 4456740) were added after total RNA extraction (1 μL to 5,000 ng RNA). Library prep was conducted using TruSeq Stranded mRNA Library Prep Kit (Illumina Cat #RS-122-2101/2) according to the manufacturer’s instructions. RNA sequencing was performed on HiSeq 2500v4 high output (50 bp, single-end reads). Tophat2 was used to align the Fastq files. We used a custom hg19 genome and transcriptome indices including ERCC transcripts. FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values were calculated and normalized using Cuffnorm. Genes that had a p < 0.05 and at least a two-fold change were considered to be significantly altered between treatments. Cutoff value for expressed genes was an FPKM value equal to or higher than 1.
Chromatin Immunoprecipitation Followed by Highly Parallel Sequencing (ChIP-Seq)
ChIP-seq was performed according to established protocols, with minor modifications. 50 × 106 cells were used for ChIP-seq of H3K27ac and 150 × 106 cells for RNAPII. Crosslinking was performed in batches of 50 × 106 cells in 50 mL tissue culture media by addition of one-tenth volume of 10X crosslinking solution (11% formaldehyde, 50 mM HEPES pH 7.3, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA ph 8.0). After 10 min of crosslinking at room temperature, formaldehyde was quenched with 125 mM glycine, cells were then washed three times in PBS pH 7.4, flash frozen in liquid nitrogen, and stored at −80°C. Frozen pellets were thawed on ice, resuspended in cold lysis buffer 1 (LB1; 5 mL per 50 × 106 cells; 50 mM HEPES pH 7.3, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100, Roche protease inhibitor cocktail (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich Cat# 4693159001), and rotated for 10 min at 4°C. LB1 was removed and pellets were resuspended in cold lysis buffer 2 (LB2; 5 mL per 50 × 106 cells; 10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA pH 8.0 and 0.5 mM EGTA pH 8.0, Roche protease inhibitor cocktail) and rotated for 10 min at 4°C. LB2 was removed and pellets were resuspended in cold sonication buffer (1.5 mL per 50 × 106 cells; 50 mM HEPES pH 7.3, 140 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, Roche protease inhibitor cocktail). Samples were divided into 1.5 mL Bioruptor Plus TPX microtubes (Diagenode, #C30010010) at 250 μL per tube and sheared at 4°C using a water bath sonicator (Bioruptor, Diagenode; 22.5 min at high output; 30 sec on, 30 sec off). Sheared lysates were clarified by centrifuging at 20,000 × g at 4°C for 10 min and supernatants were collected together, setting aside 50 μL as an input sample. For all other ChIP-seq experiments, magnetic protein G beads (Dynabeads, ThermoFisher Scientific, Cat# 10009D) were washed 3 times with, and resuspended in, 1 mL cold blocking buffer and then rotated with appropriate antibody overnight at 4°C using 100 μL of beads with 10 μg anti-H3K27ac (ABCAM ab4729) or 20 μL anti-RNAPII (Diagenode, #C15100055). Antibody:bead complexes were washed 3 times with cold blocking buffer, added to the diluted and clarified chromatin supernatant, and rotated overnight at 4°C. The bound chromatin was then washed twice with 1 mL cold sonication buffer, once with 1 mL cold sonication buffer supplemented with 500 mM NaCl, once with cold LiCl wash buffer (20 mM Tris pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate), and once with TE supplemented with 50 mM NaCl. Finally, beads were resuspended in 210 μL elution buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, and 1% SDS) and chromatin was eluted by vortexing every 5 min while incubating at 65°C for 15 min. Beads were centrifuged at 20,000 × g for 1 min and the supernatant, together with input sample was placed at 65°C overnight to reverse crosslinks. RNA was digested with 0.2 mg/mL RNase A (Roche, 10109169001) at 37°C for 2 hr and protein was digested with 0.2 mg/mL proteinase K (Life Technologies, AM2546) at 55°C for 30 min. DNA was isolated with phenol chloroform extraction and ethanol precipitation.
Libraries for Illumina sequencing were prepared using ThruPLEX DNA-seq Kit (TaKara) using 50 ng of DNA or less and amplifying according to manufacturer instructions. Amplified libraries were size-selected first using AMPure beads (Agencourt AMPure XP) and subsequently, using a 2% gel cassette in the Pippin Prep (SAGE Sciences) to capture fragments of 200–700 bp. Libraries were quantified by qPCR using the KAPA Biosystems library quantification kit, multiplexed with equimolar DNA content, and sequenced on an Illumina NextSeq 500 (single end 75 bp reads).
ChIP-Seq Data Processing
Data Accession and Code Availability
Computational code used for ChIP-seq analyses can be obtained from the Bradner Laboratory github page. Namely, Bamliquidator was used to calculate read density which can be found at http://github.com/BradnerLab/pipeline/wiki/bamliquidator; and ROSE2, was used to identify enhancers and can be found at https://github.com/BradnerLab/pipeline/(ROSE2_main.py). RNA-seq and ChIP-seq data produced for this publication can be accessed online using GEO Publication Reference ID (GSE116132).
Sequence Alignment
All datasets were aligned using Bowtie2 (version 2.2.1). All default parameters, except for –N 1 (reads that mapped uniquely to the genome with one or fewer mismatches) were used to align to human genome build NCBI37/HG19.
Identifying Enriched Regions
The MACS version 1.4.1 (Model based analysis of ChIP-Seq) 67 peak finding algorithm was used to identify regions of ChIP-Seq enrichment over background. A p value threshold of enrichment of 1e-9 was used for all datasets.
Calculating Read Density
We calculated the normalized read density of a ChIP-Seq dataset in any genomic region using the Bamliquidator (version 1.0) read density calculator (https://github.com/BradnerLab/pipeline/wiki/bamliquidator). Briefly, ChIP-Seq reads aligning to the region were extended by 200 bp and the density of reads per base pair (bp) was calculated. For ChIP-seq, the density of reads in each region was normalized to the total number of million mapped reads producing read density in units of reads per million mapped reads per bp (rpm/bp).
Mapping Typical Enhancers and Super-enhancers Using H3K27ac Enhancer Definitions
H3K27ac super-enhancers (SEs) and typical enhancers (TEs) were mapped using the ROSE2 software package that has been previously described and that is available at https://github.com/BradnerLab/pipeline/(ROSE2_main.py) (Brown et al., 2014). MACS defined peaks were considered for rank-ordering by the ROSE2 algorithm. ROSE2 optimizes a stitching parameter on a per-sample basis for combining nearby peaks. Briefly, the algorithm optimizes for the enriched fraction of stitched regions. Read density within these regions was then quantified as noted above and stitched regions were ranked by this metric. SEs were called by re-scaling both the signal values and the ranks to fall between 0 and 1 and plotting a curve with scaled ranks on the x axis and scaled signal on the y axis. The x coordinate of the intersection point of the line of slope 1 that is tangent to the curve was used to define a cutoff for SEs such that all stitched enhancers with a scaled rank greater than this cutoff were considered SEs. Default ROSE2 parameters for stitching and region filtering, including exclusion of TSS-proximal signal (within 2.5 kb), were used. ROSE2 was also used to rank-order regions disproportionately enriched for RNAPII ChIP-seq signal as described for H3K27ac ChIP-seq above, except that no peak stitching was used and without excluding ± 2.5 kb from each TSS.
cDNA Synthesis and Quantitative PCR
Total RNA was prepared as described above. cDNA was synthesized using the iScript cDNA Synthesis kit (BioRad Cat# 1708890) according to instructions of the manufacturer. Quantitative real-time PCR (qRT-PCR) was performed using SYBR™ Green PCR Master Mix (ThermoFisher Scientific) for indicated target genes and using human 18S rRNA was used as internal sample probe to correlate for inter-assay variability. Relative quantification of expression levels was performed according to the comparative threshold cycle (Ct) method assuming equal efficiency of target and housekeeping gene probes. Primer sequences are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BAK | Cell Signaling Technologies | Cat# 12105; RRID: AB_2716685 |
| BAX | Cell Signaling Technologies | Cat# 2772; RRID: AB_10695870 |
| BCL-2 | Cell Signaling Technologies | Cat# 2872; RRID: AB_10693462 |
| BIM | Cell Signaling Technologies | Cat# 2933; RRID: AB_1030947 |
| cPARP | Cell Signaling Technologies | Cat# 5625; RRID: AB_10699459 |
| NOXA | Cell Signaling Technologies | Cat# 14766; RRID: AB_2798602 |
| FoxC1 | Cell Signaling Technologies | Cat# 8758; RRID: AB_2797657 |
| BCL11A | Cell Signaling Technologies | Cat# 75432; RRID: AB_2799868 |
| IKZF1 | Cell Signaling Technologies | Cat# 14859; RRID: AB_2744523 |
| CDK7 | Cell Signaling Technologies | Cat# 2916; RRID: AB_2077142 |
| BCL-XL | Cell Signaling Technologies | Cat# 2762; RRID: AB_10694844 |
| RNA pol II CTD phospho-Ser2 | Millipore | Cat# 04-1571-I; RRID: AB_10627998 |
| RNA pol II CTD phospho-Ser5 | Millipore | Cat# 04-1572-I; RRID: AB_2801296 |
| RNA pol II CTD phospho-Ser7 | Millipore | Cat# 04-1570-I; RRID: AB_2801298 |
| Anti-Histone H3 (acetyl K27) antibody-ChIP | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Pol II monoclonal antibody | Diagenode | Cat# #C15100055; RRID: AB_2750842 |
| MYC | Abcam | Cat# ab32072; RRID: AB_731658 |
| TCF4 | Abcam | Cat# ab185736; RRID: AB_2801300 |
| IRF5 | Abcam | Cat# ab181553; RRID: AB_2801301 |
| BCL2A1 | Abcam | Cat# ab75887; RRID: AB_1523197 |
| MCL-1 | Santa Cruz Biotechnology | Cat# sc-819; RRID: AB_2144105 |
| Actin | Santa Cruz Biotechnology | Cat# sc-47778HRP; RRID: AB_2714189 |
| Biological Samples | ||
| Primary patient specimen(MCL, DHL,DLBCL) | Moffitt Cancer center | NA |
| Bacterial and Virus Strains | ||
| pVSVg | AddGene | Cat# 8454 |
| lentiCRISPR v2 | AddGene | Cat# 52961 |
| psPAX2 | AddGene | Cat# 12260 |
| pCL-Ampho | NOVUS Biologicals | Cat# NBP2-29541 |
| pMIP-BCl-2 | Homemade | |
| pcDNA/Myc TCF4 | AddGene | Cat# 16512 |
| FLAG-HA-pcDNA3.1 | AddGene | Cat# 52535 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ABT-199 | Selleckchem | Cat# S8048 |
| S63845 | ApexBio | Cat# A8737 |
| THZ1 | Dana-Farber Cancer Institute Nathanael Gray lab | NA |
| THZ531 | Dana-Farber Cancer Institute Nathanael Gray lab | NA |
| NVP2 | MedChemExpress | Cat# HY-12214A |
| AZD8055 | Selleckchem | Cat# S1555 |
| MK-2206 | Selleckchem | Cat# S1078 |
| MK-1775 | Selleckchem | Cat# S1525 |
| UNC1999 | Cayman Chemical | Cat# 14621 |
| Alisertib | Selleckchem | Cat# S1133 |
| BEZ-235 | Selleckchem | Cat# S1009 |
| AZD7762 | Selleckchem | Cat# S1532 |
| INCB053914 | Incyte Corporation (Wilmington, DE) | NA |
| INCB054329 | Incyte Corporation (Wilmington, DE) | NA |
| INCB052793 | Incyte Corporation (Wilmington, DE) | NA |
| Ibrutinib | Selleckchem | Cat# S2680 |
| JQY | Dana-Farber Cancer Institute Qi Jun lab | NA |
| (+)-JQ1 | Cayman Chemical | Cat# 11187 |
| SCH-772984 | Selleckchem | Cat# S7101 |
| Olaparib | Selleckchem | Cat# 1060 |
| I-CBP112 | Cayman Chemical | Cat# 14468 |
| ACP-196 | Selleckchem | Cat# S8116 |
| PIK-75 | Selleckchem | Cat# S1205 |
| Dinaciclib | Selleckchem | Cat# S2768 |
| BIX01294 | Selleckchem | Cat# S8006 |
| EPZ15666 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| Trametinib | Selleckchem | Cat# S2673 |
| XAV-939 | Selleckchem | Cat# S1180 |
| KU-55933 | Selleckchem | Cat# S1092 |
| A-1210477 | Selleckchem | Cat# S7790 |
| INCB050465 | Incyte Corporation (Wilmington, DE) | NA |
| SGC707 | Cayman Chemical | Cat# 17017 |
| Lenalidomide | Selleckchem | Cat# S1029 |
| Pomlidomide | Selleckchem | Cat# S1567 |
| VE-821 | Selleckchem | Cat# S8007 |
| JADA-20 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| GSK484 | Cayman Chemical | Cat# 17488 |
| DBZ | Selleckchem | Cat# S2711 |
| SGC0946 | Cayman Chemical | Cat# 13967 |
| OJI1 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| GSK-J4 | Cayman Chemical | Cat# 12073 |
| UNC0642 | Cayman Chemical | Cat# 14604 |
| Lumpib | Selleckchem | Cat# S1069 |
| INCB059872 | Incyte Corporation (Wilmington, DE) | NA |
| Merk60 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| MS275 | Selleckchem | Cat# S1053 |
| EPZ4777 | Selleckchem | Cat# S7353 |
| MS049 | Cayman Chemical | Cat# 18348 |
| JADA-19 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| WT161 | Selleckchem | Cat# S8495 |
| A-366 | Cayman Chemical | Item# 16081 |
| GSK690 | MedChemExpress | Cat# HY-117226A |
| GSK591 | Cayman Chemical | Cat# 18354 |
| A-1331852 | Selleckchem | Cat# S7801 |
| Bortezomib | Selleckchem | Cat# S1013 |
| Volasertib | Selleckchem | Cat# S2235 |
| Carfizomib | Selleckchem | Cat# S2853 |
| EZ5 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| EZ53 | Dana-Farber Cancer Institute Qi Jun lab | NA |
| EPZ5676 | Selleckchem | Cat# S7062 |
| PFI-4 | Cayman Chemical | Cat# 17663 |
| BI-9564 | Cayman Chemical | Cat# 17897 |
| ABT-263 | Selleckchem | Cat# S1001 |
| Critical Commercial Assays | ||
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K |
| Resazurin | R&D systems | Cat# AR002 |
| MethocultTM medium | STEMSTELL technologies | Cat# 4034 |
| Bovine Type I Atelo-Collagen Solution | Advanced BioMatrix | Cat# 5005-B |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 4693159001 |
| Dynabeads™ Protein G for Immunoprecipitation | ThermoFisher Scientific | Cat# 10009D |
| RNase A | Sigma-Aldrich | Cat# 10109169001 |
| Proteinase K | ThermoFisher Scientific | Cat# AM2546 |
| AMPure beads (Agencourt AMPure XP) | Beckman Coulter Life Sciences | Cat# A63882 |
| Pippin Prep | SAGE Sciences | Cat# PIP0001 |
| KAPA Biosystems library quantification kit | KAPA Biosystems | Cat# KK4824 |
| JC-1 Dye (Mitochondrial Membrane Potential Probe) | ThermoFisher Scientific | Cat# T3168 |
| Pierce™ Kinase Enrichment Kit with ATP Probe | ThermoFisher Scientific | Cat# 88310 |
| Zeba™ Spin Desalting Columns, 7K MWCO, 5 mL | ThermoFisher Scientific | Cat# 89892 |
| RNeasy Plus Mini | Qiagen | Cat# 74134 |
| Custom CGH & CGH+SNP Microarrays | Agilent | Cat# G4884A |
| BCL2 dual color break apart probe | CytoCell | Cat# LPS 028-A |
| DAPI (4′,6-diamidino-2-phenylindole) | ThermoFisher Scientific | Cat# 62247 |
| ERCC RNA Spike-In Mix | ThermoFisher Scientific | Cat# 4456740 |
| TruSeq® Stranded mRNA Library Prep (48 Samples) | Illumina | Cat# 20020594 |
| TruSeq RNA Single Indexes Set A (12 Indexes, 48 Samples) | Illumina | Cat# 20020492 |
| ThruPLEX® DNA-seq Kit | TaKaRa | Cat# R400675 |
| iScript cDNA Synthesis kit | BioRad | Cat# 1708890 |
| SYBR™ Green PCR Master Mix | ThermoFisher Scientific | Cat# 4309155 |
| Deposited Data | ||
| RNA-seq and CHIP-seq | Gene Expression Omnibus | GEO: GSE116132 |
| ABPP proteomics | Proteome Xchange | PRIDE: PXD010193 |
| Experimental Models: Cell Lines | ||
| VAL | DSMZ | Cat# ACC586 |
| Mino | ATCC | Cat# CRL-3000 |
| HBL-2 | Cellosaurus | Cat# CVCL_4216 |
| HK | Cellosaurus | Cat# CVCL_IY38 |
| autologous stromal | Homemade | NA |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD-SCID (NOD-scid IL2Rgnull) | The Jackson Laboratory | Cat# 001303 |
| Software and Algorithms | ||
| CytoGenomics Software | Aglilent | NA |
| Calcusyn | Biosoft | http://www.biosoft.com/w/calcusyn.htm |
| GraphPad Prism 7 | GraphPad Software | www.graphpad.com |
| MaxQuant | https://www.maxquant.org/ | Cox and Mann (2008) |
| ROSE2 | https://github.com/BradnerLab/pipeline/(ROSE2_main.py) | Ref: Brown et al., 2014 |
| Tophat2 | https://ccb.jhu.edu/software/tophat/index.shtml | NA |
| Cuffnorm | http://cole-trapnell-lab.github.io/cufflinks/cuffnorm/index.html | NA |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | Ref: Langmead et al., 2012 |
| MACS version 1.4.1 | https://github.com/taoliu/MACS/ | NA |
| Bamliquidator (version 1.0) | https://github.com/BradnerLab/pipeline/wiki/bamliquidator | NA |
| Gene Set Enrichment Analysis (GSEA) | http://software.broadinstitute.org/gsea/index.jsp | Ref: Subramanian et al., 2005, Mootha et al., 2003 |
| KEGG: Kyoto Encyclopedia of Genes | https://www.genome.jp/kegg/andGenomes | NA |
| PATHER | http://www.pantherdb.org/ | NA |
| Oligonucleotides | ||
| BCL2 gRNA | ACCTGACGCCCTTCACCGCG | NA |
| BCL2 gRNA | AAAGCGTCCCCGCGCGGTGA | NA |
| BCL2 gRNA | ATCAAACAGAGGCCGCATGC | NA |
| TCF4 gRNA | AATGCATCACCAACAGCGAA | NA |
| TCF4 gRNA | ACTGCTACTCGAGCTTCTCC | NA |
| TCF4 gRNA | CAGCTCTTTGTCCGTCCCTA | NA |
| PMAIP1 gRNA | TCGAGTGTGCTACTCAACTC | NA |
| PMAIP1 gRNA | ACGCTCAACCGAGCCCCGCG | NA |
| PMAIP1 gRNA | TTCTTGCGCGCCTTCTTCCC | NA |
| MCL1 gRNA | GGGTAGTGACCCGTCCGTAC | NA |
| MCL1 gRNA | GTATCACAGACGTTCTCGTA | NA |
| MCL1 gRNA | CGCGGTGACGTCGGGGACCT | NA |
| FOXC1 gRNA | CGGACCGGGCGCGTTGCCGT | NA |
| FOXC1 gRNA | ACAGTCGTAGACGAAAGCTC | NA |
| FOXC1 gRNA | CACGGAGTAGCGCGCCTGCA | NA |
| BCL2A1 gRNA | TTGAAGACGGCATCATTAAC | NA |
| BCL2A1 gRNA | TCTACGACAGCAAATTGCCC | NA |
| BCL2A1 gRNA | GCACAATCACACACCTATGC | NA |
| CDK7 gRNA | CTAGTTTGCCACCGTTTACA | NA |
| CDK7 gRNA | CTTTCTTAATGGCGACAATT | NA |
| CDK7 gRNA | TTGCAGCTCCTTGATGCTTT | NA |
| BCL11A gRNA | GATAAACAATCGTCATCCTC | NA |
| BCL11A gRNA | GGATACCAACCCGCGGGGTC | NA |
| BCL11A gRNA | GAGTGCAGAATATGCCCCGC | NA |
| IRF5 gRNA | ATGAAGCCGATCCGGCCAAG | NA |
| IRF5 gRNA | ATCGGCTTCATCCACGCCTT | NA |
| IRF5 gRNA | CCGTTGACCCATTGAAGCCC | NA |
| IKZF1 gRNA | TCATCTGGAGTATCGCTTAC | NA |
| IKZF1 gRNA | ACCTACCGGAGTGCGTCCTC | NA |
| IKZF1 gRNA | CTCCAAGAGTGACAGAGTCG | NA |
| BCL2 qRT-PCR primer | Forward: 5’-CTGCACCTGACGCCCTTCACC-3’ | NA |
| BCL2 qRT-PCR primer | Reverse: 5’-CACATGACCCCACCGAACTCAAAGA-3’ | NA |
| MCL1 qRT-PCR primer | Reverse: 5’-AGAAAGCTGCATCCAACCAT-3’ | NA |
| MCL1 qRT-PCR primer | Reverse: 5’-CCAGCTCCTACTCCAGCAAC-3’ | NA |
| PMAIP1 qRT-PCR primer | Reverse: 5’-GCTGGAAGTCGAGTGTGCTA-3’ | NA |
| PMAIP1 qRT-PCR primer | Reverse: 5’-CAGTCAGGTTCCTGAGCAGA-3’ | NA |
| FOXC1 qRT-PCR primer | Reverse: 5’-CGGGTTGGAAAGGGATATTTA-3’ | NA |
| FOXC1 qRT-PCR primer | Reverse: 5’-CAAAATGTTCTGCTCCTCTCG-3’ | NA |
| BCL2A1 qRT-PCR primer | Reverse: 5’-CATTAACTGGGGAAGGATTGTGAC-3’ | NA |
| BCL2A1 qRT-PCR primer | Reverse: 5’-GCAGAAAAGTCAGCCAGCCAGATT-3’ | NA |
| BCL11A qRT-PCR primer | Reverse: 5’-AACCCCAGCACTTAAGCAAA-3’ | NA |
| BCL11A qRT-PCR primer | Reverse: 5’-GGAGGTCATGATCCCCTTCT-3’ | NA |
| 18S qRT-PCR primer | Reverse: 5’-CCATCCAATCGGTAGTAGCG-3’ | NA |
| 18S qRT-PCR primer | Reverse: 5’-GTAACCCGTTGAACCCCATT-3’ | NA |
CNV Microarray Sample Preparation and Analysis
For copy number variation analysis, 100 ng of total genomic DNA was used to run CytoCGH_QCMT_4x array (Custom CGH & CGH+SNP Microarrays) according to the manufacturer’s protocol (Agilent Technologies). CytoGenomics software was used to analyze and generate the CNV figures.
Activity-Based Protein Profiling (ABPP)
Briefly, cell pellets were sonicated in IP/lysis buffer, desalted and then depleted of endogenous ATP with Zeba spin column, and incubated with 10 mM desthiobiotin-ATP probes at room temperature for 10 min. The labeled proteins were reduced, alkylated and trypsin digested at 37°C for 2 hr. The labeled peptides were purified with high capacity streptavidin agarose resin, washed, eluted and subjected to LC-MS/MS for peptide sequencing. The peptide identification and relative quantification were performed using MaxQuant software (Version 1.2.2.5). The procedures of ABPP were as detailed previously (Zhao et al., 2017). Fold-change distribution of the ATP binding proteome was performed by GraphPad software. GO enrichment analysis was performed on the ABPP profile using PANTHER classification system. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed on the increased protein kinases (comparing DTEP cells to parental cells) from 2 out of 3 cell lines. Raw data are available via ProteomeXchange with identifier PXD010193.
Gene Set Enrichment Analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed as described (Subramanian et al., 2005). The most differentially expressed genes ranked by Log2 fold change for each comparison were used to generate a signature for GSEA analysis. The input activated or inactivated SE gene sets were extracted from H3K27ac or RNAPII ChIP-seq data. GSEA estimates whether the members of DTEP-specific and THZ1-sensitive gene set are found at the top or bottom of the THZ1 treatment list, and if genes are specific to either parental or DTEP cells, indicating they are associated with a specific phenotype (i.e., DTEP or THZ1 treatment), rather than being distributed uniformly or randomly across the list. An enrichment score (ES) is calculated to quantify the degree to which a gene set is over-represented at the top or bottom of the entire ranked list. After calculation of the scores for a collection of gene sets, an empirical phenotype-based permutation test procedure is used to estimate p values. GSEA normalizes the ES for each gene set to account for the variation in set sizes, yielding a normalized enrichment score (NES) and a false discovery rate (FDR). The FDR gives an estimate of the probability that a set with a given NES represents a false positive finding; it is computed by comparing the tails of the observed and permutation-computed null distributions for the NES.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Unless otherwise stated, comparison and statistical significance between two groups in this paper are based on two-sided Student’s t-test. p values of less than 0.05 were considered significant. Data are shown with the mean ± SD of at least 3 experiments. Analysis of variance (ANOVA) or the Kruskal-Wallis test was used for comparing data from multiple groups.
Supplementary Material
Significance.
Drug-tolerant cell populations (DTEP) represent a reservoir of cancer cells that can survive targeted therapy or chemotherapy, which then expand and drive progression and relapse. BCL-2 family proteins have emerged as pivotal therapeutic targets for many B-cell lymphomas. Our studies identified select genetic alterations and non-mutational adaptive transcriptional responses as drug resistance mechanisms, where copy-number loss of BCL2 amplicons and CDK7-dependent transcriptional reprogramming contribute to the emergence of resistance to ABT-199, an oral BCL-2 inhibitor. Importantly, validated transcription-based combination therapy with ABT-199 prevents and overcomes drug resistance in B-cell lymphoma models and suggests a potential biomarker and strategy that could be used to prevent and override resistance in B-cell lymphoma patients treated with ABT-199.
Highlights.
Unified genetic and non-mutational mechanisms drive ABT-199 resistance in lymphoma
Rare lymphoma cells having BCL2 amplicon loss are selected during ABT-199 treatment
CDK7-dependent transcriptional reprogramming contributes to ABT-199 resistance
CDK7 inhibition prevents and overcomes ABT-199 resistance in B cell lymphoma models
ACKNOWLEDGMENTS
We sincerely thank members of the Tao, Qi, Cleveland, Sotomayor, and Shain laboratories for their support and comments, and the Proteomics, Comparative Medicine, Flow Cytometry, and Biostatistics and Bioinformatics Cores of the Moffitt Cancer Center, and the Genomics Core of Dana Farber Cancer Institute, for their outstanding service. This work was supported in part by grants from the National Cancer Institute CA179062, CA134807, and CA137123 (to J.T. and E.S.), P01-CA066996-19 and P50-CA100707-15 (to J.Q.), CA211336 (to G.G.W.), Cancer Center Support Grant P30-CA076292 to the Moffitt Cancer Center, by grants from the Lymphoma Research Foundation and Incyte Corporation (to J.T.), by an LLS-SCOR grant from the Leukemia and Lymphoma Society (to J.L.C.), and by the Cortner-Couch Chair for Cancer Research from the University of South Florida School of Medicine (to J.L.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2019.04.005.
REFERENCES
- (ANSI) (2012). Authentication of Human Cell Lines: Standardization of STR Profiling (ATCC Standards Development Organization (ATCC SDO)). [Google Scholar]
- Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, Yu L, Brown JR, Westerman D, Si EG, et al. (2016). The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 127, 3215–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bea S, Salaverria I, Armengol L, Pinyol M, Fernandez V, Hartmann EM, Jares P, Amador V, Hernandez L, Navarro A, et al. (2009). Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood 113, 3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional addiction in cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, et al. (2014). NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 56, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, Phillip J, Pera B, Yang SN, Takpradit K, et al. (2017). THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun 8, 14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceribelli M, Hou ZE, Kelly PN, Huang DW, Wright G, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL, Marcucci G, et al. (2016). A druggable TCF4-and BRD4-dependent transcriptional network sustains malignancy in blastic plasmacytoid dendritic cell neoplasm. Cancer Cell 30, 764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. (2014). CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, and Almasan A. (2015). MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 6, e1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. (2014). Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Esteve-Arenys A, Valero JG, Chamorro-Jorganes A, Gonzalez D, Rodriguez V, Dlouhy I, Salaverria I, Campo E, Colomer D, Martinez A, et al. (2018). The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 37, 1830–1844. [DOI] [PubMed] [Google Scholar]
- Fang B, Hoffman MA, Mirza AS, Mishall KM, Li J, Peterman SM, Smalley KS, Shain KH, Weinberger PM, Wu J, et al. (2015). Evaluating kinase ATP uptake and tyrosine phosphorylation using multiplexed quantification of chemically labeled and post-translationally modified peptides. Methods 81, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, and Martinez-Climent JA (2014). Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 123, 4111–4119. [DOI] [PubMed] [Google Scholar]
- Guikema JE, Amiot M, and Eldering E. (2017). Exploiting the proapoptotic function of NOXA as a therapeutic modality in cancer. Expert Opin. Ther. Targets 21, 767–779. [DOI] [PubMed] [Google Scholar]
- Hammerlindl H, and Schaider H. (2018). Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J. Cell Commun. Signal. 12, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. (2016). Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat.Med 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, and Young RA (2015). Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasili E, Burkert-Kautzsch C, Kieser A, and Strasser K. (2014). Degradation of DNA damage-independently stalled RNA polymerase II is independent of the E3 ligase Elc1. Nucleic Acids Res. 42, 10503–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, et al. (2014). An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet 46, 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, Liu F, Saunders LM, Mullally A, Abdel-Wahab O, et al. (2012). Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, and Fisher RP (2012). Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol 19, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverson JD, Sampath D, Souers AJ, Rosenberg SH, Fairbrother WJ, Amiot M, Konopleva M, and Letai A. (2017). Found in translation: how preclinical research is guiding the clinical development of the BCL2-selective inhibitor venetoclax. Cancer Discov. 7, 1376–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, Venteicher AS, Hebert CH, Carey CD, Rodig SJ, et al. (2017). Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, et al. (2003). Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A 100, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Shaffer AL 3rd, Ceribelli M, Zhang M, Wright G, Huang DW, Xiao W, Powell J, Petrus MN, Yang Y, et al. (2018). Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell 34, 286–297.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, Evans L, Ji W, Hsu CH, Thurley K, et al. (2016). Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun 7, 10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan PM, and Davies A. (2017). Current treatment of double hit and double expressor lymphoma. Hematology Am. Soc. Hematol. Educ. Program 2017, 295–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Bi C, Zhao X, Lwin T, Wang C, Yuan J, Silva AS, Shah BD, Fang B, Li T, et al. (2018). PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J. Clin. Invest 128, 5517–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruefli-Brasse A, and Reed JC (2017). Therapeutics targeting Bcl-2 in hematological malignancies. Biochem. J 474, 3643–3657. [DOI] [PubMed] [Google Scholar]
- Rusan M, Li K, Li Y, Christensen CL, Abraham BJ, Kwiatkowski N, Buczkowski KA, Bockorny B, Chen T, Li S, et al. (2018). Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 8, 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Izquierdo D, Buchonnet G, Siebert R, Gascoyne RD, Climent J, Karran L, Marin M, Blesa D, Horsman D, Rosenwald A, et al. (2003). MALT1 is deregulated by both chromosomal translocation and amplification in B-cell non-Hodgkin lymphoma. Blood 101, 4539–4546. [DOI] [PubMed] [Google Scholar]
- Schieber M, Gordon LI, and Karmali R. (2018). Current overview and treatment of mantle cell lymphoma. F1000Res 7, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Jacobson T, Meads M, Distler A, and Shain K. (2015). An organotypic high throughput system for characterization of drug sensitivity of primary multiple myeloma cells. J. Vis. Exp e53070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Silva MC, Sudalagunta P, Distler A, Jacobson T, Collins A, Nguyen T, Song J, Chen DT, Chen L, et al. (2017). An ex vivo platform for the prediction of clinical response in multiple myeloma. Cancer Res. 77, 3336–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijssen R, Slinger E, Weller K, Geest CR, Beaumont T, van Oers MH, Kater AP, and Eldering E. (2015). Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica 100, e302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, and Letai A. (2012). Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. (2015). CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, et al. (2017). Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-tefb complex. Cancer Discov. 7, 302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, Zhang L, Fu K, Bi C, Li J, et al. (2017). Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun 8, 14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li T, and Price DH (2012). RNA polymerase II elongation control. Annu. Rev. Biochem 81, 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.