

Abstract
Sepsis leads to a systemic immune response, and despite the progress of modern medicine, it is still responsible for a high mortality rate.
The immune response to sepsis is dependent on the innate and adaptive immune systems. The first line is the innate system, which requires complex and multiple pathways in order to eliminate the invading threats. The adaptive responses start after the innate response. The cell-mediated arm of CD4+ and CD8+ T and B cells is the main responsible for this response.
A coordinated cytokine response is essential for the host immune response. A dysregulated response can lead to a hyperinflammatory condition (cytokine storm). This hyperinflammation leads to neutrophils activation and may also lead to organ dysfunction. An imbalance of this response can increase the anti-inflammatory response, leading to compensatory anti-inflammatory response syndrome (CARS), persistent inflammation-immunsupression, catabolism syndrome (PICS), and, above all, an immune paralysis stat.
This immune paralysis leads to opportunistic infections, Candida species being one of the emerging microorganisms involved. The host immune response is different for bacterial or Candida sepsis.
Immune responses for bacterial and Candida sepsis are described in this paper.
Keywords: sepsis, immune response, host immune response, Candida sepsis, cytokine storm, PICS, CARS, ICU, immune paralysis
Introduction
Sepsis is the constellation of symptoms and signs occurring when an infection leads to a systemic inflammatory response, organ failure, and death. Despite progress in antibiotic therapy and in resuscitative strategies, sepsis remains the leading cause of death in the intensive care unit (ICU).
In a Rhee cohort study, in the United States of America in 2014, among 173,690 patients with sepsis, 54.7% required ICU care during hospitalization, with an associated mortality of 15%. Sepsis was present in 35% of all hospitalizations culminating in death [1]. In Europe, several studies showed that mortality due to sepsis is around 30%, higher than 50% of the cases of septic shock [2–5].
In Portugal, the community-acquired sepsis admitted to ICU (SACiUCI) study revealed that for 1 year, 778 patients had severe sepsis and septic shock, with an associated mortality of 33% [6]. In another study, the infection on admission to the ICU (INFAUCI) study, the associated mortality in septic shock was around 64.7% [6–8].
The sepsis mortality distribution has a trimodal pattern [9]: an initial early peak during the first hours/days due to inadequate fluid resuscitation and pathogen's hypervirulence, resulting in cardiac and pulmonary failure; a median peak several weeks after due to persistent organ injury; and a long-term peak, which occurs several months after the event [9].
In the third international consensus for the definitions for sepsis and septic shock (Sepsis-3), the term “severe sepsis” has been considered redundant, since it represents a misleading concept in which sepsis is considered to follow a linear continuum from the systemic inflammatory response syndrome (SIRS) through severe sepsis and septic shock. Instead, the consensus report recommends that sepsis should be defined as a life-threatening organ dysfunction caused by a dysregulated host response to an infection. The multi-organ dysfunction syndrome (MODS) can be defined as the development of potentially reversible physiologic derangement involving two or more organ systems not involved in the disorder resulting in ICU admission and arising in the wake of a potentially life-threatening physiologic insult. Moreover, septic shock is now defined as a subset of sepsis in which profound circulatory, cellular, and metabolic abnormalities are associated with a greater risk of mortality than sepsis alone [3].
The magnitude of the inflammatory response is a determinant for the sepsis outcome, since insufficient responses are not enough, whereas excessive inflammatory responses are self-destructive and may lead to microcirculatory dysfunction, causing tissue damage, MODS, and, ultimately, death. Although the pathogenicity of sepsis/septic shock involves specific (and complex) pathways, the inflammatory response to sepsis varies according to the specific causative organism, organism load, site of infection, host genotype, underlying host conditions (especially immunodepression), and the therapy used [3].
The innate immune system is the first line of the host defense for sensing and eliminating the invading microorganisms. It includes diverse cells, such as macrophages, dendritic cells (DC), neutrophils, natural killer cells, and innate lymphoid cells (ILC). The destruction and clearance of the invading pathogens and the resolution of other threats require a complex coordination of multiple innate immune pathways.
The cell-mediated arm of the adaptive immunity is orchestrated by CD4+ and CD8+ lymphocytes. Both CD4+ T-cells, also called ‘helper’, and CD4+CD25, also called ‘regulatory’ T (Treg) cells, are activated by antigen-presenting cells (APCs). The latter play a central role in controlling adaptive immunity. On the other hand, CD8+ lymphocytes or cytotoxic T-cells kill targeted infected cells, without intervention of APCs.
Two signals are required for T-cell activation: recognition of peptide antigen presented by major histocompatibility complex (signal 1) and co-stimulation through CD28, following binding to CD80 or CD86, expressed by APCs (signal 2). CD28 is the classic positive co-stimulatory receptor that, together with the T cell receptor (TCR), induces T cells to undergo proliferation and to produce cytokines such as interferon gamma (IFN-γ) and interleukin-2 (IL)-2, which are critical in controlling infection [10]. This T-cell activation is also crucial to immune paralysis phases as described later.
The adaptive immune response is normally quiescent and only comes into play after the innate response. Pathogen-specific antibodies, secreted by plasma cells that have differentiated themselves from activated B-cells, mediate the humoral element of adaptive immunity. These B-cells also differentiate into memory cells that remain quiescent but have the capacity for clonal expansion and the production of antibodies against the original pathogen, once the antigen is encountered in the future.
Sepsis may occur from any bacteria, as well as from fungal and viral organisms. Candida is an increasing cause of bloodstream infection, with significant mortality and morbidity rates [11]. The overall incidence of invasive candidiasis has increased fivefold in the past 10 years, becoming the fourth leading cause of nosocomial bloodstream infection in the United States, accounting for 8% of all bloodstream infections acquired in hospitals. Despite the availability of effective anti-fungal therapy, crude mortality in the last decade has remained high, ranging from 36 to 90% [12, 13].
In this review we will outline the immune system's role in sepsis progression, resolution, and long-term outcome, with special focus on the immune response to Candida sepsis.
Immune Dysfunction in Sepsis
Cytokines play a critical role in the host immune response to infection. A coordinated cytokine response is essential for creating an appropriate host immune response and for its resolution after the inflammatory trigger vanishes. Cytokines are essential for neutrophil activation, thrombin formation, and for the enhanced vascular permeability necessary for leukocyte extravasation into infected tissue. Serum proinflammatory cytokine levels peak within 2 h of cellular injury and return to almost undetectable levels within 6 h [14].
Imbalances of pro- and anti-inflammatory cytokines lie at the center of the MODS [15].
In fact, it is believed that a dysregulated cytokine response can lead to a hyperinflammatory condition called “cytokine storm”, which may lead to further organ injury [16].
Three decades ago, Mossman and Coffman suggested that CD4+ T cells differentiate into two subsets with reciprocal functions and patterns of cytokine secretion: termed T-helper 1 (Th1) and T-helper 2 (Th2, 17]. In 2005, a third T-cell subset, known as T-helper 17 (Th17), was identified [18, 19]. Th17 cells are characterized by the production of interleukin-17 (IL-17) and may evolve for the host protection against microbes for which Th1 or Th2 immunity are not suitable, such as extracellular bacteria and some fungi (Figure 1).
Figure 1.
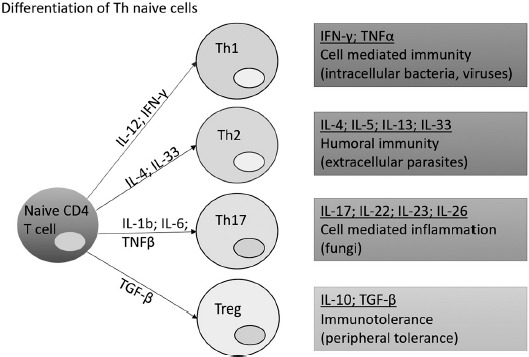
Differentiation of Th naïve cells. There are 4 subtypes of T-helper cells. For each Th activation, an induction of different immunities and associated cytokines of production is released
B lymphocyte cells (B cells) are vital for antibody production (Figure 2). There are specific B cell subtypes, regulatory B cells (Bregs), which are involved in the maintenance of homeostasis of the immune system and regulate inflammation in pathological situations. They act in synergy with T reg cells. Both cells have immunosuppressive properties, mainly through IL-10 production, and their functional impairment is associated with exacerbated and/or persistent inflammatory processes.
Figure 2.
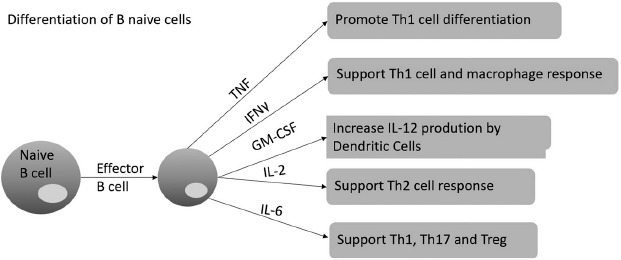
Differentiation of B naïve cells. In adaptive immune responses, activation of effector B cells induce the response of different Th response and cytokines
Anti-inflammatory cytokines (e.g., interleukin- 1 receptor A, IL-4, and IL-10) form an equally important part of the host's cytokine response to infection. Their function is to reduce the overall production of proinflammatory cytokines and to restore homeostasis once an infection is eradicated [20].
Regarding proinflammatory cytokines, a dysregulated and hyperactive anti-inflammatory response can also harm the host. Host responses during sepsis have revealed that the late phase of sepsis is dominated by a state of immune suppression. An over-exuberant IL-10 response, for example, is central to the occurrence of immunoparalysis after sepsis, and major surgery puts the host under a significant risk of developing secondary infections [21].
In the case of overwhelming compensatory anti-inflammatory response syndrome (CARS), the phenomenon of immune paralysis occurs due to immune cell apoptosis and functional impairment of lymphocytes and phagocytes, also associated with increased anti-inflammatory and decreased pro-inflammatory cytokine production. The overshooting of the anti-inflammatory response predisposes the host to secondary bacterial infection and infections to opportunistic microorganisms [22].
Besides lymphocytes, macrophages also react to external organism so as to defend the host. Macrophages represent an integral compartment of the innate immune system and are critical regulators of both normal homeostasis and pathology. Upon taking up residence in tissues, macrophages adopt either a proinflammatory M1 phenotype, or an anti-inflammatory M2 phenotype [23]. M1 represents the “classically-activated” proinflammatory phenotype of macrophages, being capable of eliminating pathogens and cells infected by viruses or that have been transformed. They produce cytokines and inducible nitric-oxide synthase (iNOS), which provides effector molecules for microbicidal activities that lead to oxidative stress, which can inhibit the proliferation of nearby cells. M2 represents an “alternatively” activated phenotype with anti-inflammatory activities that can promote wound healing [16].
Hyperinflammation
Cytokine storm syndromes are triggered by infection, and sepsis remains the most common catch-all term used to categorize these conditions. Clinical manifestations of a cytokine storm may include refractory fever, cytopenia, coagulopathy, MODS, and death [24].
The host response to bacterial and Candida infection initially occurs with its recognition by the immune system through pathogen-associated molecular patterns (PAMPs) [25].
PAMPs are expressed by both pathogenic and harmless microbes and are recognized by pathogen sensors known as pattern recognition receptors (PRRs) on immune cells. The leading families of PRRs are Toll-like receptors (TLRs), C-type lectin (CLEC)-like receptors, retinoic acid-inducible gene 1 (RIG-I)-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors, cytosolic DNA sensors, and inflammasomes. The interplay between these recognition molecule families ensures the efficient coordination of innate immune responses, either through synergistic or cooperative signaling [26].
a) Immune Response to Bacterial Sepsis
Toll-like receptors (TLRs) are transmembrane receptors present in APCs (such as macrophages or dendritic cells) that specifically recognize PAMPs present in viruses, in gram-positive bacteria (lipoteichoic acid [LTA]), in gram-negative bacteria (lipopolysaccharide [LPS]), and in Candida. They are located in the cellular membrane (TLR1, TLR2, TLR4, TLR5, and TLR6) and in intracellular membranes (TLR3, TLR7, TLR8, and TLR9), recognizing both PAMPs at the cell surface or in the cytosol, respectively. Based on their primary sequences, TLRs can be further divided into several subfamilies, each one recognizing related PAMPs [27].
Lipoproteins or lipopeptides in gram-positive bacteria are recognized by TLR2 complexed with TLR1 or TLR6, viral double-stranded RNAs by TLR3, LPS by TLR4/MD2 complex, bacterial flagellins by TLR5, viral and bacterial single-stranded RNAs by TLR7 or TLR8, and CpG-rich undermethy-lated DNAs by TLR9 [28]. The activation of most TLRs by ligand binding results in dimerization, conformational changes, and downstream signaling involving receptor/adaptor complexes, culminating with the translocation of nuclear factor (NF)-κB and/or interferon regulatory factor (IRF) 3 to the nucleus, which is followed by proinflammatory cytokine and type I interferon (IFN) transcription, respectively [29]. Most importantly, the prompt production of proinflammatory mediators is accompanied by the production and release of anti-inflammatory mediators [30].
After the antigen recognition by APC, these cells produce several cytokines (such as IL-1, IL-6, and tumor necrosis factor-α [TNF-α]) in order to control the aggression and to stimulate both innate and adaptive immune mechanisms.
Understanding the cytokine profile in patients with sepsis may be very useful for its diagnosis, as well as for the assessment of its severity, and the prediction of mortality, allowing a better patient treatment. A multiplex analysis evaluating plasma levels of 17 cytokines in patients with severe sepsis revealed that the concentrations of IL-1β, IL-6, interleukin-7, IL-8, IL-10, IL-13, IFN-γ, monocyte chemoattractant protein-1 (MCP-1), and TNF-α were significantly higher in patients with septic shock than in those with severe sepsis. Moreover, specific cytokine profiles were associated with the severity of sepsis, evolution of organ failure, and death [31].
In severe lung infections, IL-1 is a key cytokine driving proinflammatory activity in bronchoalveolar lavage fluid of patients with lung injury. The acute-response cytokines TNF and IL-1 and the chemotactic cytokines IL-8 and MCP-1 appear in the early minutes to hours after infection, followed by a more sustained increase in IL-6. Serum concentration of IL-6 in peripheral blood has been used to assess the intensity of systemic cytokine responses in patients with sepsis, as IL-6 production is stimulated by TNF and IL-1, providing an integrated signal of these two early-response cytokines [32]. In fact, serial evaluations of IL-6 and TNF-α in serum of patients with sepsis on days 1, 3, and 7 after admission showed that IL-6 levels were reduced from day 1 to 7 in the survivor group and that the TNF-α level was significantly lower on day 1 in the non-survivor group. These results suggest that a decreasing trend in IL-6 values is associated with a better prognosis in sepsis [32]. It has been reported that IL-10 paralleled the sepsis score and increased serum IL-10 and TNF-α levels, and a high IL-10 to TNF-α ratio was associated with death [33]. These results demonstrate that the sustained overproduction of the anti-inflammatory cytokine IL-10 is the main predictor of severity and fatal outcome.
Interleukin-17 is often associated with early and protective host responses to bacteria, including Staphylococcus aureus and Streptococcus pyogenes. IL-17 is a potent inducer of systemic inflammation, boosting the production or activation of inflammatory cytokines (e.g., TNF-α, IL-1b, and IL-6), chemokines, matrix metalloproteases, and transcription factors in both hematopoietic and nonhematopoietic cell types. IL-17 also acts synergistically with other inflammatory cytokines, including TNF-α, IL-1β, and IFN-γ. The rapid secretion of IL-17 not only has inflammatory effects on a wide variety of cell types throughout the body but it can also directly induce and synergize with other cytokines that are produced in the same timeframe [34]. Although IL-17 is the archetypal cytokine of the CD4+ Th17 cell lineage, it can also be quickly produced by innate-like T lymphocytes, such as γδ T cells, invariant natural killer T cells and mucosa-associated invariant T (MAIT) cells [35].
Th1 and Th2 cells are known to antagonize each other, and not surprisingly, IL-12, IFN-γ, and IL-4 can inhibit Th17 differentiation [36]. IL-17, however, does not appear to inhibit Th1 or Th2 differentiation; therefore, Th1 and Th2 cells typically master Th17 cells.
When a naïve CD4+ T cell is activated, the local cytokine milieu plays an important role in determining in which effector lineage the naïve T cell will be transformed, by inducing lineage-specific transcription factors. Naïve T cells stimulated in the presence of IL-12 become Th1 cells and express the transcription factor T-bet, while those stimulated in the presence of IL-4 become Th2 cells and express the transcription factor GATA-3 [37]. IL-23 can upregulate IL-17 in memory T cells but cannot act on naïve T cells to induce Th17 differentiation, because it is only expressed on T cells after activation. The key to Th17 differentiation in mice is the combination of transforming growth factor-β (TGF-β) and IL-6 [14]. In addition, TNF-α and IL-1β can further enhance Th17 differentiation in mice, but only in the presence of TGF-β and IL-6 [17, 38]. IL-6 and IL-23 induce a small amount of IL-17 alone and greatly enhance Th17 differentiation in the presence of IL-1β.
IL-6, IL-21, and IL-23 act sequentially: first IL-6 upregulates IL-21, then both IL-6 and IL-21 upregulate IL-23 receptor, and finally, IL-23 appears to upregulate effector function and pathogenicity in Th17 cells [39]. Th17 cells are characterized by the production of IL-17, but they also produce another inflammatory cytokines, such as IL-22 and interleukin-26. A subset of Th17 cells co-expresses IFN-γ, and other co-expresses IL-10 [40].
b) Immune Response to Candida Sepsis
During Candida infection, the (opportunistic) pathogen itself uses self-protective strategies, counteracting host immune responses provided by different immune cells subsets.
Candida species are a dimorphic fungus. The morphologic switch between the yeast phase and hyphal phase is one of the main virulence factor of Candida ssp. Moreover, hyphal formation is required for an invasive and aggressive form.
In Candida's cell wall, two layers can be distinguished: the outer layer is mainly composed of O- and N-linked glycoproteins that consist of 80–90% mannose, whereas the inner cell wall contains the skeletal polysaccharides chitin, β-1,3-glucan, and β-1,6-glucan, which confer strength and shape to the cells. These polysaccharide structures, which have been reported to differ between yeasts and hyphae, represent the main PAMPs recognized by host PRRs during an encounter with the fungus [41]. Chitin from Candida albicans has been suggested to induce the production of IL-10 via a nucleotidebinding oligomerization domain-containing protein 2 (NOD2)-dependent mechanism and, in this way, may contribute to dampening pro-inflammatory host responses during fungal infection [42].
Pyroptosis is a damage mechanism that has been identified in the damage of macrophages by Candida albicans [43]. It is an inflammatory programmed cell death that depends on the NLRP3, ASC, and caspase 1-proteins. These proteins form an inflammasome protein complex that activates caspase-1, allowing it to cleave gasdermin D, resulting in a gasdermin fragment that can form pores in the host cell membrane [44]. There are multiple stages of the macrophage-Candida interaction that need to occur for pyroptosis to be activated. The first step for macrophages is to recognize and phagocytize the fungal cell. A key host receptor that mediates phagocytosis is dectin-1. Dectin-1 can interact with TLR2 and can induce intracellular signaling via SYK- and RAF1-dependent pathways [45]. Within the phagosome, the fungus encounters limited nutrients, alterations in osmolarity, and antimicrobial defense mechanisms such as oxidative stress. Third, both the fungus and the host need to initiate the transcriptional programs that enable pyroptosis.
On the host side, macrophages need to prime the inflammasome by increasing transcription of the inflammasome components, such as NRLP3 and IL-1β.
Using opsonization or unopsonization, polymorphonuclear cells have two distinct mechanisms for killing Candida. Unopsonized Candida albicans is recognized via CR3 and is CR3 and CARD9-dependent [46]. For this, recognition receptor dectin-1 is not required. In contrast, opsonized C. albicans is recognized via FcγR, and PKC and NADPH oxidase activity constitute the main killing elements. Combinations of killing mechanisms are in play, which occur independently of pattern recognition receptors like TLR and dectin-1 dominating the activation of monocytic cells, and that can compensate for each other under deficiency conditions [47]. A new cluster of C-type lectin receptors (CLRs) involved in antifungal immunity was characterized recently, and members of this family include clec6a (dectin-2), clec4d (MCL), and clec4e (macrophage-inducible C-type lectin or Mincle) [48]. Dectin-2 – the prototypical member of this family – contains 1 CRD in the extracellular domain, which can recognize α-mannans and O-linked mannoproteins in a Ca2+-dependent manner. This interaction can modulate cytokine and reactive oxygen species (ROS) production, as well as fungal killing [49]. All the receptors of this family together with the immunoreceptor tyrosine-based activation motif (ITAM)-containing Fc receptor γ-chain (FcRγ) chain combine to drive intracellular signaling via the Syk-CARD9 pathway. Signaling through these receptors can often synergize or antagonize other CLRs, including dectin-1, TLRs, and inflammasomes [50]. In vivo, dectin-2 has been shown to exert a protective role against Candida albicans, Candida glabrata, and Trichophyton rubrum infections, and, together with MCL, they have been suggested to promote Th17 responses [51].
Neutrophils use oxidative and nonoxidative killing mechanisms against fungal pathogens. Oxidative mechanisms include ROS production, mediated by enzymes NADPH oxidase and myeloperoxidase, while nonoxidative mechanisms include the release of granules containing proteins with antimicrobial and degradative properties, including defensins, lysozyme, lactoferrin, gelatinases, elastase, and cathepsin-G [52].
Among the most prominent mechanisms, we can find the rapid formation of reactive oxygen intermediates (ROI) termed ‘oxidative burst’. Upon activation, the neutrophilic NADPH oxidase-complex is assembled on the cytoplasmic membrane to release superoxide into the extracellular space, or on the phagosomal membrane to release oxidants into phagosome [46]. Aside from inducing oxidative stress, ROI are required for the formation of the so-called neutrophilic extracellular traps (NETs) [53]. NET formation seems to be CR3 dependent and ROI independent in tissues [54]. NETs provide a barrier past, which a pathogen cannot easily pass, and instead becomes entangled in a mesh of cytotoxic compounds. These are structures formed of released neutrophil chromatin, decorated with anti-microbial substances, mainly calprotectin, which are normally stored within neutrophilic granules and can be formed within 10 min of activation. NETs have been shown to entrap free bacteria in the bloodstream and therefore prevent dissemination in an Escherichia coli model of sepsis [55]. They may form upon direct contact with a pathogen, including Candida albicans, after which both filamentous and yeast forms are trapped and killed. The relevance of NETs to Candida sepsis may be suggested by increased susceptibility of mice deficient in calprotectin, a key component of NETs, to systemic candidiasis [46, 56].
Together, these signaling pathways induce the secretion of cytokines and chemokines and initiate phagocytosis to clear Candida infections [57].
In Candida infections, different TLRs can activate specific arms in collaboration with CLRs such as dectin-1 and man-nose receptors (Figure 3). Intracellular TLRs, including TLR3, TLR7, and TLR9, are involved in antifungal immunity. Aspergillus fumigatus can activate the phagocytosis-dependent TLR9/BTK/calcineurin/NFAT signaling pathway, leading to the production of proinflammatory cytokines, such as TNF-α, neutrophil recruitment, and fungal clearance [58]. There is some controversy regarding the role of TLR7 in fungal infection. Phagocytosis of Candida glabrata by DCs induces a deleterious TLR7-dependent type-I IFN response that promotes fungal persistence in host tissues. Another study showed that mice lacking TLR7 were highly susceptible to systemic Candida albicans infection, as TLR7 was required on DCs for sensing Candida-derived RNA and subsequent IL-12 production [59].
Figure 3.
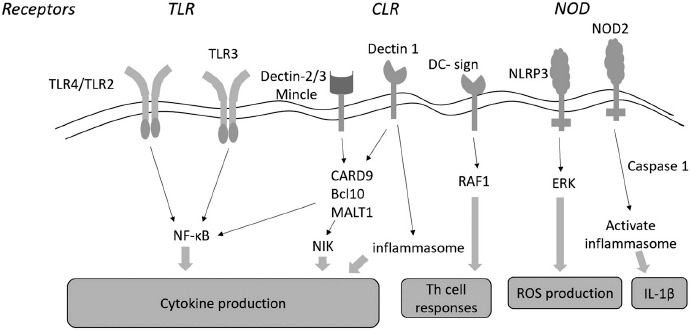
Recognition of Candida species is mediated by Toll-like receptors (TLRs), C-type lectin receptors (CLRs) and nucleotide-binding oligomerization domain (NOD) receptors. Both TLR4/TLR2 can induce production of proinflammatory signals and cytokines through the MyD88-dependent NF-κ B pathway. However, TLR3-mediated recognition is carried out through activation of the IRF3 transcription factor. The CLR receptors, such as dectin-1, dectin-2, dectin-3, and Mincle, stimulate the release of inflammatory cytokines by activating T cell lineage-specific tyrosine kinase (Syk) and downstream complex (CARD9-Bcl10-MALT1). Dectin-1 promotes maturation of inflammasome, so as to induce production of cytokines. DC-specific intracellular adhesion molecule-grabbing non-integrin (DC-SIGN) receptor can induce TH cell responses through Raf1 activation. In addition, NLRP3 stimulates reactive oxygen species (ROS) production through an Erk dependent pathway, and NOD2 receptor activates inflammasome by affecting maturation of IL-1β via caspase-1
DCs exposed to fungal pathogens exhibit augmented expression of proinflammatory cytokines and activation markers, which results in their migration to local lymph nodes, where they can stimulate naïve T cells toward distinct effector T-cell subsets or induce tolerance. Dectin-1-mediated activation of plasmacytoid DCs has been suggested to promote Th2 responses, while dectin-1 activation on myeloid DCs decreases Th2 responses upon β-glucan challenge [60]. Specific DC subsets have also been shown to act in a complementary fashion during fungal infections, so as to induce appropriate T-cell responses. Tissue-resident Flt3L-dependent DCs and CCR2-dependent monocyte-derived DCs can collaborate in antigen presentation and T-cell priming during mucosal candidiasis [61]. On the other hand, TNF-α from monocyte-derived DCs induces neutrophilia versus eosinophilia during persistent fungal infection in the airways, by modulating the balance between IL-17 and interleukin-5 production [62]. The ability of DC to shape the type of effector T-cell responses depends on several factors, including the nature of fungal organisms, the site of infection, and the host susceptibility. Fungal morphology can also determine T-helper cell differentiation. Yeast forms of Candida albicans can induce Th17 responses through dectin-1 interaction on Langerhans cells and subsequent IL-6 production. Conversely, filamentous forms induced Th1 responses due to the absence of dectin-1 ligation [63].
During Candida infections, releases of TNF-α, IL-6, IL-12, and IL-1β are stimulated via several pathways, whereas the secretion of IL-23 and IL-17 depend on the stimulation by IL-23 rather than through lectin receptors. Th17 cells secrete special cytokines, including IL-17A, IL-17F, IL-21, and IL-22. Cytokines IL-1β, IL-6, and TGF-β induce the development of Th17 cells, while IL-23 is an essential cytokine for the maintenance of Th17 cells. Among the abovementioned cytokines, the serum TGF-β and IL-23 levels were found to have increased and to be able to differentiate patients with candidemia from patients with bacteremia. On the other hand, IL1-β levels were only higher in comparison with healthy control subjects (not concerning the comparison between Candida and bacterial infections) [15]. The protective role of Th17 responses in the antifungal host defense was first established in IL-17 receptor-deficient (IL-17RA) mice, which showed increased susceptibility to a disseminated Candida albicans infection. It has been demonstrated that IL17RA mice showed an increased mortality and higher fungal loads in kidneys in a model of disseminated candidiasis, which was partially caused by a lower neutrophil recruitment in the IL-17RA mice [64]. However, in one study of IL-17RA mice, it was suggested that IL-22 mediates protection, whereas IL-17A contributes to disease susceptibility [65]. All these observations strongly indicate that Th17 responses are important, not only for systemic but also for mucosal host defenses against Candida infections in humans. Heather et al. showed that IL-17, IL-23, and TGF-β serum levels were significantly increased in candidemia when compared with bacteremia. Conversely, the specificity of IL17 is higher than the specificities of IL-23 and TGF-β in patients with candidemia [66]. In addition, several studies have shown that IL-23 and IL-17 play an important role in the development of the inflammatory response against the infectious agent [67].
Immune Suppression
Immune resolution often starts about 3–4 days after septic shock onset. Homeostasis, as a status of balance among the different mechanisms between cell proliferation and cell death, is the final goal, which sometimes is not achieved. With the advances in modern medicine, the introduction of supportive therapies leads to a decrease in the mortality rate in the first phase of sepsis (hyperinflammation). However, immune inbalance in septic patients is also a path, and the immunosuppressed phase is the consequence.
This immunosuppressed phase can progress to an upsurge in the release of anti-inflammatory cytokines that can progress to a state of a quite profound immune suppression, the so-called CARS. This syndrome contributes to the increased risk of secondary and opportunistic infections, leading to late deaths associated with sepsis and septic shock. A decrease of the total lymphocyte count and the reactivation of latent virus and/or bacterial infections are indicative of this immunosuppression phase. With this status of immunity, malnutrition, changes in microbiota, and loss of natural barriers, the risk for nosocomial infections increases. Moreover, reactivation of latent microorganisms, re-infections, and amplification of inflammation lead to a suppression of the adaptive immunity [67, 68] (Figure 4).
Figure 4.
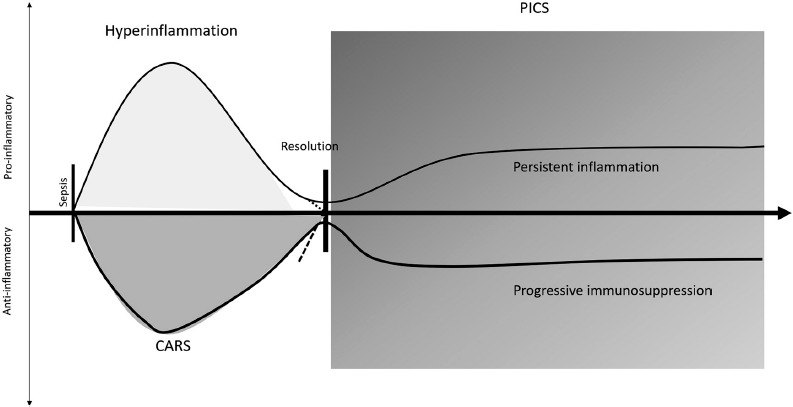
After sepsis, an hyperacute phase of hyperinflammation starts. At the same time, an immunosuppression response begins. Following this response, critically ill patients can return to homeostasis and have an immune resolution and recovery, or develop persistent inflammationimmunsupression and catabolism syndrome (PICS), resulting in persistent inflammation and progressive immunosuppression
This immunosuppressed state is characterized by the production of inhibitory cytokines such as IL-10 and IL-4, which limit the intensity of immune cell activation and negatively modulate inflammation.
IL-10 is an anti-inflammatory cytokine produced by several different cell types (Th2 cells, Tregs, and Bregs). Nevertheless, Th1 cells have also been found to secrete IL-10 under certain conditions. TGF-β and IL-6 synergize to induce IL-17 production, also synergize to induce IL-10.
IL-10 produced by Th17 cells may have an important protective function by limiting inflammation and tissue damage normally caused by IL-17 [69]. Alike Th subsets, B cells subsets can also be distinguished by distinct cytokines' secretion profile. An example is the subset characterized by upregulation of CD1+ expression and enhanced production of IL-10 [70].
After CARS, there is another recently defined immunosupression phase that occurs when patients survive their initial sepsis insult and become chronically critically ill – the so-called “persistent inflammation-immunsupression and catabolism syndrome” (PICS) [67]. PICS can be defined as an inflammatory persistent status with organ failure, with the need of suportive therapy, as well as an augmentation of protein catabolism (leading to dificult assessement of nutrition needs) and compromised wound healing.
PICS can be identified in critically ill patients with admission in ICU ≥ 10 days, with persistent inflammation defined as high C-reactive protein concentration and low level of retinol binding protein, immunosupression characterized by a low lymphocyte count, and a catabolic state defined by serum albumin, creatinine height index, and weight loss or low body mass index during hospitalization [71].
Defects in antigen-presenting cell function, including endotoxin tolerance and impaired cytokine production, reduce the ability of APCs to stimulate lymphocyte driven immune functions following sepsis. The apoptosis of lymphocytes and APCs (dendritic cells, macrophages, and B cells) is considered a hallmark of septic immune suppresion [72, 73]. Autophagy provides a way to eliminate DAMPs and PAMPs by packaging pathogen components, damaged organelles, and cellular proteins into vesicles targeted for lysosomal degradation, resulting in reduced inflammation and cellular activation. Other late features of septic shock are the expansion of regulatory T cells and myeloid suppressor cells, Th2 cell polarization, and reprogramming of macrophages to an M2 phenotype, all of which contribute to decreasing inflammation and impaired phagocytosis [74]. In addition to a diminished innate immune function, adaptive immunity is similarly impaired. Splenocytes collected from deceased patients with sepsis exhibited reduced numbers of CD4+ and CD8+ lymphocytes that emanated from substantial apoptosis. Reduced lymphocyte numbers do not just reflect the risk for viral reactivation following sepsis. In fact, lymphopenia, 4 days after sepsis onset, is associated with the development of secondary infection and is predictive of long-term mortality, 1 year after sepsis [75].
Sepsis-induced immune suppression is mediated by lymphocyte exhaustion, increased rates of B and T cell apoptosis, and immunoparalysis (or endotoxin tolerance) [76].
An important cytokine in immunoparalysis is IL-33, an IL-1 cytokine family member. Endogenous IL-33, released in response to severe tissue damage, activates ILC2s, which produce IL-4 and IL-13 that drive M2 polarization of macrophages, resulting in the expansion of Treg cell population via the production of IL-10. However, IL-33-induced IL-10-secreting M2 macrophages increase the expansion of Treg cell population, therefore contributing to the development of long-term sepsis-induced immunosuppression [67, 69].
CD28 is a major T cell costimulatory receptor, and the coengament of which can prevent anergy and cell death. The CD28 receptor is a member of a complex family of polypetides that includes at least two receptors and two ligands. Cytotoxic lymphocyte-associated molecule-4 (CTLA-4, CD 152) is the second member of the CD28 receptor family. The ligands or counterreceptors for these two proteins are the B7 family members, CD80 (B7–1) and CD86 (B7–2) [77].
Anergic T cells have limited effector function. CTLA-4 expression interferes with T-cell activation by reducing CD28 costimulation. CTLA-4 is structurally similar to CD28 and binds with CD80 and CD86 at a higher affinity than CD28. CTLA-4, through the binding with CD80/86, limits costimulation through CD28, leading to T-cell anergy. Anti-CTLA-4 neutralizing antibodies inhibit CTLA-4 binding to CD80/86, promoting T-cell activation [74].
The characterization of the “paralyzed monocyte” by studing HLA-DR or CD80/CD86 expression on CD 14+ is now a reality in laboratory test. The identification of T-cell suppressor molecules is probably the future of immunoparalysis diagnosis [71].
Overall, these specific cellular alterations coalesce into a chronic state of immune suppression, characterized by persistent, recurrent, secondary, and nosocomial infectious complications often resulting in hospital readmissions and poor long-term survival.
Conclusion
Immune responses to bacterial and Candida sepsis are complex and involve several players. More importantly, the balance between protective immunity and harmful hyper-inflammation is hard to define and several protective inflammatory reactions have also been shown to contribute to the sepsis pathology. The understanding of these mechanisms may offer new insight into the pathophysiology of these infections, as well as open new avenues for news tests that may allow early diagnosis for bacterial and fungal sepsis.
The knowledge acquired from the characterization of the immune response in immunosuppression and immunoparalysis states, could lead to different forms of management septic patients.
As the immune response in sepsis comprise several immune markers, a multiparameter test to evaluate several cytokines at the same time, in combination with other clinical risk scores, is likely to provide a more accurate diagnosis.
The combination of these eventual tests could help clinics to start an early goal-directed immune modulatory therapy and provide optimal clinical benefit to those patients, who survive after initial sepsis.
Footnotes
Abbreviations
APC: antigen-presenting cell
B cells: lymphocyte B
Breg: regulatory lymphocyte B cells
CARD9: CARD9 gene
CARS: compensatory anti-inflammatory response syndrome
CD: cluster of differentiation
CLEC: C-type lectin
CLR: C-type lectin receptors
CRD: carbohydrate recognition domain
CTLA-4: cytotoxic lymphocyte-associated molecule-4
DC: dendritic cells
DNA: deoxyribonucleic acid
FcRγ: Fc receptor gamma-chain
HLA-DR: Human leukocyte antigen – DR
ICU: intensive care unit
IFN: interferon
IFN-γ: interferon-gamma
IL: interleukin
ILC: innate lymphoid cells
iNOS: inducible nitric-oxide synthase
IRF: interferon regulatory factor
LPS: lipopolysaccharide
LTA: lipoteichoic acid
M1: macrophage 1 phenotype
M2: macrophage 2 phenotype
MCP-1: monocyte chemoattractant protein-1
Mincle: macrophage-inducible C-type lectin
MODS: multi-organ disfunction syndrome
NADPH: nicotinamide adenine dinucleotide phosphate
NET: neutrophil extracellular traps
NF: nuclear factor
NLRP3: NLRP3 gene
NOD: nucleotide-binding oligomerization domain
NOD2: nucleotide-binding oligomerization domain 2
PAMP: pathogen-associated molecular patterns
PICS: persistent inflammation-immunosuppression and catabolism syndrome
PRR: pattern recognition receptors
RIG-1: retinoic acid-inducible gene 1
RNA: ribonucleic acid
ROI: reactive oxygen intermediates
ROS: reactive oxygen species
SIRS: systemic Inflammatory Response Syndrome
T cells: lymphocyte T
TGF-β: transforming growth factor-beta
Th1: lymphocyte T helper 1 cells
Th2: lymphocyte T helper 2 cells
Th17: lymphocyte T helper 17 cells
TLR: Toll-like receptors
TNF: tumor necrosis factor
TNF-α: tumor necrosis factor alfa
Treg: regulatory T cells
Funding Sources
Nothing to declare.
Authors' Contributions
P.P. conceptualized and designed the paper. P.P., J.A.P., and L.M.B. drafted and critically revised the manuscript and approved its final version.
Conflict of Interest
The authors declare no conflict of interest
References
- 1.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA [Internet]. 2017. October 3;318(13):1241 Available from: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.2017.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleischmann C, Thomas-Rueddel DO, Hartmann M, Hartog CS, Welte T, Heublein S, et al. Hospital Incidence and Mortality Rates of Sepsis: An Analysis of Hospital Episode (DRG) Statistics in Germany From 2007 to 2013. Dtsch Aerzteblatt Online [Internet]. 2016. March 11;113(10):159–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27010950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA [Internet]. 2016. February 23;315(8):801 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26903338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving Sepsis Campaign. Crit Care Med [Internet]. 2017. March;45(3):486–552. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28098591. [DOI] [PubMed] [Google Scholar]
- 5.Bassetti M, Merelli M, Righi E, Diaz-Martin A, Rosello EM, Luzzati R, et al. Epidemiology, species distribution, antifungal susceptibility, and outcome of candidemia across five sites in Italy and Spain. J Clin Microbiol [Internet]. 2013. December;51(12):4167–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24108614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardoso T, Carneiro AH, Ribeiro O, Teixeira-pinto A, Costa- A. Reducing mortality in severe sepsis with the implementation of a core 6-hour bundle : results from the Portuguese community-acquired sepsis study (SACiUCI study). Crit Care. 2010;14(R83). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonc J. Impact of infection on admission and of the process of care on mortality of patients admitted to the Intensive Care Unit : the INFAUCI study. Clin Microbiol Infect. 2014;20:1308–15. [DOI] [PubMed] [Google Scholar]
- 8.Pereira JM, Gonçalves-Pereira J, Ribeiro O, Baptista JP, Froes F, Paiva JA. Impact of antibiotic therapy in severe community-acquired pneumonia: Data from the Infauci study. J Crit Care [Internet]. 2018. February 1;43:183–9. Available from: https://www.sciencedirect.com/science/article/pii/S088394411730480X?via%3Dihub. [DOI] [PubMed] [Google Scholar]
- 9.Delano MJ, Ward PA. The Immune System’s Role in Sepsis Progression, Resolution and. Immunol Rev. 2017;274(1):330–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol [Internet]. 2013. April;13(4):227–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23470321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfaller MA, Diekema DJ, Turnidge JD, Castanheira M, Jones RN. Twenty Years of the SENTRY Antifungal Surveillance Program: Results for Candida Species From 1997–2016. Open Forum Infect Dis [Internet]. 2019. March 15;6(Supplement_1):S79–94. Available from: https://academic.oup.com/ofid/article/6/Supplement_1/S79/5381621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzman JA, Tchokonte R, Sobel JD, Guzman Guzman JA, Tchokonte R, et al. Septic Shock Due to Candidemia: Outcomes and Predictors of Shock Development. J Clin Med Res [Internet]. 2011. April 4;3(2):65–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21811532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clancy CJ, Nguyen MH. The end of an era in defining the optimal treatment of invasive candidiasis. Clin Infect Dis. 2012;54(8):1123–5. [DOI] [PubMed] [Google Scholar]
- 14.Bonavia Anthony, Groff A, Karamchandani Kunal, Singbartl K. Clinical Utility of Extracorporeal Cytokine Hemoadsorption Therapy: A Literature Review. Blood Purif. 2018;46:337–49. [DOI] [PubMed] [Google Scholar]
- 15.Akin H, Akalin H, Budak F, Ener B, Ocakoğlu G, Gürcüoğlu E, et al. Alterations of serum cytokine levels and their relation with inflammatory markers in candidemia. Med Mycol [Internet]. 2015. April 1;53(3):258–68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25627661. [DOI] [PubMed] [Google Scholar]
- 16.Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol [Internet]. 2017. July 29;39(5):517–28. Available from: http://link.springer.com/10.1007/s00281-017-0639-8. [DOI] [PubMed] [Google Scholar]
- 17.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol [Internet]. 1986. April 1;136(7):2348–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2419430. [PubMed] [Google Scholar]
- 18.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol [Internet]. 2005. November 2;6(11):1123–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16200070. [DOI] [PubMed] [Google Scholar]
- 19.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol [Internet]. 2005. November 2;6(11):1133–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16200068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget [Internet]. 2018. January 23;9(6):7204 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29467962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Xu MM, Wang K, Adler AJ, Vella AT, Zhou B, et al. Macrophage polarization and Metainflammation HHS Public Access. Transl Res [Internet]. 2018;191:29–44. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5776711/pdf/nihms922278.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arens C, Bajwa SA, Koch C, Siegler BH, Schneck E, Hecker A, et al. Sepsis-induced long-term immune paralysis - results of a descriptive, explorative study. Crit Care [Internet]. 2016;20(1):10–3. Available from: 10.1186/s13054-016-1233-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep [Internet]. 2014;6:13 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24669294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG, et al. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev [Internet]. 2012;76(1):1092–2172. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3294426/pdf/zmr16.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson JP, Moyes DL. Adaptive immune responses to Candida albicans infection. Virulence. 2015;6(4):327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agier J, Pastwińska J, Brzezińska-Błaszczyk E. An overview of mast cell pattern recognition receptors. Inflamm Res [Internet]. 2018. September;67(9):737–46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29909493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses [Internet]. 2011;3(6):920 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3186011/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roveran K, Shimada T, Boyd H, Walley R, Genga KR, Shimada T, et al. The Understanding and Management of Organism Toxicity in Septic Shock. J Innate Immun [Internet]. 2018;6(5–6):1–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29763894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kopitar-Jerala N. Innate Immune Response in Brain, NF-Kappa B Signaling and Cystatins. Front Mol Neurosci [Internet]. 2015. December 9;8:73 Available from: http://journal.frontiersin.org/Article/10.3389/fnmol.2015.00073/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Buechler NL, Woodruff AG, Long DL, Zabalawi M, Yoza BK, et al. Sirtuins and Immuno-Metabolism of Sepsis. Int J Mol Sci. 2018;19(2738):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, et al. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care [Internet]. 2007;11(2):R49 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17448250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus Anti-inflammatory Cytokine Profile in Patients with Severe Sepsis: A Marker for Prognosis and Future Therapeutic Options. J Infect Dis [Internet]. 2000. January 1;181(1):176–80. Available from: https://academic.oup.com/jid/article-lookup/doi/10.1086/315214. [DOI] [PubMed] [Google Scholar]
- 33.Mansour Haeryfar Welch SM, Young HA, Singh B, Kyoungok Kim JK, DB O, Hess DA, et al. Rapid and Rigorous IL-17A Production by a Distinct Subpopulation of Effector Memory T Lymphocytes Constitutes a Novel Mechanism of Toxic Shock Syndrome Immunopathology. J Immunol [Internet]. 2017;198:2805–18. Available from: http://www.jimmunol.org/content/198/7/2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect [Internet]. 2013. September;2(9):e60 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26038490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Netea MG, Joosten LAB, van der Meer JWM, Kullberg B-J, van de Veerdonk FL. Immune defence against Candida fungal infections. Nat Rev Immunol [Internet]. 2015. October 21;15(10):630–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26388329. [DOI] [PubMed] [Google Scholar]
- 36.Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev [Internet]. 2013. March;252(1):89–103. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23405897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A [Internet]. 2009. October 20;106(42):17876–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19805038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opal SM. Immunotherapy for Sepsis: a new approach against an ancient foe. N Engl J Med 2010. July 1;363(1):87–89. doi101056/NEJMcibr1004371 NIH NIH. 2010;363(1):87–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol [Internet]. 2007. September;8(9):967–74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17581537. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, Tian J, Wang S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front Immunol [Internet]. 2018. May 28;9:1112 Available from: https://www.frontiersin.org/article/10.3389/fimmu.2018.01112/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brock M. Application of bioluminescence imaging for in vivo monitoring of fungal infections. Int J Microbiol. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gow NAR, Hube B. Importance of the Candida albicans cell wall during commensalism and infection. Curr Opin Microbiol [Internet]. August, 2012:406–12. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1369527412000483. [DOI] [PubMed]
- 43.Krysan DJ, Sutterwala FS, Wellington M. Catching Fire: Candida albicans, Macrophages, and Pyroptosis. Goldman WE, editor. PLoS Pathog [Internet]. 2014. June 26;10(6):e1004139 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24967821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Meara TR, Cowen LE. Insights into the host-pathogen interaction: C. albicans manipulation of macrophage pyroptosis. Microb cell (Graz, Austria) [Internet]. 2018. November 12;5(12):566–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30533421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gow N A R, van de Veerdonk FL, AJP Brown, Netea MG. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev Microbiol [Internet]. 2011;10(2):112–22. Available from: 10.1038/nrmicro2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duggan S, Leonhardt I, Hünniger K, Kurzai O. Host response to Candida albicans bloodstream infection and sepsis. Virulence [Internet]. 2015;6(4):316–26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25785541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gazendam RP, van Hamme JL, Tool ATJ, van Houdt M, Verkuijlen PJJH, Herbst M, et al. Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. Blood [Internet]. 2014. July 24;124(4):590–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24948657. [DOI] [PubMed] [Google Scholar]
- 48.Lionakis MS, Levitz SM. Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu Rev Immunol [Internet]. 2018. April 26;36(1):157–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29237128. [DOI] [PubMed] [Google Scholar]
- 49.Becker KL, Ifrim DC, Quintin J, Netea MG, van de Veerdonk FL. Antifungal innate immunity: recognition and inflammatory networks. Semin Immunopathol [Internet]. 2015. March 20;37(2):107–16. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25527294. [DOI] [PubMed] [Google Scholar]
- 50.Salazar F, Brown GD. Antifungal Innate Immunity: A Perspective from the Last 10 Years. J Innate Immun [Internet]. 2018;10(5–6):373–97. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29768268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 Recognition of α-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity [Internet]. 2010. May 28;32(5):681–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20493731. [DOI] [PubMed] [Google Scholar]
- 52.Brown GD. Innate Antifungal Immunity: The Key Role of Phagocytes. Annu Rev Immunol [Internet]. 2011. April 23;29(1):1–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20936972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol [Internet]. 2007. January 15;176(2):231–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17210947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida albicans. J Immunol [Internet]. 2013;190(8):4136–48. Available from: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.1202671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular Neutrophil Extracellular Traps Capture Bacteria from the Bloodstream during Sepsis. Cell Host Microbe [Internet]. 2012. September 13;12(3):324–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22980329. [DOI] [PubMed] [Google Scholar]
- 56.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. Levitz SM, editor. PLoS Pathog [Internet]. 2009. October 30;5(10):e1000639 Available from: https://dx.plos.org/10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Da A, Dantas S, Lee KK, Raziunaite I, Schaefer K, Wagener J, et al. Cell biology of Candida albicans-host interactions. Curr Opin Microbiol [Internet]. 2016;34:111–8. Available from: 10.1016/j.mib.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med [Internet]. 2015. March 1;7(3):240–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25637383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biondo C, Malara A, Costa A, Signorino G, Cardile F, Midiri A, et al. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur J Immunol [Internet]. 2012. October;42(10):2632–43. Available from: http://doi.wiley.com/10.1002/eji.201242532. [DOI] [PubMed] [Google Scholar]
- 60.Joo H, Upchurch K, Zhang W, Ni L, Li D, Xue Y, et al. Opposing Roles of Dectin-1 Expressed on Human Plasmacytoid Dendritic Cells and Myeloid Dendritic Cells in Th2 Polarization. J Immunol [Internet]. 2015. August 15;195(4):1723–31. Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.1402276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trautwein-Weidner K, Gladiator A, Kirchner FR, Becattini S, Rülicke T, Sallusto F, et al. Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis. Noverr MC, editor. PLOS Pathog [Internet]. 2015. October 2;11(10):e1005164 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26431538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fei M, Bhatia S, Oriss TB, Yarlagadda M, Khare A, Akira S, et al. TNF- from inflammatory dendritic cells (DCs) regulates lung IL-17A/IL-5 levels and neutrophilia versus eosinophilia during persistent fungal infection. Proc Natl Acad Sci [Internet]. 2011. March 29;108(13):5360–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21402950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kashem SW, Igyártó BZ, Gerami-Nejad M, Kumamoto Y, Mohammed J, Jarrett E, et al. Candida albicans Morphology and Dendritic Cell Subsets Determine T Helper Cell Differentiation. Immunity [Internet]. 2015. February 17;42(2):356–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25680275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papon N, Courdavault V, Lanoue A, Clastre M, Brock M. Illuminating Fungal Infections with Bioluminescence. PLoS Pathog. 2014;10(7):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting Edge: IL-17–Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J Immunol [Internet]. 2013;190(2):521–5. Available from: http://jimmunol.org/content/190/2/521.abstract. [DOI] [PubMed] [Google Scholar]
- 66.Hünniger K, Lehnert T, Bieber K, Martin R, Figge MT, Kurzai O. A Virtual Infection Model Quantifies Innate Effector Mechanisms and Candida albicans Immune Escape in Human Blood. PLoS Comput Biol. 2014;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brakenridge SC, Efron PA, Mira JC, Moore FA, Mohr AM, Gentile LF, et al. Sepsis Pathophysiology, Chronic Critical Illness, and Persistent Inflammation-Immunosuppression and Catabolism Syndrome. Crit Care Med [Internet]. 2016;45(2):253–62. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5243156/pdf/nihms802865.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev [Internet]. 2008;223:87–113. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3299089/pdf/nihms-356858.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garraud O, Smits HH, Mageed RA, Brouard S, Brosseau C, Durand M, et al. CD9 + Regulatory B Cells Induce T Cell Apoptosis via IL-10 and Are Reduced in Severe Asthmatic Patients. Front Immunol [Internet]. 2018;9:3034 Available from: www.frontiersin.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB, et al. ARTICLE IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun [Internet]. 2017;8 Available from: www.nature.com/naturecommunications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gentile LF, Cuenca AG, Efron PA, Ang D, Mckinley BA, Moldawer LL, et al. Persistent inflammation and immunosuppression: A common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg [Internet]. 2012;72(6):1491–501. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3705923/pdf/nihms474435.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med [Internet]. 1999. July;27(7):1230–51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10446814. [DOI] [PubMed] [Google Scholar]
- 73.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of Dendritic Cells, But Not Macrophages, in Patients with Sepsis. J Immunol [Internet]. 2002;168(5):2493–500. Available from: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 74.Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47(5):765–79. [DOI] [PubMed] [Google Scholar]
- 75.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA [Internet]. 2011. December 21;306(23):2594 Available from: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sundar KM, Sires M. Sepsis induced immunosuppression: Implications for secondary infections and complications. Indian J Crit Care Med [Internet]. 2013. May;17(3):162–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24082613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spec A, Shindo Y, Burnham C-AD, Wilson S, Ablordeppey EA, Beiter ER, et al. T cells from patients with Candida sepsis display a suppressive immunophenotype. Crit Care [Internet]. 2015;20(1):15 Available from: http://ccforum.com/content/20/1/15. [DOI] [PMC free article] [PubMed] [Google Scholar]