

Abstract
Macrophages play a central role in the development of atherosclerotic cardiovascular disease (ASCVD), which encompasses coronary artery disease, peripheral artery disease, cerebrovascular disease, and aortic atherosclerosis. In each vascular bed, macrophages contribute to the maintenance of the local inflammatory response, propagate plaque development, and promote thrombosis. These central roles, coupled with their plasticity, makes macrophages attractive therapeutic targets in stemming the development of and stabilizing existing atherosclerosis. In the context of ASCVD, classically activated M1 macrophages initiate and sustain inflammation, and alternatively activated M2 macrophages resolve inflammation. However, this classification is now considered an oversimplification, and a greater understanding of plaque macrophage physiology in ASCVD is required to aid in the development of therapeutics to promote ASCVD regression. Reviewed herein are the macrophage phenotypes and molecular regulators characteristic of ASCVD regression, and the current murine models of ASCVD regression.
Keywords: atherosclerosis, coronary artery disease, inflammation, macrophages, thrombosis
Highlights.
Macrophages in atherosclerotic cardiovascular disease play a central role in the development of plaques.
Classically activated M1 macrophages are implicated in initiating and sustaining inflammation, and alternatively activated or M2 macrophages are linked to inflammation resolution.
Macrophage plasticity makes them attractive therapeutic targets to stem the development of and stabilize existing atherosclerosis.
Understanding the basis of metabolic and epigenetic reprogramming of macrophage polarization is anticipated to translate to new therapeutic opportunities to promote atherosclerotic cardiovascular disease regression to reduce residual inflammatory risk.
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality worldwide.1 Vascular inflammation, even after robust cholesterol lowering, is considered an important contributor to the risk of recurrent atherothrombotic events, and macrophages represent a likely contributor to residual inflammatory risk. Initiated by the retention of apoB (apolipoprotein B)-containing lipoproteins in the arterial wall, ASCVD represents a failure to resolve the inflammatory response.2–4 Vascular lipid deposits activate the immune system leading to the local accumulation of both innate and adaptive immune cells, which facilitates the formation of lipid-rich lesions. Plaques may form in any number of vascular beds, with ASCVD most commonly referring to either coronary artery disease, peripheral artery disease, cerebrovascular disease, or aortic atherosclerosis. Atherosclerotic lesions grow slowly over the years, eventually impeding blood flow and leading to the clinical manifestation of stable angina or claudication. However, obstructive and nonobstructive lesions may also erode or abruptly rupture, resulting in the local accumulation of tissue factor and platelet activation, culminating in rapid thrombotic vascular occlusion and life-threatening conditions, such as myocardial infarction, stroke, acute limb ischemia, and cardiovascular death.
Please see www.ahajournals.org/atvb/atvb-focus for all articles published in this series.
Macrophages in ASCVD Progression
Predominantly derived from circulating monocytes and local proliferation,5–8 macrophage numbers increase up to 20-fold within mouse aortae during atherogenesis.2,9,10 Additionally, there is evidence that vascular smooth muscle cells can dedifferentiate to a plaque macrophage-like state.11–14 Recruitment of monocytes into the intimal space is a process that occurs early in life, with the initial stages evident in infants less than a year old,15,16 and atherosclerotic plaques are prevalent in adolescents and young adults.17–19 ASCVD represents a chronic inflammatory process that continues throughout adulthood, and for many culminates in a major adverse cardiac event.
A key feature of ASCVD is lipoprotein ingestion and accumulation by arterial macrophages, which gives rise to foam cells. Foam cell buildup contributes to plaque lipid storage and sustained plaque growth.10,20 Macrophages contribute to the maintenance of the local inflammatory response by secreting proinflammatory cytokines, chemokines, and producing reactive oxygen and nitrogen species. Additionally, macrophages engage in crosstalk with vascular smooth muscle cells, amplifying the inflammatory cycle by producing additional proinflammatory cytokines and extracellular matrix components, further promoting the retention of lipoproteins.2,21 Plaque macrophages have a decreased ability to migrate, impeding inflammation resolution, promoting the progression of lesions into complicated, rupture-prone plaques. Moreover, this persistent inflammation drives macrophage apoptosis, and in the absence of efficient efferocytosis, leads to the accumulation of debris and apoptotic cells, facilitating necrotic core formation in atherosclerotic plaque.2,10,22
A defining feature of macrophages is their plasticity, which allows them to produce a tailored response to local microenvironment stimuli.23–26 During inflammation, macrophages may act to either promote inflammation or resolve it during wound and tissue repair.24,27 The classical model of macrophage activation defines both pro- and anti-inflammatory macrophages with distinct physiological roles and activators. At the broadest level, macrophages are classified as either M1, classically activated, or M2, alternatively activated.23,28 In vitro, M1 macrophages polarize in response to toll-like receptor ligands, interferons, pathogen-associated molecular complexes, lipopolysaccharides, and lipoproteins. Fueled primarily by glycolysis,29 M1 macrophages contribute to tissue destruction and secrete pro-inflammatory factors including high levels of IL (interleukin)-1β, IL-6, and TNF-α (tumor necrosis factor-α).26,30,31 Consistent with their inflammatory phenotype, they express pro-inflammatory transcription factors, including nuclear factor-κB and STAT (signal transducer and activator of transcription)-1. M2 macrophages are at the other end of the spectrum with a fatty acid oxidation dependent-phenotype and anti-inflammatory properties.32 M2 macrophages are polarized in response to the cytokines IL-4 and IL-13 and secrete anti-inflammatory factors such as the IL-1 receptor agonist, IL-10 and collagen. M2 macrophages are characterized by their expression of CD163 (cluster of differentiation 163), mannose receptor 1, resistin like-β, and high levels of arginase-1.
In the context of plaques, macrophages adhering to both the classically activated and alternatively activated subsets are present in human and mouse lesions, with M1 as the predominant subtype.33–37 In human lesions, macrophages expressing proinflammatory markers are in rupture-prone, unstable regions, and M2-like macrophages in stable regions and the adventitia.38–43 However, recent evidence suggests that macrophages exist on an activation continuum and that the M1/M2 classification system is an oversimplification of macrophage heterogeneity and their diverse functions.36,43,44
In the context of murine ASCVD, several alternative macrophage classifications are described.36,45 These alternate phenotypes include hemorrhage-residing Mhem macrophages, which phagocytize and use erythrocyte remnants and hemoglobin deposits.46–48 This subset is atheroprotective and resistant to foam cell formation, attributed to their high expression of the cholesterol transporters ABCA1 (ATP-binding cassette transporter A1) and ABCG1 (ATP-binding cassette transporter G1) and the nuclear receptors, LXR (liver X receptor)-α and LXR-β.49 Mox macrophages, a proatherogenic subset induced by oxidized phospholipids that protect from oxidative stress through nuclear factor erythroid-derived 2-related factor 2–mediated expression of antioxidant enzymes such as heme oxygenase 1, thioredoxin reductase 1, and sulfiredoxin-1. In hypercholesterolemic mice, Mox macrophages are reported to account for 30% of plaque macrophages, with M1 and M2 subsets making up 40% and 20% of the remaining cohort, respectively.50 Finally, M4 macrophages are a subset polarized by platelet factor 4.51 This population is found in human lesions52 and characterized by high expression of matrix metalloprotease 7 and S100A8.53 M4 macrophages are defined as atherogenic based on their production of proinflammatory cytokines (IL-6 and TNF-α) and defective phagocytic properties.51,52
Macrophage heterogeneity in plaques was first appreciated using immunohistochemistry and, at the molecular level, by laser capture microdissection.54,55 Technological advances, including mass cytometry time of flight and single-cell RNA sequencing, have further expanded our knowledge of macrophage heterogeneity in progressing plaques.56,57 These technologies have assisted in characterizing the heterogeneous nature of plaque macrophages, and have identified a new previously unreported subset identified.58–60 Termed triggering receptor expressed on myeloid cells 2 (TREM)hi macrophages, this subset expresses high levels of the genes Trem2, Cd9, Ctsd, and Spp1 and low expression of inflammatory cytokines, with ascribed biological functions of lipid metabolism and cholesterol efflux.58–60 This unique population is proposed to be cholesterol-enriched and represents foamy macrophages.59 Altogether, TREMhi macrophages provide an alternate hypothesis in which macrophages subsets in plaques are inflammatory.
As described above, there are various modes of macrophage activation. Collectively, they demonstrate that macrophages in plaques may have only a partial resemblance to M1 and M2 macrophage phenotypes. Further research is necessary to identify gene-expression profiles and transcriptional pathways that underlie the identity and diversity of macrophages in ASCVD. Additionally, whether results in mice are translatable to human plaques, which have distinct phenotypic differences (eg, hemorrhage and rupture), is essential to determine for the development of therapies to reduce macrophage-associated residual inflammatory risk.
Atherosclerosis Regression
Macrophages are the hallmarks of ASCVD contributing to plaque development, local inflammation, and the promotion of thrombosis. This central role, coupled with their plasticity, makes macrophages attractive therapeutic targets to stem the progression of plaques and stabilize existing atherosclerosis.
Studies in the 1970s undertaken in nonhuman primates and pigs made the initial observations of macrophages contributing to atherosclerosis regression.61–63 These seminal studies employed atherogenic high-fat, high-cholesterol diets to induce atherosclerosis progression and subsequent low-fat, low-cholesterol diets to reduce hypercholesterolemia. In both models, 4 to 6 months of regression diet feeding decreased aortic lesion macrophage foam cells, reduced necrotic plaque area, and increased the thickness and density of fibrous caps. An ASCVD regression review in 1985 stated, “it is obvious that the role of macrophages in regression may be very complex and a comprehensive study of such is unattainable by a single experiment by one or a small group of investigators”.61 Since then, the generation of hyperlipidemic mouse models,64–66 extensively used to model human ASCVD,67,68 which allow for the rapid, reproducible development of plaques, has further increased the field’s understanding of the regulators of plaque progression. In 2001, in response to the need for further murine atherosclerosis model development basic research into the mechanisms that govern ASCVD regression or stabilization was stimulated by the establishment of an aortic transplantation approach.64
Clinical trials in humans have demonstrated that robust cholesterol reduction prevents major adverse cardiovascular events.65,66 Imaging studies using intravascular ultrasound and optical coherence tomography suggest that dramatic LDL (low-density lipoprotein) lowering (ie, statins, PCSK9 [proprotein convertase subtilisin/kexin type 9] inhibition) prevents plaque progression and may even induce plaque regression.67–70 The development of LDL-C (LDL cholesterol)-lowering therapies that facilitate unprecedented reductions in LDL-C, relative to traditional statins, are likely to provide further insight into the role of residual inflammatory risk and plaque progression and regression.22,71–74 Advances in imaging techniques provide insight into the compositional changes in remodeling plaques.75 Optical coherence tomography allows detailed visualization of plaques and provides information on plaque composition (eg, lipids and calcification) and thickness of the fibrous cap, a classical marker of plaque inflammation and vulnerability. Given that plaque lipid concentrations are positively associated with macrophage accumulation, this relationship provides indirect evidence for reduced plaque macrophage count during human ASCVD regression.76
Evidence for monocyte and macrophage phenotypes associated with plaque vulnerability are derived from plaques taken from subjects with different stages of atherosclerosis.41–43,77 However, translation of macrophage studies in mice to human ASCVD regression carries the caveat that responses of monocyte-derived macrophages from mice and humans are still needed to be compared side-by-side.24 Further imaging advancements indicate that monitoring of plaque macrophage content and phenotype may one day be a possibility in humans as it is in mice.78 These data are likely to further our understanding of human and murine lesions during ASCVD regression.79,80
Reviewed below are the current murine models of ASCVD regression, the well-described contribution of macrophages, and preclinical efforts to develop macrophage-targeted therapeutics to suppress plaque growth and inflammation.
Mouse Models of ASCVD Regression
Relative to baseline plaques, murine ASCVD regression encompasses one or more of the following, a reduction in plaque (1) size, (2) cholesterol content, or (3) macrophage content. All models begin with a progression phase to establish a baseline plaque in which plasma apoB lipoprotein levels are high (LDL-C >300 mg/dL), followed by a phase of low atherogenic lipoprotein levels (VLDL [very-low–density lipoprotein] and LDL-C) to induce regression. Preclinical models of ASCVD regression are considered analogous to high-intensity statin treatment in humans.81
Plaque Transplantation
Representing the first ASCVD model of regression, the plaque transplant model involves transplanting an atherosclerotic thoracic arch64 or aortic arch segment82,83 from a hyperlipidemic donor mouse (eg, ApoE−/− or Ldlr−/−) into a normolipidemic recipient mouse (ie, C57Bl/6J). The rapid change in atherogenic apoB lipoproteins induces plaque regression, characterized by decreased lesion, macrophage, and lipid areas over a short period.34,64,74,84,85 This original model has been instrumental in elucidating mechanisms that contribute to regression and the assessment of ASCVD-reducing therapies.30,74,86,87
Elimination of Atherogenic Lipoprotein Production
Reversa mice are LDLR (LDL receptor) deficient and genetically modified via the introduction of a conditional allele of Mttp (microsomal triglyceride transfer protein) in the liver (Ldlr−/−Apob100/100Mttpfl/flMx1-Cre+/+) and can serve as a reversible ASCVD model.88 Inhibition of microsomal triglyceride transfer protein (IFN-α [interferon-alpha], IFN-β, or synthetic double-stranded RNA [eg, polyinosinic:polycytidylic acid, pIpC]), in conjunction with a switch to a chow diet, results in reduced VLDL and LDL (apoB lipoprotein cholesterol; >1000 mg/dL to <150 mg/dL) and rapid regression of plaques.89 Similar changes in atherogenic lipoproteins and plaque regression can also be achieved in Ldlr−/− mice treated with the microsomal triglyceride transfer protein inhibitor, such as BMS 212122.90 Additionally, the elimination of apoB production with an Apob antisense oligonucleotide promotes ASCVD regression in Ldlr−/− mice.91
Modulation of LDL Receptor or ApoE Expression
Early lipid lowering approaches in mice used adeno-associated virus therapies to induce hepatic overexpression of Apoe in ApoE−/− mice and Ldlr in Ldlr−/− mice.92–94 Recent studies in wild-type mice have also described the utility of LDLR antisense oligonucleotides in raising apoB lipoprotein cholesterol levels and withdrawal of the antisense oligonucleotide and subsequent antagonism with sense oligonucleotides to accelerate the reduction of lipid levels and facilitate regression.95 Similarly, increasing PCSK9 levels, a protein that directs hepatic LDLR for degradation,96 elevates circulating LDL-C and induces atherosclerosis in wild-type mice.37,97–99 The PCSK9 adeno-associated virus model can be used for regression studies by a diet switch to chow, with lipid lowering further accelerated by the inclusion of an microsomal triglyceride transfer protein inhibitor to the diet.37 Given that they override the necessity for complicated and time-consuming backcrosses, ASCVD models that are genotype-independent will facilitate the relatively rapid assessment of factors that regulate ASCVD regression. Further, mechanistic work will benefit from the rapid induction of gene-specific phenotypes in adult mice with established lesions to test candidates that may regulate regression.100
Reduction in Dietary Cholesterol
A simple switch in diet from an atherogenic high-fat, high-cholesterol diet to a low-fat, low-cholesterol diet in some atherosclerosis-prone mouse models is sufficient to induce ASCVD regression and comparable to the use of statins or beneficial dietary changes in humans. However, this method of regression is slower and, in some mice apoB lipoprotein cholesterol levels (eg, ApoE−/− mice) may not normalize to sufficient levels to achieve regression.37,101,102
Macrophages in ASCVD Regression
Plaque macrophage content is determined by monocyte recruitment and macrophage proliferation, emigration, and death.2 Historically, atherosclerosis studies have placed a significant emphasis on understanding mechanisms of monocyte recruitment into the vascular wall and devising strategies to block their influx into plaques.2 However, recent studies show that there are also factors that determine macrophage retention within plaques,10 and it is hypothesized that if these processes are favorably modulated, plaque macrophage content may be reduced and ASCVD regression achieved.
Broad changes in the plaque macrophage transcriptome are characteristic of ASCVD regression, most commonly distinguished by enrichment of M2-associated transcripts.54,85,86,103–105 The dynamic change in plaque macrophage phenotype raises the possibility that inducing of macrophage polarization potential in vivo will represent a viable therapeutic option for ASCVD regression and suppress residual inflammatory risk. Detailed below are known factors that modulate plaque macrophage content and phenotype in the context of ASCVD regression.
Factors That Regulate Plaque Macrophage Regression
Macrophage Trafficking
Plaque macrophage emigration is characteristic of regressing lesions in several murine models of regression.86,103,106 Consistent with macrophage motility, cytoskeletal-binding, and Rho GTPase genes are among the top enriched transcripts in plaque macrophages in response to lipid lowering.107 One mechanism for decreased plaque macrophage content shown in the aortic transplantation model is increased macrophage egress to lymph nodes, mediated via CCR7 (C-C chemokine receptor type 7).87,106 Macrophage CCR7 transcript expression is, in part, regulated by a sterol response element in its promoter. Thus, it is hypothesized that lipid lowering induces plaque macrophage CCR7 expression and migration.85,108 In mice deficient in low-density lipoprotein receptor-related protein 1, a separate group independently presented evidence for a role of CCR7 in regression. During regression, LRP-1 (low-density lipoprotein receptor-related protein 1) deficiency increased macrophage cholesterol efflux and CCR7 expression and promoted macrophage emigration to lymph nodes and plaque regression.109
Epigenome-guided analysis of the transcriptome of plaque macrophages during ASCVD regression revealed activation of the Wnt signaling pathway.107 Given that in macrophages, Wnt signaling promotes cell motility through a β-catenin-dependent mechanism,110 and β-catenin knockdown promotes atherosclerosis progression,111 this pathway may represent an unexplored regulator of ASCVD regression. Additionally, the macrophage retention factors, netrin 1 and semaphorin 3E, transcripts differentially regulated in progressing and regressing plaques, also hold promise as potential mediators of plaque macrophage content given their ability to modulate macrophage retention and migration in progressing plaques.2,54,112,113
Monitoring plaque macrophage flux is an essential component in the assessment of the effectiveness of therapies designed to promote ASCVD regression. The most readily used technique in murine regression studies takes advantage of the ability of monocytes to take up fluorophore-labeled beads (eg, Rahman et al,74 Nagareddy et al,86 Distel et al,103 and Potteaux et al114). The monocyte bead labeling technique can be used to monitor plaque monocyte entry and macrophage egress.89 To monitor entry, mice are injected intravenously with beads before harvest (typically 24–72 hours), and to monitor macrophage egress, mice are injected with beads before the induction of regression (typically 24–96 hours). Quantification of bead-bearing cells in plaques at the time of harvest allows for the assessment of monocyte and macrophage trafficking.
This technique represents a relatively rapid and straightforward labeling procedure and does not alter the phenotype of bead-bearing cells.115 The predominant drawback of this approach is the relatively low incorporation of beads into circulating monocytes (≈5%–10%) and the selectivity of Ly6Clo monocytes in taking up the beads, the monocyte subset with reduced capacity to enter lesions. Prior injection of mice with clodronate liposomes, to deplete all circulating monocytes, can skew bead labeling to the Ly6Chi subset.116 However, this comes with the inherent drawback of potential depletion of plaque-residing macrophages given their ability to deplete macrophage populations in the spleen, bone marrow, and liver.116,117 Alternatively, Ly6Chi monocytes may be labeled with the modified thymidine analog EdU (5-ethynyl-2´-deoxyuridine) that at time points less than 72 hours is selectively incorporated in the Ly6Chi subset.74,114 Ly6Chi monocytes that enter plaques are determined by staining for EdU. Dual staining with the proliferation marker Ki67 allows for the distinction between recruited monocytes, and macrophages proliferating in situ.
Fluorescent reporter lines (eg, GFP-CD68 [green fluorescent protein CD68], CX3CR1-GFP [CX3C chemokine receptor 1 green fluorescent protein]) and congenic mice (eg, CD45.1, CD45.2) also allow for monitoring plaque monocyte/macrophage trafficking.104,116 The utility of these models is best suited to regression studies undertaken in the transplant model, as this allows for differential labeling of either donor plaque cells or recipient circulating cells to assess regression-mediated changes. The utilization of inducible reporter lines (eg, CreloxP or FLP-FRT [flippase/flippase recognition target] system) that could be triggered at the time of regression also represents a viable method to track myeloid cell trafficking in transplant-free regression models. Fluorescent reporter lines represent a useful model to monitor monocyte/macrophage flux via intravital imaging. However, the relatively short monitoring window and the need to expose the site of interest to detect signals limit this approach.
Macrophage Polarization
Macrophage polarization is considered a dynamic process,23,26 and editing of this potential is an emerging therapeutic area for a variety of inflammation-based disorders.23,118 M1 macrophages characterize progression lesions while regressing plaques are enriched in M2 macrophages.69 M2 macrophage enrichment in plaque regression is consistent with the view that M1 macrophages are pro-atherogenic and promote an unstable plaque, while M2 macrophages promote tissue repair and plaque stability. In vitro, the phenotypes of M1 and M2 macrophages are reversible, and there is evidence that this may occur in mice in vivo.119–121 However, whether macrophage interconversion occurs in the context of ASCVD regression, in humans or mice, is largely unknown and is an area of active research. Alternate possibilities for plaque M2 macrophage enrichment during regression are (1) egress of M1 macrophages from plaques, (2) entry of monocytes and their polarization to M2 macrophages, and (3) proliferation of resident yolk-sac-derived M2 macrophages.6,122
In atherogenic mice, the deletion of NR4A1, a transcription factor that regulates the Ly6Clo monocyte phenotype and favors M2 macrophage differentiation,123 results in plaque M1 macrophage enrichment and accelerates atherosclerosis.124,125 Similarly, the deletion of the transcription factor KLF4 (Krüppel-like factor 4), which promotes M2 and inhibits M1 macrophage polarization,126 enhances pro-inflammatory M1 macrophage activation, foam cell formation, and accelerates atherosclerosis in ApoE−/− mice.127 Likewise, stimulation of the PPAR-γ (peroxisome proliferator-activated receptor-gamma) pathway, which promotes M2 macrophage polarization,128 decreases atherosclerosis development in the ApoE−/− and Reversa mice.89,129 Similarly, loss of Akt2 enhances the ability of macrophages to polarize to the M2 state and suppresses atherogenesis.130 Treatment of Ldlr−/− mice with the M2-polarizing cytokine IL-13 promotes a plaque M2 macrophage phenotype, along with an increase in plaque collagen content, and a reduction in monocyte recruitment in lesions and macrophage content.131 However, it remains to be established whether the mechanisms that promote or hinder macrophage M2 polarization under conditions of hypercholesterolemia can be applied to regressing plaques.
A recent study reported that after reversal of hypercholesterolemia, the recruitment of Ly6Chi monocytes and their STAT6-dependent conversion to M2 macrophages is essential for reducing plaque macrophage content and suppression of ACSVD inflammation during regression.74 To date, the factors that regulate STAT6-signaling to mediate this change are unknown, but given IL-4 and IL-13 facilitate M2 polarization through a STAT6-dependent pathway, it is hypothesized that local production of these cytokines (eg, by basophils, eosinophils) during regression may mediate this process. Important to note, however, are findings in an alternate model of plaque regression, which found that suppression of monocyte recruitment is essential for plaque macrophage regression.114 This is consistent with studies in diabetic mice showing impaired regression after lipid lowering because of increased monocyte recruitment.103 These findings in different murine models of regression reveal that many different pathways positively affect the regression of established plaques. They also highlight that despite robust apoB lipoprotein lowering, plaques did not regress completely, providing evidence for additional unidentified mechanisms that contribute to residual inflammatory risk during regression exist.
A defined function of macrophages is their efferocytotic capacity, an essential process for the resolution of inflammation and plaque stabilization by reducing necrotic core area.132,133 Macrophage efferocytosis, referring to their ability to clear apoptotic cells and debris, is mediated through receptors including MERTK (tyrosine-protein kinase MER), LRP-1 and CD47 and is regarded as a protective anti-inflammatory function of M2 macrophages.134,135 Thus, enrichment of M2 macrophages, or enhancement of macrophage efferocytotic capacity (eg, via PPAR-γ activation136), may represent a viable strategy to promote ASCVD regression and plaque stabilization.
Recent studies demonstrate that macrophage inflammatory responses and their metabolism are codependent.137 Classically activated M1 macrophages shift to anabolic metabolism by upregulating either glycolysis or the pentose-phosphate pathway, while M2 macrophages are fueled by oxidative phosphorylation and fatty acid oxidation.138 In the context of ASCVD, environmental signals including hyperlipidemia, hypoxia, and hyperglycemia skew macrophage polarization toward a glycolytic inflammatory M1-like phenotype, a macrophage phenotype of both unstable murine and human plaques.77,86,103,139–142 How metabolic shifts in macrophages contribute to lesion progression and stability and the changes that occur after LDL-C lowering are currently unknown. However, preclinical studies provide insight into how reprogramming of macrophages to an anti-inflammatory M2-like phenotype suppresses plaque progression. Antagonism of miR-33, a microRNA elevated in macrophages in progressing lesions, promotes regression105 and skews macrophages towards an M2 state, as evidenced by increased mRNA expression of genes encoding AMP kinase and fatty acid oxidation, elevated mitochondrial respiration, and decreased glycolysis.102 Additionally, the treatment of Ldlr−/− mice with the M2-polarizing cytokine IL-13 inhibits atherosclerosis progression, in part by its ability to skew plaque macrophage phenotype towards an M2 state.131
Cholesterol Efflux
During ASCVD plaque regression, reductions in atherogenic apoB lipoproteins result in an improved HDL-C (high-density lipoprotein cholesterol) to LDL-C ratio. Increased concentrations of functional HDL particles are likely to represent a significant contributor to ASCVD regression, given their ability to mediate cholesterol efflux, facilitate foam cell migration, and induce M2 polarization.85,120,143 In vitro, treatment of macrophages with HDL enhances the expression of M2 macrophage markers (arginase 1 and retnlb) in a STAT6-dependent process120 and induces the transcriptional regulator ATF3, a repressor of inflammation.144 Given that ATF3 activates Wnt/β-catenin signaling in macrophages to mediate migration,145 a process that is promoted by HDL in cholesterol-loaded macrophages,146 these data may provide further mechanistic insight into the beneficial effects of HDL during regression.
In vivo studies further support a role of raising levels of functional HDL particles during the regression phase, as demonstrated by studies where HDL was raised in transplant recipients (by transgenic overexpression of human apoA-I), or by infusion of HDL, and accelerated plaque regression.85,147 Further, in ApoE−/− mice with established lesions, intervention with injections of apoA-I or statin-containing HDL particles suppressed plaque progression.148–150 Antagonism of miR-33, a negative regulator of circulating HDL levels, as noted above, also represents another option to promote plaque M2 macrophage enrichment and plaque regression.102,103,105
The beneficial role of cholesterol efflux in ASCVD regression is supported by preclinical LXR-focused studies. LXR is a transcription factor that induces the expression of genes involved in cholesterol transport and efflux151,152 and is essential for retarding atherosclerosis progression and promoting atherosclerosis regression.84,153 Additionally, the nonspecific efflux molecule, cyclodextrin, promotes atherosclerosis regression via LXR-mediated macrophage reprogramming to improve cholesterol efflux and exert anti-inflammatory effects.154
The lymphatic vasculature, localized in the adventitia, may also represent an underappreciated pathway contributing to ASCVD regression.155 In murine models, the lymphatic system was shown to be a critical component of reverse cholesterol transport, facilitating the effective removal of cholesterol effluxed from plaque macrophages.155,156 In the aortic transplant model, lymphatic-mediated reverse cholesterol transport accounted for 50% of cholesterol delivery from cholesterol-loaded macrophages into the plasma compartment and was essential for ASCVD regression.156 In mice, hypercholesterolemia is proposed to induce lymphatic dysfunction and drive atherosclerosis.157,158 While it remains to be established whether lymphatic function is restored after cholesterol-lowering in mice, experimental evidence indicates that during the regression phase, plaque cholesterol content is reduced and monocyte-derived cells exit plaques with some reaching lymph nodes.156 However, a recent report indicates that the proximity of a macrophage to the lymphatic vasculature before lipid lowering determines its egress capacity, rather than functional changes to the cell.115 Whether lymphatic-mediated cholesterol mobilization represents a significant plaque stabilizing or regression-inducing pathway in humans is unknown.159
Diabetes Mellitus Impairs Plaque Macrophage Inflammation Resolution
Despite advances in therapies to reduce CVD risk, patients with diabetes mellitus have a 2- to 4-fold higher risk of ASCVD and associated morbidity and mortality.66,160,161 Notably, diabetes mellitus not only increases CVD events but also impairs the resolution or regression of ASCVD.161 Consistent with the clinical data, diabetic mice have impaired atherosclerosis regression, as measured by the quantity and inflammatory state of plaque macrophages after aggressive lipid lowering.103,104,162,163 Diabetes mellitus in mice also increases monocytosis by activation of myelopoiesis, enhancing monocyte infiltration and plaque macrophage content, and impairs the polarization of plaque macrophages to the M2 state despite lipid lowering.86,104,164 Recently, we considered the outcome of raising functional HDL in diabetic mice and established that HDL can overcome diabetes mellitus–mediated impairments to regression by promoting a plaque M2 macrophage phenotype and suppressing aberrant myelopoiesis on lipid lowering.103,143 In the context of regression, we ascribe the beneficial effects of raising functional HDL to its cholesterol efflux capacity and anti-inflammatory functions—pathways dysregulated in both diabetic patients and mice.143
Therapeutic Targeting of Plaque Macrophages
Even with potent cholesterol reduction, many patients experience a major adverse cardiac event. This is highlighted by data from the FOURIER trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects with Elevated Risk), which demonstrate that patients with ASCVD randomized to a PCSK9 inhibitor (in conjunction with statin therapy) reached a median LDL-C of 30 mg/dL; 10% of all patients still experienced a cardiovascular event during a median follow-up of 26 months.165 These data, along with other trials, of potent LDL-C lowering, demonstrate that lowering cholesterol alone is not sufficient to completely reduce ASCVD-associated morbidity and mortality.70,166–170 While robust LDL-C lowering is maintained with PCSK9 inhibitors and may induce ASCVD regression, the degree of regression is limited.70 Residual inflammatory risk has emerged as a mechanism predisposing individuals to cardiovascular events, which remains even after aggressive LDL-C lowering therapies in humans.22,72,73 Additionally, even in mice, aggressive LDL-C lowering does not, in most cases, lead to the complete regression of lesions (eg, 74,143). Indeed, maximal plaque regression did not occur if a decrease in macrophage inflammation was prevented at the same time lipid levels were lowered.74 In combination with lipid lowering therapies, specific targeting of plaque macrophage-mediated inflammation may represent a viable option to reduce residual inflammatory CVD risk in humans and accelerate plaque regression in both humans and mice. Namely, therapies that reduce plaque macrophage content by promoting macrophage efferocytosis, emigration, or polarization to a pro-resolving phenotype are likely to have beneficial clinical outcomes when coupled with optimal medical therapies.
Current preclinical efforts have included the targeted delivery of LXR agonists to reduce plaque macrophage inflammation and promote cholesterol efflux (eg, Guo et al171 and Yu et al172). While the benefits of LXR-pathway activation are appreciated, preclinical studies have not translated well clinically, as synthetic LXR ligands strongly activate sterol regulatory element-binding protein 1c, inducing hypertriglyceridemia.173 Recently, however, desmosterol and synthetic desmosterol mimetics were shown in vivo to specifically target LXR pathways in macrophages and have minimal effects on hepatocytes, providing a potential new therapeutic strategy.174 In addition to reducing plaque lipid content, increased cholesterol efflux would be expected to favorably affect the inflammatory state of macrophages and enhance their ability to emigrate.89,146
Preclinical and clinical observations indicate that the reprogramming of plaque macrophages to an anti-inflammatory M2 phenotype will promote ASCVD regression and plaque stabilization. Treatment of Ldlr−/−mice with helminth-derived antigens, a eukaryotic parasitic worm that strongly induces anti-inflammatory, immune responses, was found to suppress myeloid cell activation, intraplaque inflammation (TNF-α, MCP-1) and reduce the recruitment of macrophages to lesions.175 These studies raise the interesting hypothesis that helminth-derived components or alternate strategies to induce M2 polarization may provide novel opportunities to mediate ASCVD regression and reduce systemic inflammation.176
During ASCVD regression, the balance of M1 and M2 macrophages switches, with M2-like macrophages more predominant. The role of metabolic shifts in determining the phenotype of macrophages in lesions and how this alters in response to cholesterol-lowering is an area of active research. Further research to decipher what induces metabolic and epigenetic reprogramming in plaque macrophages, and factors that promote or inhibit macrophage polarization in vivo (ie, mitochondrial function, inducible glycolysis inhibitors177,178) will likely translate to new therapeutic opportunities to promote ASCVD regression.
Vaccine-Mediated Suppression of Proinflammatory Immune Responses
Plaque macrophages and dendritic cells can function as APC (antigen-presenting cells) to activate members of the adaptive immune system including T and B cells, and evidence of APC-T cell interaction suggests antigen-specific immune activation through immune synapses in the plaque.179 The interaction of plaque macrophages with T cells is increasingly recognized to alter macrophage and plaque inflammation, as factors secreted by T helper type 1, Th2, and regulatory T cells (Tregs) can differentially skew macrophage phenotype.180 Tregs can dampen effector T cell responses by secretion of anti-inflammatory cytokines,181–183 promote the polarization of M1 macrophages to M2 macrophages by secretion of IL-10, and reduce macrophage lipid accumulation.184,185
The beneficial effects of Tregs to influence macrophage phenotype indicate that therapies promoting endogenous and antigen-specific Treg activity may alleviate plaque inflammation. In the context of atherosclerosis, the primary antigens identified to be responsible for triggering T cell activation are epitopes of oxLDL (oxidized low-density lipoprotein),186 apoB-100,187 and HSP (heat shock protein) 60/65.188 Research into immunization with antigenic proteins and peptides for the resolution of ASCVD by balancing pro- and anti-atherogenic T cell responses is currently confined to preclinical studies.189,190 This approach, however, holds promise as atherosclerosis-relevant antigens have been shown to induce antigen-specific Tregs and be atheroprotective in mice.191 These studies provide optimism that a therapy that restores tolerance to autoantigens may represent a viable strategy to reduce residual inflammatory risk in humans with ASCVD and promote ASCVD regression by polarizing macrophages to a tissue reparative state.
Conclusions
We increasingly recognize that on lipid lowering, macrophages can modulate plaque progression, regression, rupture, erosion, or stabilization. Detailed knowledge of how macrophage physiology contributes to clinical outcomes is essential to understand processes that promote macrophage pro-resolving characteristics. Further research is necessary to identify gene-expression profiles and transcriptional regulators of macrophage phenotype and function and reconcile how divergent plaque macrophage phenotypes (ie, M1, M2, Mhem, Mox, M4, and Tremhi) contribute to ASCVD stability. Currently, pro-resolving macrophages that participate in efficient efferocytosis, tissue remodeling, migration, and suppression of inflammatory processes are considered optimal for atherosclerotic regression (Figure). Thus, the development of therapeutics to enrich this phenotype may reduce cardiovascular morbidity and mortality by resolution of residual inflammatory risk.
Figure.
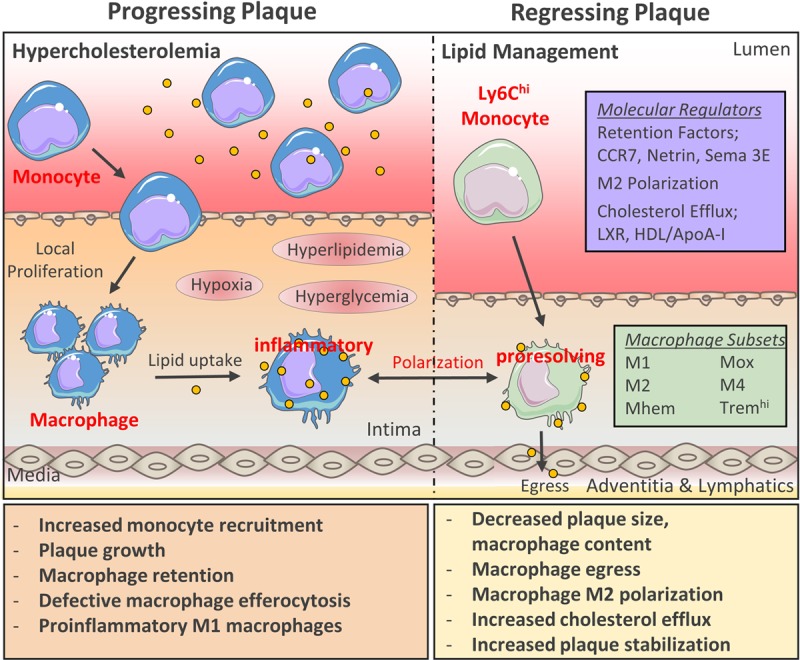
Dynamics of macrophage plasticity and trafficking in atherosclerosis. Atherosclerotic lesions are characterized by proinflammatory macrophages which sustain lesion growth by contributing to local and systemic plaque inflammation. Atherosclerosis development and macrophage dysfunction is accelerated during hypercholesterolemia (high LDL-C [lowdensity lipoprotein-cholesterol), and hyperglycemia. Atherogenic lipid-lowering remodels lesions towards a stable phenotype, a process driven mainly by macrophages. Broad changes in the plaque macrophage transcriptome are characteristic of atherosclerosis regression, most commonly distinguished by enrichment of M2-associated transcripts. The dynamic change in plaque macrophage phenotype raises the possibility that inducing of macrophage polarization potential in vivo will represent a viable therapeutic option for atherosclerotic cardiovascular disease regression and suppress residual inflammatory risk.
Additionally, despite preclinical models playing a crucial role in expanding our understanding of the heterogeneous nature of plaque macrophages, it is essential to ascertain how these findings translate to human pathophysiology. Translation to humans is essential given that the events that precipitate myocardial infarction, stroke, and acute limb ischemia (eg, plaque erosion and rupture), do not occur in current murine models. Recent advancements in lipid lowering therapies, which facilitate robust and sustained LDL-C reductions, are likely to increase our understanding of changes to plaque composition during regression. Additionally, an increased understanding of the role of macrophages in human ASCVD through omics studies and characterization of human tissue are anticipated to reconcile murine and human ASCVD and facilitate the development of strategies to promote ASCVD regression.
Acknowledgments
Sincere thanks to Edward A. Fisher and Jeffrey S. Berger and for their support and guidance and critical reading of this work. Additional thanks to the Reviewers of this paper and my fellow past and present lab members who have contributed to my knowledge and understanding of macrophages.
Sources of Funding
T.J. Barrett is supported by an American Heart Association Career Development Award (18CDA34110203AHA) and an American Society of Hematology Award (18-A0-00-1001884).
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- APC
- antigen-presenting cell
- apoB
- apolipoprotein B
- ASCVD
- atherosclerotic cardiovascular disease
- CCR7
- C-C chemokine receptor type 7
- FOURIER
- Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects with Elevated Risk
- HDL-C
- high-density lipoprotein cholesterol
- HSP
- heat shock protein
- IL
- interleukin
- KLF4
- Krüppel-like factor 4
- LDL
- low-density lipoprotein
- LDL-C
- LDL cholesterol
- LDLR
- LDL receptor
- LXR
- liver X receptor
- MERTK
- tyrosine-protein kinase MER
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- STAT
- signal transducer and activator of transcription
- TNF-α
- tumor necrosis factor-α
- Tregs
- regulatory T cell
- TREM
- termed triggering receptor expressed on myeloid cells 2
For Sources of Funding and Disclosures, see page 28.
References
- 1.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon D, Reidy MA, Benditt EP, Schwartz SM. Cell proliferation in human coronary arteries. Proc Natl Acad Sci U S A. 1990;87:4600–4604. doi: 10.1073/pnas.87.12.4600. doi: 10.1073/pnas.87.12.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 9.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–1282. doi: 10.1084/jem.20052205. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore KJ, Koplev S, Fisher EA, Tabas I, Björkegren JLM, Doran AC, Kovacic JC. Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: JACC macrophage in CVD series (Part 2). J Am Coll Cardiol. 2018;72:2181–2197. doi: 10.1016/j.jacc.2018.08.2147. doi: 10.1016/j.jacc.2018.08.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–667. doi: 10.1161/CIRCRESAHA.115.304634. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.HOLMAN RL, McGILL HC, Jr, STRONG JP, GEER JC. The natural history of atherosclerosis: the early aortic lesions as seen in New Orleans in the middle of the of the 20th century. Am J Pathol. 1958;34:209–235. [PMC free article] [PubMed] [Google Scholar]
- 16.Stary HC. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis. 1987;64:91–108. doi: 10.1016/0021-9150(87)90234-6. doi: 10.1016/0021-9150(87)90234-6. [DOI] [PubMed] [Google Scholar]
- 17.Knoflach M, Kiechl S, Penz D, Zangerle A, Schmidauer C, Rossmann A, Shingh M, Spallek R, Griesmacher A, Bernhard D, et al. Cardiovascular risk factors and atherosclerosis in young women: atherosclerosis risk factors in female youngsters (ARFY study). Stroke. 2009;40:1063–1069. doi: 10.1161/STROKEAHA.108.525675. doi: 10.1161/STROKEAHA.108.525675. [DOI] [PubMed] [Google Scholar]
- 18.McMahan CA, Gidding SS, McGill HC., Jr Coronary heart disease risk factors and atherosclerosis in young people. J Clin Lipidol. 2008;2:118–126. doi: 10.1016/j.jacl.2008.02.006. doi: 10.1016/j.jacl.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Stary HC. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am J Clin Nutr. 2000;72(5 suppl):1297S–1306S. doi: 10.1093/ajcn/72.5.1297s. doi: 10.1093/ajcn/72.5.1297s. [DOI] [PubMed] [Google Scholar]
- 20.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bäck M, Yurdagul A, Jr, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406. doi: 10.1038/s41569-019-0169-2. doi: 10.1038/s41569-019-0169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 26.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 29.Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15:813–826. doi: 10.1016/j.cmet.2012.04.023. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 32.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–855. doi: 10.1038/ni.2956. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamson S, Leitinger N. Phenotypic modulation of macrophages in response to plaque lipids. Curr Opin Lipidol. 2011;22:335–342. doi: 10.1097/MOL.0b013e32834a97e4. doi: 10.1097/MOL.0b013e32834a97e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436–445. doi: 10.1016/j.ijcard.2015.03.055. doi: 10.1016/j.ijcard.2015.03.055. [DOI] [PubMed] [Google Scholar]
- 35.de Gaetano M, Crean D, Barry M, Belton O. M1- and M2-type macrophage responses are predictive of adverse outcomes in human atherosclerosis. Front Immunol. 2016;7:275. doi: 10.3389/fimmu.2016.00275. doi: 10.3389/fimmu.2016.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Paoli F, Staels B, Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J. 2014;78:1775–1781. doi: 10.1253/circj.cj-14-0621. doi: 10.1253/circj.cj-14-0621. [DOI] [PubMed] [Google Scholar]
- 37.Peled M, Nishi H, Weinstock A, Barrett TJ, Zhou F, Quezada A, Fisher EA. A wild-type mouse-based model for the regression of inflammation in atherosclerosis. PLoS One. 2017;12:e0173975. doi: 10.1371/journal.pone.0173975. doi: 10.1371/journal.pone.0173975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barlis P, Serruys PW, Devries A, Regar E. Optical coherence tomography assessment of vulnerable plaque rupture: predilection for the plaque ‘shoulder’. Eur Heart J. 2008;29:2023. doi: 10.1093/eurheartj/ehn085. doi: 10.1093/eurheartj/ehn085. [DOI] [PubMed] [Google Scholar]
- 39.Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. doi: 10.1038/nrcardio.2014.173. doi: 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]
- 40.Cho KY, Miyoshi H, Kuroda S, Yasuda H, Kamiyama K, Nakagawara J, Takigami M, Kondo T, Atsumi T. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J Stroke Cerebrovasc Dis. 2013;22:910–918. doi: 10.1016/j.jstrokecerebrovasdis.2012.11.020. doi: 10.1016/j.jstrokecerebrovasdis.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Lee CW, Hwang I, Park CS, Lee H, Park DW, Kang SJ, Lee SW, Kim YH, Park SW, Park SJ. Macrophage heterogeneity of culprit coronary plaques in patients with acute myocardial infarction or stable angina. Am J Clin Pathol. 2013;139:317–322. doi: 10.1309/AJCP7KEYGN3OBGQX. doi: 10.1309/AJCP7KEYGN3OBGQX. [DOI] [PubMed] [Google Scholar]
- 42.Shaikh S, Brittenden J, Lahiri R, Brown PA, Thies F, Wilson HM. Macrophage subtypes in symptomatic carotid artery and femoral artery plaques. Eur J Vasc Endovasc Surg. 2012;44:491–497. doi: 10.1016/j.ejvs.2012.08.005. doi: 10.1016/j.ejvs.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Williams JW, Giannarelli C, Rahman A, Randolph GJ, Kovacic JC. Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD aeries (Part 1). J Am Coll Cardiol. 2018;72:2166–2180. doi: 10.1016/j.jacc.2018.08.2148. doi: 10.1016/j.jacc.2018.08.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, Mason JC, Haskard DO. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res. 2012;110:20–33. doi: 10.1161/CIRCRESAHA.111.247577. doi: 10.1161/CIRCRESAHA.111.247577. [DOI] [PubMed] [Google Scholar]
- 47.Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166–177. doi: 10.1016/j.jacc.2011.10.852. doi: 10.1016/j.jacc.2011.10.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, Haskard DO. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. 2009;174:1097–1108. doi: 10.2353/ajpath.2009.080431. doi: 10.2353/ajpath.2009.080431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyle JJ. Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr Opin Lipidol. 2012;23:453–461. doi: 10.1097/MOL.0b013e328356b145. doi: 10.1097/MOL.0b013e328356b145. [DOI] [PubMed] [Google Scholar]
- 50.Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gleissner CA, Shaked I, Erbel C, Böckler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. 2010;106:203–211. doi: 10.1161/CIRCRESAHA.109.199505. doi: 10.1161/CIRCRESAHA.109.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184:4810–4818. doi: 10.4049/jimmunol.0901368. doi: 10.4049/jimmunol.0901368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, Linden F, Wolf A, Doesch A, Lasitschka F, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 2015;21:255–265. doi: 10.1177/1753425914526461. doi: 10.1177/1753425914526461. [DOI] [PubMed] [Google Scholar]
- 54.Feig JE, Vengrenyuk Y, Reiser V, Wu C, Statnikov A, Aliferis CF, Garabedian MJ, Fisher EA, Puig O. Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PLoS One. 2012;7:e39790. doi: 10.1371/journal.pone.0039790. doi: 10.1371/journal.pone.0039790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trogan E, Choudhury RP, Dansky HM, Rong JX, Breslow JL, Fisher EA. Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2002;99:2234–2239. doi: 10.1073/pnas.042683999. doi: 10.1073/pnas.042683999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cole JE, Park I, Ahern DJ, Kassiteridi C, Danso Abeam D, Goddard ME, Green P, Maffia P, Monaco C. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. 2018;114:1360–1371. doi: 10.1093/cvr/cvy109. doi: 10.1093/cvr/cvy109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winkels H, Ehinger E, Ghosheh Y, Wolf D, Ley K. Atherosclerosis in the single-cell era. Curr Opin Lipidol. 2018;29:389–396. doi: 10.1097/MOL.0000000000000537. doi: 10.1097/MOL.0000000000000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509. doi: 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 59.Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 2018;123:1127–1142. doi: 10.1161/CIRCRESAHA.118.312804. doi: 10.1161/CIRCRESAHA.118.312804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. 2018;122:1675–1688. doi: 10.1161/CIRCRESAHA.117.312513. doi: 10.1161/CIRCRESAHA.117.312513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daoud AS, Fritz KE, Jarmolych J, Frank AS. Role of macrophages in regression of atherosclerosis. Ann N Y Acad Sci. 1985;454:101–114. doi: 10.1111/j.1749-6632.1985.tb11848.x. doi: 10.1111/j.1749-6632.1985.tb11848.x. [DOI] [PubMed] [Google Scholar]
- 62.Strong JP, Eggen DA, Stary HC. Reversibility of fatty streaks in rhesus monkeys. Primates Med. 1976;9:300–320. [PubMed] [Google Scholar]
- 63.Tucker CF, Catsulis C, Strong JP, Eggen DA. Regression of early cholesterol-induced aortic lesions in rhesus monkeys. Am J Pathol. 1971;65:493–514. [PMC free article] [PubMed] [Google Scholar]
- 64.Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–547. doi: 10.1067/mva.2001.115963. doi: 10.1067/mva.2001.115963. [DOI] [PubMed] [Google Scholar]
- 65.Cholesterol Treatment Trialists C. Efficacy and safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet. 2019;393:407–415. doi: 10.1016/S0140-6736(18)31942-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, et al. Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown G, Albers JJ, Fisher LD, Schaefer SM, Lin JT, Kaplan C, Zhao XQ, Bisson BD, Fitzpatrick VF, Dodge HT. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med. 1990;323:1289–1298. doi: 10.1056/NEJM199011083231901. doi: 10.1056/NEJM199011083231901. [DOI] [PubMed] [Google Scholar]
- 68.Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, Crowe T, Howard G, Cooper CJ, Brodie B, et al. Investigators R. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–1080. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 69.Rahman K, Fisher EA. Insights from pre-clinical and Clinical Studies on the role of innate inflammation in atherosclerosis regression. Front Cardiovasc Med. 2018;5:32. doi: 10.3389/fcvm.2018.00032. doi: 10.3389/fcvm.2018.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV Randomized Clinical Trial. JAMA. 2016;316:2373–2384. doi: 10.1001/jama.2016.16951. doi: 10.1001/jama.2016.16951. [DOI] [PubMed] [Google Scholar]
- 71.Larsen LE, Stoekenbroek RM, Kastelein JJP, Holleboom AG. Moving targets: recent advances in lipid-lowering therapies. Arterioscler Thromb Vasc Biol. 2019;39:349–359. doi: 10.1161/ATVBAHA.118.312028. doi: 10.1161/ATVBAHA.118.312028. [DOI] [PubMed] [Google Scholar]
- 72.Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016;37:1720–1722. doi: 10.1093/eurheartj/ehw024. doi: 10.1093/eurheartj/ehw024. [DOI] [PubMed] [Google Scholar]
- 73.Ridker PM. Canakinumab for residual inflammatory risk. Eur Heart J. 2017;38:3545–3548. doi: 10.1093/eurheartj/ehx723. doi: 10.1093/eurheartj/ehx723. [DOI] [PubMed] [Google Scholar]
- 74.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. doi: 10.1172/JCI75005. doi: 10.1172/JCI75005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raggi P, Baldassarre D, Day S, de Groot E, Fayad ZA. Non-invasive imaging of atherosclerosis regression with magnetic resonance to guide drug development. Atherosclerosis. 2016;251:476–482. doi: 10.1016/j.atherosclerosis.2016.06.028. doi: 10.1016/j.atherosclerosis.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 76.Felton CV, Crook D, Davies MJ, Oliver MF. Relation of plaque lipid composition and morphology to the stability of human aortic plaques. Arterioscler Thromb Vasc Biol. 1997;17:1337–1345. doi: 10.1161/01.atv.17.7.1337. doi: 10.1161/01.atv.17.7.1337. [DOI] [PubMed] [Google Scholar]
- 77.Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354. doi: 10.1084/jem.20150900. doi: 10.1084/jem.20150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swirski FK, Nahrendorf M. Imaging macrophage development and fate in atherosclerosis and myocardial infarction. Immunol Cell Biol. 2013;91:297–303. doi: 10.1038/icb.2012.72. doi: 10.1038/icb.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fayad ZA, Swirski FK, Calcagno C, Robbins CS, Mulder W, Kovacic JC. Monocyte and macrophage dynamics in the cardiovascular system: JACC macrophage in CVD series (Part 3). J Am Coll Cardiol. 2018;72:2198–2212. doi: 10.1016/j.jacc.2018.08.2150. doi: 10.1016/j.jacc.2018.08.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye YX, Calcagno C, Binderup T, Courties G, Keliher EJ, Wojtkiewicz GR, Iwamoto Y, Tang J, Pérez-Medina C, Mani V, et al. Imaging macrophage and hematopoietic progenitor proliferation in atherosclerosis. Circ Res. 2015;117:835–845. doi: 10.1161/CIRCRESAHA.115.307024. doi: 10.1161/CIRCRESAHA.115.307024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaneda H, Terashima M, Yamaguchi H. The role of intravascular ultrasound in the determination of progression and regression of coronary artery disease. Curr Atheroscler Rep. 2012;14:175–185. doi: 10.1007/s11883-012-0234-3. doi: 10.1007/s11883-012-0234-3. [DOI] [PubMed] [Google Scholar]
- 82.Chereshnev I, Trogan E, Omerhodzic S, Itskovich V, Aguinaldo JG, Fayad ZA, Fisher EA, Reis ED. Mouse model of heterotopic aortic arch transplantation. J Surg Res. 2003;111:171–176. doi: 10.1016/s0022-4804(03)00039-8. doi: 10.1016/s0022-4804(03)00039-8. [DOI] [PubMed] [Google Scholar]
- 83.Li W, Luehmann HP, Hsiao HM, Tanaka S, Higashikubo R, Gauthier JM, Sultan D, Lavine KJ, Brody SL, Gelman AE, et al. Visualization of monocytic cells in regressing atherosclerotic plaques by intravital 2-photon and positron emission tomography-based imaging-brief report. Arterioscler Thromb Vasc Biol. 2018;38:1030–1036. doi: 10.1161/ATVBAHA.117.310517. doi: 10.1161/ATVBAHA.117.310517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feig JE, Pineda-Torra I, Sanson M, Bradley MN, Vengrenyuk Y, Bogunovic D, Gautier EL, Rubinstein D, Hong C, Liu J, et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415–4424. doi: 10.1172/JCI38911. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–7171. doi: 10.1073/pnas.1016086108. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, Hamilton RL, Fisher EA, Young SG. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation. 2003;107:1315–1321. doi: 10.1161/01.cir.0000054781.50889.0c. doi: 10.1161/01.cir.0000054781.50889.0c. [DOI] [PubMed] [Google Scholar]
- 89.Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG, Fisher EA. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation. 2011;123:989–998. doi: 10.1161/CIRCULATIONAHA.110.984146. doi: 10.1161/CIRCULATIONAHA.110.984146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hewing B, Parathath S, Mai CK, Fiel MI, Guo L, Fisher EA. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis. 2013;227:125–129. doi: 10.1016/j.atherosclerosis.2012.12.026. doi: 10.1016/j.atherosclerosis.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res. 2015;117:933–942. doi: 10.1161/CIRCRESAHA.115.307182. doi: 10.1161/CIRCRESAHA.115.307182. [DOI] [PubMed] [Google Scholar]
- 92.Harris JD, Graham IR, Schepelmann S, Stannard AK, Roberts ML, Hodges BL, Hill V, Amalfitano A, Hassall DG, Owen JS, et al. Acute regression of advanced and retardation of early aortic atheroma in immunocompetent apolipoprotein-E (apoE) deficient mice by administration of a second generation [E1(-), E3(-), polymerase(-)] adenovirus vector expressing human apoE. Hum Mol Genet. 2002;11:43–58. doi: 10.1093/hmg/11.1.43. doi: 10.1093/hmg/11.1.43. [DOI] [PubMed] [Google Scholar]
- 93.Kassim SH, Li H, Vandenberghe LH, Hinderer C, Bell P, Marchadier D, Wilson A, Cromley D, Redon V, Yu H, et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One. 2010;5:e13424. doi: 10.1371/journal.pone.0013424. doi: 10.1371/journal.pone.0013424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Craeyveld E, Gordts SC, Nefyodova E, Jacobs F, De Geest B. Regression and stabilization of advanced murine atherosclerotic lesions: a comparison of LDL lowering and HDL raising gene transfer strategies. J Mol Med (Berl) 2011;89:555–567. doi: 10.1007/s00109-011-0722-x. doi: 10.1007/s00109-011-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Basu D, Hu Y, Huggins LA, Mullick AE, Graham MJ, Wietecha T, Barnhart S, Mogul A, Pfeiffer K, Zirlik A, et al. Novel reversible model of atherosclerosis and regression using oligonucleotide regulation of the LDL receptor. Circ Res. 2018;122:560–567. doi: 10.1161/CIRCRESAHA.117.311361. doi: 10.1161/CIRCRESAHA.117.311361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, et al. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J. 2007;406:203–207. doi: 10.1042/BJ20070664. doi: 10.1042/BJ20070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bjørklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 98.Lu H, Howatt DA, Balakrishnan A, Graham MJ, Mullick AE, Daugherty A. Hypercholesterolemia induced by a PCSK9 gain-of-function mutation augments angiotensin II-induced abdominal aortic aneurysms in C57BL/6 mice-brief report. Arterioscler Thromb Vasc Biol. 2016;36:1753–1757. doi: 10.1161/ATVBAHA.116.307613. doi: 10.1161/ATVBAHA.116.307613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, Muriana FJ, Fuster V, Ibáñez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. doi: 10.1161/ATVBAHA.114.303617. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 100.Shrestha E, Voisin M, Barrett TJ, Nishi H, Cantor DJ, Hussein MA, David G, Pineda-Torra I, Fisher EA, Garabedian MJ. Phosphorylation of LXRα impacts atherosclerosis regression by modulating monocyte/macrophage trafficking. 2018. p. 363366.
- 101.Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, DeKemp EM, Beanlands RA, Perisic L, Maegdefessel L, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. doi: 10.1126/sciadv.1600224. doi: 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348. doi: 10.1172/JCI81676. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res. 2014;115:759–769. doi: 10.1161/CIRCRESAHA.115.304164. doi: 10.1161/CIRCRESAHA.115.304164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ, Fisher EA. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes. 2011;60:1759–1769. doi: 10.2337/db10-0778. doi: 10.2337/db10-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ramsey SA, Vengrenyuk Y, Menon P, Podolsky I, Feig JE, Aderem A, Fisher EA, Gold ES. Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the Wnt signaling pathway. PLoS Genet. 2014;10:e1004828. doi: 10.1371/journal.pgen.1004828. doi: 10.1371/journal.pgen.1004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feig JE, Shang Y, Rotllan N, Vengrenyuk Y, Wu C, Shamir R, Torra IP, Fernandez-Hernando C, Fisher EA, Garabedian MJ. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One. 2011;6:e28534. doi: 10.1371/journal.pone.0028534. doi: 10.1371/journal.pone.0028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, Linton MF, Fazio S. Deletion of macrophage low-density Lipoprotein Receptor-Related Protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C Chemokine Receptor Type 7 (CCR7) expression in plaque macrophages. Circulation. 2018;138:1850–1863. doi: 10.1161/CIRCULATIONAHA.117.031702. doi: 10.1161/CIRCULATIONAHA.117.031702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, Ellis P, Langford C, Vannberg FO, Knight JC. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet. 2012;44:502–510. doi: 10.1038/ng.2205. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang F, Liu Z, Park SH, Gwag T, Lu W, Ma M, Sui Y, Zhou C. Myeloid β-catenin deficiency exacerbates atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2018;38:1468–1478. doi: 10.1161/ATVBAHA.118.311059. doi: 10.1161/ATVBAHA.118.311059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ, Parathath S, Distel E, Feig JL, Alvarez-Leite JI, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143. doi: 10.1038/ni.2205. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wanschel A, Seibert T, Hewing B, Ramkhelawon B, Ray TD, van Gils JM, Rayner KJ, Feig JE, O’Brien ER, Fisher EA, et al. Neuroimmune guidance cue semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler Thromb Vasc Biol. 2013;33:886–893. doi: 10.1161/ATVBAHA.112.300941. doi: 10.1161/ATVBAHA.112.300941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe-/- mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Williams JW, Martel C, Potteaux S, Esaulova E, Ingersoll MA, Elvington A, Saunders BT, Huang LH, Habenicht AJ, Zinselmeyer BH, et al. Limited macrophage positional dynamics in progressing or regressing murine atherosclerotic plaques-brief report. Arterioscler Thromb Vasc Biol. 2018;38:1702–1710. doi: 10.1161/ATVBAHA.118.311319. doi: 10.1161/ATVBAHA.118.311319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 117.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828. doi: 10.1182/blood-2009-11-253534. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- 118.Schultze JL. Reprogramming of macrophages–new opportunities for therapeutic targeting. Curr Opin Pharmacol. 2016;26:10–15. doi: 10.1016/j.coph.2015.09.007. doi: 10.1016/j.coph.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 119.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanson M, Distel E, Fisher EA. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One. 2013;8:e74676. doi: 10.1371/journal.pone.0074676. doi: 10.1371/journal.pone.0074676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. 2011;12:778–785. doi: 10.1038/ni.2063. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hamers AA, Vos M, Rassam F, Marinković G, Marincovic G, Kurakula K, van Gorp PJ, de Winther MP, Gijbels MJ, de Waard V, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res. 2012;110:428–438. doi: 10.1161/CIRCRESAHA.111.260760. doi: 10.1161/CIRCRESAHA.111.260760. [DOI] [PubMed] [Google Scholar]
- 125.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–2749. doi: 10.1172/JCI45444. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sharma N, Lu Y, Zhou G, Liao X, Kapil P, Anand P, Mahabeleshwar GH, Stamler JS, Jain MK. Myeloid krüppel-like factor 4 deficiency augments atherogenesis in apoE-/- mice–brief report. Arterioscler Thromb Vasc Biol. 2012;32:2836–2838. doi: 10.1161/ATVBAHA.112.300471. doi: 10.1161/ATVBAHA.112.300471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 129.Calkin AC, Forbes JM, Smith CM, Lassila M, Cooper ME, Jandeleit-Dahm KA, Allen TJ. Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler Thromb Vasc Biol. 2005;25:1903–1909. doi: 10.1161/01.ATV.0000177813.99577.6b. doi: 10.1161/01.ATV.0000177813.99577.6b. [DOI] [PubMed] [Google Scholar]
- 130.Babaev VR, Hebron KE, Wiese CB, Toth CL, Ding L, Zhang Y, May JM, Fazio S, Vickers KC, Linton MF. Macrophage deficiency of Akt2 reduces atherosclerosis in Ldlr null mice. J Lipid Res. 2014;55:2296–2308. doi: 10.1194/jlr.M050633. doi: 10.1194/jlr.M050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, Wagner O, Stangl H, Soehnlein O, Binder CJ. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med. 2012;4:1072–1086. doi: 10.1002/emmm.201201374. doi: 10.1002/emmm.201201374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–1095. doi: 10.1189/jlb.0209115. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. doi: 10.1038/nature18935. doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yurdagul A, Jr, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86. doi: 10.3389/fcvm.2017.00086. doi: 10.3389/fcvm.2017.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19:526–537. doi: 10.1038/s41590-018-0113-3. doi: 10.1038/s41590-018-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aarup A, Pedersen TX, Junker N, Christoffersen C, Bartels ED, Madsen M, Nielsen CH, Nielsen LB. Hypoxia-inducible factor-1α expression in macrophages promotes development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:1782–1790. doi: 10.1161/ATVBAHA.116.307830. doi: 10.1161/ATVBAHA.116.307830. [DOI] [PubMed] [Google Scholar]
- 140.Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, de Graaf J, Joosten LA, Netea MG, Gomes ME, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. 2016;254:228–236. doi: 10.1016/j.atherosclerosis.2016.10.019. doi: 10.1016/j.atherosclerosis.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 141.Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1β production in activated human macrophages. Circ Res. 2014;115:875–883. doi: 10.1161/CIRCRESAHA.115.304437. doi: 10.1161/CIRCRESAHA.115.304437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tomas L, Edsfeldt A, Mollet IG, Perisic Matic L, Prehn C, Adamski J, Paulsson-Berne G, Hedin U, Nilsson J, Bengtsson E, et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur Heart J. 2018;39:2301–2310. doi: 10.1093/eurheartj/ehy124. doi: 10.1093/eurheartj/ehy124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Barrett TJ, Distel E, Murphy AJ, Hu J, Garshick MS, Ogando Y, Liu J, Vaisar T, Heinecke JW, Berger JS, et al. Apolipoprotein AI) promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation. 2019;140:1170–1184. doi: 10.1161/CIRCULATIONAHA.119.039476. doi: 10.1161/CIRCULATIONAHA.119.039476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 2014;15:152–160. doi: 10.1038/ni.2784. doi: 10.1038/ni.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sha H, Zhang D, Zhang Y, Wen Y, Wang Y. ATF3 promotes migration and M1/M2 polarization of macrophages by activating tenascin–C via Wnt/β-catenin pathway. Mol Med Rep. 2017;16:3641–3647. doi: 10.3892/mmr.2017.6992. doi: 10.3892/mmr.2017.6992. [DOI] [PubMed] [Google Scholar]
- 146.Pagler TA, Wang M, Mondal M, Murphy AJ, Westerterp M, Moore KJ, Maxfield FR, Tall AR. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ Res. 2011;108:194–200. doi: 10.1161/CIRCRESAHA.110.228619. doi: 10.1161/CIRCRESAHA.110.228619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C, Otten MJ, Zaidi N, Lobatto ME, van Rijs SM, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi: 10.1038/ncomms4065. doi: 10.1038/ncomms4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hewing B, Parathath S, Barrett T, Chung WK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MS, Didonato JA, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789. doi: 10.1161/ATVBAHA.113.303044. doi: 10.1161/ATVBAHA.113.303044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shah PK, Yano J, Reyes O, Chyu KY, Kaul S, Bisgaier CL, Drake S, Cercek B. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–3050. doi: 10.1161/hc2501.092494. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]
- 151.Hussein MA, Shrestha E, Ouimet M, Barrett TJ, Leone S, Moore KJ, Hérault Y, Fisher EA, Garabedian MJ. LXR-mediated ABCA1 expression and function are modulated by high glucose and PRMT2. PLoS One. 2015;10:e0135218. doi: 10.1371/journal.pone.0135218. doi: 10.1371/journal.pone.0135218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shrestha E, Hussein MA, Savas JN, Ouimet M, Barrett TJ, Leone S, Yates JR, 3rd, Moore KJ, Fisher EA, Garabedian MJ. Poly(ADP-ribose) polymerase 1 represses liver X receptor-mediated ABCA1 expression and cholesterol efflux in macrophages. J Biol Chem. 2016;291:11172–11184. doi: 10.1074/jbc.M116.726729. doi: 10.1074/jbc.M116.726729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee SD, Tontonoz P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis. 2015;242:29–36. doi: 10.1016/j.atherosclerosis.2015.06.042. doi: 10.1016/j.atherosclerosis.2015.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek A, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra50. doi: 10.1126/scitranslmed.aad6100. doi: 10.1126/scitranslmed.aad6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang LH, Elvington A, Randolph GJ. The role of the lymphatic system in cholesterol transport. Front Pharmacol. 2015;6:182. doi: 10.3389/fphar.2015.00182. doi: 10.3389/fphar.2015.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013;123:1571–1579. doi: 10.1172/JCI63685. doi: 10.1172/JCI63685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, Angeli V. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol. 2009;175:1328–1337. doi: 10.2353/ajpath.2009.080963. doi: 10.2353/ajpath.2009.080963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Ohman M, Heinonen SE, Samaranayake H, Heikura T, Alitalo K, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1162–1170. doi: 10.1161/ATVBAHA.114.302528. doi: 10.1161/ATVBAHA.114.302528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in cardiovascular medicine. Circ Res. 2016;118:515–530. doi: 10.1161/CIRCRESAHA.115.306544. doi: 10.1161/CIRCRESAHA.115.306544. [DOI] [PubMed] [Google Scholar]
- 160.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 161.Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, et al. JAPAN-ACS Investigators. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome–serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial). Circ J. 2010;74:1165–1174. doi: 10.1253/circj.cj-09-0766. doi: 10.1253/circj.cj-09-0766. [DOI] [PubMed] [Google Scholar]
- 162.Gaudreault N, Kumar N, Olivas VR, Eberlé D, Stephens K, Raffai RL. Hyperglycemia impairs atherosclerosis regression in mice. Am J Pathol. 2013;183:1981–1992. doi: 10.1016/j.ajpath.2013.08.019. doi: 10.1016/j.ajpath.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yuan C, Hu J, Parathath S, Grauer L, Cassella CB, Bagdasarov S, Goldberg IJ, Ramasamy R, Fisher EA. Human aldose reductase expression prevents atherosclerosis regression in diabetic mice. Diabetes. 2018;67:1880–1891. doi: 10.2337/db18-0156. doi: 10.2337/db18-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Willecke F, Yuan C, Oka K, Chan L, Hu Y, Barnhart S, Bornfeldt KE, Goldberg IJ, Fisher EA. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in LDL receptor-deficient mice. PLoS One. 2015;10:e0128996. doi: 10.1371/journal.pone.0128996. doi: 10.1371/journal.pone.0128996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 166.Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol. 2005;46:1225–1228. doi: 10.1016/j.jacc.2005.07.006. doi: 10.1016/j.jacc.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 167.Du H, Li X, Su N, Li L, Hao X, Gao H, Kwong JS, Vandvik PO, Yang X, Nemeth I, et al. Proprotein convertase subtilisin/kexin 9 inhibitors in reducing cardiovascular outcomes: a systematic review and meta-analysis. Heart. 2019;105:1149–1159. doi: 10.1136/heartjnl-2019-314763. doi: 10.1136/heartjnl-2019-314763. [DOI] [PubMed] [Google Scholar]
- 168.Waters DD, Hsue PY. PCSK9 inhibition to reduce cardiovascular risk: tempering expectations. Circ Res. 2017;120:1537–1539. doi: 10.1161/CIRCRESAHA.117.311015. doi: 10.1161/CIRCRESAHA.117.311015. [DOI] [PubMed] [Google Scholar]
- 169.Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. doi: 10.1056/NEJMoa1110874. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- 170.Toth PP, Worthy G, Gandra SR, Sattar N, Bray S, Cheng LI, Bridges I, Worth GM, Dent R, Forbes CA, et al. Systematic review and network meta-analysis on the efficacy of evolocumab and other therapies for the management of lipid levels in hyperlipidemia. J Am Heart Assoc. 2017;6:e005367. doi: 10.1161/JAHA.116.005367. doi: 10.1161/JAHA.116.005367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Guo Y, Yuan W, Yu B, Kuai R, Hu W, Morin EE, Garcia-Barrio MT, Zhang J, Moon JJ, Schwendeman A, et al. Synthetic high-density lipoprotein-mediated targeted delivery of liver X receptors agonist promotes atherosclerosis regression. EBioMedicine. 2018;28:225–233. doi: 10.1016/j.ebiom.2017.12.021. doi: 10.1016/j.ebiom.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yu M, Amengual J, Menon A, Kamaly N, Zhou F, Xu X, Saw PE, Lee SJ, Si K, Ortega CA, et al. Targeted nanotherapeutics encapsulating liver X receptor agonist GW3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in Ldlr(-/-) mice. Adv Healthc Mater. 2017;6 doi: 10.1002/adhm.201700313. doi: 10.1002/adhm.201700313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, Seidman JS, Henke A, Roland JT, Ozeki KA, et al. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc Natl Acad Sci U S A. 2018;115:E4680–E4689. doi: 10.1073/pnas.1714518115. doi: 10.1073/pnas.1714518115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wolfs IM, Stöger JL, Goossens P, Pöttgens C, Gijbels MJ, Wijnands E, van der Vorst EP, van Gorp P, Beckers L, Engel D, et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014;28:288–299. doi: 10.1096/fj.13-235911. doi: 10.1096/fj.13-235911. [DOI] [PubMed] [Google Scholar]
- 176.Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-targeted therapeutics for metabolic disease. Trends Pharmacol Sci. 2018;39:536–546. doi: 10.1016/j.tips.2018.03.001. doi: 10.1016/j.tips.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. 2017;8:289. doi: 10.3389/fimmu.2017.00289. doi: 10.3389/fimmu.2017.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MC, Ahmed M, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17:684–696. doi: 10.1016/j.celrep.2016.09.008. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 179.Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, von Vietinghoff S, Galkina E, Miller YI, Acton ST, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012;122:3114–3126. doi: 10.1172/JCI61758. doi: 10.1172/JCI61758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Spitz C, Winkels H, Bürger C, Weber C, Lutgens E, Hansson GK, Gerdes N. Regulatory T cells in atherosclerosis: critical immune regulatory function and therapeutic potential. Cell Mol Life Sci. 2016;73:901–922. doi: 10.1007/s00018-015-2080-2. doi: 10.1007/s00018-015-2080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kita T, Yamashita T, Sasaki N, Kasahara K, Sasaki Y, Yodoi K, Takeda M, Nakajima K, Hirata K. Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc Res. 2014;102:107–117. doi: 10.1093/cvr/cvu002. doi: 10.1093/cvr/cvu002. [DOI] [PubMed] [Google Scholar]
- 182.Laurat E, Poirier B, Tupin E, Caligiuri G, Hansson GK, Bariéty J, Nicoletti A. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2001;104:197–202. doi: 10.1161/01.cir.104.2.197. doi: 10.1161/01.cir.104.2.197. [DOI] [PubMed] [Google Scholar]
- 183.Steffens S, Burger F, Pelli G, Dean Y, Elson G, Kosco-Vilbois M, Chatenoud L, Mach F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation. 2006;114:1977–1984. doi: 10.1161/CIRCULATIONAHA.106.627430. doi: 10.1161/CIRCULATIONAHA.106.627430. [DOI] [PubMed] [Google Scholar]
- 184.Lin J, Li M, Wang Z, He S, Ma X, Li D. The role of CD4+CD25+ regulatory T cells in macrophage-derived foam-cell formation. J Lipid Res. 2010;51:1208–1217. doi: 10.1194/jlr.D000497. doi: 10.1194/jlr.D000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. doi: 10.1161/ATVBAHA.108.179697. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 187.Fredrikson GN, Hedblad B, Berglund G, Alm R, Ares M, Cercek B, Chyu KY, Shah PK, Nilsson J. Identification of immune responses against aldehyde-modified peptide sequences in apoB associated with cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23:872–878. doi: 10.1161/01.ATV.0000067935.02679.B0. doi: 10.1161/01.ATV.0000067935.02679.B0. [DOI] [PubMed] [Google Scholar]
- 188.Wick G, Jakic B, Buszko M, Wick MC, Grundtman C. The role of heat shock proteins in atherosclerosis. Nat Rev Cardiol. 2014;11:516–529. doi: 10.1038/nrcardio.2014.91. doi: 10.1038/nrcardio.2014.91. [DOI] [PubMed] [Google Scholar]
- 189.Kimura T, Tse K, Sette A, Ley K. Vaccination to modulate atherosclerosis. Autoimmunity. 2015;48:152–160. doi: 10.3109/08916934.2014.1003641. doi: 10.3109/08916934.2014.1003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yamashita T, Sasaki N, Kasahara K, Hirata K. Anti-inflammatory and immune-modulatory therapies for preventing atherosclerotic cardiovascular disease. J Cardiol. 2015;66:1–8. doi: 10.1016/j.jjcc.2015.02.002. doi: 10.1016/j.jjcc.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 191.Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2015;35:280–287. doi: 10.1161/ATVBAHA.114.303568. doi: 10.1161/ATVBAHA.114.303568. [DOI] [PMC free article] [PubMed] [Google Scholar]