

Supplemental Digital Content is available in the text.
Keywords: aneurysm, dissecting; animals; interleukin-17; mice; risk factors
Objective:
Aortic dissection (AD) is a fatal disease that occurs suddenly without preceding clinical signs or symptoms. Although high salt intake is a proposed risk factor for cardiovascular diseases, the relationship between AD and high salt intake has not been clarified. We examined the effect of high-salt challenge on a mouse AD model.
Approach and Results:
AD was induced in male mice by continuous infusion of β-aminopropionitrile and Ang II (angiotensin II). High-salt challenge exacerbated aortic wall destruction in AD. Deletion of Il17a (IL-17KO [IL (interleukin)-17A knockout]) did not affect the AD phenotype at baseline, but it abolished the high salt–induced worsening of the aortic destruction. Unexpectedly, aortas of IL-17KO mice exhibited global changes in ECM (extracellular matrix)-related genes without alteration of proinflammatory genes, altered architecture of collagen fibers, and reduced stiffness before AD induction. The aortas of IL-17KO mice were less sensitive to AD-inducing stimuli, as shown by the induction of phenotypic modulation markers SMemb and vimentin, suggesting a reduced stress response. The aortas of IL-17KO mice had a higher population of smooth muscle cells with nuclear-localized phosphorylated Smad2, indicative of TGFβ (transforming growth factor-beta) signal activation. Consistently, pretreatment of smooth muscle cells in culture with IL-17A blunted the activation of Smad2 by TGFβ1.
Conclusions:
These findings indicate that high salt intake has a worsening effect on AD in the context of high aortic wall stiffness, which is under the control of IL-17A through ECM metabolism. Therefore, salt restriction may represent a low-cost and practical way to reduce AD risk.
Highlights.
Aortic dissection is a sudden, unpredictable, and fatal aortic destruction of which disease mechanism is largely unknown.
IL (interleukin)-17A interfered with TGFβ (transforming growth factor-beta) signal, altered extracellular matrix metabolism, and made aortic walls stiffer.
High-salt challenge exacerbated aortic wall destruction in aortic dissection only when IL-17A was intact and aortic wall was stiff.
Salt restriction may represent a low-cost and practical way to reduce aortic dissection risk for those who have stiffened aortic walls.
Aortic dissection (AD) is an abrupt tearing of the aortic wall, which is accompanied by severe pain and has largely unknown pathogenesis. AD is a serious medical emergency with high mortality, reaching 75% in 2 weeks without appropriate treatment because of rapid destruction of the aortic walls.1 Despite recent advances in diagnostic modalities and surgical techniques, mortality associated with emergency surgery remains high (10%–35%) at experienced centers. In addition to the high mortality of in-hospital patients, ≈20% of patients die before reaching the hospital. No medical therapy is currently available to stabilize the injured aortic wall after AD develops. Although AD prevention is desirable, it is currently impractical because the condition has few, if any, warning signs before onset. In addition, because the annual incidence of AD in the general population is ≈6 in 100 000,2 medical intervention in the general population is considered impractical for preventing AD. Ambiguity about the pathogenesis of AD hampers the development of effective preventive and therapeutic interventions.
See accompanying editorial on page 17
Recent studies using mouse models of AD have emphasized the importance of inflammatory molecules, including IL (interleukin)-6, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, IL-17, Cxcl1 (C-X-C motif chemokine ligand 1), and Ccl2 (C-C motif chemokine ligand 2).3–6 We have also reported that tenascin C7 and myeloid cell Socs38 ameliorate AD by suppressing excessive proinflammatory responses and promoting adaptive metabolism in the ECM (extracellular matrix). However, how the inflammatory response and ECM metabolism are related in the context of AD pathogenesis remains unclear.
High salt intake is associated with a high incidence of cardiovascular disease, including hypertension,9 and aortic wall stiffening.10 Recent studies have demonstrated that high salt intake induces pathogenic Th17 cells—an IL-17–producing cell type,11,12 providing a link between high salt intake and inflammation. However, a link between high salt intake and AD has not been established.
In the current study, we explored the effect of high salt intake on AD development and the involvement of the IL-17 pathway. To investigate the molecular events preceding AD, we developed a new mouse model in which AD was induced by simultaneous infusion of Ang II (angiotensin II) and β-aminopropionitrile (BAPN)—a collagen/elastin cross-link inhibitor—for 14 days. As the AD developed in a quantitatively and chronologically predictable manner, this model allowed us to analyze the molecular events preceding AD development.
Methods
The data, analytical methods, and study materials will be available to other researchers for the purpose of reproducing the results or replicating the procedure as long as the situation allows. The transcriptome dataset has been deposited in Gene Expression Omnibus of the National Center for Biotechnology Information (accession No. GSE116434). The study materials are commercially available except for human aortic tissue samples, which will not be publicly available. For the sources of animals, cell lines, and antibodies, please see the Major Resources Table in the online-only Data Supplement.
Animal Experiments
All animal protocols were approved by the Animal Experiments Review Boards of Kurume University. All mice were fed normal laboratory diet and allowed access to freely available drinking water unless otherwise stated. All of the animal experiments were performed using male mice aged 11 to 14 weeks because AD predominantly affects men.13 AD was induced by simultaneous administration of BAPN (150 mg/kg per day) and Ang II (1000 ng/kg per min) using osmotic minipumps (Alzet model 1002). Two pumps, one for BAPN and another for Ang II, were implanted while the mice were under anesthesia with 2% isoflurane. The mice were killed by pentobarbital overdose at the indicated time points to collect blood and tissue samples (Figure I in the online-only Data Supplement). The aortic tissue was excised either immediately for protein and mRNA expression analysis or after perfusion and fixation with 4% paraformaldehyde in PBS at physiological pressure for histological analysis. To analyze protein and mRNA expression, a 10-mm length of aorta was excised above the branching point of the right renal artery, quickly frozen in liquid nitrogen, and stored at −80°C until sample extraction.
High-Salt Challenge and Genetic Modification of Mice
High-salt challenge was achieved by substituting 1% NaCl for drinking water for the indicated time period. We used mice with a genetic deletion of Il17a (IL-17KO [IL-17A knockout])14 backcrossed with C57BL/6J mice for >8 generations to investigate the role of IL-17 in AD pathogenesis. WT (wild type) C57BL/6J mice (Charles River Laboratories, Japan) served as a control.
Quantitative Assessment of AD Lesions
From a clinical perspective, the severity of AD is assessed by the extent and location of aortic wall destruction, which affects life-threatening complications, such as rupture, cardiac tamponade, and distal ischemia.15,16 A detailed 3-dimensional analysis of our mouse model by propagation-based phase-contrast synchrotron imaging indicated progressive disruption of multiple medial layers, a hallmark of AD, associated with the formation of intramural hematoma and increased aortic diameter.17 Visually, the AD lesions with a hematoma had an increased diameter. Accordingly, the lesions with aortic wall destruction due to AD were defined in this study by a diameter at least 1.5-fold greater than the reference diameter. The normal aortic diameter differs in different segments of the aorta; therefore, we divided the aorta into 4 segments (Figure II in the online-only Data Supplement): the aortic arch, from the aortic root to just distal of the left subclavian artery; the descending thoracic aorta to just proximal of the celiac trunk; the suprarenal aorta to just proximal of the left renal artery; and the infrarenal aorta to the iliac bifurcation. The reference diameter was measured at the most distal part of each segment in 8 mice of the relevant genotype and the mean value used. We also characterized the severity of AD by the rate of thoracic or abdominal aortic rupture.
Expression Analysis
To analyze protein expression, frozen aortic samples were pulverized using the SK Mill (TOKKEN, Japan) and proteins extracted with radioimmunoprecipitation buffer (No. 08714-04; Nacalai Tesque, Japan). Immunoblotting was performed with antibodies as indicated in the Major Resources Table in the online-only Data Supplement. Plasma levels of IL-17A were measured using a bead-based assay (Bio-Plex, No. 171-G5013M; Bio-Rad). To analyze mRNA expression, we isolated total RNA from the same part of the aorta as the protein expression analysis using the RNeasy kit (Qiagen). We performed transcriptome analyses using the SurePrint G3 Mouse Gene Expression v2 8x60K Microarray Kit (Agilent). The dataset has been deposited to the Gene Expression Omnibus of the National Center for Biotechnology Information (accession No. GSE116434). Biological process–focused gene enrichment analysis was performed using the Database for Annotation, Visualization, and Integrated Discovery (https://david.ncifcrf.gov/)18 with the Gene Ontology terms set to GOTERM_BP_FAT. Expression of the indicated genes was measured by quantitative reverse transcription polymerase chain reaction using commercially available probes (Qiagen).
Histological Analysis
We performed Elastica van Gieson or hematoxylin-eosin staining on 5-μm sections. Imaging cytometric analysis of mouse aortas was performed using ArrayScan XTI (Thermo Fisher Scientific) and FlowJo 10 software (FlowJo). Two aortic tissue sections were obtained from each mouse in the WT (n=8) and IL-17KO (n=8) groups, stained as indicated in the Major Resources Table in the online-only Data Supplement. Microscopic images were obtained using a computerized bright-field/wide-field fluorescence microscope (BZ-9000; Keyence, Japan) or laser scanning confocal microscope (FV-1000; Olympus, Japan).
Collagen Deposition in Aortic Walls
For the histological analysis of collagen deposition, we stained the aortic tissue sections with Picrosirius Red. We obtained the tissue sections from descending thoracic aorta just distal to left subclavian artery, because this segment is frequently affected by AD and was used for the measurement of mechanical properties. Bright-field and polarized light-illuminated histological images were used. The area between the innermost and outermost elastic lamellae, the aortic media, was traced manually on the bright-field images. The collagen deposition area, determined by Picrosirius Red staining in the media or adventitia, and the medial area were measured using ImagePro Plus software (version 7.0.1; Media Cybernetics). The percent collagen deposition in the media or adventitia was calculated by dividing the Picrosirius Red–positive area by the medial area of the corresponding section.
Stiffness of the Aortic Walls
To measure the mechanical properties of the aorta, we used a device that we developed (Muromachi Kikai, Japan). Briefly, we cut aortic rings with a width of 0.6 mm from the descending thoracic aorta (Figure III in the online-only Data Supplement), as this segment is frequently affected by AD. Also, this segment is devoid of major aortic branches that affect the mechanical properties. For the same reason, aortic rings with intercostal arteries were excluded from the measurement. During the preparation of the aortic rings, the ventral side of the aorta was painted with dye to identify the orientation. The aortic ring was immersed in calcium-free PBS containing 10 mmol/L 2,3-butanedione monoxim to suppress smooth muscle cell (SMC) contraction19 and mounted on 2 tungsten rods (0.25 mm diameter) oriented to the lateral sides of the aorta. The tungsten rods were pulled apart at a speed of 0.1 mm/s until the aortic ring was broken. This was recorded by video and the passive force documented. Because we used aortic rings, rather than linearized strips, half of the measured force reflects the wall stress. We simultaneously measured the force development and the displacement of the aortic ring from the maximal length without force development. As an index of aortic wall stiffness, we calculated the mean force/displacement ratio from 0 to 5 mN, which corresponds to ≈0 to 80 mm Hg of blood pressure provided that the aortic ring diameter, the wall thickness, and the width are 0.8, 0.075, and 0.6 mm, respectively.
Cell Culture Experiments
Mouse aortic SMCs (No. JCRB0150) were obtained from the National Institutes of Biomedical Innovation, Health and Nutrition (Tokyo, Japan) and maintained in DMEM with high glucose and fetal bovine serum. Confluent SMCs were serum starved for 24 hours before starting the experiments. Serum-starved SMCs were stimulated with or without 25 ng/mL recombinant mouse IL-17A (R&D Systems, Minneapolis, MN) for 24 hours, followed by 10 ng/mL recombinant mouse TGFβ (transforming growth factor-beta)-1 (R&D Systems) for 1 hour. Cellular proteins were solubilized in radioimmunoprecipitation buffer and subjected to immunoblotting.
Data Acquisition and Statistical Analysis
Animals were randomly assigned to the experimental groups. The data were acquired by researchers or technicians who were blinded to the genetic modification and experimental intervention of the mice. All data are expressed as means±SEs. Statistical analyses were performed with GraphPad PRISM 5 (GraphPad Software). When the data passed the D’Agostino and Pearson normality test and Bartlett test for equal variances, we performed an unpaired t test for the 2 experimental groups or 1-way ANOVA to compare ≥3 groups, followed by Bonferroni multiple comparison correction. For non-normal data distributions, we performed the Mann-Whitney U test for 2 experimental groups or Kruskal-Wallis test followed by Bonferroni multiple comparisons test for ≥3 groups. Significance was indicated by a 2-sided P<0.05.
Results
Mouse Model of AD
In this study, we intended to analyze the molecular changes preceding AD development and during aortic wall destruction after AD development. We modified a previously reported AD model20 to induce more gradual changes. We simultaneously infused BAPN and Ang II using osmotic minipumps, which allowed us to examine the time course of molecular and pathological changes from normal aorta to AD over 14 days of BAPN+Ang II administration (Figure 1). Histological analysis of AD in this model revealed disruption of the tunica media with intramural hematoma, a hallmark of human AD, and infiltration of inflammatory cells mainly in the tunica adventitia. The incidence of AD and aortic wall lesions due to AD increased with BAPN dose (Figure 1B), and we deliberately chose 150 mg/kg per day BAPN, which caused AD in ≈90% of mice and aortic rupture in ≈30% of mice, allowing the detection of either a better or worse outcome. Three-dimensional analysis of this model revealed progressive destruction of the aortic walls compatible with AD.17 Transcriptome analysis 3 days after starting BAPN+Ang II, but before the onset of AD, revealed the induction of 1722 (6.03%) probes and the suppression of 1207 (4.23%) probes among the 28 562 probes with reliable signals (Figure 1C). To test the relevance of this mouse AD model to human AD, we performed annotation analysis for the previously reported transcriptome for aortas with congenital bicuspid aortic valve,21 as patients with bicuspid aortic valve are prone to AD. The bicuspid aortic valve aorta exhibited significant induction of 366 (1.58%) genes and the suppression of 477 (1.22%) genes among 30 121 probes with reliable signals compared with aortic samples with normal tricuspid aortic valves (Figure 1C). We analyzed the functional annotation of these datasets using Database for Annotation, Visualization, and Integrated Discovery (Tables I and II in the online-only Data Supplement).18 Our BAPN+Ang II model shared common gene annotations with bicuspid aortic valve: cell proliferation, cell migration, morphogenesis, and smooth muscle regulation (Table 1). These gene expression profiles in AD model were also consistent with those observed in human AD,22 indicating that this model shares various aspects of human AD. In addition, BAPN+Ang II caused an inflammatory response, which is likely to contribute to the rapid development of AD in this model.20
Figure 1.
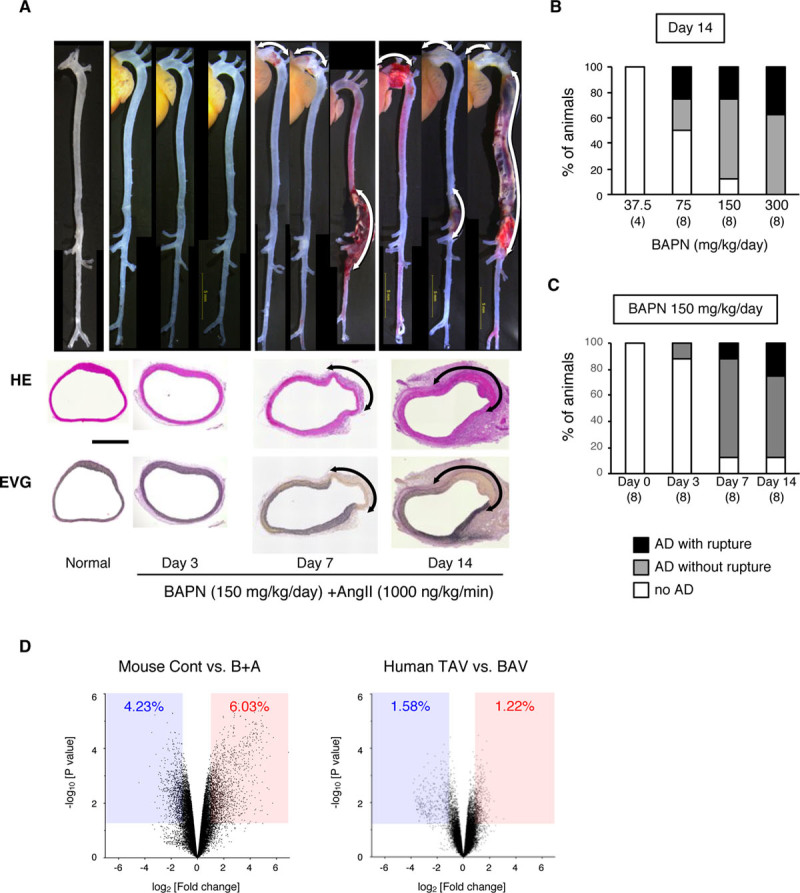
Mouse model of aortic dissection (AD). The mouse AD model was created by continuous infusion of β-aminopropionitrile (BAPN) and Ang II (angiotensin II) for 14 d. A, Representative macroscopic and histological images from hematoxylin-eosin (HE) and Elastica van Gieson (EVG) staining taken before (normal) and after BAPN+Ang II (B+A) treatment. Scale bar=500 μm. B, The incidence of AD for the indicated doses of B+A (1000 ng/kg per min). C, The incidence of AD by 150 mg/kg per d BAPN and 1000 ng/kg per min Ang II. The numbers of mice in each group are shown in parentheses. D, Volcano plots comparing the control (Cont; without any challenge) and B+A AD models, and human aortas with a normal tricuspid aortic valve (TAV) and congenitally abnormal bicuspid aortic valve (BAV). The blue-shaded area and the red-shaded area indicate the genes with significant (P<0.05) suppression (fold change, <0.5) and induction (fold change, >2), respectively.
Table 1.
Enrichment Analysis of GO Terms in Transcriptomes of the Mouse AD Model and Human BAV Aorta

Effect of High-Salt Challenge on AD and the Involvement of IL-17
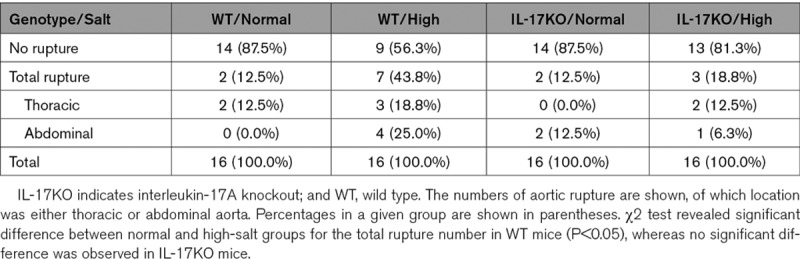
We tested the effect of high salt intake by giving AD mice 1% NaCl as drinking water 1 week before and during the BAPN+Ang II infusion and made comparisons with the normal-salt-intake group. The high-salt challenge did not affect the systolic blood pressure or pulse rate of the mice (Figure IV in the online-only Data Supplement). The length of aortic wall destruction due to AD was significantly greater in the aortic arch, descending thoracic aorta, and total aorta in the high-salt group compared with the normal-salt group (Figure 2A and 2B; Figure V in the online-only Data Supplement). Recent studies have proposed that high salt intake activates IL-17 via the induction of pathogenic Th17 cells.11,12 In the normal-salt condition, the length of aortic wall destruction was comparable between WT and IL-17KO mice. However, the worsening of AD by high-salt challenge was abolished in IL-17KO mice (Figure 2A and 2B; Figure V in the online-only Data Supplement). The aortic rupture was also increased by high-salt challenge only in WT mice (Table 2). These findings indicate that the worsening effect of high salt on AD was dependent on IL-17A.
Figure 2.
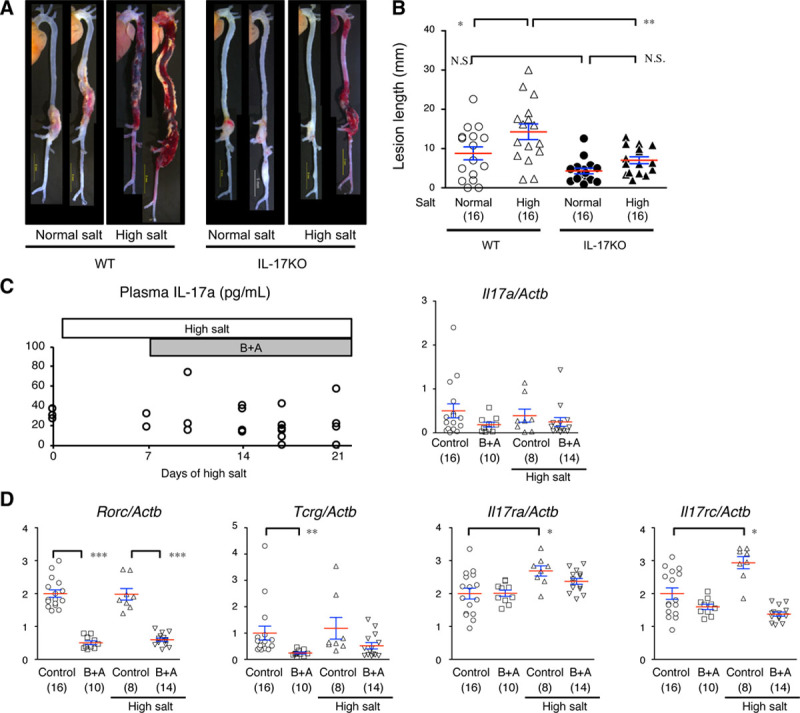
Effect of high-salt intake on an aortic dissection (AD) model in WT (wild type) or IL-17KO (IL [interleukin]-17A knockout) mice. A, Representative macroscopic images for the WT and IL-17KO mice given normal drinking water or high-salt water (1% NaCl, high-salt challenge). B, The AD lesion lengths (mm). Data are from 16 mice in each experimental group. C, The plasma concentration of IL-17A (pg/mL) and Il17a mRNA expression levels in the AD model. D, The mRNA levels of Il17ra, Il17rc, Rorc, and Tcrg determined by quantitative reverse transcription polymerase chain reaction before and 1 d after starting the high-salt challenge and β-aminopropionitrile+angiotensin II (B+A) treatment. The mRNA expression levels were normalized for Actab and are shown relative to the control without any experimental interventions. The number of mice in each group is shown in parentheses. The means (red lines)±SEs (blue lines) are shown. *P<0.05, **P<0.01, ***P<0.001. N.S. indicates not significant.
Table 2.
Effect of High Salt on Fatal Aortic Rupture
To examine whether high-salt challenge activated the IL-17 pathway, we examined the plasma concentration of IL-17A and mRNA expression of Il17a in the aortic tissue (Figure 2C). However, neither high salt nor BAPN+Ang II induced the expression of IL-17A. Next, we examined the mRNA expression levels of Rorc, a marker of Th17, and Tcrg, a marker of γδT cells, and the IL-17A cognate receptor genes Il17ra and Il17rc in the mouse aorta (Figure 2D). High-salt challenge showed no effect, and BAPN+Ang II suppressed these markers in IL-17–producing cell types. High-salt challenge induced IL-17 receptors, but BAPN+Ang II suppressed this expression. These findings indicate that high-salt or BAPN+Ang II challenge did not induce IL-17–producing cells in the aortic tissue or increase the plasma IL-17A concentration, but it modulated the gene expression of IL-17 receptors.
Effect of High-Salt Challenge on IL-17 Downstream Signaling and Transcriptome
We examined whether the modulation of the expression of IL-17 receptors by high-salt challenge resulted in activation of the NFκB (nuclear factor-kappa B) pathway, canonical downstream signaling in the IL-17 pathway. High-salt challenge for 10 days did not result in changes to the expression of total or phosphorylated (activated) NFκB in the aorta (Figure 3A). In the transcriptome analyses, high-salt challenge caused changes in the expression of only 0.5% of the detected genes with or without 3 days of BAPN+Ang II or IL-17KO (Figure 3B). Considering that BAPN+Ang II—an AD-inducing stimuli—resulted in changes in the expression of ≈10% of the genes (Figure 1C), it was unlikely that high-salt challenge caused fundamental changes in the aortic tissue response to the AD-inducing stimuli. The annotation analysis for the high salt–sensitive genes revealed only weak enrichment of xenobiotic metabolism (enrichment score, 2.04; Table 3), which may not have direct involvement in AD pathogenesis or IL-17 signaling. Therefore, although high-salt challenge exacerbated AD in an IL-17A–dependent manner and modulated the expression of IL-17 receptors, it was unlikely to have a direct effect on the downstream signaling or aortic tissue response in the IL-17 pathway.
Figure 3.
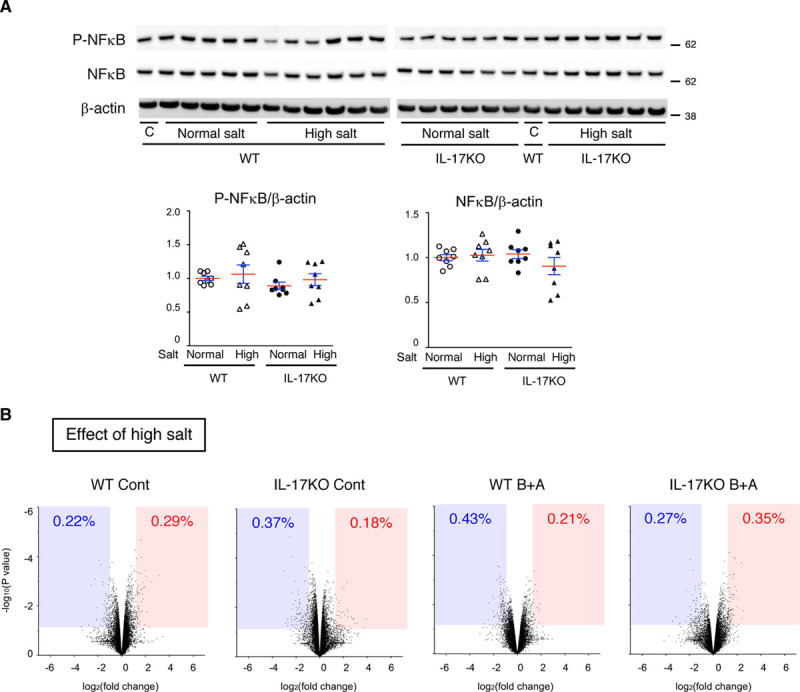
Effect of high-salt challenge on NFκB (nuclear factor-kappa B) and the transcriptome. A, Representative immunoblots and quantitative analyses for activated (phosphorylated) NFκB (P-NFκB), total NFκB, and β-actin, which served as the internal loading control. WT (wild type) and IL-17KO (IL [interleukin]-17A knockout) mice were given normal (normal salt) or 1% NaCl (high salt). An identical sample (C) was loaded on both electrophoresis gels, for WT samples and IL-17KO samples, so that the relative expression levels could be determined. The means (red lines)±SEs (blue lines) are shown. B, Volcano plots comparing the normal and high-salt conditions in WT mice without (WT Cont) or with β-aminopropionitrile (BAPN)+Ang II (angiotensin II; WT B+A) and IL-17KO mice without (IL-17KO Cont) or with BAPN+Ang II (IL-17KO B+A). The blue-shaded area and the red-shaded area indicate the genes with significant (P<0.05) suppression (fold change, <0.5) and induction (fold change, >2), respectively.
Table 3.
Enrichment Analysis of Transcriptomes With High-Salt Challenge and IL-17KO

Impact of IL-17KO on the Aorta Gene Expression Profile
We examined whether IL-17KO has an impact on the aortic tissue in the transcriptome analysis. Unexpectedly, IL-17KO alone resulted in changes to the expression of 14.0% of the detected genes (Figure 4A). We performed the gene annotation analysis for the differentially expressed genes (Table 3) and revealed enrichment of morphogenesis-related genes in the aortas of IL-17KO mice (Table 3), suggesting that IL-17A plays a role in maintaining tissue architecture. Because ECM is essential for the aortic tissue architecture and abnormalities in the ECM participate in AD pathogenesis, we assessed 550 genes with Gene Ontology terms that include “extracellular matrix.” As indicated by the heat map of the hierarchical clustering analysis (Figure 4B; Table III in the online-only Data Supplement), aorta from IL-17KO mice exhibited differential expression of 237 of 550 ECM-related genes compared with aortas from WT mice at baseline. These findings suggest that IL-17A may be involved in ECM metabolism.
Figure 4.
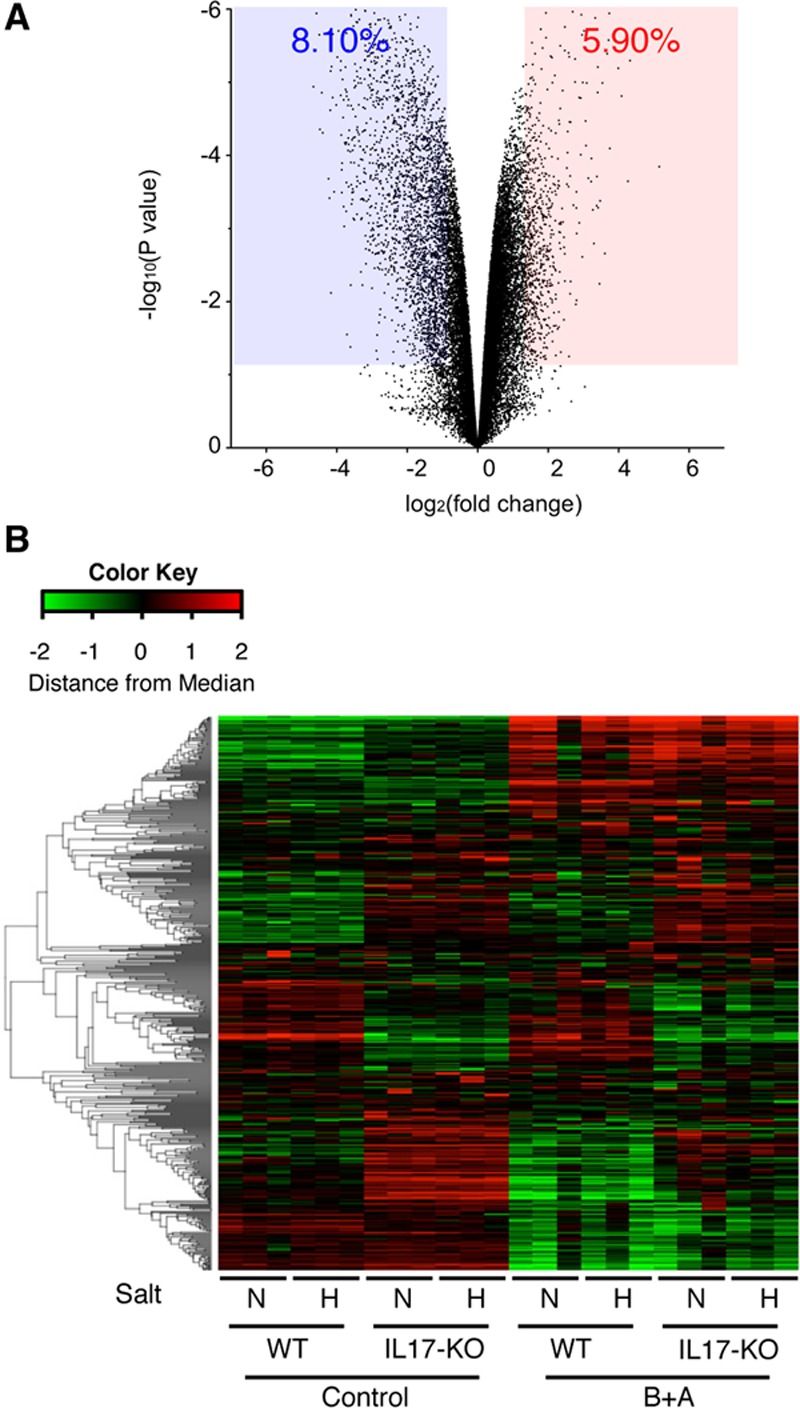
Expression of ECM (extracellular matrix) genes in the thoracic aorta. A, A volcano plot of the effect of Il17a deletion (IL-17KO [IL (interleukin)-17A knockout]) comparing WT (wild type) and IL-17KO mouse aorta without any challenge. The blue-shaded area and the red-shaded area indicate the genes with significant (P<0.05) suppression (fold change, <0.5) and induction (fold change, >2), respectively. B, Heat map of the hierarchical clustering analysis for genes with the annotation of extracellular matrix. After log conversion of the signal value, the distance from the median is shown in green, black, and red when lower, intermediate, and higher than those of other samples, respectively, within a given gene. The samples are from WT or IL-17KO mice treated with (H) or without (N) high-salt challenge and with or without β-aminopropionitrile+angiotensin II (B+A) challenge. None of the samples had aortic dissection.
Involvement of IL-17A in ECM Metabolism
To further investigate the impact of IL-17A on ECM metabolism before AD induction, we examined collagen fibers in the aorta using Picrosirius Red stain (Figure 5A and 5B). The aortic media in IL-17KO mice had higher collagen content than WT mice, regardless of the high-salt challenge. The observation of Picrosirius Red–stained aorta with polarized illumination revealed bright yellow to red color, indicative of the mature collagen in the adventitia of the aortas of WT mice (Figure 5C and 5D). Mature collagen was less abundant in the adventitia and more abundant in the media of IL-17KO mice. In contrast to collagen fibers, elastin fibers as detected by Elastica van Gieson staining were not different between WT and IL-17KO aortas (Figure 5E and 5F). These findings underscore the importance of IL-17 in ECM metabolism, especially of collagen fibers in the aorta.
Figure 5.
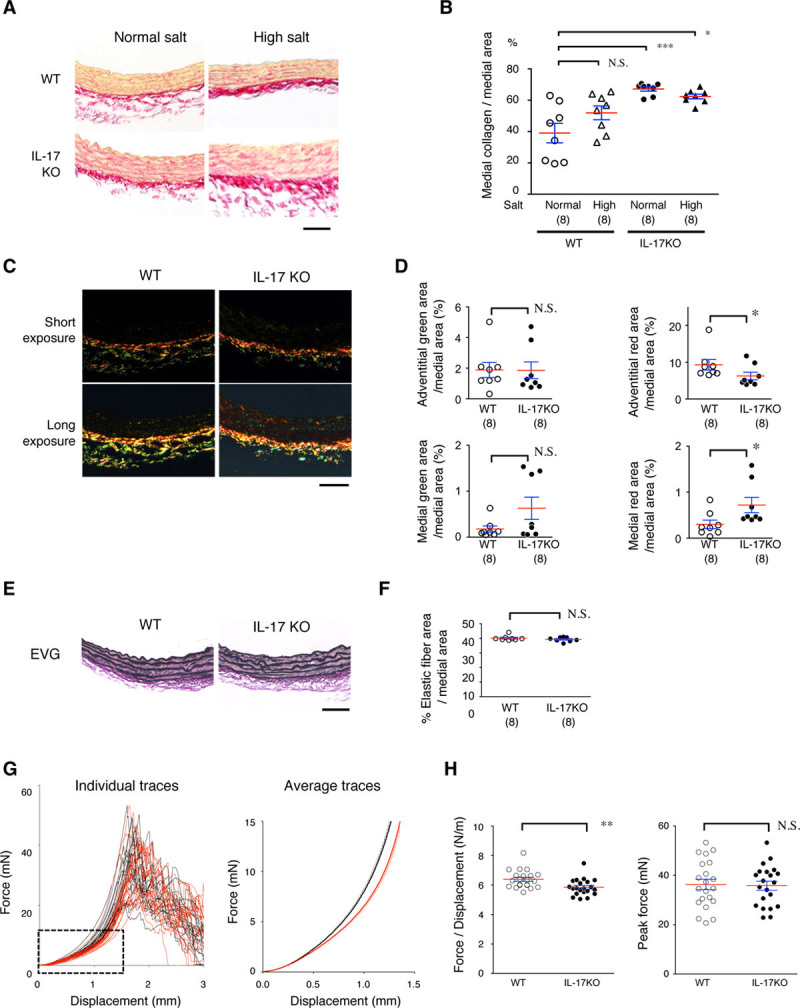
Histological and biomechanical analyses of aortic walls. A–D, Aortic samples were stained for collagen fibers by Picrosirius Red and observed by bright-field illumination (A and B) or polarized light illumination (C and D). Representative photomicrographs are shown with the luminal side of the aortic section facing up (A and C). Quantitative analysis of Picrosirius Red staining is shown for bright-field (B) and polarized light illumination (D) as the mean (red lines)±SE (blue lines). *P<0.05, ***P<0.001. E and F, Elastic lamellae were analyzed by Elastica van Gieson (EVG) staining (E) and their area shown relative to the medial area (F). Scale bars=50 μm. G and H, Biomechanical analysis of the excised aortic rings. G, Force-displacement curves are shown for the descending aorta from WT (wild type; black lines) and IL-17KO (IL [interleukin]-17A knockout) mice (red lines). The average force-displacement curve corresponds to the dotted rectangle in the plot of the individual traces. H, The force/displacement ratios, as an indicator of aortic wall stiffness, and the peak force (mN) for the breakage of aortas are shown. Data represent the mean (red lines)±SE (blue lines) of 21 observations in each experimental group. **P<0.01. N.S. indicates not significant.
We examined the mechanical properties of aortic rings excised from the descending aortas of WT and IL-17KO mice. We measured the passive force-displacement relationship as an index of the aortic wall stiffness and the peak force when the aortic rings broke (Figure 5G and 5H; Figure III in the online-only Data Supplement). The individual and averaged force-displacement curves of IL-17KO samples had a rightward shift compared with WT samples (Figure 5G). We calculated the mean force/displacement ratio from 0 to 5 mN, which approximately corresponds to 0 to 80 mm Hg of blood pressure. The mean force/displacement ratio was significantly lower in the aortas of IL-17KO mice compared with WT mice (Figure 5H). On the contrary, the peak force development, corresponding to the aortic ring breakage, did not differ between WT and IL-17KO mice. Therefore, the stiffness of aortas was significantly lower in IL-17KO mice than WT mice (0–80 mm Hg), but the ultimate tensile strength in the aorta did not differ between WT and IL-17KO mice.
Effect of IL-17KO on TGFβ Signaling in the Aorta
Next, we examined the Smad pathway, which plays a central role in ECM metabolism (Figure 6A and 6B). P-Smad2 or P-Smad3 did not show statistically significant difference among the experimental groups. As for Smad7, an inhibitory Smad, IL-17KO mice had significantly lower expression of Smad7 compared with WT animals in high-salt condition. Expression of total Smad2 or Smad3 did not change with high-salt challenge or in IL-17KO. In contrast, expression of Smad4 was significantly higher in IL-17KO aorta regardless of the high-salt challenge.
Figure 6.
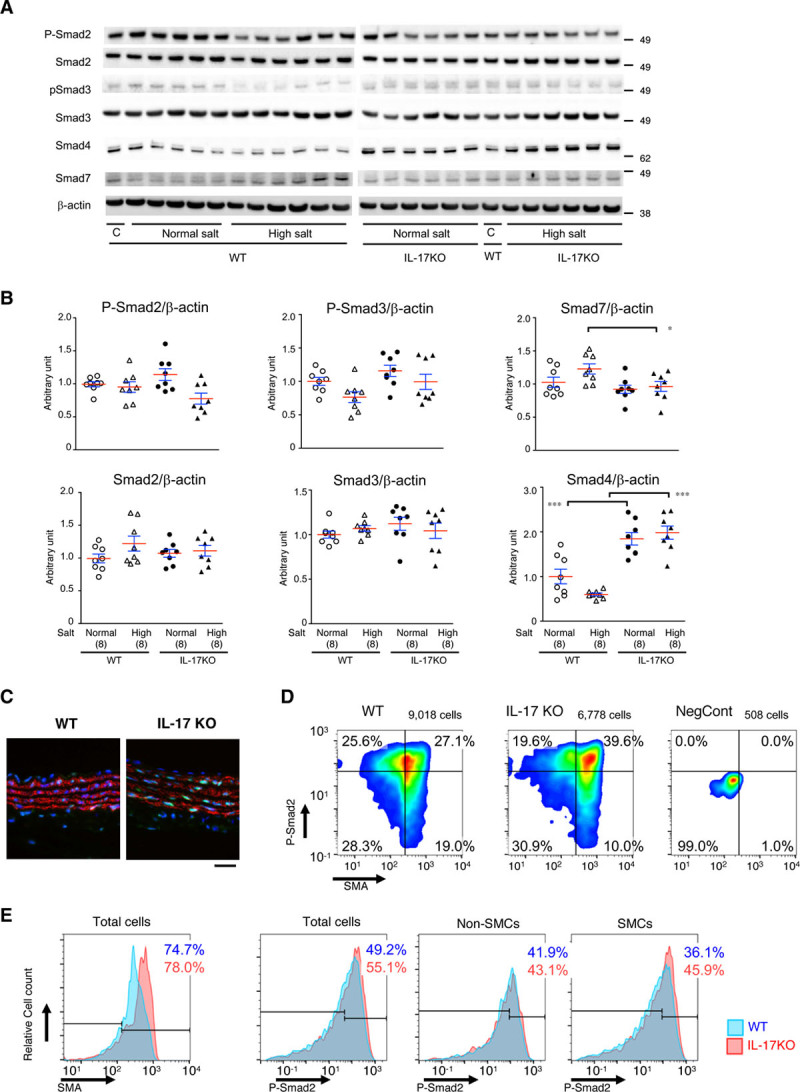
TGFβ (transforming growth factor-beta) signaling in the aortic walls. A, Representative immunoblots for total and activated (phosphorylated) Smad2 and Smad3, total Smad4, and Smad7, as well as β-actin, which served as the internal loading control. WT (wild type) and IL-17KO (IL [interleukin]-17A knockout) mice were given normal (normal salt) or 1% NaCl (high salt). C, An identical sample was loaded on both electrophoresis gels, for WT samples and IL-17KO samples, so the relative expression levels could be determined. B, Quantitative analyses for P-Smad2, total Smad2, P-Smad3, total Smad3, Smad4, and Smad7. *P<0.05, ***P<0.001. Data are shown for individual samples with the mean (red lines)±SE (blue lines) of 8 independent observations for each experimental group. C–E, TGFβ signaling at the single-cell level. Representative immunofluorescence images for 5-μm paraffin-embedded sections (C), the results of imaging cytometry as scattergrams (D), and histograms of SMA (smooth muscle α-actin) and P-Smad2 (E) are shown for sections of proximal descending aorta from WT or IL-17KO mice and for the negative control samples (NegCont) in which primary antibodies were omitted. Eight mice were assigned to each experimental group to obtain 2 aortic sections from a single mouse. The total cell population was divided into SMA-negative non–smooth muscle cells (SMCs) and SMA-positive SMCs by an arbitrarily determined SMA gate that was applied to all samples, followed by the analysis for P-Smad2. Blue and red numbers indicate the percentages of cell populations with high P-Smad2 signal from WT and IL-17KO aortas, respectively. Scale bar=50 μm.
To understand Smad signaling at the single-cell level, we performed immunofluorescent staining and imaging cytometry for nuclear P-Smad2 and SMA (smooth muscle α-actin)—a marker of SMCs (Figure 6C and 6D; Figures VII and VIII in the online-only Data Supplement). The cell population with nuclear P-Smad2 was 49.2% for aortas from WT mice and 55.1% for aortas from IL-17KO mice. For SMA-negative non-SMCs, the P-Smad2–positive cell population was comparable between WT (41.9%) and IL-17KO (43.1%) mice, whereas for SMA-positive SMCs, the P-Smad2–positive cell population was higher in IL-17KO (45.9%) than in WT (36.1%) mice. These findings suggest that IL-17A interferes with Smad signaling in SMCs.
Cross Talk of IL-17 and TGFβ Pathways in Aortic SMCs
We examined the cross talk between the IL-17A and TGFβ pathways in aortic SMCs in culture (Figure 7). Administration of IL-17A resulted in the transient activation of Jnk, indicating that SMCs were responsive to exogenous IL-17A. Administration of TGFβ1 caused prominent phosphorylation of both Smad2 and Smad3 in SMCs. Pretreatment of aortic SMCs with IL-17A for 24 hours resulted in no significant change in the phosphorylation of Smad2 or Smad3. However, IL-17A pretreatment significantly suppressed the subsequent activation of Smad2 and Smad3 by TGFβ1. Administration of IL-17A or TGFβ1 caused no significant changes in the expression of Smad2, Smad3, or Smad4. These findings demonstrate the suppressive effect of IL-17A on TGFβ signaling in aortic SMCs.
Figure 7.
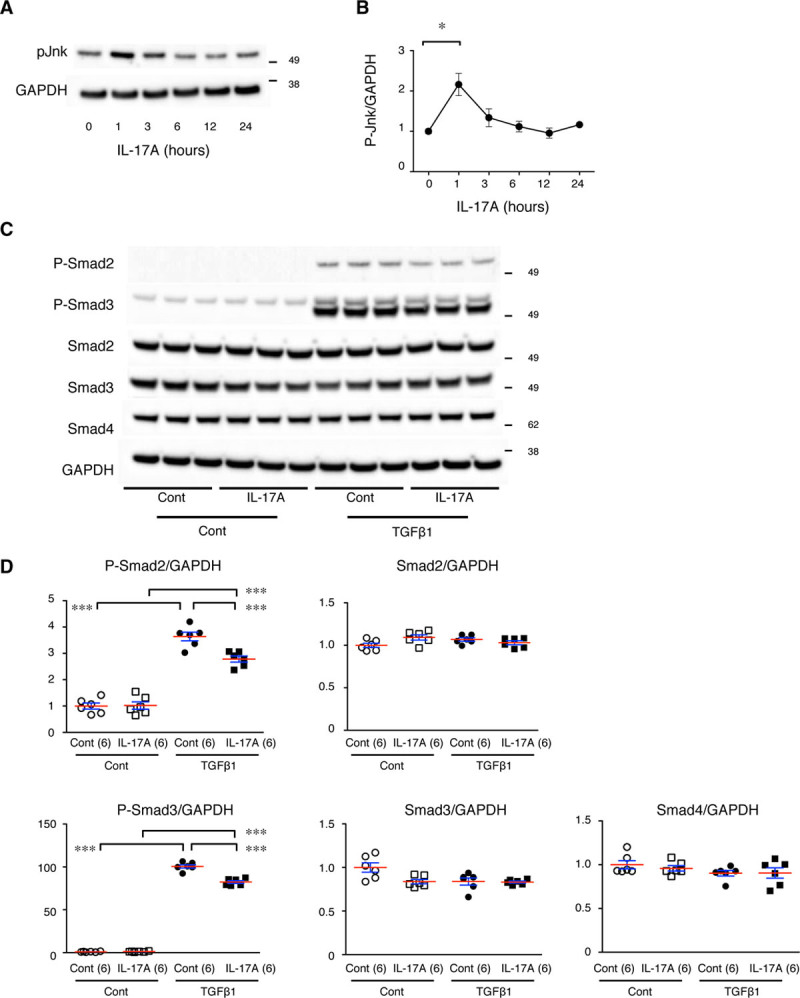
Cross talk between the IL (interleukin)-17 and TGFβ (transforming growth factor-beta) pathways in aortic smooth muscle cells (SMCs) in culture. A, Representative immunoblots for the time course of activated (phosphorylated) Jnk (P-Jnk) after IL-17A stimulation in aortic SMCs in culture. B, Quantitative analysis of P-Jnk. *P<0.05. C, Representative immunoblots for activated (phosphorylated) Smad2 (P-Smad2), P-Smad3, total Smad2, and total Smad3 with and without IL-17 pretreatment and TGFβ1 stimulation. GAPDH served as an internal loading control (Cont). D, Quantitative analyses of the immunoblots in C are shown as the mean (red lines)±SE (blue lines). ***P<0.001.
Phenotypic Modulation of SMCs and the Inflammatory Response in the Aorta
Vascular SMCs change their phenotype in response to various stress stimuli for homeostasis, adaptation, and tissue remodeling.23 Accordingly, we examined the expression of SMC phenotype markers SMemb and vimentin as synthetic phenotype markers and SM2 and calponin-1 as contractile phenotype markers (Figure 8). In WT mice, expression of SMemb tended to increase with high-salt challenge, BAPN+Ang II challenge, or both, and the induction was significant only with both challenges. In IL-17KO mice, the increase in SMemb expression on high-salt and BAPN+Ang II challenge was less significant compared with WT mice. SMemb expression was higher in IL-17KO aorta than in WT aorta at baseline. Expression of vimentin also increased with high-salt and BAPN+Ang II challenge in WT mice (P<0.01, ANOVA) but not IL-17KO mice (P=0.71, ANOVA). Expression of SM2 and calponin-1 did not significantly change with high-salt or BAPN+Ang II challenge or between WT and IL-17KO mice.
Figure 8.
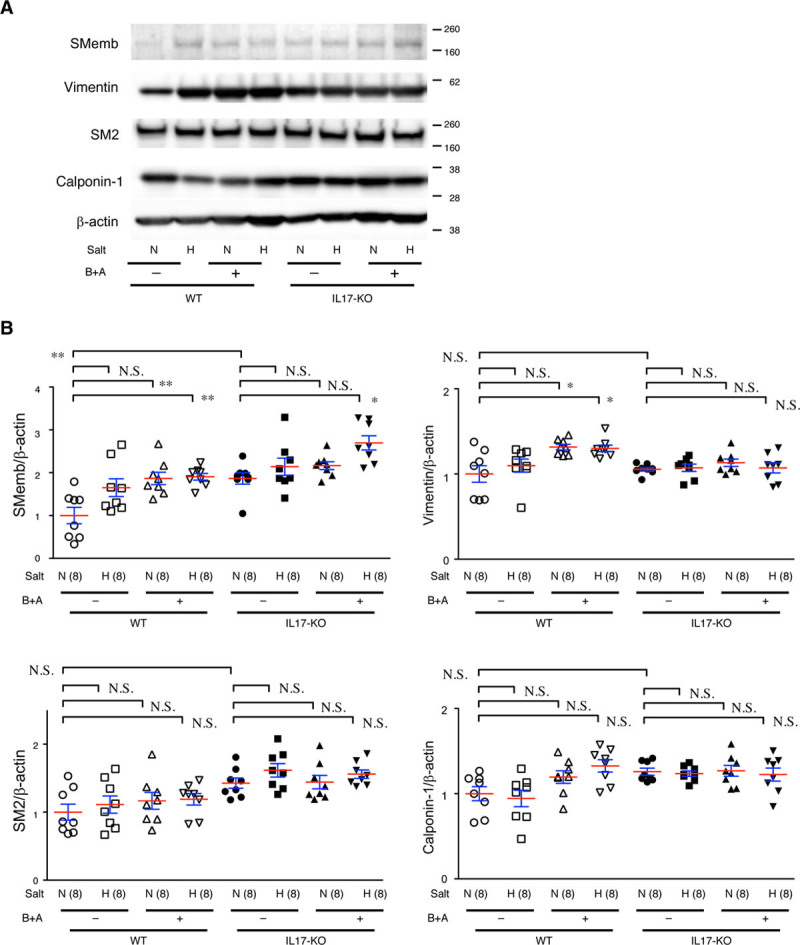
Smooth muscle cell (SMC) differentiation status in the aorta. Aortic samples from WT (wild type) and IL-17KO (IL [interleukin]-17A knockout) mice given normal (N) or high (H) salt and β-aminopropionitrile+angiotensin II (B+A) were immunoblotted for SMC differentiation markers, synthetic phenotype markers SMemb and vimentin, and contractile phenotype markers SM2 and calponin-1. β-actin served as the internal loading control. A, Representative images and (B) the quantitative analysis are shown. The means (red lines)±SEs (blue lines) are shown for 8 independent experiments for each group. *P<0.05, **P<0.01. N.S. indicates not significant.
We also examined the activation (phosphorylation) of Stat3 and Jnk—important signal mediators for aortopathies (Figure 9).3–6,20,24 Hig- salt challenge alone caused no significant changes, whereas BAPN+Ang II caused a slight increase in P-Stat3 or P-Jnk. Combination of high-salt and BAPN+Ang II challenge caused a significant increase in P-Stat3 in both WT and IL-17KO mice. As for P-Jnk, BAPN+Ang II and high-salt challenges caused significant increase in WT aorta but not in IL-17KO aorta. These findings suggest that while combination of BAPN+Ang II and high-salt challenges exaggerated proinflammatory response as shown by P-Stat3 and P-Jnk in the aorta, IL-17KO rendered the aorta less sensitive to the challenges in terms of the phenotypic modulation of SMCs.
Figure 9.
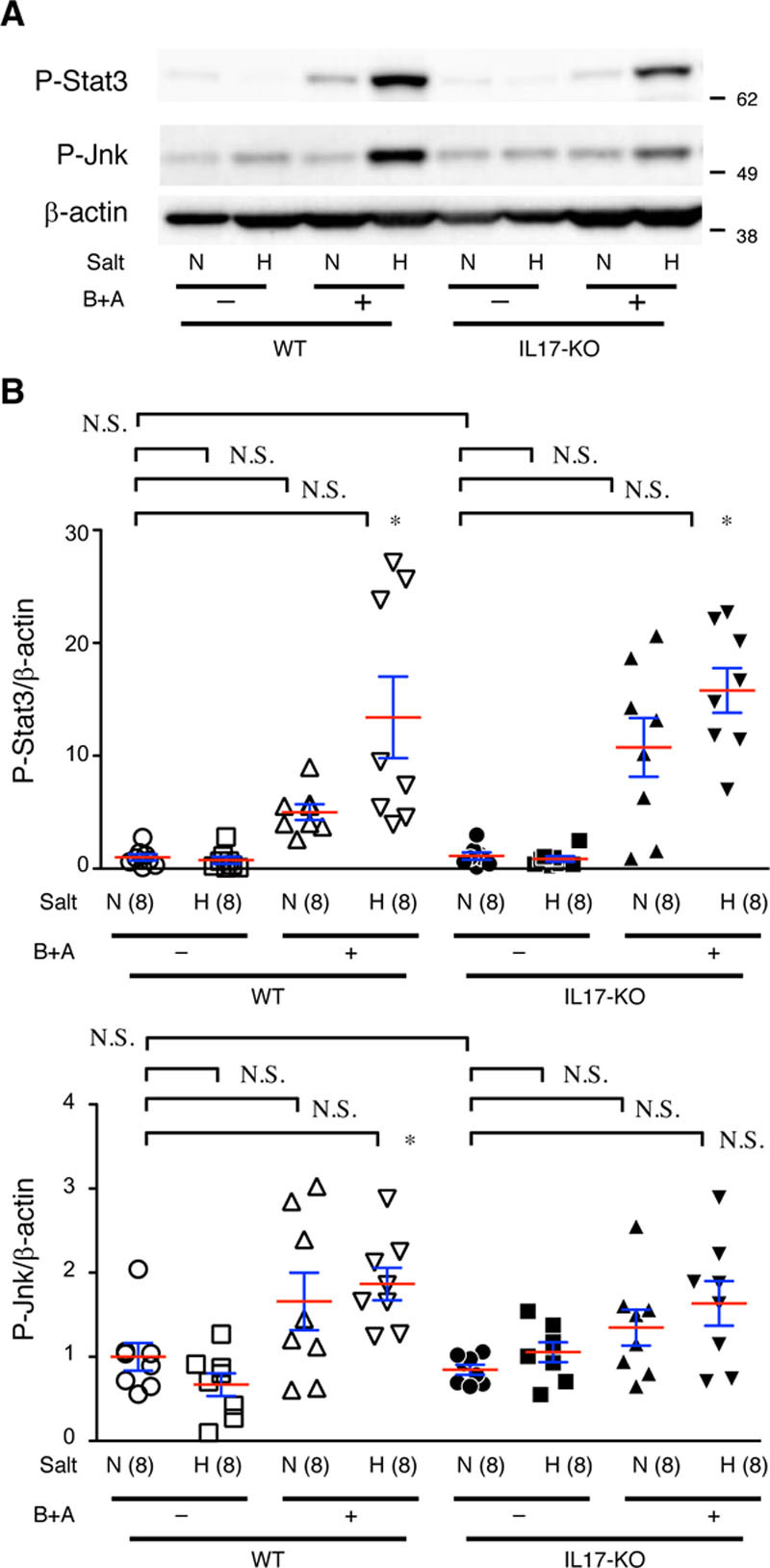
Proinflammatory signaling in the aorta. A, Representative images from immunoblotting for P-Stat3 and P-Jnk. B, Quantitative analysis. β-actin served as an internal loading control. WT (wild type) and IL-17KO (IL [interleukin]-17A knockout) mice were treated with (H) and without (N) high salt and β-aminopropionitrile+angiotensin II (B+A). The means (red lines)±SEs (blue lines) are presented for 8 independent experiments in each group. *P<0.05, **P<0.01. N.S. indicates not significant.
Discussion
In this study, we demonstrated that high-salt challenge worsened AD, which was dependent on the presence of IL-17A. Unexpectedly, IL-17KO resulted in less aortic stiffness, which was associated with a lesser stress response as assessed by phenotypic markers of SMCs without detectable changes in the inflammatory response in our experimental condition. High-salt challenge did not seem to alter the gross aortic response as assessed by transcriptome analysis. However, high-salt challenge augmented the BAPN+Ang II–induced activation of Jnk and Stat3 and phenotypic changes in SMCs, suggesting an enhanced stress on the aortic wall.
IL-17 plays a critical role in acute inflammation25 and is reported to participate in AD pathogenesis by promoting inflammation.4 However, IL-17KO did not alter the inflammatory response 3 days after BAPN+Ang II challenge in our AD model. This could be due to differences in the AD models; we induced AD by BAPN+Ang II infusion in young mice, whereas Brasier et al used Ang II infusion alone in aged mice,3,4 which may express higher levels of IL-17.26,27 Another possibility is that IL-17A may participate in inflammation in later stages of AD. Instead of regulating inflammation, the data indicated that IL-17A regulates nearly half of the 550 ECM-related genes. Although IL-17A has been reported to regulate matrix metalloproteinases in the context of inflammation,28–30 this is the first report that IL-17A participates in global ECM metabolism in normal physiology without obvious inflammation.
Another novel finding in this study is the cross talk between the IL-17 and TGFβ pathways. Pretreatment of cultured SMCs with IL-17A resulted in a blunted response of Smad2 and Smad3 activation by TGFβ1. In the chronic in vivo situation without IL-17A, nuclear localization of P-Smad2 was observed preferentially in SMCs. The nuclear localization of P-Smad2 may be explained by the altered expression of Smad4 that is essential for nuclear localization of Smad2/3.31 Alternatively, the TGFβ signaling may undergo direct interference by IL-17A, as observed in the cell culture experiments. Because TGFβ is a main regulator of ECM metabolism and SMC differentiation,32 altered TGFβ signaling may explain such changes in the ECM metabolism of IL-17KO aortas.
The global changes in gene expression and altered architecture of ECM are likely the basis for the reduced stiffness of the aortic wall in IL-17KO mice. Aortic stiffness is a risk factor for cardiovascular events33 and is associated with genetic and nongenetic aortopathies, including AD, in humans.34 In patients with Marfan syndrome, the stiffness predicts aortic dilation.35 Aortic stiffness is increased in the mouse Marfan model,36 and the increase in aortic stiffness is associated with increased propensity for AD.7 In this regard, it is noteworthy that the beneficial effect of IL-17KO was observed only in mice with high-salt challenge—a situation in which augmented hemodynamic stress has been reported because of an increase in plasma volume.10
High salt intake is proposed to be a risk factor for cardiovascular diseases, including hypertension37—a risk factor for AD.38 However, whether high salt intake is truly a risk factor for cardiovascular diseases is unclear.39 In addition, high salt intake has not been demonstrated to be a risk factor for AD. This could be because of the confounding factors and variability in individual physiology.39 In this regard, it is of interest that high salt intake promoted the AD model only in WT mice in this study but not in IL-17KO mice in which ECM metabolism and aortic stiffness are different. This finding can be interpreted as high salt intake being a risk factor for AD is dependent on the physiological conditions of the aorta, such as wall stiffness.
Taken together, our findings indicate that high salt intake worsened AD only in the presence of intact IL-17A expression. IL-17A plays a pivotal role in aortic wall homeostasis, possibly by modulating TGFβ signaling and ECM metabolism, resulting in stiffening of the aorta (Figure IX in the online-only Data Supplement). Further studies are required for risk stratification and management for AD predisposition, taking aortic wall stiffness and high salt intake into account.
Acknowledgments
We thank Kiyohiro, Watanabe, Yo, Koga, Inuzuka, Nishigata, Nakao, Shiramizu, Nakayama, and Yamamoto for their technical assistance.
Sources of Funding
This work was funded, in part, by grants from the Japan Society for the Promotion of Science (25861236 and 16K19973 to N. Nishida; 21390367, 24390334, 24659640, 26670621, and 16H05428 to H. Aoki); Daiichi Sankyo Foundation of Life Science, the Uehara Memorial Foundation (to H. Aoki), the Vehicle Racing Commemorative Foundation (to H. Aoki), Bristol-Myers Squibb (to H. Aoki); and TaNeDS grants from Daiichi Sankyo (to H. Aoki).
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- AD
- aortic dissection
- Ang II
- angiotensin II
- BAPN
- β-aminopropionitrile
- ECM
- extracellular matrix
- IL
- interleukin
- IL-17KO
- interleukin-17A knockout
- NFκB
- nuclear factor-kappa B
- SMA
- smooth muscle α-actin
- SMC
- smooth muscle cell
- TGFβ
- transforming growth factor-beta
- WT
- wild type
For Sources of Funding and Disclosures, see page 204.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.119.313336.
References
- 1.Crawford ES. The diagnosis and management of aortic dissection. JAMA. 1990;264:2537–2541. [PubMed] [Google Scholar]
- 2.Nienaber CA, Clough RE. Management of acute aortic dissection. Lancet. 2015;385:800–811. doi: 10.1016/S0140-6736(14)61005-9. doi: 10.1016/S0140-6736(14)61005-9. [DOI] [PubMed] [Google Scholar]
- 3.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ju X, Ijaz T, Sun H, Ray S, Lejeune W, Lee C, Recinos A, 3rd, Guo DC, Milewicz DM, Tilton RG, et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2013;33:1612–1621. doi: 10.1161/ATVBAHA.112.301049. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anzai A, Shimoda M, Endo J, Kohno T, Katsumata Y, Matsuhashi T, Yamamoto T, Ito K, Yan X, Shirakawa K, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 6.Son BK, Sawaki D, Tomida S, Fujita D, Aizawa K, Aoki H, Akishita M, Manabe I, Komuro I, Friedman SL, et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat Commun. 2015;6:6994. doi: 10.1038/ncomms7994. doi: 10.1038/ncomms7994. [DOI] [PubMed] [Google Scholar]
- 7.Kimura T, Shiraishi K, Furusho A, Ito S, Hirakata S, Nishida N, Yoshimura K, Imanaka-Yoshida K, Yoshida T, Ikeda Y, et al. Tenascin C protects aorta from acute dissection in mice. Sci Rep. 2014;4:4051. doi: 10.1038/srep04051. doi: 10.1038/srep04051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohno-Urabe S, Aoki H, Nishihara M, Furusho A, Hirakata S, Nishida N, Ito S, Hayashi M, Yasukawa H, Imaizumi T, et al. The role of macrophage Socs3 in the pathogenesis of aortic dissection. J Am Heart Assoc. 2018;7:e007389. doi: 10.1161/JAHA.117.007389. doi: 10.1161/JAHA.117.007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strazzullo P, D’Elia L, Kandala NB, Cappuccio FP. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ. 2009;339:b4567. doi: 10.1136/bmj.b4567. doi: 10.1136/bmj.b4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordaillat M, Reboul C, Gaillard V, Lartaud I, Jover B, Rugale C. Plasma volume and arterial stiffness in the cardiac alterations associated with long-term high sodium feeding in rats. Am J Hypertens. 2011;24:451–457. doi: 10.1038/ajh.2010.260. doi: 10.1038/ajh.2010.260. [DOI] [PubMed] [Google Scholar]
- 11.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nienaber CA, Fattori R, Mehta RH, Richartz BM, Evangelista A, Petzsch M, Cooper JV, Januzzi JL, Ince H, Sechtem U, et al. International Registry of Acute Aortic Dissection. Gender-related differences in acute aortic dissection. Circulation. 2004;109:3014–3021. doi: 10.1161/01.CIR.0000130644.78677.2C. doi: 10.1161/01.CIR.0000130644.78677.2C. [DOI] [PubMed] [Google Scholar]
- 14.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol. 2010;55:e27–e129. doi: 10.1016/j.jacc.2010.02.015. doi: 10.1016/j.jacc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Cronenwett JL, Johnston KW. Vascular Surgery. Philadelphia: Elsevier Health Sciences; 2014. [Google Scholar]
- 17.Logghe G, Trachet B, Aslanidou L, Villaneuva-Perez P, De Backer J, Stergiopulos N, Stampanoni M, Aoki H, Segers P. Propagation-based phase-contrast synchrotron imaging of aortic dissection in mice: from individual elastic lamella to 3D analysis. Sci Rep. 2018;8:2223. doi: 10.1038/s41598-018-20673-x. doi: 10.1038/s41598-018-20673-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Waurick R, Knapp J, Van Aken H, Bokník P, Neumann J, Schmitz W. Effect of 2,3-butanedione monoxime on force of contraction and protein phosphorylation in bovine smooth muscle. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:484–492. doi: 10.1007/pl00005380. doi: 10.1007/pl00005380. [DOI] [PubMed] [Google Scholar]
- 20.Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, Itoh H, Hori S, Aikawa N, Okada Y. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126:3070–3080. doi: 10.1161/CIRCULATIONAHA.112.097097. doi: 10.1161/CIRCULATIONAHA.112.097097. [DOI] [PubMed] [Google Scholar]
- 21.Hirata Y, Aoki H, Shojima T, Takagi K, Takaseya T, Akasu K, Tobinaga S, Fukumoto Y, Tanaka H. Activation of the AKT pathway in the ascending aorta with bicuspid aortic valve. Circ J. 2018;82:2485–2492. doi: 10.1253/circj.CJ-17-1465. doi: 10.1253/circj.CJ-17-1465. [DOI] [PubMed] [Google Scholar]
- 22.Müller BT, Modlich O, Prisack HB, Bojar H, Schipke JD, Goecke T, Feindt P, Petzold T, Gams E, Müller W, et al. Gene expression profiles in the acutely dissected human aorta. Eur J Vasc Endovasc Surg. 2002;24:356–364. doi: 10.1053/ejvs.2002.1731. doi: 10.1053/ejvs.2002.1731. [DOI] [PubMed] [Google Scholar]
- 23.Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018;52:48–64. doi: 10.1016/j.cellsig.2018.08.019. doi: 10.1016/j.cellsig.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 25.von Vietinghoff S, Ley K. Interleukin 17 in vascular inflammation. Cytokine Growth Factor Rev. 2010;21:463–469. doi: 10.1016/j.cytogfr.2010.10.003. doi: 10.1016/j.cytogfr.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang JY, Lee SY, Rhee CK, Kim SJ, Kwon SS, Kim YK. Effect of aging on airway remodeling and muscarinic receptors in a murine acute asthma model. Clin Interv Aging. 2013;8:1393–1403. doi: 10.2147/CIA.S50496. doi: 10.2147/CIA.S50496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Angulo A, Faris R, Cavazos D, Jolly C, Daniel B, DeGraffenried L. Age-related alterations in T-lymphocytes modulate key pathways in prostate tumorigenesis. Prostate. 2013;73:855–864. doi: 10.1002/pros.22631. doi: 10.1002/pros.22631. [DOI] [PubMed] [Google Scholar]
- 28.Camargo LDN, Righetti RF, Aristóteles LRCRB, Dos Santos TM, de Souza FCR, Fukuzaki S, Cruz MM, Alonso-Vale MIC, Saraiva-Romanholo BM, Prado CM, et al. Effects of anti-IL-17 on inflammation, remodeling, and oxidative stress in an experimental model of asthma exacerbated by LPS. Front Immunol. 2017;8:1835. doi: 10.3389/fimmu.2017.01835. doi: 10.3389/fimmu.2017.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakashima T, Jinnin M, Yamane K, Honda N, Kajihara I, Makino T, Masuguchi S, Fukushima S, Okamoto Y, Hasegawa M, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. 2012;188:3573–3583. doi: 10.4049/jimmunol.1100591. doi: 10.4049/jimmunol.1100591. [DOI] [PubMed] [Google Scholar]
- 30.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe M, Masuyama N, Fukuda M, Nishida E. Regulation of intracellular dynamics of Smad4 by its leucine-rich nuclear export signal. EMBO Rep. 2000;1:176–182. doi: 10.1093/embo-reports/kvd029. doi: 10.1038/sj.embor.embor608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-beta family signaling in connective tissue and skeletal diseases. Cold Spring Harb Perspect Biol. 2017;9:a022269. doi: 10.1101/cshperspect.a022269. doi: 10.1101/cshperspect.a022269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavalcante JL, Lima JA, Redheuil A, Al-Mallah MH. Aortic stiffness: current understanding and future directions. J Am Coll Cardiol. 2011;57:1511–1522. doi: 10.1016/j.jacc.2010.12.017. doi: 10.1016/j.jacc.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Grillo A, Pini A, Marelli S, Gan L, Giuliano A, Trifiro G, Santini F, Salvi L, Salvi P, Viecca F, et al. 5b.05: Marfan syndrome: assessment of aortic dissection risk by analysis of aortic viscoelastic properties. J Hypertens. 2015;33(suppl 1):e67. [Google Scholar]
- 35.Nollen GJ, Groenink M, Tijssen JG, Van Der Wall EE, Mulder BJ. Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur Heart J. 2004;25:1146–1152. doi: 10.1016/j.ehj.2004.04.033. doi: 10.1016/j.ehj.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 36.Marque V, Kieffer P, Gayraud B, Lartaud-Idjouadiene I, Ramirez F, Atkinson J. Aortic wall mechanics and composition in a transgenic mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2001;21:1184–1189. doi: 10.1161/hq0701.092136. doi: 10.1161/hq0701.092136. [DOI] [PubMed] [Google Scholar]
- 37.Whelton PK. Sodium, blood pressure, and cardiovascular disease: a compelling scientific case for improving the health of the public. Circulation. 2014;129:1085–1087. doi: 10.1161/CIRCULATIONAHA.114.008138. doi: 10.1161/CIRCULATIONAHA.114.008138. [DOI] [PubMed] [Google Scholar]
- 38.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8:103–113. doi: 10.1038/nrcardio.2010.187. doi: 10.1038/nrcardio.2010.187. [DOI] [PubMed] [Google Scholar]
- 39.O’Donnell MJ, Mente A, Smyth A, Yusuf S. Salt intake and cardiovascular disease: why are the data inconsistent? Eur Heart J. 2013;34:1034–1040. doi: 10.1093/eurheartj/ehs409. doi: 10.1093/eurheartj/ehs409. [DOI] [PubMed] [Google Scholar]