

Supplemental Digital Content is available in the text.
Keywords: angiotensin II, glucagon-like peptide-1 receptor, hypertension, inflammation, liraglutide, oxidative stress
Objective:
Cardiovascular outcome trials demonstrated that GLP-1 (glucagon-like peptide-1) analogs including liraglutide reduce the risk of cardiovascular events in type 2 diabetes mellitus. Whether GLP-1 analogs reduce the risk for atherosclerosis independent of glycemic control is challenging to elucidate as the GLP-1R (GLP-1 receptor) is expressed on different cell types, including endothelial and immune cells.
Approach and Results:
Here, we reveal the cardio- and vasoprotective mechanism of the GLP-1 analog liraglutide at the cellular level in a murine, nondiabetic model of arterial hypertension. Wild-type (C57BL/6J), global (Glp1r−/−), as well as endothelial (Glp1rflox/floxxCdh5cre) and myeloid cell–specific knockout mice (Glp1rflox/floxxLysMcre) of the GLP-1R were studied, and arterial hypertension was induced by angiotensin II. Liraglutide treatment normalized blood pressure, cardiac hypertrophy, vascular fibrosis, endothelial dysfunction, oxidative stress, and vascular inflammation in a GLP-1R–dependent manner. Mechanistically, liraglutide reduced leukocyte rolling on the endothelium and infiltration of myeloid Ly6G−Ly6C+ and Ly6G+Ly6C+ cells into the vascular wall. As a consequence, liraglutide prevented vascular oxidative stress, reduced S-glutathionylation as a marker of eNOS (endothelial NO synthase) uncoupling, and increased NO bioavailability. Importantly, all of these beneficial cardiovascular effects of liraglutide persisted in myeloid cell GLP-1R-deficient (Glp1rflox/floxxLysMcre) mice but were abolished in global (Glp1r−/−) and endothelial cell–specific (Glp1rflox/floxxCdh5cre) GLP-1R knockout mice.
Conclusions:
GLP-1R activation attenuates cardiovascular complications of arterial hypertension by reduction of vascular inflammation through selective actions requiring the endothelial but not the myeloid cell GLP-1R.
Highlights.
In the present study, we reveal in a mouse model of arterial hypertension:
that GLP-1 (glucagon-like peptide-1) analogs reduce blood pressure and protect endothelial function independent of glucose control;
that GLP-1 analogs avoid uncoupling of eNOS (endothelial NO synthase) by reduction of vascular inflammation and oxidative stress;
that these protective effects by GLP-1 analogs require the endothelial but not myeloid cell GLP-1 receptor.
Arterial hypertension is one of the leading risk factors for atherosclerosis, which culminates in thrombo-embolic events such as myocardial infarction or stroke and thereby represents a leading cause of global mortality and morbidity with severe disability.1 Basic research and clinical trials provide substantial evidence for a key role of inflammation in arterial hypertension.2,3 In experimental studies, depletion of inflammatory cells in ATII (angiotensin II)-infused mice prevented increased blood pressure and endothelial dysfunction.4,5 Furthermore, the CANTOS trial (Canakinumab Antiinflammatory Thrombosis Outcome Study) revealed that anti-inflammatory treatment of cardiovascular disease with the monoclonal antibody canakinumab, targeting IL (interleukin)-1β, reduces major adverse cardiovascular events by 15% compared with placebo in patients with atherosclerotic cardiovascular disease.3 Thus, anti-inflammatory therapy has the potential to significantly lower rates of recurrent cardiovascular events.
GLP-1 (glucagon-like peptide-1) is a peptide hormone with known anti-inflammatory properties that was first discovered as a regulator of insulin release from β-cells in the pancreas, and synthetic analogs of GLP-1 are used for the treatment of type 2 diabetes mellitus. Clinical trials of GLP-1 analogs were designed to test the cardiovascular safety in patients with diabetes mellitus. Unexpectedly, long-acting GLP-1R (GLP-1 receptor) agonists reduced the risk of cardiovascular events in subjects with type 2 diabetes mellitus, with similar risk reduction (eg, LEADER trial [Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results] hazard ratio, 0.87 [95% CI 0.78–0.97] P=0.01) as observed for IL-1β blockade by canakinumab (hazard ratio, 0.85 [95% CI. 0.74–0.98] P=0.21).3,6,7 Other clinical trials investigating the effects of GLP-1 on cardiovascular disease demonstrated cardiovascular protective effects after myocardial infarction,8 reduction of blood pressure,9 and an improvement of endothelial dysfunction.10 Although the mechanisms are not fully understood, reduction of inflammation has been suggested to play an important role for cardiovascular protection by GLP-1 analogs.
Multiple experimental studies have shown that native GLP-1 and pharmacological GLP-1R agonists reduce cardiac and vascular inflammation (for review, see Drucker et al11). Our group demonstrated that the GLP-1 analog liraglutide improves survival in lipopolysaccharide-induced endotoxemia by improving vascular function and reducing systemic and vascular inflammation12 and that these protective effects are lost in global Glp1r−/− mice with sepsis.13 In cell culture experiments, Krasner et al14 showed that liraglutide lowers the induction of VCAM-1 (vascular cell adhesion molecule-1) and E-cadherin by lipopolysaccharide and reduces adhesion of THP-1 (Tohoku hospital pediatrics-1) monocytes to human aortic endothelial cells. Furthermore, animal studies revealed that GLP-1R agonists, including native GLP-1, exendin-4, and liraglutide, attenuate the development of atherosclerosis in murine models.11 GLP-1R agonists are known to reduce white blood cell infiltration and adhesion to the vascular wall in mouse models of atherosclerosis and cardiac dysfunction.15–17 However, the precise mechanism(s) mediating the cardiovascular protective effects of GLP-1 remains poorly understood, and their elucidation is complicated by the fact that the canonical GLP-1R is expressed on several cell types in the vasculature, heart, and on immune cells.15,18,19 Additionally, analysis of GLP-1R expression is complicated by poor specificity and sensitivity of many antibodies (for review see Pujadas et al20). Previous studies reported an essential role of myeloid cells4 and endothelial activation together with eNOS (endothelial NO synthase) dysfunction for the pathogenesis of hypertension.21 To pursue the underlying mechanisms, we have now studied genetic mouse models inactivating the Glp1r in myeloid or endothelial cell lineages.
Here, we show that liraglutide confers antioxidant and anti-inflammatory effects through mechanisms requiring the endothelial GLP-1R. These findings may have relevance for ongoing efforts directed at understanding clinical observations linking GLP-1R agonists to reduction of major adverse cardiovascular events.
Materials and Methods
The authors declare that all supporting data are available within the article and its online-only Data Supplement.
Animals and In Vivo Treatment
All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the US National Institutes of Health and approval was granted by the Ethics Committee of the University Hospital Mainz and the Landesuntersuchungsamt Rheinland-Pfalz (Koblenz, Germany; permit number: 23 177-07/G 17-1-050). The studies were performed in male C57BL/6J, global Glp1r−/−,22 and endothelial (Glp1rflox/floxxCdh5cre, Glp1r ec−/−) as well as myelomonocytic (Glp1rflox/floxxLysMcre, Glp1r my−/−) cell–specific GLP-1R knockout mice (8–12 weeks old).23,24 According to Payne et al,23 a nonspecific cre recombinase activity has been noted in hematopoietic cells for the Cdh5-cre alleles. In the recent study, unintended deletion of the GLP-1R in Glp1r ec−/− mice other than endothelial cells was excluded by evaluation of Glp1r mRNA in mouse white blood cells (Figure IIIF in the online-only Data Supplement). Animals were housed under a 12-hour light/dark cycle in the institutional animal facility with free access to standard diet and water. Effects of sexual hormones on vascular function are known.25 To exclude the latter effects in the present study, experiments were performed in male mice only. For detailed description, see online-only Data Supplement.
Detection of Oxidative Stress
Oxidative stress was detected in whole blood, heart, and vascular tissue.26,27 For detailed description, see online-only Data Supplement.
Noninvasive Blood Pressure Recordings
Noninvasive blood pressure recording was performed by tail-cuff to determine systolic blood pressure with a Coda Monitor System (Kent Scientific), which has been validated against telemetry monitoring.28 For detailed description, see online-only Data Supplement.
S-Glutathionylation of eNOS and Western Blotting
eNOS was immunoprecipitated, and the precipitate was subsequently immunoblotted for S-glutathionylation.29 For detailed description, see online-only Data Supplement.
Electron Paramagnetic Resonance Spectroscopy
Electron paramagnetic resonance spectroscopy was used to assess aortic NO synthesis/bioavailability using colloid Fe(II)-diethyldithiocarbamate (Fe[DETC]2) as spin trap.30 For detailed description, see online-only Data Supplement.
Determination of Nitrate and Nitrite
Nitrite and nitrate were measured by ENO-20 NOx analyzer (Eicom Corporation, Tokyo, Japan), based on the liquid chromatography method with postcolumn derivatization with Griess reagent.31 For detailed description, see online-only Data Supplement.
Intravital Fluorescence Microscopy
A real-time imaging software (NIS Elements, Nikon) was used for image acquisition and analysis. Cell recruitment was quantified in 6 fields of view (100μm×150μm) per femoral vein.32 For detailed description, see online-only Data Supplement.
Isometric Tension Recordings
For vascular reactivity studies, 4 mm thoracic aortic rings free of perivascular fat were mounted on force transducers in organ bath chambers and preconstricted with prostaglandin F2α (2 μM) to reach ≈80% of the maximal tone induced by KCl bolus. Concentration-relaxation curves were recorded in response to the endothelium-dependent vasodilator acetylcholine (1 nM–3.3 μM).33
Reverse Transcription Polymerase Chain Reaction
The acid guanidinium thiocyanate-phenol-chloroform extraction method was used for RNA isolation.34 For detailed description see online-only Data Supplement.
Statistics
Data are expressed as means±SEM. Statistical calculations were performed with GraphPad Prism 7 (GraphPad Software, Inc). The unpaired Student t test was used after testing for normality and equal variance (Shapiro-Wilk test) to compare 2 groups. One-way ANOVA and Bonferroni or Tukey post hoc test were used to compare multiple groups if the data followed a normal distribution, otherwise nonparametric Kruskal-Wallis and Dunn post hoc tests were used. Two-way ANOVA (with Bonferroni correction for comparison of multiple means) was used for comparisons of vasodilator potency and efficacy in response to the endothelium-dependent vasodilator acetylcholine. A limitation of the study is a reduced sample size in some experiments in which only 3 to 4 animals were used. P<0.05 were considered statistically significant and marked by asterisks (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001).
Results
Liraglutide Improves Blood Pressure and Cardiac Hypertrophy in ATII-Induced Arterial Hypertension Independent of Glucose/Insulin Modulation
Chronic ATII infusion in C57BL/6J mice increased systolic blood pressure associated with development of cardiac hypertrophy. These effects of ATII infusion were completely normalized by administration of the GLP-1 analog liraglutide (Figure 1A through 1C). Liraglutide led to a significant reduction in body weight after 7 days of treatment, whereas ATII infusion alone showed no significant effect on body weight (Figure 1D). Neither ATII infusion nor liraglutide treatment had any significant effect on glucose homeostasis, as assessed by unaltered blood glucose and plasma insulin levels after an oral glucose tolerance test (Figure 1E and 1F, Figure IH and II in the online-only Data Supplement). Expression of Glp1r mRNA in aorta was not affected by ATII but was significantly elevated in response to liraglutide treatment (Figure IA in the online-only Data Supplement). Thus, liraglutide reduces blood pressure without impacting glucose homeostasis in nondiabetic mice with ATII-induced arterial hypertension, leading to amelioration of cardiac hypertrophy.
Figure 1.
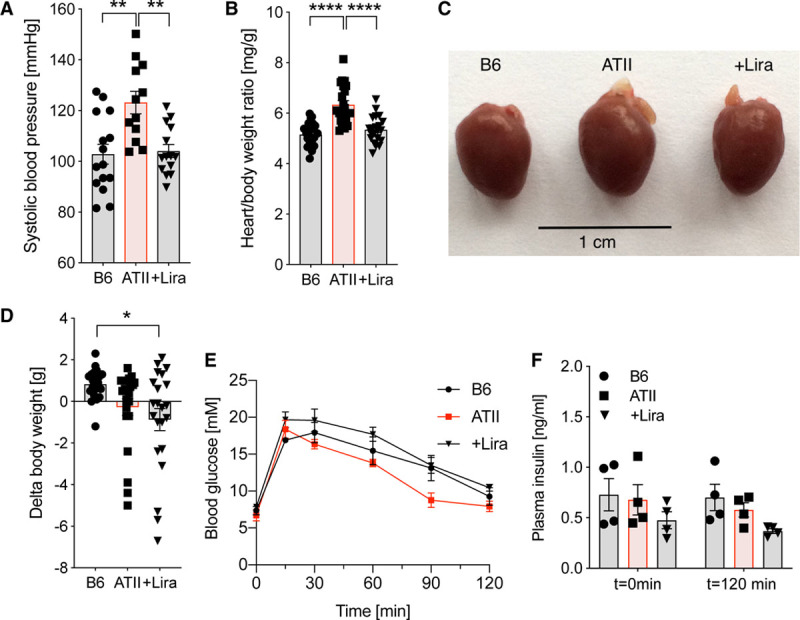
Liraglutide (Lira) improves blood pressure and cardiac hypertrophy in ATII (angiotensin II)-induced arterial hypertension independent of glucose/insulin modulation. A, In wild-type mice (C57BL/6J), systolic blood pressure was measured by noninvasive blood pressure recording on day 6 after beginning of ATII infusion. B, Heart/body weight ratio is shown next to (C) representative pictures of mouse hearts to demonstrate ATII-induced cardiac hypertrophy. D, Animals were weighed before and after 7 d of treatment to determine Δbody weight. E, Fasting blood glucose levels and (F) insulin levels after oral glucose tolerance testing were evaluated in whole blood/plasma after 7 d of treatment. One-way ANOVA and Bonferroni multiple comparison test; N=12–15 (A), 21–22 (B and D), 4 (E and F) animals per group. Data are means±SEM. *P<0.05; **P<0.01; ****P<0.0001.
Liraglutide Reduces Cardiac and Whole Blood Oxidative Stress by Suppression of Inflammation and NADPH Oxidase Activity in ATII-Induced Arterial Hypertension
Hypertensive mice had significantly higher levels of reactive oxygen species in whole blood and cardiac tissue, accompanied by elevated levels of 3-NT (3-nitrotyrosine) positive proteins in the heart, all of which were reduced by liraglutide treatment (Figure 2A through 2C). In addition, liraglutide significantly decreased ATII-induced oxidative stress in cardiac tissue caused by an increase of Nox (NADPH oxidase) activity (Figure 2D), reduced elevated levels of ADMA (asymmetrical dimethylarginine) known to promote dysregulation of the eNOS (Figure 2E), and attenuated F4/80 expression in cardiac tissue as an indicator for infiltration of immune cells into the heart (Figure 2F). Thus, liraglutide reduces cardiac oxidative stress in ATII-induced hypertensive heart disease.
Figure 2.
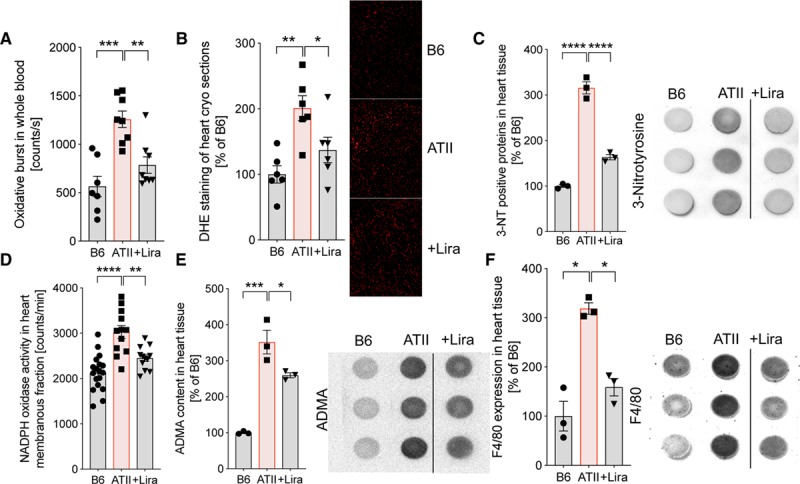
Liraglutide (Lira) reduces cardiac and whole blood oxidative stress by suppression of inflammation and NADPH oxidase activity in ATII (angiotensin II)-induced arterial hypertension. A, In wild-type mice (C57BL/6J), whole blood oxidative burst was determined by chemiluminescence (8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H)dione sodium salt [L-012]) after zymosan A stimulation. B and C, Cardiac formation of reactive oxygen species was visualized by dihydroethidium (DHE) staining in cryosections of the left ventricle and dot blot analysis of cardiac tissue (left ventricle) using a specific antibody against 3-nitrotyrosine (3-NT). Representative photomicrographs (red fluorescence, superoxide formation) and representative dot blot images are shown beside the densitometric analysis. D, The activity of NADPH oxidase in isolated membranous fraction of cardiac tissue was examined by lucigenin-enhanced chemiluminescence. E, Asymmetrical dimethylarginine (ADMA) as an indicator for eNOS (endothelial NO synthase) dysfunction and (F) F4/80 levels as a readout for inflammation were determined by dot blot analysis in cardiac tissue. Representative dot blot images are shown beside the densitometric analysis. One-way ANOVA and Bonferroni multiple comparison test; N=7–8 (A), 6 (B), 3 (C), 12–18 (D) and 3 (E and F) animals per group. Data are means±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Liraglutide Improves ATII-Induced Endothelial Dysfunction and Vascular Fibrosis by Reduction of Vascular Oxidative Stress and Recoupling of eNOS
ATII-infused mice exhibited impaired endothelial function, and Sirius red staining revealed significant vascular fibrosis, a contributor to vascular stiffness. Treatment with liraglutide reduced the development of these adverse vascular effects in ATII-treated mice (Figure 3A and 3D), whereas liraglutide treatment had no effect on vascular function in nonhypertensive animals (Figure IB in the online-only Data Supplement). ATII-infused mice showed eNOS uncoupling as revealed by a marked increase in S-glutathionylation of the enzyme and further supported by L-NAME (L-NG-nitroarginine methyl ester) inhibitable endothelial dihydroethidium staining (Figure 3B and 3C), consistent with the extent of endothelial dysfunction (Figure 3A) and impaired NO bioavailability (Figure 3E). These adverse effects of ATII infusion were all normalized by liraglutide treatment (Figure 3A through 3E). eNOS protein expression was not different between the groups (Figure IG in the online-only Data Supplement). Plasma levels of nitrate (NO3−) were increased in hypertensive mice, indicative of pronounced oxidative break-down of NO and/or iNOS (inducible NO synthase) activity, which was further supported by increased mRNA expression of Nox2 and iNOS (or Nos2) in the aorta of ATII-infused mice (Figure 3F through 3H). Liraglutide administration significantly reduced the inflammatory burden reflected by lowered Nox2 and Nos2 mRNA levels in aorta and attenuated nitrate plasma levels (Figure 3F through 3H). Thus, the GLP-1 analog liraglutide displays antioxidant and anti-inflammatory properties that help to prevent eNOS uncoupling, resulting in higher NO bioavailability and vascular protection in arterial hypertension.
Figure 3.
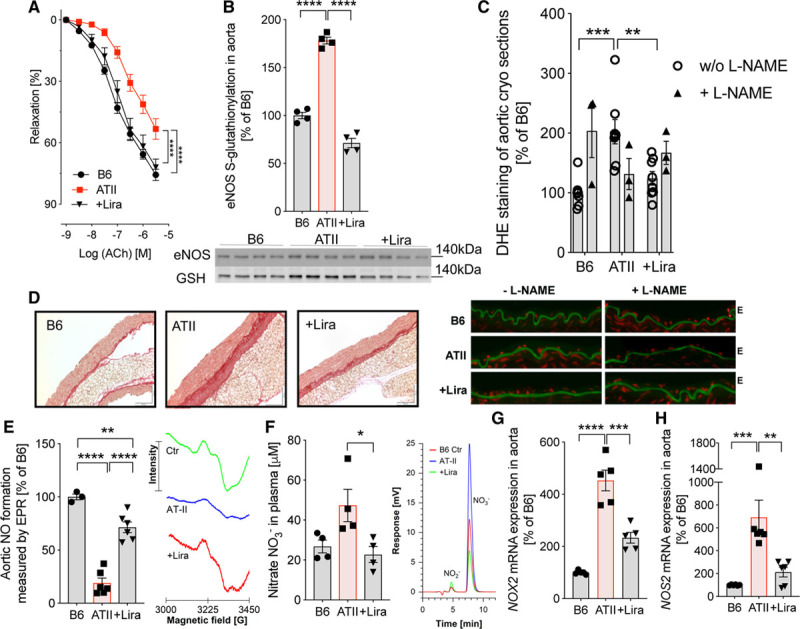
Liraglutide (Lira) improves ATII (angiotensin II)-induced endothelial dysfunction and vascular fibrosis by recoupling of eNOS (endothelial NO synthase) and reduction of vascular oxidative stress. A, In wild-type mice (C57BL/6J), vascular function was evaluated by endothelium (E)-dependent (acetylcholine [ACh]) relaxation of aortic rings (isometric tension studies). Two-way ANOVA and Bonferroni multiple comparison test; N=16–28 animals per group. B, S-glutathionylation of eNOS as a readout for uncoupling of the enzyme was measured by using a specific glutathione antibody (GSH) after immunoprecipitation of eNOS. Representative Western blot images are shown below the densitometric analysis. C, The specific NOS-inhibitor L-NAME (L-NG-nitroarginine methyl ester) was used in aortic cryosections stained with dihydroethidium (DHE) to detect reactive oxygen species (ROS) and to uncover eNOS uncoupling. Representative photomicrographs are shown below the densitometric analysis. Green, laminae (autofluorescence); red fluorescence, superoxide formation. D, Representative (out of 3–4) Sirius red stainings of aorta are shown to demonstrate vascular fibrosis. E, Electron paramagnetic resonance spectroscopy revealed NO levels in aorta. Representative electron paramagnetic resonance spectroscopy (EPR) spectra showing the resulting iron-nitrosyl triplet signal are shown besides the quantification bar graph. F, High-performance liquid chromatography (HPLC) was used to detect nitrate (NO3−) in plasma. Representative chromatograms are shown beside the quantification bar graph. G and H, NADPH oxidase 2 (Nox2) and inducible NO synthase (iNOS/Nos2) mRNA expression was measured by quantitative reverse transcription polymerase chain reaction in aortic tissue. One-way ANOVA and Bonferroni multiple comparison test; N=4 (B), 8 without and 3 with L-NAME (C), 3–6 (E), 4 (F), and 4–6 (G and H). Data are means±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Liraglutide Ameliorates ATII-Induced Inflammation of the Vasculature by Prevention of Ly6G−Ly6C+- and Ly6G+ Ly6C+ Cell Infiltration to the Vessel Wall
Intravital microscopy visualized a trend of increased rolling of leukocytes at the inner vascular wall in ATII-infused mice. Liraglutide administration significantly suppressed rolling of leukocytes in arterial hypertension (Figure 4A). Representative videos of intravital microscopy are available in the online-only Data Supplement. In support of an enhanced inflammatory phenotype in the aorta of hypertensive mice, nuclear factor κb (NF-κb; Figure 4B), tumor necrosis factor alpha (Tnfa) mRNA, and IL-1β protein expression, as well as transforming growth factor beta (Tgfb) and interferon gamma (Infg) mRNA expression (Figure IC through IF in the online-only Data Supplement) were markedly upregulated upon ATII infusion. Furthermore, mRNA transcripts corresponding to Vcam1, intercellular adhesion molecule-1 (Icam1) and Selp, encoding adhesion molecules (VCAM-1, ICAM-1 and P-selectin) and known downstream targets of NF-κb, were strongly upregulated in the aortas of ATII-infused mice (Figure 4C through 4E). This proinflammatory cascade responsible for leukocyte adhesion and migration was mitigated by liraglutide treatment (Figure 4B through 4E, Figure IC through IF in the online-only Data Supplement). Immunohistochemical staining of blood vessels revealed high expression of Ly6B.2 as an indicator for vascular inflammation in ATII-infused mice, which was normalized by liraglutide treatment (Figure 4F). Using flow cytometry, we identified specific subsets of immune cells, namely Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils, that infiltrated into the vascular wall of hypertensive animals, findings attenuated by liraglutide administration (Figures 4G and 4H). Thus, liraglutide ameliorates ATII-induced rolling and infiltration of leukocytes by downregulation of central proinflammatory mediators (NF-κb, TNF-α, and IL-1β) with concomitant reduced expression of adhesion molecules (VCAM-1, ICAM-1, and P-selectin).
Figure 4.
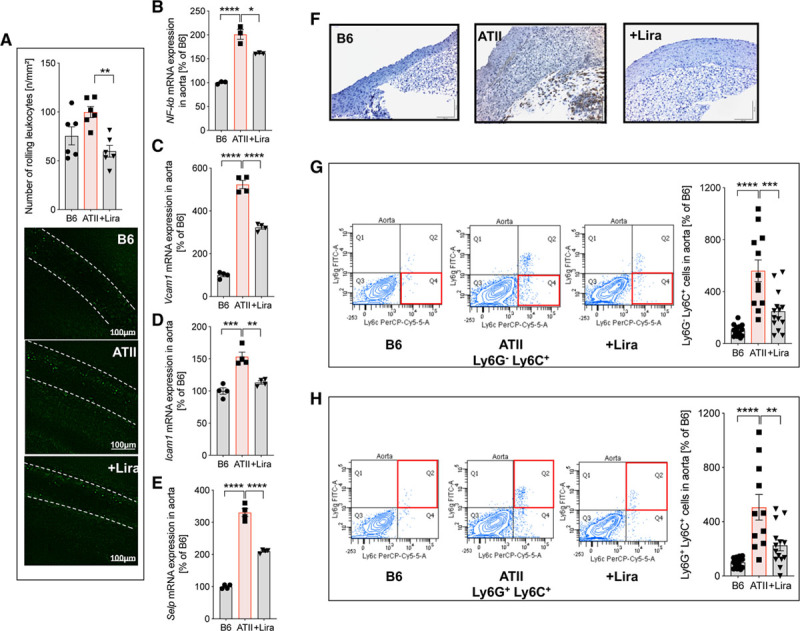
Liraglutide (Lira) ameliorates ATII (angiotensin II)-induced inflammation of the vasculature by prevention of Ly6G−Ly6C+- and Ly6G+ Ly6C+ cell infiltration to the vascular wall. A, In wild-type mice (C57BL/6J), rolling of leukocytes at the vascular wall (V. femoralis) was quantified by intravital microscopy. Nucleated cells were visualized by staining with acridine orange. Representative images at day 7 are shown below the quantification bar graph. Representative videos of intravital microscopy experiments are available in the online-only Data Supplement. B–E, mRNA expression of genes encoding for nuclear factor-κb (NF-κb) and adhesion molecules vascular cell adhesion molecule-1 (Vcam1), intercellular adhesion molecule-1 (Icam1), and P-selectin (Selp) in aorta were measured by quantitative reverse transcription polymerase chain reaction. F–H, Vascular inflammation was further characterized by immunohistochemistry and flow cytometry of aortic tissue. Representative pictures of 3–4 independent aortic Ly6 (Ly6B.2) immunohistochemical stainings (F; for IgG isotype control see Figure V in the online-only Data Supplement) and flow cytometry of Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils in aortic cell suspensions (G and H) are shown. Representative original plots of Ly6G−Ly6C+ and Ly6G+Ly6C+ cells are shown beside the quantification bar graph (for gating strategy, see Figure IV in the online-only Data Supplement). One-way ANOVA and Bonferroni multiple comparison test; N=6 (A), 3 (B–E) and 12-14 (G and H) animals per group. Data are means±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Canonical GLP-1R Expressed on Endothelial Cells, Not on Myeloid Cells, Is Essential for Cardiovascular Protection by Liraglutide.
Global Glp1r−/− mice exhibited impaired endothelial function compared to C57BL/6J mice, which was further deteriorated by ATII infusion; treatment with liraglutide did not improve acetylcholine-dependent vasodilation in the absence of a functional canonical GLP-1R (Figure 5A). Similarly, liraglutide did not reduce vascular oxidative stress in ATII-infused Glp1r−/− mice (Figure 5B). Furthermore, liraglutide failed to normalize and even deteriorated levels of aortic Nox2, Nos2, and Tnfa mRNA as well as cardiac Nox2 activity in ATII-infused Glp1r−/− mice (Figures 5C through 5F). Collectively, these findings suggest that the cardiovascular protection seen in C57BL/6J mice is mediated via canonical GLP-1R activation.
Figure 5.
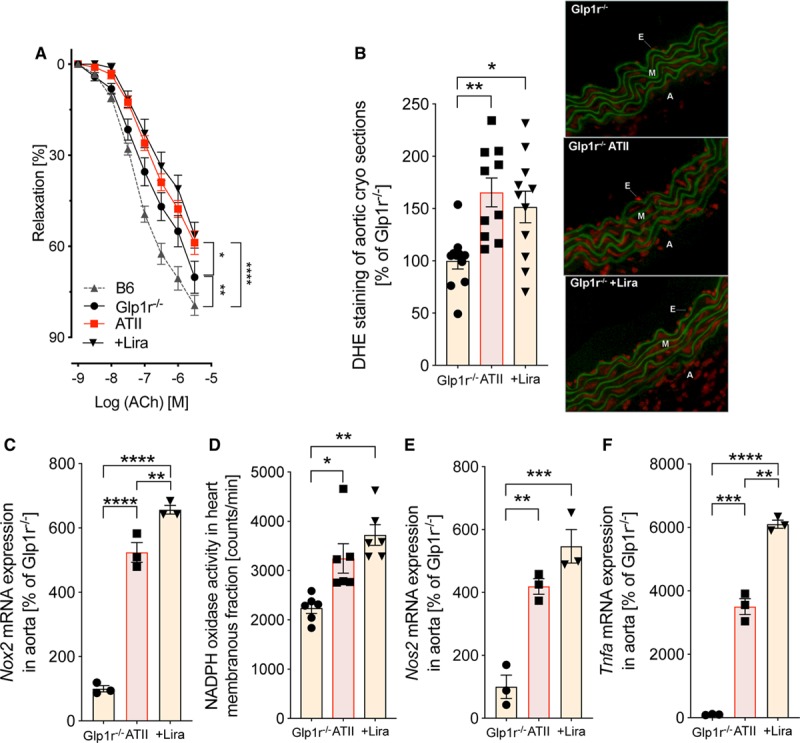
The canonical GLP-1 (glucagon-like peptide-1) receptor is essential for vascular protection by liraglutide (Lira) in mice with arterial hypertension. A, In global GLP-1 receptor knockout mice (Glp1r−/−), vascular function was evaluated by endothelium (E)-dependent (acetylcholine [ACh]) relaxation of aortic rings (isometric tension studies). Two-way ANOVA and Bonferroni multiple comparison test; N=8–10 animals per group. B, Aortic cryosections stained with dihydroethidium (DHE) were used to detect reactive oxygen species in the vascular wall. Representative photomicrographs are shown below the densitometric analysis. Green, laminae (autofluorescence); red fluorescence, superoxide formation. C, Aortic mRNA expression of NADPH oxidase 2 (Nox2) was evaluated by quantitative reverse transcription polymerase chain reaction (rtPCR). D, Activity of all NADPH oxidase isoforms was measured by chemiluminescence (lucigenin) in cardiac membranous fraction. E and F, Inducible NO synthase (iNOS/Nos2) and Tnfa mRNA expression were measured by quantitative rtPCR in aortic tissue. One-way ANOVA and Bonferroni multiple comparison test; N=10–11 (B), 3 (C), 6 (D) and 3 (E and F) animals per group. Data are means±SEM. A indicates adventitia; ATII, angiotensin II; and M, media. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
To better define the cellular target of vascular-protective liraglutide signaling through GLP-1R, we generated endothelial cell–specific (Glp1r ec−/−) and myeloid cell–specific (Glp1r my−/−) GLP-1R knock-out mice. Glp1r mRNA in bone marrow–derived macrophages of myeloid-specific knockout mice (Glp1r my−/−) was not detectable compared to the background strain (LysMcre mice; Figure 6A). Moreover, ATII infusion and liraglutide treatment did not alter nonfasting blood glucose in Glp1r ec−/− and Glp1r my−/− mice (Figures IIA and IIIA in the online-only Data Supplement). However, liraglutide reduced body weight in both mouse strains (Figures IIB and IIIB in the online-only Data Supplement).
Figure 6.
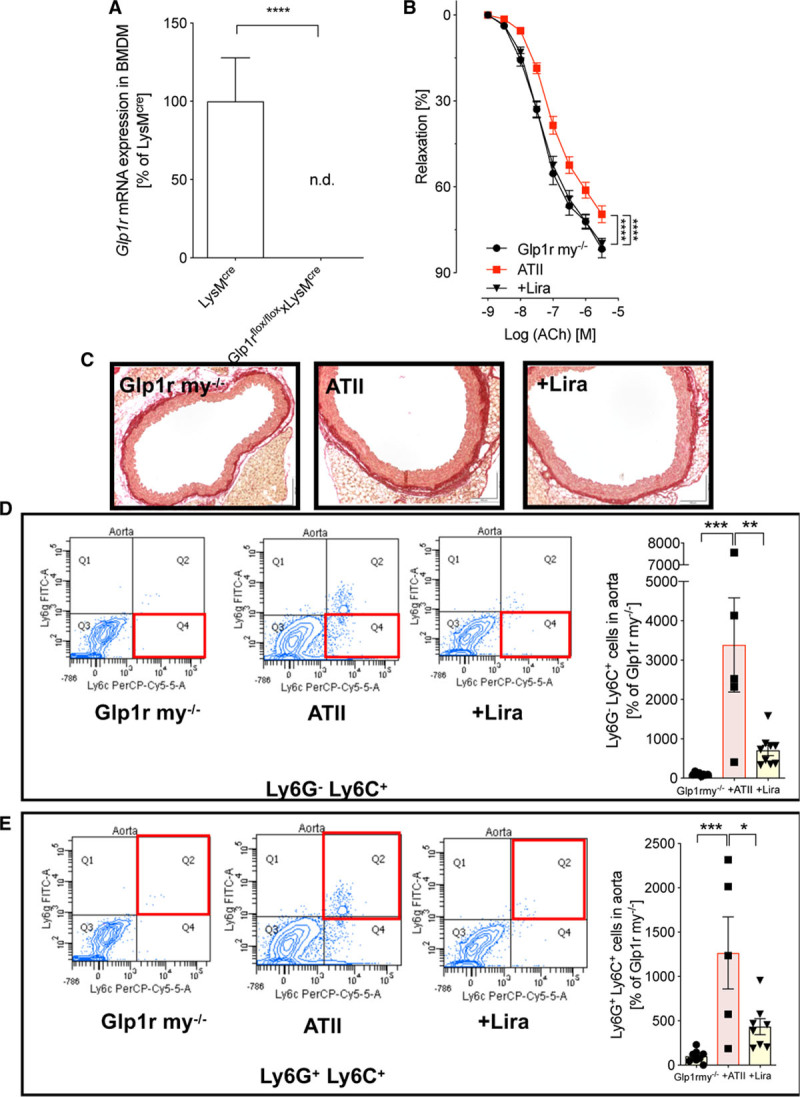
The canonical GLP-1 (glucagon-like peptide-1) receptor expressed on myeloid cells (LysM+) is not responsible for cardiovascular protection by liraglutide (Lira). A, In myeloid cell (LysM+)-specific GLP-1 receptor knockout mice (Glp1rflox/floxxLysMcre, Glp1r my−/−), mRNA expression of the Glp1r was measured in bone marrow–derived macrophages (BMDM) and expression levels are shown in relation to the background strain (LysMcre mice). Student t test; N=3 animals per group. B, Vascular function was evaluated by endothelium-dependent (acetylcholine [ACh]) relaxation of aortic rings (isometric tension studies). Two-way ANOVA and Bonferroni multiple comparison test; N=16–22 animals per group. C, Representative (out of 3–4) Sirius red staining of aortic sections to assess vascular fibrosis. D and E, Flow cytometry of aortic cell suspensions identified Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils inside the vascular wall. Representative original plots of Ly6G−Ly6C+ and Ly6G+Ly6C+ cells are shown beside the quantification bar graph (for gating strategy, see Figure IV in the online-only Data Supplement). One-way ANOVA and Bonferroni multiple comparison test; N=5–9 animals per group. Data are means±SEM. ATII indicates angiotensin II; FITC, fluorescein isothiocyanate; and n.d., not detectable. *P<0.05; **P<0.01; ***P<0.001, ****P<0.0001.
Given the demonstrated reduction of leukocyte infiltration by liraglutide treatment in the wild-type mice, we first analyzed the response to ATII infusion in Glp1r my−/− mice. Unlike findings in the global Glp1r−/−, ATII-induced vascular dysfunction was evident in Glp1r my−/− mice, and endothelial responsiveness was restored by liraglutide (Figure 6B). In addition, cardiac hypertrophy (Figure IIC in the online-only Data Supplement), vascular fibrosis (Figure 6C), and myeloid cell infiltration of the vessel wall induced by ATII infusion was markedly reduced by liraglutide treatment (Figure 6D and 6E). Hence, GLP-1R expression in myeloid cells is not required for the cardiovascular protective effect of liraglutide.
Mouse lung endothelial cells of Glp1r ec−/− (Glp1rflox/floxxCdh5cre) mice contained about 90% less Glp1r mRNA compared with the background strain (Cdh5cre mice), and a knockdown of Glp1r in white blood cells was ruled out (Figure 7A, Figure IIIF in the online-only Data Supplement). In contrast to Glp1r my−/− mice, liraglutide failed to improve endothelial function (Figure 7B) or reduce blood pressure (Figure 7C) in hypertensive Glp1r ec−/− mice. Vascular fibrosis (Figure 7D) and cardiac hypertrophy (Figure IIIC in the online-only Data Supplement) was aggravated by ATII infusion and not significantly improved by liraglutide treatment in Glp1r ec−/− mice, in contrast to findings in Glp1r my−/− mice. Furthermore, liraglutide failed to prevent upregulation of Nox2, Vcam1, and Tnfa mRNA (Figure 7E and Figure IIID and IIIE in the online-only Data Supplement) as well as higher nitrate levels indicative of oxidative NO break-down and iNOS activity (Figure 7F) in hypertensive Glp1r ec−/− mice. Associated with these signs of persistent endothelial dysfunction, the infiltration of Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils was not significantly reduced by liraglutide treatment of ATII-infused Glp1r ec−/− mice (Figure 7G and 7H). Thus, the cardiovascular protection of liraglutide in mice with ATII-induced vascular dysfunction is predominantly mediated via the endothelial GLP-1R.
Figure 7.
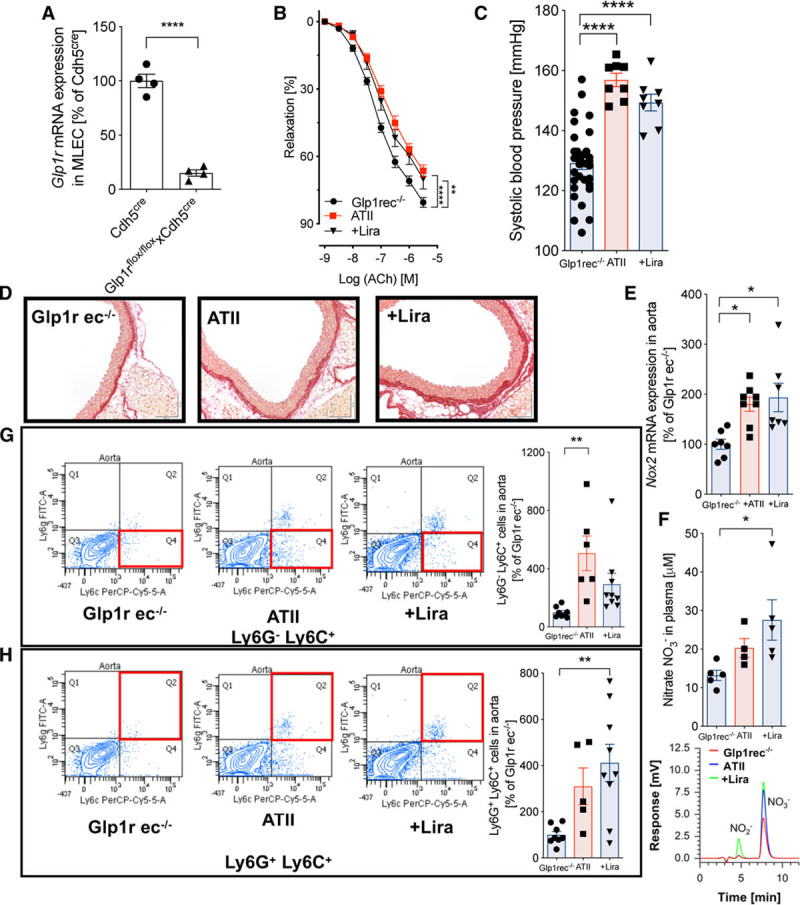
The canonical GLP-1 (glucagon-like peptide-1) receptor expressed on endothelial cells is essential for cardiovascular protection by liraglutide (Lira). A, In endothelial cell–specific GLP-1 receptor knockout mice (Glp1rflox/floxxCdh5cre, Glp1r ec−/−), mRNA expression of the Glp1r was measured in isolated mouse lung endothelial cells (MLECs) and expression levels are shown in relation to the background strain (Cdh5cre mice). Student t test; N=4 animals per group. B, Vascular function was evaluated by endothelium-dependent (acetylcholine [ACh]) relaxation of aortic rings (isometric tension studies). Two-way ANOVA and Bonferroni multiple comparison test; N=20–30 animals per group. C, Systolic blood pressure in mice was determined by noninvasive blood pressure recording on day 6 of ATII (angiotensin II) infusion. D, Representative (out of 3–4) Sirius red staining of aortas reveal vascular fibrosis. E, NADPH oxidase 2 (Nox2) mRNA expression was measured by quantitative reverse transcription polymerase chain reaction in aortic tissue. F, high-performance liquid chromatography (HPLC) was used to detect nitrate (NO3−) in plasma. Representative chromatograms are shown below the quantification bar graph. G and H, Flow cytometry of aortic tissue identified Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils inside the vascular wall. Representative original plots of Ly6G−Ly6C+ and Ly6G+Ly6C+ cells are shown beside the quantification bar graph (for gating strategy, see Figure IV in the online-only Data Supplement). One-way ANOVA and Bonferroni multiple comparison test; N=8–31 (C), 6–8 (E), 4–5 (F), and 5–9 (G and H) animals per group. Data are means±SEM. *P<0.05; **P<0.01; ***P<0.001, ****P<0.0001.
Discussion
Here, we show that the GLP-1 analog liraglutide prevents the adverse cardiovascular effects of ATII-induced arterial hypertension. Liraglutide exerts its cardiovascular protective actions through the GLP-1R and normalizes vascular inflammation, oxidative stress, endothelial function, blood pressure, and cardiac hypertrophy in this animal model of arterial hypertension. Mechanistically, GLP-1R activation by liraglutide attenuates vessel wall infiltration by Ly6G−Ly6C+ inflammatory monocytes and Ly6G+Ly6C+ neutrophils, which in turn lowers oxidative stress and prevents uncoupling of eNOS in hypertensive mice. NO bioavailability is thereby maintained, endothelium-mediated vasorelaxation is preserved, and vascular remodeling and fibrosis are prevented. Consistent with the known actions of GLP-1, we found no effects of liraglutide on glycemia or insulin levels, supporting a glucose-independent mechanism. We identify that the canonical GLP-1R expressed on endothelial cells, but not on myeloid cells, is required for the cardiovascular protection by the GLP-1 analog liraglutide.
GLP-1R agonists lower blood pressure in preclinical studies and humans. One potential mechanism was proposed by Kim et al19 who showed that GLP-1R activation by liraglutide promotes the secretion of ANP (atrial natriuretic peptide) with a subsequent increase of cGMP-mediated vasorelaxation and urinary sodium secretion leading to reduction of blood pressure in ATII-infused mice. The effect was dependent on the canonical GLP-1R and absent in global GLP-1R knockout mice. With the present studies, we found a comparable blood pressure reduction by liraglutide, which resulted in a reduction of cardiac hypertrophy and reduced heart/body weight ratio (both parameters indicative of hypertensive heart disease), which were associated with a reduction of body weight but independent of glucose-lowering. Liraglutide also reduced systemic and cardiac oxidative stress and inflammation. Studies using a cardiomyocyte-specific GLP-1R knockout strain (Glp1rCM−/−) found no relevant role for the GLP-1R expressed on cardiomyocytes in ischemic cardiomyopathy,24 which excludes that direct liraglutide effects on cardiomyocytes are responsible for the cardioprotective effects. Thus, improvements of cardiac hypertrophy, cardiac oxidative stress, and inflammation likely result from blood pressure lowering effects and prevention of infiltration of immune cells by liraglutide. The blood pressure reduction by liraglutide was consistent with previous data19 but clearly of greater magnitude compared with human studies in which a blood pressure reduction of 2 to 4 mm Hg is reported.35 The latter represents a limitation of the present study with respect to translation of our animal data and their clinical relevance.
Vascular inflammation is a crucial component in the development of arterial hypertension2 with sequential steps of inflammatory cell infiltration into the vascular wall, resulting in high levels of vascular oxidative stress and eNOS uncoupling and finally atherosclerosis.2,4,5,36–38 GLP-1R activation on endothelial cells prevented these effects and improved vascular relaxation and NO bioavailability in ATII-infused mice. Mechanistically, liraglutide suppressed an inflammatory cascade involving NF-κb and downregulated mRNA encoding vascular adhesion molecules VCAM-1, ICAM-1, and P-selectin in the arteries, which explains the observed reduction in leukocyte rolling and vessel wall infiltration.
In a selective ablation model of lysozyme M-positive (LysM+) myelomonocytic cells, it has been shown that infiltrating proinflammatory monocytes and macrophages cause vascular oxidative stress and mediate ATII-induced vascular dysfunction.4 Consistent with these findings, reduced leukocyte infiltration in liraglutide-treated animals was associated with attenuated vascular reactive oxygen species production, restoration of the function of eNOS and suppression of Nos2/Nox2 mRNA expression. Under conditions of high oxidative stress, eNOS uncouples and converts into a source of reactive oxygen species, which is specifically detectable by reduced aortic NO bioavailability, eNOS S-glutathionylation, and eNOS-dependent reactive oxygen species formation by using the selective NOS-inhibitor L-NAME in dihydroethidium cryosections.36,37 In contrast, increased iNOS activity was reflected by higher plasma nitrate levels, and both eNOS uncoupling as well as iNOS overactivation were linked in the setting of hypertension, as also previously demonstrated for the ATII hypertension model.39 Liraglutide treatment interrupted this detrimental cascade in hypertensive mice by prevention of leukocyte influx and vascular inflammation.
Our data are in agreement with cell culture experiments of Hattori et al40 demonstrating that liraglutide causes a dose-dependent increase of NO release in human umbilical vein endothelial cells via eNOS activation and downregulation of NFKB1, TNFA, VCAM1, and ICAM1 mRNA expression. Similar observations have been made in ApoE−/− mice treated with GLP-1 analog for 28 days, where exendin-4 reduced numbers of Mac-2+ cells adhering to the endothelium and decreased levels of Vcam1 and Icam1 mRNA.15 Similar evidence for a mechanistic involvement of the GLP-1R was demonstrated using the GLP-1R antagonist exendin (9-39) to diminish liraglutide’s protective effect in atherosclerosis-prone ApoE−/− mice.41
Anti-inflammatory effects of GLP-1 analogs are well established.11 Nevertheless, it still remains unclear whether GLP-1R agonists modify immune cell function through direct effects on immune cells or indirect mechanisms (eg, suppression of endothelial cell activation) and whether these effects require the canonical GLP-1R. Immunoreactive GLP-1R protein and Glp1r mRNA transcripts have been detected in murine monocytes and macrophages, but due to low sensitivity and specificity of GLP-1R antibodies, the precise cellular and tissue expression of the immune GLP-1R remains uncertain.42,43 Endothelial cells also express the canonical GLP-1R; however, the importance of the endothelial GLP-1R for the cardiovascular protection by GLP-1R agonists has been unexplored.44 Nevertheless, multiple studies have demonstrated effects of GLP-1 analogs on endothelial cells and vascular function ex vivo and in vivo in mice and humans (for review see Drucker et al11). GLP-1R agonists are thought to exert cardiovascular protection dependent on the known GLP-1R. However, GLP-1(9-36)amide, generated by DPP-4-mediated cleavage of native GLP-1, exerts cardioprotective actions in the ischemic mouse heart. Moreover, GLP-1(9-36) also exerts direct vasodilatory actions on murine blood vessels and induces vasodilation in mesenteric vessels.45 These actions persisted in blood vessels of Glp1r−/− mice. Hence, we cannot fully exclude a potential contribution of GLP-1R independent effects of liraglutide and its metabolites,11 which represents a limitation of our present study.
Here, we demonstrated that the canonical GLP-1R expressed on endothelial cells, but not on myeloid cells, is required to improve endothelial dysfunction by reduction of vascular inflammation and oxidative stress in a murine model of arterial hypertension. Glp1r−/− mice exhibit endothelial dysfunction, which was only modestly exacerbated by ATII, suggesting a role of the endogenous GLP-1R system in vascular function. The latter hypothesis is supported by a recent report by He et al46 who demonstrated that increased endogenous GLP-1 bioavailability protects against atherosclerosis. Importantly, liraglutide failed to normalize endothelial function, vascular oxidative stress, cardiac Nox activity, and mRNA expression of proinflammatory genes (Nox2, Nos2, Tnfa) in hypertensive Glp1r−/− mice, clearly highlighting the essential role of GLP-1R signaling for cardiovascular protection by GLP-1 analogs. However, blood pressure and cardiac hypertrophy trended lower in liraglutide-treated Glp1r ec−/− mice, consistent with previous findings in mice invoking a role for ANP in the blood pressure reduction produced by GLP-1R agonists.19
In conclusion, clinical trials revealed that the GLP-1 analog liraglutide and multiple structurally distinct GLP-1R agonists improve cardiovascular outcomes through incompletely understood mechanisms, in which atherosclerosis might play an essential role.6 The present study provides evidence that liraglutide prevents endothelial dysfunction and lowers blood pressure in normoglycemic mice with experimental arterial hypertension, a major risk factor for atherosclerosis and subsequent cardiovascular disease. This was in concert with a reduction of vascular inflammation, and subsequent normalization of vascular oxidative stress. The GLP-1R expressed on endothelial, but not on myeloid cells, plays an indispensable role in the cardiovascular protection induced by liraglutide in mice with ATII-induced experimental hypertension. Given the major role of renin-angiotensin-aldosterone system activation for cardiovascular and renal complications observed in patients with diabetes mellitus,47 our present data obtained in a model of ATII-triggered hypertension may contribute to a better understanding of the cardioprotective effects of liraglutide reported in clinical trials.6,7
Acknowledgments
We are indebted to Jessica Rudolph, Jörg Schreiner, Bettina Mros, Nicole Glas, and Angelica Karpi for expert technical assistance. This article contains results that are part of the doctoral thesis of Johanna Helmstädter. We are grateful to Randy Seeley, University of Michigan, providing the Glp1rflox/flox mouse and to Christoph Reinhardt (Platform Intravital Microscopy of the Center for Thrombosis and Hemostasis (Mainz, Germany) for providing the intravital microscopy platform.
Sources of Funding
This study was supported by a grant of the German Research Foundation (DFG STE2528/2-1) to S. Steven, who holds a Virchow-Fellowship from the Center of Thrombosis and Hemostasis (Mainz, Germany) funded by the Federal Ministry of Education and Research (BMBF 01EO1003). K. Frenis and S. Kalinovic hold PhD stipends of the TransMed PhD Program at the University Medical Center Mainz, and their positions are funded by a vascular biology research grant within the collaborative research group “Novel and neglected cardiovascular risk factors” from the Boehringer Ingelheim Foundation. P. Wenzel is supported DFG (WE4361/7-1) and BMBF (01EO1003). D.J. Drucker is supported by a CIHR (Canadian Institutes of Health Research) Grant 154321, a BBDC (Banting & Best Diabetes Centre)-Novo Nordisk Chair in Incretin Biology and operating grant support from Novo Nordisk, Inc. S. Chlopicki is support by FNP (Foundation for Polish Science) Team Tech–Core Facility program. We thank the Mainz Heart Foundation for continuous support. T. Muünzel, P. Wenzel, and W. Ruf are Principal Investigator of the DZHK (German Center for Cardiovascular Research), Partner Site Rhine-Main, Mainz, Germany.
Disclosures
D.J. Drucker has been a consultant to Forkhead Biotherapeutics, Intarcia, Merck, Novo Nordisk, Pfizer, and Sanofi, Inc and received operating grant support from Merck, Novo Nordisk, and Shire/Takeda Inc. The other authors report no conflicts.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- ATII
- angiotensin II
- CANTOS
- Canakinumab Antiinflammatory Thrombosis Outcome Study
- eNOS
- endothelial NO synthase
- GLP-1
- glucagon-like peptide-1
- Glp1r ec−/−
- Glp1rflox/floxxCdh5cre, endothelial GLP-1R knockout
- Glp1r my−/−
- Glp1rflox/floxxLysMcre, myeloid GLP-1R knockout
- GLP-1R
- glucagon-like peptide-1 receptor
- ICAM-1
- intercellular adhesion molecule-1
- IL
- interleukin
- iNOS
- inducible NO synthase
- LEADER
- Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results
- Ly6C
- lymphocyte antigen 6 complex, locus C1
- Ly6G
- lymphocyte antigen 6 complex, locus G
- LysM
- lysozyme M
- NFκb
- nuclear factor-kappa-B
- Nox
- NADPH oxidase
- TNF-α
- tumor necrosis factor alpha
- VCAM-1
- vascular cell adhesion molecule-1
- WT
- wild-type
For Sources of Funding and Disclosures, see page 157.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/atv.0000615456.97862.30.
References
- 1.Murray CJ, Atkinson C, Bhalla K, Birbeck G, Burstein R, Chou D, Dellavalle R, Danaei G, Ezzati M, Fahimi A, et al. U.S. Burden of Disease Collaborators. The state of US health, 1990-2010: burden of diseases, injuries, and risk factors. JAMA. 2013;310:591–608. doi: 10.1001/jama.2013.13805. doi: 10.1001/jama.2013.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116:307–311. doi: 10.1161/CIRCRESAHA.116.301313. doi: 10.1161/CIRCRESAHA.116.301313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 4.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 5.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, et al. LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–322. doi: 10.1056/NEJMoa1603827. doi: 10.1056/NEJMoa1603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, et al. SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–1844. doi: 10.1056/NEJMoa1607141. doi: 10.1056/NEJMoa1607141. [DOI] [PubMed] [Google Scholar]
- 8.Lønborg J, Kelbæk H, Vejlstrup N, Bøtker HE, Kim WY, Holmvang L, Jørgensen E, Helqvist S, Saunamäki K, Terkelsen CJ, et al. Exenatide reduces final infarct size in patients with ST-segment-elevation myocardial infarction and short-duration of ischemia. Circ Cardiovasc Interv. 2012;5:288–295. doi: 10.1161/CIRCINTERVENTIONS.112.968388. doi: 10.1161/CIRCINTERVENTIONS.112.968388. [DOI] [PubMed] [Google Scholar]
- 9.Fonseca VA, Devries JH, Henry RR, Donsmark M, Thomsen HF, Plutzky J. Reductions in systolic blood pressure with liraglutide in patients with type 2 diabetes: insights from a patient-level pooled analysis of six randomized clinical trials. J Diabetes Complications. 2014;28:399–405. doi: 10.1016/j.jdiacomp.2014.01.009. doi: 10.1016/j.jdiacomp.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koska J, Schwartz EA, Mullin MP, Schwenke DC, Reaven PD. Improvement of postprandial endothelial function after a single dose of exenatide in individuals with impaired glucose tolerance and recent-onset type 2 diabetes. Diabetes Care. 2010;33:1028–1030. doi: 10.2337/dc09-1961. doi: 10.2337/dc09-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. 2016;24:15–30. doi: 10.1016/j.cmet.2016.06.009. doi: 10.1016/j.cmet.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Steven S, Hausding M, Kröller-Schön S, Mader M, Mikhed Y, Stamm P, Zinßius E, Pfeffer A, Welschof P, Agdauletova S, et al. Gliptin and GLP-1 analog treatment improves survival and vascular inflammation/dysfunction in animals with lipopolysaccharide-induced endotoxemia. Basic Res Cardiol. 2015;110:6. doi: 10.1007/s00395-015-0465-x. doi: 10.1007/s00395-015-0465-x. [DOI] [PubMed] [Google Scholar]
- 13.Steven S, Jurk K, Kopp M, Kröller-Schön S, Mikhed Y, Schwierczek K, Roohani S, Kashani F, Oelze M, Klein T, et al. Glucagon-like peptide-1 receptor signalling reduces microvascular thrombosis, nitro-oxidative stress and platelet activation in endotoxaemic mice. Br J Pharmacol. 2017;174:1620–1632. doi: 10.1111/bph.13549. doi: 10.1111/bph.13549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krasner NM, Ido Y, Ruderman NB, Cacicedo JM. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS One. 2014;9:e97554. doi: 10.1371/journal.pone.0097554. doi: 10.1371/journal.pone.0097554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, Fujitani Y, Hirose T, Kawamori R, Watada H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. 2010;59:1030–1037. doi: 10.2337/db09-1694. doi: 10.2337/db09-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagashima M, Watanabe T, Terasaki M, Tomoyasu M, Nohtomi K, Kim-Kaneyama J, Miyazaki A, Hirano T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011;54:2649–2659. doi: 10.1007/s00125-011-2241-2. doi: 10.1007/s00125-011-2241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noyan-Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li RK, Volchuk A, Robinson LA, Billia F, Drucker DJ, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127:74–85. doi: 10.1161/CIRCULATIONAHA.112.091215. doi: 10.1161/CIRCULATIONAHA.112.091215. [DOI] [PubMed] [Google Scholar]
- 18.Jensen EP, Poulsen SS, Kissow H, Holstein-Rathlou NH, Deacon CF, Jensen BL, Holst JJ, Sorensen CM. Activation of GLP-1 receptors on vascular smooth muscle cells reduces the autoregulatory response in afferent arterioles and increases renal blood flow. Am J Physiol Renal Physiol. 2015;308:F867–F877. doi: 10.1152/ajprenal.00527.2014. doi: 10.1152/ajprenal.00527.2014. [DOI] [PubMed] [Google Scholar]
- 19.Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, Simpson JA, Drucker DJ. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013;19:567–575. doi: 10.1038/nm.3128. doi: 10.1038/nm.3128. [DOI] [PubMed] [Google Scholar]
- 20.Pujadas G, Drucker DJ. Vascular biology of glucagon receptor superfamily peptides: mechanistic and clinical relevance. Endocr Rev. 2016;37:554–583. doi: 10.1210/er.2016-1078. doi: 10.1210/er.2016-1078. [DOI] [PubMed] [Google Scholar]
- 21.Douglas G, Hale AB, Patel J, Chuaiphichai S, Al Haj Zen A, Rashbrook VS, Trelfa L, Crabtree MJ, McNeill E, Channon KM. Roles for endothelial cell and macrophage Gch1 and tetrahydrobiopterin in atherosclerosis progression. Cardiovasc Res. 2018;114:1385–1399. doi: 10.1093/cvr/cvy078. doi: 10.1093/cvr/cvy078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL, Drucker DJ. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med. 1996;2:1254–1258. doi: 10.1038/nm1196-1254. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 23.Payne S, De Val S, Neal A. Endothelial-specific cre mouse models. Arterioscler Thromb Vasc Biol. 2018;38:2550–2561. doi: 10.1161/ATVBAHA.118.309669. doi: 10.1161/ATVBAHA.118.309669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ussher JR, Baggio LL, Campbell JE, Mulvihill EE, Kim M, Kabir MG, Cao X, Baranek BM, Stoffers DA, Seeley RJ, et al. Inactivation of the cardiomyocyte glucagon-like peptide-1 receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Mol Metab. 2014;3:507–517. doi: 10.1016/j.molmet.2014.04.009. doi: 10.1016/j.molmet.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pabbidi MR, Kuppusamy M, Didion SP, Sanapureddy P, Reed JT, Sontakke SP. Sex differences in the vascular function and related mechanisms: role of 17β-estradiol. Am J Physiol Heart Circ Physiol. 2018;315:H1499–H1518. doi: 10.1152/ajpheart.00194.2018. doi: 10.1152/ajpheart.00194.2018. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuhmacher S, Wenzel P, Schulz E, Oelze M, Mang C, Kamuf J, Gori T, Jansen T, Knorr M, Karbach S, et al. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension. 2010;55:897–904. doi: 10.1161/HYPERTENSIONAHA.109.149542. doi: 10.1161/HYPERTENSIONAHA.109.149542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng M, Whitesall S, Zhang Y, Beibel M, D’Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens. 2008;21:1288–1291. doi: 10.1038/ajh.2008.301. doi: 10.1038/ajh.2008.301. [DOI] [PubMed] [Google Scholar]
- 29.Kröller-Schön S, Steven S, Kossmann S, Scholz A, Daub S, Oelze M, Xia N, Hausding M, Mikhed Y, Zinssius E, et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal. 2014;20:247–266. doi: 10.1089/ars.2012.4953. doi: 10.1089/ars.2012.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleschyov AL, Mollnau H, Oelze M, Meinertz T, Huang Y, Harrison DG, Munzel T. Spin trapping of vascular nitric oxide using colloid Fe(II)-diethyldithiocarbamate. Biochem Biophys Res Commun. 2000;275:672–677. doi: 10.1006/bbrc.2000.3361. doi: 10.1006/bbrc.2000.3361. [DOI] [PubMed] [Google Scholar]
- 31.Kus K, Walczak M, Maslak E, Zakrzewska A, Gonciarz-Dytman A, Zabielski P, Sitek B, Wandzel K, Kij A, Chabowski A, et al. Hepatoselective Nitric Oxide (NO) Donors, V-PYRRO/NO and V-PROLI/NO, in nonalcoholic fatty liver disease: a comparison of antisteatotic effects with the biotransformation and pharmacokinetics. Drug Metab Dispos. 2015;43:1028–1036. doi: 10.1124/dmd.115.063388. doi: 10.1124/dmd.115.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, Hörmann N, Reiss C, Subramaniam S, Wilms E, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood. 2017;130:542–553. doi: 10.1182/blood-2016-11-754416. doi: 10.1182/blood-2016-11-754416. [DOI] [PubMed] [Google Scholar]
- 33.Münzel T, Giaid A, Kurz S, Stewart DJ, Harrison DG. Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc Natl Acad Sci U S A. 1995;92:5244–5248. doi: 10.1073/pnas.92.11.5244. doi: 10.1073/pnas.92.11.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 35.Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation. 2017;136:849–870. doi: 10.1161/CIRCULATIONAHA.117.028136. doi: 10.1161/CIRCULATIONAHA.117.028136. [DOI] [PubMed] [Google Scholar]
- 36.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knorr M, Hausding M, Kröller-Schuhmacher S, Steven S, Oelze M, Heeren T, Scholz A, Gori T, Wenzel P, Schulz E, et al. Nitroglycerin-induced endothelial dysfunction and tolerance involve adverse phosphorylation and S-Glutathionylation of endothelial nitric oxide synthase: beneficial effects of therapy with the AT1 receptor blocker telmisartan. Arterioscler Thromb Vasc Biol. 2011;31:2223–2231. doi: 10.1161/ATVBAHA.111.232058. doi: 10.1161/ATVBAHA.111.232058. [DOI] [PubMed] [Google Scholar]
- 38.Oelze M, Warnholtz A, Faulhaber J, Wenzel P, Kleschyov AL, Coldewey M, Hink U, Pongs O, Fleming I, Wassmann S, et al. NADPH oxidase accounts for enhanced superoxide production and impaired endothelium-dependent smooth muscle relaxation in BKbeta1 −/− mice. Arterioscler Thromb Vasc Biol. 2006;26:1753–1759. doi: 10.1161/01.ATV.0000231511.26860.50. doi: 10.1161/01.ATV.0000231511.26860.50. [DOI] [PubMed] [Google Scholar]
- 39.Kossmann S, Hu H, Steven S, Schönfelder T, Fraccarollo D, Mikhed Y, Brähler M, Knorr M, Brandt M, Karbach SH, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. doi: 10.1074/jbc.M114.604231. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattori Y, Jojima T, Tomizawa A, Satoh H, Hattori S, Kasai K, Hayashi T. A glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates nitric oxide production and exerts anti-inflammatory action in endothelial cells. Diabetologia. 2010;53:2256–2263. doi: 10.1007/s00125-010-1831-8. doi: 10.1007/s00125-010-1831-8. [DOI] [PubMed] [Google Scholar]
- 41.Gaspari T, Welungoda I, Widdop RE, Simpson RW, Dear AE. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE( −/−) mouse model. Diab Vasc Dis Res. 2013;10:353–360. doi: 10.1177/1479164113481817. doi: 10.1177/1479164113481817. [DOI] [PubMed] [Google Scholar]
- 42.Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia. 2010;53:730–740. doi: 10.1007/s00125-009-1643-x. doi: 10.1007/s00125-009-1643-x. [DOI] [PubMed] [Google Scholar]
- 43.Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, Streutker C, Holland D, Cao X, Baggio LL, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE( −/−) mice. Endocrinology. 2013;154:127–139. doi: 10.1210/en.2012-1937. doi: 10.1210/en.2012-1937. [DOI] [PubMed] [Google Scholar]
- 44.Ussher JR, Drucker DJ. Cardiovascular actions of incretin-based therapies. Circ Res. 2014;114:1788–1803. doi: 10.1161/CIRCRESAHA.114.301958. doi: 10.1161/CIRCRESAHA.114.301958. [DOI] [PubMed] [Google Scholar]
- 45.Erdogdu O, Nathanson D, Sjöholm A, Nyström T, Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol Cell Endocrinol. 2010;325:26–35. doi: 10.1016/j.mce.2010.04.022. doi: 10.1016/j.mce.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 46.He S, Kahles F, Rattik S, Nairz M, McAlpine CS, Anzai A, Selgrade D, Fenn AM, Chan CT, Mindur JE, et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature. 2019;566:115–119. doi: 10.1038/s41586-018-0849-9. doi: 10.1038/s41586-018-0849-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, Mustonen J Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351:1952–1961. doi: 10.1056/NEJMoa042274. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]