

Abstract
Objective
To define the natural history of the C9orf72 amyotrophic lateral sclerosis (C9ALS) patient population, develop disease biomarkers, and characterize patient pathologies.
Methods
We prospectively collected clinical and demographic data from 116 symptomatic C9ALS and 12 non–amyotrophic lateral sclerosis (ALS) full expansion carriers across 7 institutions in the United States and the Netherlands. In addition, we collected blood samples for DNA repeat size assessment, CSF samples for biomarker identification, and autopsy samples for dipeptide repeat protein (DPR) size determination. Finally, we collected retrospective clinical data via chart review from 208 individuals with C9ALS and 450 individuals with singleton ALS.
Results
The mean age at onset in the symptomatic prospective cohort was 57.9 ± 8.3 years, and median duration of survival after onset was 36.9 months. The monthly change was −1.8 ± 1.7 for ALS Functional Rating Scale–Revised and −1.4% ± 3.24% of predicted for slow vital capacity. In blood DNA, we found that G4C2 repeat size correlates positively with age. In CSF, we observed that concentrations of poly(GP) negatively correlate with DNA expansion size but do not correlate with measures of disease progression. Finally, we found that size of poly(GP) dipeptides in the brain can reach large sizes similar to that of their DNA repeat derivatives.
Conclusions
We present a thorough investigation of C9ALS natural history, providing the basis for C9ALS clinical trial design. We found that clinical features of this genetic subset are less variant than in singleton ALS. In addition, we identified important correlations of C9ALS patient pathologies with clinical and demographic data.
A hexanucleotide (G4C2) repeat expansion in chromosome 9 open reading frame 72 (C9orf72) causes 39% of familial and 7% of nonfamilial or singleton amyotrophic lateral sclerosis (SALS), representing the largest genetically identified subgroup of amyotrophic lateral sclerosis (ALS) to date.1–3 In addition, the same expansion can cause frontotemporal lobar degeneration (FTLD) in isolation or comorbidly with ALS.1,4,5 While this locus typically has under 30 repeats, individuals with ALS with C9orf72 expansion mutations (C9ALS) have hundreds to thousands.4,6–8 In addition to the common ALS pathology of TDP-43 aggregation,9 presence of this large expansion leads to the accumulation of 2 C9ALS-specific pathologies: nuclear RNA foci consisting of repeat-derived RNA, and dipeptide repeat proteins (DPRs), which are translated through repeat associated, non-ATG (RAN) translation.10–13 For C9ALS G4C2 repeats, RAN translation yields 5 distinct species of DPRs: poly(GP), poly(GA), poly(PR), poly(GR), and poly(PA), each shown to aggregate in CNS tissues of individuals with C9ALS/FTLD.10–14 Mounting evidence suggests these expansion-related pathologies are toxic and may be directly involved in disease pathogenesis.15 As such, therapeutics targeted at reducing or eliminating RNA foci and DPRs are in development, with multiple rapidly approaching human trials (NCT03626012),16–20 the design of which will be determined by the natural history of the C9ALS patient population.
Here we present a prospective natural history study of individuals with C9ALS in the United States and the Netherlands. We documented a wide array of demographic and clinical data in a cohort of 116 symptomatic and 12 non-ALS full expansion carriers. In addition, we collected blood and CSF samples from these individuals for analysis of DNA repeat size and C9ALS biomarkers, respectively. As the expansion in C9orf72 is somatically unstable,7 we examined potential relationships between repeat size and clinical and demographic data. Previous studies suggest that CSF poly(GP) levels, while not strongly correlated with disease characteristics, are steady over time and responsive to C9ALS therapeutics, providing promise for use as a pharmacodynamics biomarker.21–23 Thus, we examined poly(GP) levels in CSF and correlated these measurements with DNA repeat expansion size and clinical characteristics. Finally, we investigated the size of poly(GP) dipeptides in postmortem CNS tissues.
Methods
Participant identification and enrollment
Participants for the prospective natural history study were enrolled at 7 institutions: Washington University (WU), Massachusetts General Hospital (MGH), University Medical Center Utrecht (Utrecht), University of Massachusetts, Columbia University Medical Center, Cedars-Sinai Medical Center, and Johns Hopkins University. Data management was handled by the Neurologic Clinical Research Institute (NCRI) at MGH. The C9ALS subgroup (n = 116) required a minimum diagnosis of possible ALS based on the most recent revision of the El Escorial Criteria, while a separate non-ALS subgroup (n = 12) included individuals without ALS-related motor symptoms. Of the 12 participants without ALS, all received cognitive assessment with the ALS Cognitive Behavioral Screen (ALS-CBS). C9orf72 expansions of greater than 50 repeats were confirmed for all participants via Clinical Laboratory Improvement Amendments–approved testing provided by Prevention Genetics (Marshfield, WI).
A separate cohort of 208 individuals with C9ALS and 450 individuals with SALS from WU and Utrecht was assessed retrospectively for C9orf72 natural history, dating back to 2006. All individuals were first delineated by the presence or absence of a C9orf72 expansion mutation, confirmed via in-house genetic testing. Those without an expansion were then screened for the presence or absence of a family history of ALS, and those with a family history were further excluded to reduce potential presence of other ALS-causing mutations.24 The remaining individuals were categorized as SALS.
Standard protocol approvals, registrations, and patient consents
This study was approved by each recruiting center's Institutional Review Board and written informed consent was provided by all participants.
Data and patient sample collection
Clinical data for the prospective study were collected in person or via telephone interviews with individuals or caregivers, with the exception of slow vital capacity (SVC), which was assessed via handheld electronic spirometer. Clinical assessments included the ALS Functional Rating Scale–Revised (ALSFRS-R), ALS-CBS, and ALS Caregiver Behavioral Questionnaire (ALSCBQ). Severe cognitive impairment consistent with a diagnosis of FTLD was defined as an initial ALS-CBS score of 10 or less out of a total score of 20, and moderate cognitive impairment as 11–15. Family history of ALS, dementia, or both was assessed with criteria defined in Byrne et al.25 Additional data included family and personal medical history, medication use, ALS-related history (onset site, onset date, timeline of symptom progression), physical and neurologic examination, vital signs, and demographics. Biological specimens from prospective study participants were also collected, including whole blood, serum, CSF, and urine. Participants were enrolled over a 44-month period and data were collected until study closing. In addition, participants were followed for survival outcomes after study closing, up until the date of this report. Study site personnel were trained on good clinical practices and study outcome measures. Data were collected by the sites and recorded on source documents, then subsequently captured in the NeuroBANK patient-centric platform (neurobank.org/) and monitored remotely for consistency and completeness.
Longitudinal data were acquired through follow-up visits or phone calls. In total, we were able to acquire at least 2 longitudinal measures from 88 participants for ALSFRS-R, 53 for SVC, 42 for ALS-CBS, and 29 for ALSCBQ. The average time (and range) between measures was 2.6 months (0.6–14.5) for ALSFRS-R, 7.6 (0.7–26.6) for SVC, 7.8 (0.7–26.6) for ALS-CBS, and 3.9 (0.6–18.1) for ALSCBQ. A total of 21 participants in the prospective C9ALS subgroup reached mortality within 6 months of study enrollment and 12 more between 6 and 12 months. In addition, 21 C9ALS participants enrolled less than 6 months before study closing. Of the 42 participants enrolled in the study for 6 months or less, 16 were still able to provide longitudinal data for ALSFRS-R, 12 for SVC, 10 for ALS-CBS, and 7 for ALSCBQ. Other reasons for lack of follow-up are mixed and include inability to perform tasks due to disease progression, time constraints during clinic visits, and inability to travel to the study site.
In the non-ALS subgroup (n = 12 individuals), longitudinal ALS-CBS measures were available for 7 individuals, with an average (range) of 9.2 months (5.3–18.2) between measures. No individuals without ALS reached mortality during the study. Five participants without ALS were enrolled within 9 months of study closure, the average time between measures in this group, 4 of whom did not provide longitudinal data. One other individual conducted their follow-up remotely via phone and thus was unable to provide a longitudinal ALS-CBS assessment.
Clinical data for the retrospective cohort were collected through chart review and recorded to a central database.
Blood-derived DNA samples from unexpanded individuals were from the Knight Alzheimer's Disease Research Center at WU and the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (loni.ucla.edu\ADNI; for up-to-date information, see adni-info.org). As there is no association between repeat size and dementia of the Alzheimer type status,26 all individuals were grouped together for analysis. This study was approved by each recruiting center's institutional review board and was carried out in accordance with the approved protocol.
Autopsy samples used in poly(GP) size assessments were from a separate cohort of individuals with C9ALS from WU. All samples were collected between 6 and 32 hours of death, flash frozen in liquid nitrogen, and stored at −80°C until use.
DNA repeat size measurement
Presence or absence of an expanded repeat was assessed with repeat primed PCR (rpPCR) as previously described26 using published primer concentrations and sequences.2 PCR products were analyzed with an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) and GeneMapper software. rpPCR was also used to quantify repeat sizes in unexpanded individuals.
G4C2 repeat size assessment of full expansions was performed with Southern blot. DNA was isolated from whole blood samples by a core facility at WU and sent to Dr. Robert H. Brown's laboratory for analysis. DNA was digested with AluI and DdeI, followed by gel electrophoresis and probing with a (GGGGCC)5-DIG probe. Bands were visualized using an anti-DIG antibody and a chemiluminescent protocol. For quantification, densitometry plots were generated for each lane using GelEval software (FrogDance Software; version 1.37). Density peaks for each ladder band were used to create a standard curve, from which sample density peak sizes were interpolated (GraphPad Prism; version 7.0). Due to the upper limit of the ladder, this method could not accurately distinguish expansion sizes greater than 3,855 repeats. Sixteen samples were measured twice, with a median difference of 318 repeats between measurements. All Southern blot size assessments were blinded to eliminate experimenter bias. Prior to unblinding, samples with technical abnormalities (e.g., gel band too faint, presence of nonspecific artifact) were excluded. In total, samples from 100 individuals with C9ALS and participants without ALS were analyzed, with 11 being excluded, leaving 89 for further analysis.
Haplotype analysis
Unexpanded individuals were genotyped for a 24 single nucleotide polymorphism (SNP) at-risk haplotype associated with expanded C9orf72 repeats27 using an Illumina 610 array. Stringent quality control criteria were applied to remove low-quality SNPs.28 The SNPs reported previously as part of the risk haplotype for C9orf7227 were extracted and haplotype analysis was performed using PLINK. Haplotype carrier status was defined as individuals with at least 21/24 matching SNPs.
Poly(GP) measurement and size assessment
Relative concentrations of poly(GP) were measured via immunoassay. A monoclonal mouse anti-poly(GP) antibody (Biogen, Boston, MA) was incubated on goat anti-mouse plates (Meso Scale Diagnostics, Rockville, MD) for capture. Consecutive incubations with a polyclonal rabbit anti-poly(GP) antibody (Biogen) followed by a sulfo-tagged goat anti-rabbit antibody (Meso Scale Diagnostics) were used for detection. “Relative concentration” designations refer to the amount of poly(GP) signal present in an equivalent amount of DPR-containing tissue or cell lysate. Thus, axes display ng/mL of total standard lysate rather than absolute concentration of poly(GP).
For assessment of poly(GP) in CSF, undiluted samples were measured in triplicate (45 μL/well). Any sample displaying less than twice the average signal in blank wells was considered a zero value and all non-zero samples achieved a coefficient of variation of less than 15.
For poly(GP) size assessment, samples were first separated with size exclusion chromatography (SEC) and then quantified with the poly(GP) immunoassay. Autopsy samples were homogenized in TEN buffer (10 mM Tris pH 8.0, 1 mM EDTA, 100 mM NaCl) with 2% sodium dodecyl sulfate (SDS) and fresh protease inhibitors (Sigma Aldrich, St. Louis, MO), sonicated briefly, and centrifuged at 100,000 g for 30 minutes at 4°C to remove insoluble protein species. Resultant supernatants were normalized by volume, loaded into a Superdex 200 10/300 GL SEC column (GE Healthcare, Chicago, IL), and separated into 1 mL fractions at a flow rate of 0.75 mL/min in TEN buffer + 0.5% SDS. SEC fractions were measured with the poly(GP) immunoassay in triplicate.
Statistical analyses
Statistical analyses were performed in GraphPad Prism version 7.0. Central tendency markers represent mean, while error bars represent SD. All correlations were analyzed with linear regression. All reported p values were corrected for multiple comparisons across the entire study (false discovery rate).
Survival analyses for the prospective C9ALS cohort included 88 individuals with definitive survival endpoints, while the remaining 28 participants who were either alive or lost to follow-up were right-censored on last known date of contact. Censored data points are marked with tick marks in figure 1A. Survival comparisons of retrospective C9ALS and SALS were done with log-rank (Mantel-Cox) test.
Figure 1. Individuals with amyotrophic lateral sclerosis (ALS) with chromosome 9 open reading frame 72 expansion mutations (C9ALS) natural history: Descriptive characteristics.

(A) Distribution of ages at onset for the prospective C9ALS cohort (n = 116 individuals; mean age at onset 57.9 ± 8.3 years). (B) Mortality in the prospective C9ALS cohort (n = 88 individuals reaching survival endpoint; n = 28 individuals alive or lost to follow-up; right-censored on last known date of contact; marked with tick marks; median survival 36.9 months). (C) Distribution of onset locations in the prospective C9ALS cohort. (D) Family history of ALS, dementia, or both in the prospective cohort (n = 128 individuals). (E) Distribution of ages at onset for retrospective C9ALS (n = 208 individuals; mean age at onset 59.0 ± 9.3 years) and singleton amyotrophic lateral sclerosis (SALS) (n = 450 individuals; mean age at onset 59.3 ± 12.1 years) cohorts. (F) Mortality in retrospective C9ALS (n = 195 individuals; median survival 29.9 months) and SALS cohorts (n = 214 individuals; median survival 30 months). Upper quartile survival displays significantly decreased disease duration in slow-progressing C9ALS (n = 48 individuals; median survival 55 months) as compared to SALS (n = 53 individuals; median survival 73 months) (log-rank [Mantel-Cox] test: p < 0.001).
Data availability
All data from this study are stored in the NeuroBANK™ (neurobank.org/) data repository at the MGH NCRI and are linked with biospecimen repositories. We will share deidentified datasets with researchers who want to advance understanding of neurologic disease. A limited amount of deidentified biofluid samples (DNA, serum, peripheral blood mononuclear cells, urine, and CSF) collected from this study are stored at the Northeast ALS (NEALS) Biorepository (neals.org/for-als-researchers/neals-sample-repository/) and are publicly available to researchers around the world. All requests for data and biofluid samples will go through the NEALS Sample Repository through an application request system for qualified researchers. All biofluid samples will be available until they are depleted. Data will be made public 1 year postpublication.
Results
C9ALS natural history: Prospective disease onset and survival
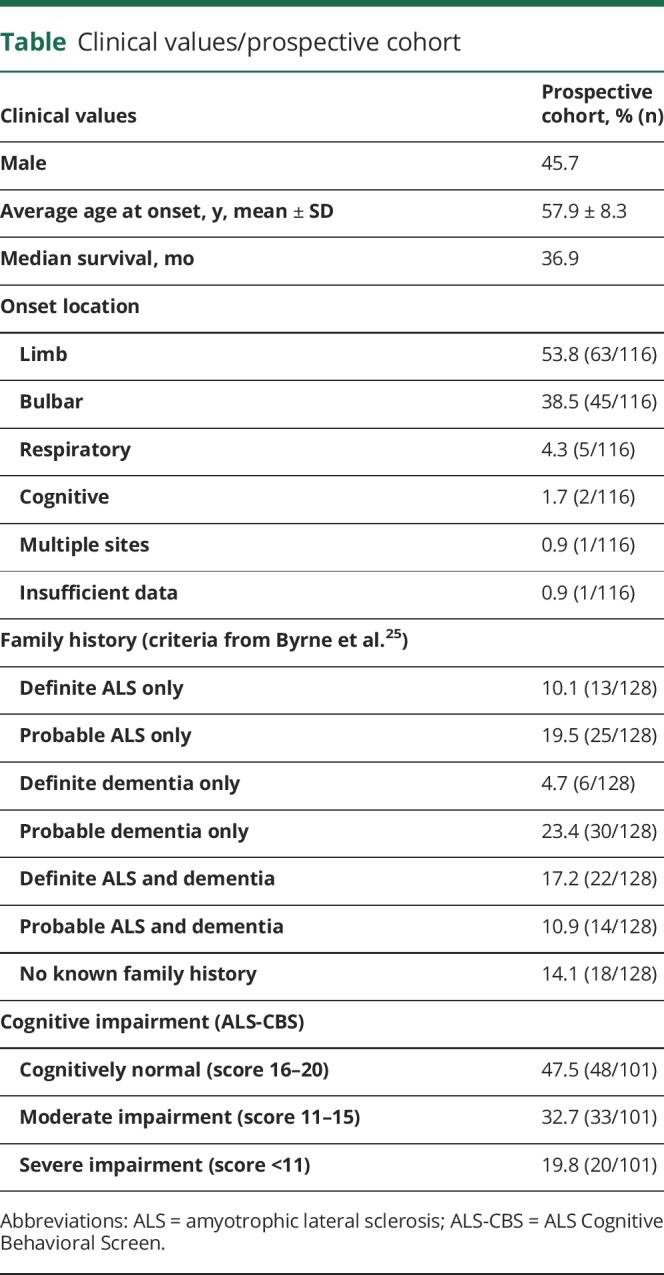
Data were collected prospectively from 116 symptomatic C9ALS and 12 non-ALS full expansion carriers. Of the symptomatic individuals, 53 were male and 63 female. Average age at disease onset was 57.9 ± 8.3 years (figure 1A and table). At the time of this report, 88 individuals had reached a survival endpoint (either mortality or initiation of invasive ventilation) and 28 were either still alive or lost to follow-up. Median survival in this population was 36.9 months after disease onset (figure 1B and table). Site of disease onset and patient-reported family histories (following criteria defined by Byrne et al.25) are described in figure 1, C and D, and the table.
Table.
Clinical values/prospective cohort
C9ALS natural history: Retrospective disease onset and survival
In addition to the prospective study, we collected retrospective survival and age at onset information from a separate cohort of 208 individuals with C9ALS and 450 individuals with SALS. Mean age at onset from the retrospective cohort was similar to the prospective cohort: 59.0 ± 9.3 years for C9ALS and 59.3 ± 12.1 years for SALS (figure 1E). Median survival was 29.9 and 30 months for C9ALS and SALS, respectively; however, notably, the C9ALS group had a significantly smaller fraction of slow-progressing individuals (figure 1F).
C9ALS natural history: Prospective ALSFRS-R, SVC, ALS-CBS, and ALSCBQ
Longitudinal data for individuals with C9ALS were available for ALSFRS-R (n = 88), SVC (n = 53), ALS-CBS (n = 42), and ALSCBQ (n = 29). In addition, baseline ALS-CBS data were collected from 101 C9ALS participants. We observed an average monthly decline in ALSFRS-R of −1.8 ± 1.7 and SVC of −1.4% ± 3.24% (figure 2, A and B). At first visit, 47.5% (48/101) of individuals with available data presented as cognitively normal, 32.7% (33/101) as moderately impaired, and 19.8% (20/101) as severely impaired (table). We observed little change in ALS-CBS or ALSCBQ total scores (figure 2, C and D) or individual subscores (figure 2, E–L) over time in this cohort. In addition, we obtained baseline ALS-CBS data for all 12 participants in the non-ALS cohort. Of these, 6/12 presented as cognitively normal, 3/12 as moderately impaired, and 3/12 as severely impaired. During the course of this study, no change in ALS-CBS impairment status was observed in the 7 individuals without ALS for whom longitudinal measures were available.
Figure 2. Individuals with amyotrophic lateral sclerosis (ALS) with chromosome 9 open reading frame 72 expansion mutations (C9ALS) natural history: Measures of disease progression.

(A) ALS Functional Rating Scale–Revised (ALSFRS-R), (B) slow vital capacity (SVC), (C) ALS Cognitive Behavioral Screen (ALS-CBS), and (D) ALS Caregiver Behavioral Questionnaire (ALSCBQ) rates of decline for individuals with C9ALS with at least 2 longitudinal data points. (ALSFRS-R: n = 88; SVC: n = 53; ALS-CBS: n = 42; ALSCBQ: n = 29.) (E–H) ALSFRS-R (n = 88) and (I–L) ALS-CBS (n = 42) subscore rates of change.
Relationship of expansion size and age in prospective cohort
We probed G4C2 repeat size in blood DNA from 89 individuals in the prospective C9ALS and non-ALS cohorts. The average repeat size was 2,789 ± 757 repeats, noting that 10 individuals' repeat sizes were within 318 repeats of the upper limit of detection. We observed a significant positive correlation of age at disease onset and repeat size (figure 3A). As expected, we also found a positive correlation between repeat size and age at sample collection (figure 3, B and C). We observed no significant correlations between repeat size and survival after onset or ALSFRS-R rate of decline (figure 3, D and E).
Figure 3. Relationship of G4C2 repeat size and age.

G4C2 repeat size in blood, as measured by southern blot, has a significant positive correlation with (A) age at disease onset (n = 79; R = 0.254; p = 0.05) and (B) age at sample collection (n = 89; R = 0.281; p = 0.02). (C) When individuals are separated by median repeat size, age at collection is significantly higher in the top half than the bottom half (n = 89; Mann-Whitney U test: p < 0.01**). Repeat size does not correlate with (D) survival (n = 61; R = 0.031; p = 0.91) after onset or (E) ALS Functional Rating Scale–Revised (ALSFRS-R) rates of decline (n = 66; R = 0.053; p = 0.90). (F) In an unexpanded (<30 repeats) population, haplotype-carrying individuals have a significantly larger average repeat size in blood than noncarriers (n = 244 haplotype carriers, n = 430 noncarriers; Mann-Whitney U test: p < 0.0001). (G) In individuals without ALS carrying the chromosome 9 open reading frame 72 (C9orf72)–associated risk haplotype with large, yet unexpanded repeat sizes (upper quintile; 13–25 repeats), repeat size is significantly correlated with age at sample collection (n = 47; R = 0.434; p < 0.01). (H) Repeat size is significantly higher in the top half of large, unexpanded repeat carriers than the bottom half (n = 47; Mann-Whitney U test: p < 0.01**). All p values are corrected for multiple comparisons (false discovery rate).
Relationship of expansion size and age in unexpanded individuals
We hypothesized that repeat size and age correlations we observed in the prospective cohort may represent dynamic G4C2 expansion over time. We extended our analyses to blood-derived DNA samples from a large (n = 674) cohort of individuals with unexpanded repeat sizes (<30 repeats) from WU and ADNI. Within this population, 36% (244/674) of individuals were carriers of the C9orf72 risk haplotype. Mirroring previously described populations,5,29 haplotype carriers in this cohort have a significantly larger repeat size than noncarriers, suggesting that this genetic background may be permissive to expansion (figure 3F).
Inherited repeat size is a known risk factor for active expansion in similar repeat disorders.30 We hypothesized that repeat size would be age-dependent in unexpanded individuals with large repeat sizes. We defined large, unexpanded repeats as the upper quintile of repeat sizes in the haplotype carrier group (upper 47 of 244 individuals). Indeed, within this group, we observed a significant positive correlation with age at sample collection (figure 3, G and H).
Poly(GP) dipeptides are large and CSF levels negatively correlate with DNA repeat size
We measured relative concentrations of poly(GP) in CSF of C9ALS and non-ALS carriers from the prospective cohort, as well as individuals with SALS from WU, confirming C9-specificity of the immunoassay (figure 4A). Next, we examined poly(GP) in longitudinal CSF draws from a subset of individuals and observed that poly(GP) levels remain steady over time (figure 4B), consistent with previous reports.22 We found no significant correlations between poly(GP) CSF levels and ALS history measures, such as age at onset, survival, and ALSFRS-R rate of change (figure 4C–E). Interestingly, we observed a significant negative correlation between DNA repeat size and poly(GP) levels (figure 4F).
Figure 4. Poly(GP) in individuals with amyotrophic lateral sclerosis (ALS) with chromosome 9 open reading frame 72 expansion mutations (C9ALS) CSF and size in autopsy tissue.

(A) Poly(GP) in CSF is highly C9-specific, with signal only observed in full expansion carriers (n = 32 C9ALS; n = 4 singleton ALS [SALS]; n = 6 C9+ carriers without ALS [AC]). (B) Poly(GP) levels are consistent between draws for 9 out of 10 individuals with longitudinal CSF draws. (C–F) CSF poly(GP) correlations with (C) ALS age at onset (n = 30 individuals; R = 0.040; p = 0.91), (D) survival (n = 20 individuals; R = 0.023; p = 0.92), (E) ALS Functional Rating Scale–Revised (ALSFRS-R) average monthly rate of change (n = 25 individuals; R = 0.111; p = 0.90), and (F) blood DNA repeat size, as measured by southern blot (n = 34 individuals; R = 0.371; p = 0.05). (G) size exclusion chromatography (SEC) standard proteins (Bio-Rad, Hercules, CA) separated by a superdex 10/300 GL SEC column used in poly(GP) size assessments, demonstrating efficient separation of relevant protein sizes. (H) Poly(GP) is observed in large SEC-separated C9ALS autopsy CNS samples. All samples are normalized to fraction 9 (the largest SEC fraction after void volume). Dotted line represents SEC standard proteins as shown in G. Matched patient autopsies were used for cerebellum and frontal cortex analyses. All p values are corrected for multiple comparisons (false discovery rate).
Finally, we investigated DPR size in human CNS autopsy tissue. We fractionated soluble protein from C9ALS CNS autopsy tissues by size and measured relative poly(GP) concentration in various size ranges. This assay efficiently separated protein sizes covering the expected range of DPR repeats (figure 4G). We observed poly(GP) signal only in large fractions of both cerebellum and frontal cortex (figure 4H), indicative of full length repeat peptides.
Discussion
Immense progress has been made in understanding the pathobiology of C9ALS since its discovery in 2011. Of particular importance has been the discovery of RNA foci and DPR pathologies, which numerous laboratory studies have now linked to toxicity in human and animal models.15 Correspondingly, much focus has been recently placed on repeat RNA-lowering therapeutics such as antisense oligonucleotides (ASOs), which effectively reduce both RNA foci and DPR pathologies and have been shown to alleviate defects in C9ALS cell and animal models.16–19 These promising treatments are rapidly progressing toward clinical trials (for example, NCT03626012), following the path of similar ASO therapies for spinal muscular atrophy (SMA)31 and SOD1 ALS.32 The data herein will help facilitate design of these trials by providing an evidence-based description of the natural history and pathologic features of C9ALS and further supporting poly(GP) as a pharmacodynamic biomarker.
In our prospective analysis of C9ALS clinical characteristics, we found a mean age at onset of 57.9 ± 8.3 years and a median survival of 36.9 months (figure 1, A and B). Interestingly, our retrospective C9ALS cohort presented with similar onset (59.0 ± 9.3 years) but a shorter survival (29.9 months) than the prospective cohort. While the reasons for these disparities are unclear, this may reflect inherent differences in prospective and retrospective data collection. A number of retrospectively collected C9ALS natural history datasets have been reported previously; however, clinical characteristics in these studies have been notably variable, for reasons difficult to pinpoint.1,4,5,33–37 In addition, one small prospective study has described C9ALS disease progression38; however, this study included only 21 symptomatic individuals with C9ALS, of whom only 9 reached a survival endpoint during the study period. By prospectively collecting clinical data from a large, multicenter C9ALS population, this report definitively describes C9ALS natural history, which will aid upcoming trials in accurately determining study length and other measures, such as power calculations, that are dependent on expectations of participants' survival.
In our retrospective analysis, we identified less variance in age at onset and fewer very slowly progressing cases in C9ALS compared to SALS (figure 1E, F). In measures of disease progression from prospective individuals with C9ALS, we observed an ALSFRS-R rate of decline of −1.8 ± 1.7 and SVC of −1.4% ± 3.24% of predicted (figure 2, A and B). In comparison, the PRO-ACT dataset consisting of over 8,600 individuals with ALS compiled from 16 ALS clinical trials demonstrated more variation in rates of decline, at −1.02 ± 2.3 and −2.3% ± 6.9% for ALSFRS-R and forced vital capacity, respectively,39 but why differences are observed between the mean values of these measures between these 2 datasets is not yet understood. The C9ALS population presented here displays a relatively more homogenous clinical description than the non-C9ALS population. Finally, we assessed utility of ALS-CBS and ALSCBQ in evaluating longitudinal cognitive changes in the prospective C9ALS cohort. In either measure, we were unable to observe changes over time (figure 2, C and D), though it is unclear whether these results represent a true lack of decline or an inability of these measures to accurately assess longitudinal cognitive function. With all measures of disease progression presented here, it is important to consider possible ascertainment bias due to unavailability of follow-up and longitudinal data, which may be more likely to occur in individuals with quickly progressing disease. In addition, while rare, a small percentage of the C9ALS individuals may carry a second, unrelated disease-linked mutation, which may affect progression rates in an undetermined way. This was not assessed in this population. Nevertheless, collectively these data provide a template for C9ALS clinical trial design moving forward, and it is expected that the relative homogeneity of the C9ALS population will decrease the number of participants required to observe a treatment effect.
Importantly, during our prospective study, we collected C9ALS biofluids, which are now housed in the NEALS Biorepository and are available for C9orf72-related research (neals.org/for-als-researchers/neals-sample-repository/). We analyzed blood DNA repeat size in full expansion carriers, which we found to correlate with individuals' ages at sample collection (figure 3, B and C), a result that mirrors several previously published reports in C9ALS populations,4,7,40–43 including one recent report showing a similar relationship in a large cohort of presymptomatic expansion carriers.43 Interestingly, we also observed a positive correlation of repeat size and age in a separate population of unexpanded individuals without ALS (figure 3, G and H). One possible interpretation of these data is that the C9orf72 repeat is dynamic and able to expand over time. Similar genomic instability has been demonstrated in other expansion disorders.30,44–48 Further, an abundance of SALS is attributed to C9orf72 expansions1 and somatic mosaicism is commonly observed in C9ALS expansions,4,6,7 both suggesting dynamic expansion is possible. Alternatively, it is possible that presence of larger repeats is in some way protective and is thus over-represented in older populations. Indeed, we observed a positive correlation between repeat size and age at onset in the C9ALS prospective cohort (figure 3A). In support of this, it is known that presence of large repeats decreases RNA levels of C9orf72,2,3,19,49 which could potentially provide a protective effect by lowering levels of nascent RNA foci or DPRs. In addition, sequestration of larger repeat RNAs into RNA foci in the nucleus could result in less cytoplasmic RAN translation, as supported by one study that found RNA foci and poly(GP) inclusions rarely occurring in the same cells.11 Consistent with this, in this study we observed a negative correlation between DNA repeat size and poly(GP) levels in individuals with C9ALS (figure 4F), which could be attributed to these or other yet unexplored mechanisms. It should be noted that these results were obtained from blood-derived DNA and may not accurately represent CNS expansion characteristics. It is also important to note that one previous report found a significantly older age at onset and lower methylation of an upstream CpG island in short expansion carriers (55–100 repeats) than in full expansion carriers, supporting a possible inverse relationship of repeat size and onset age,8 while 2 other reports were unable to find any relationship of repeat size with onset age,6,50 albeit with smaller sample sizes than in this study. Future studies in cell culture and animal models will help to refine the interpretation of these interesting correlations.
As C9ALS-targeted therapies begin to move toward clinical trials, it becomes increasingly important to develop methods to measure drug target engagement. To this end, C9ALS pharmacodynamic biomarkers have begun to be explored, such as poly(GP).22 We sought to provide insight into the characteristics of poly(GP) in both CSF and autopsy tissue to further investigate use of this DPR as a C9ALS biomarker. While we found no correlations between CSF poly(GP) and ALS natural history measures (figure 4, C–E), we were able to confirm that CSF poly(GP) is C9-specific and its levels are consistent over time (figure 4, A and B), supporting its continued use as a pharmacodynamics biomarker for expansion-targeted therapeutics. However, the lack of correlation between poly(GP) levels and clinical measures, which is consistent with previous findings,22 suggests that this measure likely does not inform on disease status and perhaps other pathologies are involved in disease pathogenesis, though these analyses could be confounded by low sample sizes in some cases. Finally, we sought to examine DPR size in individuals with C9ALS. In human CNS samples, we observed very large poly(GP) dipeptides, including those upwards of 2,000 repeats (figure 4H). While we did not observe smaller poly(GP) species here, it is possible that these are below the sensitivity limits of our immunoassay; as the epitope for poly(GP) antibodies is inherently repetitive, this assay likely has greater sensitivity for larger species. In addition, while we only used soluble lysates in these analyses and ran this experiment under highly denaturing conditions, we cannot rule out the possibility that these sizes could be obscured by some degree of secondary structure or protein interactions. Nevertheless, this experiment highlights 2 important findings: large DPRs are synthesized in human brains and poly(GP) being measured with immunoassays is likely only representing large species. Our results suggest that when possible, large DPRs should be used in mechanistic studies.
We have presented a comprehensive clinical description of C9ALS and provided a template for upcoming C9ALS clinical trials. In addition, through collection of longitudinal biofluid samples and comparison to our prospectively collected clinical and demographic datasets, we have highlighted important pathobiological correlations within this population. Together, these results establish baseline clinical and pathologic characteristics for C9ALS and provide a reliable resource for future clinical and translational studies.
Acknowledgment
The authors thank the participants, families, and caregivers involved in this study; members of the Northeast ALS Consortium (NEALS) for referrals to the study; the study coordinators at each site: Mark Levine-Weinberg, Jessica Zhu, Leah Miller, Julia Yasek, Tiina Jie Huan Xu, Daniela Grasso, Helen Mejia-Santana, Jessica Singleton, Natalia Leontovich, Kristen Riley, Carolyn Prina, Dana Fine, Peggy Allred, Vy Nguyen, Koral Wheeler, Tommy Bunte, and Katherine Fetterman; and the group from NCRI: Leonard Friedman, Priyadarshini Vader, Lucia Alvarado-Balderrama, Amanda Nichols, Haining Li, and Ysura Wahab.
Glossary
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- ALS
amyotrophic lateral sclerosis
- ALS-CBS
ALS Cognitive Behavioral Screen
- ALSCBQ
ALS Caregiver Behavioral Questionnaire
- ALSFRS-R
ALS Functional Rating Scale–Revised
- ASO
antisense oligonucleotide
- C9ALS
individuals with amyotrophic lateral sclerosis with chromosome 9 open reading frame 72 expansion mutations
- C9orf72
chromosome 9 open reading frame 72
- DPR
dipeptide repeat protein
- FTLD
frontotemporal lobar degeneration
- MGH
Massachusetts General Hospital
- NCRI
Neurologic Clinical Research Institute
- NEALS
Northeast Amyotrophic Lateral Sclerosis Consortium
- RAN
repeat associated, non-ATG
- rpPCR
repeat primed PCR
- SALS
singleton amyotrophic lateral sclerosis
- SDS
sodium dodecyl sulfate
- SEC
size exclusion chromatography
- SMA
spinal muscular atrophy
- SNP
single nucleotide polymorphism
- SVC
slow vital capacity
- WU
Washington University
Appendix. Authors
Study funding
Funding provided by Biogen Inc., the ALS Association, the Muscular Dystrophy Association, the Knight Alzheimer's Disease Research Center at Washington University, and C9ID foundation. Other support provided by USPHS grant 5UL1 RR024992-02. Unexpanded control data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. A.J.C. was supported by grant 5T32GM008151-32 and by the Lucille P. Markey Special Emphasis Pathway in Human Pathobiology at Washington University. R.H. Baloh was supported by NIH R01 NS097545 and the Robert and Louis Schwab Family. R.H. Brown receives support from NINDS (R01 NS088689, R01 NS073873, R01 NS104022), the ALS Association, ALS Finding a Cure, ALS ONE, the Angel Fund for ALS Research, and Project ALS. T.M.M. was supported by NINDS R01 NS078398. Industry sponsor: Biogen Inc.
Disclosure
A. Cammack reports no disclosures relevant to the manuscript. N. Atassi has consulted for MT Pharma, Neuropore, Chronos, Boston Pharmaceuticals, Denali, GSK, and Anelixis. T. Hyman reports no disclosures relevant to the manuscript. L. van den Berg has received personal fees from Shire, Biogen, Cytokinetics, and Treeway, outside the submitted work. M. Harms has grant funding from Biogen. R. Baloh has consulted for Kite Pharmaceuticals, Maze Therapeutics, and Acurastem. R. Brown, M. van Es, J. Veldink, and B. de Vries report no disclosures relevant to the manuscript. J. Rothstein is a consultant for Glaxo Smith Kline and Expansion Therapeutics. C. Drain, J. Jockel-Balsarotti, A. Malcolm, and S. Boodram report no disclosures relevant to the manuscript. A. Salter has received consulting fees for statistical reviews in Circulation: Cardiovascular Imaging. N. Wightman, H. Yu, A. Sherman, T. Esparza, D. McKenna-Yasek, M. Owegi, and C. Douthwright report no disclosures relevant to the manuscript. A. McCampbell is an employee of Biogen. T. Ferguson is an employee of Biogen. C. Cruchaga receives research support from Biogen, EISAI, Alector, and Parabon and is a member of the advisory board of ADx Healthcare, Halia Therapeutics, and Vivid Genomics. M. Cudkowicz is a consultant for Biohaven, Takeda, Avexis, and Biogen and was chair of DSMB for Lilly. T. Miller has served on medical advisory boards for Biogen and Ionis Pharmaceuticals and is a consultant for Cytokinetics. T.M. Miller and Washington University have a licensing agreement with Ionis Pharmaceuticals and with C2N. Go to Neurology.org/N for full disclosures.
References
- 1.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dols-Icardo O, García-Redondo A, Rojas-García R, et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet 2014;23:749–754. [DOI] [PubMed] [Google Scholar]
- 7.Nordin A, Akimoto C, Wuolikainen A, et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet 2015;72:1–10. [DOI] [PubMed] [Google Scholar]
- 8.Gijselinck I, Van Mossevelde S, Van Der Zee J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 2016;21:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacKenzie IR, Arzberger T, Kremmer E, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 2013;126:859–879. [DOI] [PubMed] [Google Scholar]
- 10.Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013;77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gendron TF, Bieniek KF, Zhang YJ, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013;126:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori K, Weng S, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013;339:1335–1339. [DOI] [PubMed] [Google Scholar]
- 13.Zu T, Liu Y, Bañez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA 2013;110:E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackenzie IR, Frick P, Grässer F, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 2015;130:845–861. [DOI] [PubMed] [Google Scholar]
- 15.Gendron TF, Petrucelli L. Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb Perspect Med 2018;8:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013;80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides: targeting GGGGCC-containing RNAs. Neuron 2016;90:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA 2013;110:E4530–E4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013;5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Q, Lehmer C, Michaelsen M, et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol Med 2017;9:687–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Z, Zhang Y, Gendron TF, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014;83:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med 2017;9:eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmer C, Oeckl P, Weishaupt JH, et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med 2017;9:859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner MR, Al-Chalabi A, Chio A, et al. Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry 2017;88:1042–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byrne S, Bede P, Elamin M, et al. Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011;12:157–159. [DOI] [PubMed] [Google Scholar]
- 26.Harms MB, Cady J, Zaidman C, et al. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:2234.e13–2234.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mok K, Traynor BJ, Schymick J, et al. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging 2012;33:209.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wijsman EM, Pankratz ND, Choi Y, et al. Genome-wide association of familial late-onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith BN, Newhouse S, Shatunov A, et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur J Hum Genet 2013;21:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YH, Kuhl DPA, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991;67:1047–1058. [DOI] [PubMed] [Google Scholar]
- 31.Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017;377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 32.Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 2013;12:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart H, Rutherford NJ, Briemberg H, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012;123:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 2012;11:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irwin DJ, McMillan CT, Brettschneider J, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Mossevelde S, van der Zee J, Gijselinck I, et al. Clinical evidence of disease anticipation in families segregating a C9orf72 repeat expansion. JAMA Neurol 2017;74:445–452. [DOI] [PubMed] [Google Scholar]
- 38.Floeter MK, Traynor BJ, Farren J, et al. Disease progression in C9orf72 mutation carriers. Neurology 2017;89:234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atassi N, Berry J, Shui A, et al. The PRO-ACT database. Neurology 2014;83:1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013;92:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hübers A, Marroquin N, Schmoll B, et al. Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexanucleotide repeat in different motor neuron diseases. Neurobiol Aging 2014;35:1214.e1–1214.e6. [DOI] [PubMed] [Google Scholar]
- 42.Suh ER, Lee EB, Neal D, et al. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol 2015;130:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fournier C, Barbier M, Camuzat A, et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol Aging 2019;74:234.e1–234.e8. [DOI] [PubMed] [Google Scholar]
- 44.Cossée M, Schmitt M, Campuzano V, et al. Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA 1997;94:7452–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ditch S, Sammarco MC, Banerjee A, Grabczyk E. Progressive GAA-TTC repeat expansion in human cell lines. PLoS Genet 2009;5:28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Møllersen L, Rowe AD, Larsen E, Rognes T, Klungland A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet 2010;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du J, Campau E, Soragni E, et al. Role of mismatch repair enzymes in GAA·TTC triplet-repeat expansion in Friedreich Ataxia induced pluripotent stem cells. J Biol Chem 2012;287:29861–29872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMurray C. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 2010;11:786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014;507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, Lin Z, Chen X, et al. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2015;38:217.e15–217.e22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data from this study are stored in the NeuroBANK™ (neurobank.org/) data repository at the MGH NCRI and are linked with biospecimen repositories. We will share deidentified datasets with researchers who want to advance understanding of neurologic disease. A limited amount of deidentified biofluid samples (DNA, serum, peripheral blood mononuclear cells, urine, and CSF) collected from this study are stored at the Northeast ALS (NEALS) Biorepository (neals.org/for-als-researchers/neals-sample-repository/) and are publicly available to researchers around the world. All requests for data and biofluid samples will go through the NEALS Sample Repository through an application request system for qualified researchers. All biofluid samples will be available until they are depleted. Data will be made public 1 year postpublication.