

Abstract
Cholesterol catabolism plays an important role in Mycobacterium tuberculosis’s (Mtb’s) survival and persistence in the host. Mtb exploits three β-oxidation cycles to fully degrade the side-chain of cholesterol. Five cistronic genes in a single operon encode three enzymes, 3-oxo-4-pregnene-20-carboxyl-CoA dehydrogenase (ChsE1-ChsE2), 3-oxo-4,17-pregnadiene-20-carboxyl-CoA hydratase (ChsH1-ChsH2), and 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA retro-aldolase (Ltp2) to perform the last β-oxidation cycle in this pathway. Among these three enzymes, ChsH1-ChsH2 and Ltp2 form a protein complex that is requisite for catalysis of carbon-carbon bond cleavage. In this work, we report the structure of full length ChsH1-ChsH2-Ltp2 complex based on small-angle X-ray scattering (SAXS) and single particle electron microscopy data. Mutagenesis experiments confirm the requirement for Ltp2 to catalyze the retro-aldol reaction. The structure illustrates how acyl transfer between enzymes may occur. Each protomer of ChsH1-ChsH2-Ltp2 contains three protein components: a chain of ChsH1, a chain of ChsH2, and a chain of Ltp2. Two protomers dimerize at the interface of Ltp2 to form a heterohexameric structure. This unique heterohexameric structure of ChsH1-ChsH2-Ltp2 provides entry to further understand the mechanism of cholesterol catabolism in Mtb.
Keywords: thiolase, reverse Claisen, enolate anion, thioester, MaoC-like hydratase, Rv3542c, Rv3541c, Rv3540c
Graphical Abstract
Introduction
Mycobacterium tuberculosis (Mtb) infects approximately 1.7 billion people worldwide.1 Tuberculosis (TB) is the leading cause of human death from a single infectious agent. In 2017, 10 million people developed active TB disease. Among them, half a million people were estimated to be infected with multi-drug resistant (MDR) TB.2 The rising level of drug resistant Mtb infection highlights the unmet need for further TB drug discovery. Identifying novel and essential metabolic pathways in Mtb is one avenue to deliver new drug targets.3 Upon being engulfed by a macrophage, Mtb manipulates the host immune system and triggers increased uptake of lipids into the macrophage, which leads to the formation of foamy macrophages with accumulated lipid bodies that form one shell of cells in the lung granuloma.4 Furthermore, the necrotic core of a matured granuloma is full of accumulated lipid-rich caseum.5 Mtb can catabolize these lipids, including fatty acids and cholesterol, for its survival and persistence in the host.6, 7
Mtb’s cholesterol catabolism provides a feedstock of important central metabolic intermediates like acetate (two carbons) and propionate (three carbons) for biosynthesis of secondary metabolites.8, 9 More importantly, several genes for cholesterol uptake and catabolism are essential for the survival, persistence and virulence of Mtb in infected mice models.6, 10–12 Therefore, Mtb’s cholesterol metabolism pathway plays a significant role in its pathogenesis.
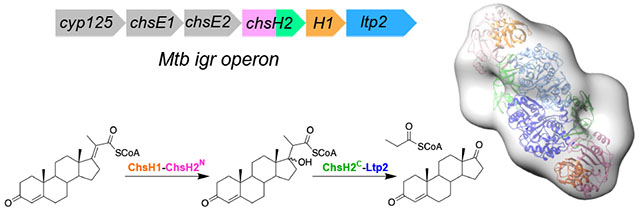
An operon of genes under the regulation of KstR1 was identified to be required for Mtb’s survival in macrophages.10 The Mtb intracellular growth operon (igr) knock-out mutant was unable to grow on cholesterol in vitro and showed attenuated growth in a mouse model of infection during early stage of infection.11 The igr operon contains 6 genes, cyp125 (Rv3545c), chsE1 (Rv3544c), chsE2 (Rv3543c), chsH2(Rv3542c), chsH1 (Rv3541c), and ltp2 (Rv3540c) (Figure 1A). Cyp125 catalyzes the oxidation of the terminal C26 methyl of cholesterol and deletion of cyp125 can be rescued by cyp142 (Rv3518c). The remaining five genes in the igr operon encode enzymes that catalyze the last β-oxidation cycle in the cholesterol side-chain degradation pathway (Figure 1B).13–15 These five cistronic genes are conserved in other cholesterol-degrading actinobacteria, whereas, cyp125 is not conserved in the operon.
Figure 1. Biochemical function of igr genes.

(A) Organization of the igr operon in Mtb from Rv3545c (cyp125) to Rv3540c (ltp2). (B) The chemical reactions catalyzed by the gene products encoded by the igr operon. The substrates are 3-OPC-CoA: 3-oxo-4-pregnene-20-carboxyl-CoA; 3-OPDC-CoA: 3-oxo-4,17-pregnadiene-20-carboxyl-CoA; 17-HOPC-CoA: 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA; AD: androstenedione, and propionyl-CoA.
In the igr operon, two pairs of genes each encode a heterotetrameric enzyme. One pair, chsE1-chsE2, encodes an acyl-CoA dehydrogenase and the other pair, chsH1-chsH2, encodes an enoyl-CoA-hydratase (Figure 1).13, 14 These enzyme complexes form α2β2 heterotetramers unique to actinobacterial cholesterol catabolism pathways. The heterotetrameric protein structure of ChsH1-ChsH2 was characterized by X-ray crystallography of a truncated version in which the C-terminal domain of ChsH2 was absent.14 Sequence analysis indicated that the C-terminus belongs to the DUF35/DUF35_N family. A bioinformatic study of DUF35 homologs revealed that this motif tends to be fused with protein domains belonging to thiolases, short chain acyl-CoA dehydrogenases, and sterol-carrier proteins, where the DUF35 domain is likely to function as a noncovalent acyl-CoA delivery protein in different acyl-CoA utilization reactions.16
The final gene in the igr operon, ltp2, encodes a retro-aldolase which has been demonstrated to cleave the acyl hydroxyl CoA three-carbon side-chain completely from the steroid ring of 17-HOPC-CoA (Figure 1B).15 Recently, Ltp2 has been shown to form a protein complex with ChsH1-ChsH2, the enoyl-CoA hydratase, upon co-expression.15 Intriguingly, Ltp2 specifically interacts with the DUF35 (ChsH2C) domain of ChsH2 and can form a complex with the DUF35 (ChsH2C) domain alone. A crystal structure of DUF35-Ltp2 from Thermomonospora curvata was recently solved and the complex forms an αββα heterotetrameric structure.17 Two Ltp2 monomers homodimerize and the DUF35 monomer is positioned at each end of the Ltp2 dimer.17 This structure suggests that the ChsH1-ChsH2 heterotetramer can be extended beyond the DUF35 domain to form a larger complex.
Here, we characterize the full length ChsH1-ChsH2-Ltp2 complex structurally and biochemically. Small-angle X-ray scattering (SAXS) and negative staining electron microscopy provide low resolution structural information of ChsH1-ChsH2-Ltp2 complex. Analytical ultracentrifugation (AUC) demonstrates the complex exists predominately as a diprotomer with each protomer containing a chain of ChsH1, a chain of ChsH2 and a chain of Ltp2. A model of ChsH1-ChsH2-Ltp2 is created through protein-protein docking based on the EM map. The structure reveals a different protomer dimerization interface than previous structures suggested. The dimerization interface of ChsH1-ChsH2N is disrupted to form the ChsH1-ChsH2-Ltp2 full protein complex, although at high concentrations, maintenance of this dimerization interface may lead to fibril formation. Furthermore, the structure illustrates how acyl transfer may occur. Through mutagenesis experiments, we validate the importance of Ltp2 for catalyzing the retro-aldol reaction.
Materials and Methods
Materials, strains, media, and general methods.
Mtb H37Rv total genomic DNA was obtained from the TB Research Materials Facility at Colorado State University (NIAD NO1-AI40091). Coenzyme A was purchased from Sigma-Aldrich (St. Louis, MO). 3-Oxo-pregna-4,17-diene-20-carboxyl-CoA (3-OPDC-CoA) was synthesized as reported.9 Isopropyl β-D-1-thiogalactopyranoside (IPTG) was purchased from Denville Scientific (Metuchen, NJ). Kanamycin was purchased from IBI Scientific (Peoria, IA). Ampicillin was purchased from Roche Diagnostics (Indianapolis, IN). iProof DNA polymerase and IMAC nickel resin were from Bio-Rad (Hercules, CA). Restriction endonucleases, Quick Ligase, and protein ladder were from New England Biolabs (Beverly, MA). Superdex 200 HiLoad 16/60 columns were from GE Healthcare Biosciences Corp. (Piscataway, NJ). Oligonucleotides were from Eurofins Genomics (Louisville, KY) or Integrated DNA Technologies (Coralville, IA). Sanger DNA sequencing (performed by GENEWIZ LLC) was used to verify the coding sequence of the expression plasmids. All other chemicals were obtained from Fisher Scientific (Hampton, NH), unless otherwise stated. BL21(DE3) E. coli was obtained from BioRad. Rhodococcus jostii RHA1 strain is a gift from Lindsay Eltis (Department of Microbiology and Immunology, University of British Columbia). pTipQC1 and pTipQC2 vectors are gifts from Tomohiro Tamura (National Institute of Advanced Industrial Science and Technology, Sapporo, Japan). ChsH1-ChsH2 (Uniprot IDs: I6XHI0-I6YGF8, and ChsH1-ChsH2N (amino acid residues 1–186) were purified from a heterologous expression system using BL21(DE3) E. coli as previously reported.9, 13, 14 The 2xYT media is composed of 16 g tryptone, 10 g yeast extract and 5 g NaCl per liter. The LB media is composed of 25 g/L Luria broth. Buffer A: 20 mM Tris-HCl pH 8.5, 300 mM NaCl, and 10 mM imidazole. Buffer B: 20 mM Tris-HCl pH 8.5, 300 mM NaCl, and 500 mM imidazole. Buffer C: 50 mM Tris-HCl buffer pH 8.5, and 200 mM NaCl, 5% glycerol.
Expression plasmid construction.
chsH1, chsH2, and ltp2 were amplified from Mtb H37Rv total genomic DNA by PCR (Table S1). The PCR products were digested with appropriate restriction endonucleases and ligated into pET28b, pTipQC1, or pTipQC2 vectors. DNA sequencing of the plasmids confirmed that the sequences were correct and that no mutations were introduced during PCR amplification. Mutants were prepared using QuickChange site-directed mutagenesis. The mutations were confirmed by plasmid DNA sequencing.
Protein preparation using heterologous expression in Rhodococcus jostii RHA1.
Construct pigr-3-RHA1, pigr-3_Ltp2c85A-RHA1, pigr-3_Ltp2H344A-RHA1, or pigr-3_Ltp2C85A,H344A-RHA1 was used for the cistronic expression of ChsH1-ChsH2-Ltp2 (Uniprot IDs: I6XHI0-I6YGF8-I6Y3T7) complex in Rhodococcus jostii RHA1. The appropriate plasmid was transformed into Rhodococcus jostii RHA1 cells through electroporation. Single colonies were selected on LB plates containing 30 μg/mL chloramphenicol and cultured in LB media at 30 °C. Expression was induced at OD600 ~ 0.6-0.8 by the addition of 20 μg/mL thiostrepton, and cells were grown at 25 °C for 24 h.
Cells were harvested by centrifugation at 5000 rpm for 15 min at 4 °C and all subsequent steps were conducted at 4 °C. The cells were suspended in Buffer A and lysed by cell disruption (4 passages at 27,000 psi). Cellular debris was removed by ultracentrifugation at 40,000 rpm for 1 h. The supernatant was loaded onto IMAC nickel resin, washed with 10 column volumes of Buffer A, and eluted with Buffer B. Protein solutions were immediately desalted by dialysis with Buffer C.
Isolated proteins in Buffer C were concentrated by ultrafiltration (MWCO 10 kDa) to less than 2 mL and purified by size-exclusion chromatography (SEC) on a Superdex 200 column equilibrated in Buffer C. The column was equilibrated in Buffer C at a flow rate of 1.0 mL/min and monitored at 280 nm. Fractions were collected and analyzed by reducing SDS-PAGE analysis. The fractions containing desired proteins were concentrated by ultrafiltration (MWCO 10 kDa).
Hydratase and retro-aldolase assay.
ChsH1-ChsH2, ChH1-ChsH2N, ChsH1-ChsH2-Ltp2, ChsH1-ChsH2-Ltp2C85A, ChsH1-ChsH2-Ltp2H344A, and ChsH1-ChsH2-Ltp2C85A H344A were assayed for catalytic activity with 3-OPDC-CoA as the substrate and the reaction mixture was monitored by MALDI-TOF mass spectrometry. Reactions containing 100 μM substrate 3-OPDC-CoA, 100 mM HEPES at pH 7.4, and 10 μM enzyme were incubated at 25 °C for 1 h, after which the reactions were quenched by adding 10% TFA. All the collected samples were subjected to zip-tip U-C18 (Millipore) extraction. The samples were washed with 0.1% trifluoroacetic acid (TFA) solution and eluted with 50% acetonitrile in 0.1% TFA solution from zip-tip, and then subjected to MALDI-TOF mass spectrometry. Matrices for MALDI-TOF mass spectrometry were prepared by dissolving 2,5-dihydroxybenzoic acid (20 mg/mL) in a 7:3 (v/v) mixture of 0.1% (v/v) trifluoroacetic acid and acetonitrile. MALDI-TOF mass spectra were acquired on a Bruker Autoflex II TOF/TOF operated in the reflectron negative mode. Mass spectral data were analyzed using Bruker flexAnalysis software (version 3.0).
SAXS of ChsH1-ChsH2 and ChsH1-ChsH2-Ltp2 complexes in solution.
Buffer C was used as the background solution for data collection. SAXS data sets were collected for ChH1-ChsH2N at 136 μM, ChsH1-ChsH2 at 50 μM, and ChsH1-ChsH2-Ltp2 at 30 μM at the National Synchrotron Light Source (NSLS II) at Brookhaven National Laboratory (Upton, NY) on Beamline X9 using an energy of 13.5 keV. Solutions were transferred to a 96-well plate and placed in a water-thermostatted sample holder (10 °C). Throughout the course of the experiment (about 30 sec per run), samples were flowed continuously through a 0.9 mm quartz capillary tube to minimize damage from X-ray radiation (~ 20 μL).
The scattering contribution of the ChsH1-ChsH2 or ChsH1-ChsH2-Ltp2 protein complex was obtained by subtracting the buffer scattering profile from the protein solution scattering profile. Data processing was performed using a Python-based package developed at X9. The data were fit in GNOM to produce a well-behaved P(r) curve. Twenty protein bead models were reconstructed in DAMMIF, and then aligned and compared in DAMSEL to determine the most probable model. DAMSUP was used to align all models with the most probable model, and all the aligned models were averaged in DAMAVER to compute a probability map. DAMFILT was applied to filter the average model at a preset cut-off volume. The “damfilt” models were used to yield the final SAXS envelopes.
Negative stain EM sample preparation and image acquisition.
ChsH1-ChsH2-Ltp2 complex (20 ng/μL) was applied onto a negatively glow-discharged carbon-coated grid (400 mesh, copper grid) for 60 s and excess protein was removed by blotting using filter paper. Uranyl acetate at 2% (Electron Microscopy Science) was applied on the grid after the protein was removed for 60 s and then blotted. The grid was dried for at least 30 minutes before subjection to the electron microscope. Images were collected by SerialEM using a FEI Tecnai G2 F20 Transmission Electron Microscope (TEM) at 200 kV equipped with a Gatan Ultrascan 4000 CCD camera (model 895), at 62,000x magnification and a 1.8 Å/pixel sampling rate. Micrographs were recorded at varying defocus values.
EM data processing and image analysis.
The collected images were imported into Relion3.0 for data processing. In Relion, the contrast transfer functions (CTF) were estimated using gctf.18 Around 1,000 particles were manually picked from 20 micrographs, which were used to generate 2D classes for autopicking templates. Eventually, 255888 particles were autopicked and extracted. 2D classification was performed in Relion3.0 using all the particles and good 2D classes were selected. The resulting particles after 2D classification selection were submitted for following steps. First, an initial model was calculated in Relion using a Stochastic Gradient Descent (SGD) algorithm from the selected particles under C2 symmetry. The initial model was used as a reference for 3D classifications. 3D classes were selected based on their consistency with the initial model and number of particles they contain. 3D refinement was performed using the initial model after low-pass filtering it to 60 A with C2 symmetry. For resolution determination, post-processing was run to get a gold-standard Fourier shell correlation (FSC) curve. For comparison of the SAXS and EM data, EM2DAM (part of the ATSAS software suite) was used to convert the EM model to a beads model. A theoretical X-ray scattering curve from the EM beads model was generated and compared with the experimental SAXS scattering curve in Chimera.
Analytical Ultracentrifugation.
Sedimentation velocity AUC experiments were performed at 20 °C using a Beckman Coulter XL-I analytical ultracentrifuge using an An-60Ti rotor at 325,000 rpm for 12 h with scans performed every 60 s. A double-sector cell, equipped with a 12-mm Epon centerpiece and sapphire windows, was loaded with 400 and 420 μl of sample and gel filtration buffer, respectively. Samples at concentrations of 0.425 mg/ml, 0.850 mg/ml, and 1.7 mg/ml were monitored with UV at 280 nm. The data were analyzed with Sedfit version 1501b19 using a continuous c(s) distribution. Numerical values for the solvent density, viscosity, and the partial specific volume were determined using Sednterp.20 Residual and c(s) distribution graphs were plotted using GUSSI.21
Rosetta protein-protein docking.
Protein-protein docking was used to build an atomic model for full length ChsH1-ChsH2-Ltp2 complex. Two different models were constructed and compared. For the first construction, hetero-tetrameric ChsH1-ChsH2N was placed in the center of the EM map in Chimera. A copy of heterodimeric DUF35 (ChsH2C)-Ltp2T.curvata was computationally isolated from the crystal structure (UniProt IDs: D1AB77-D1AB74; PDB code: 6OK1) and placed manually in the remaining EM density on one end with ChsH2C and ChsH2N in close proximity. Rosetta local docking was performed on the manually combined structure, and the best pose was selected in terms of the docking energy and fitting in the EM map. Then, Rosetta local refine was performed on the selected model. The other copy of heterodimeric DUF35 (ChsH2C)-Ltp2 was generated in Pymol based on the C2 symmetry of central hetero-tetrameric ChsH1-ChsH2N. Thus, the first model of ChsH1-ChsH2-Ltp2 complex was created.
For the second construction, hetero-tetrameric DUF35 (ChsH2C)-Ltp2T. curvata was placed in the center of the EM map in Chimera. A copy of hetero-dimeric ChsH1-ChsH2N was computationally isolated from the crystal structure (PDB code: 4W78) and placed manually in the remaining EM density on one end with ChsH2C and ChsH2N in close proximity. Rosetta local docking was performed on the manually combined structure, and the best pose was selected in terms of the docking energy and fitting in the EM map. The other copy of heterodimeric ChsH1-ChsH2N was generated in Pymol based on the C2 symmetry of central hetero-tetrameric DUF35 (ChsH2C)-Ltp2. Thus, the second model of ChsH1-ChsH2-Ltp2 complex was generated. The two different constructions were fitted into the SAXS models and compared in Chimera.
Results
Early attempts to express chsH1, chsH2, and ltp2 in a cistronic construct (pigr3) only yielded soluble ChsH1-ChsH2 protein complex, but Ltp2 was insoluble and captured in inclusion bodies.9, 14 ChsH1-ChsH2 was assayed using 3-OPDC-CoA as a substrate, (Figure S1). Intriguingly, the formation of propionyl-CoA was observed consistently in conjunction with formation of the hydrated acyl-CoA 17-HOPC-CoA, despite IMAC nickel resin-purification of ChsH1-ChsH2. In contrast, when assaying ChsH1-ChsH2N in which the C-terminus is truncated,14 the formation of propionyl-CoA was not observed under the same conditions and only 17-HOPC-CoA was obtained (Figure S1). This observation indicated that the DUF35 domain of ChsH2 is important for the carbon-carbon bond cleavage reaction, and suggested that ChsH1-ChsH2 and Ltp2 might form a protein complex through the C-terminus of ChsH2 although a stoichiometric protein complex was not formed under the conditions used, and soluble Ltp2 was never identified. During the course of our experiments, Seah et al. demonstrated soluble proteins could be obtained by heterologous expression of chsH1, chsH2, and ltp2 in Rhodococcus jostii RHA1.15 They confirmed that Ltp2 catalyzes the retro-aldol conversion of 17-HOPC-CoA to propionyl-CoA and androstenedione. We used the same expression system to obtain soluble ChsH1-ChsH2-Ltp2 complex (Figure S2) and characterized the protein structurally and biochemically.
SAXS analysis revealed structural information of full length ChsH1-ChsH2 and ChsH1-ChsH2-Ltp2 complex.
To structurally characterize ChsH1-ChsH2 and ChsH1-ChsH2-Ltp2, we turned to SAXS, which has been used successfully to study proteins that are difficult to crystallize. We collected scattering profiles of full length ChsH1-ChsH2 and ChsH1-ChsH2-Ltp2. In order to easily compare, we also obtained SAXS data on ChsH1-ChsH2N, for which the crystal structure is known.14 SAXS analysis of the three protein complexes confirmed they are homogeneously dispersed and there is no aggregation detected in Guinier plots (Figure 2A). The radii of gyration for ChsH1-ChsH2N, ChsH1-ChsH2, and ChsH1-ChsH2-Ltp2, calculated from the slope of Guinier plots at low angles, were estimated to be 28.49 Å, 41.48 Å, and 58.66 Å, respectively (Figure 2A and Table S2). Dmax values and Porod volumes calculated from the distance distribution curves for the three proteins are consistent with their relative sizes (Figure 2B and Table S2).
Figure 2. SAXS analysis for ChsH1-ChsH2N, ChsH1-ChsH2, and ChsH1-ChsH2-Ltp2.

(A) Guinier analysis of data showed the proteins are not aggregated. The regions used for Guinier analysis are shown as an inset. (B) Distance distribution plots from the scattering data calculated with GNOM. (C) (D) (E) Molecular envelopes generated from bead models calculated with the solution scattering data for (C) ChsH1-ChsH2N, (D) full length ChsH1-ChsH2, and (E) ChsH1-ChsH2-Ltp2 complex. In (C), the crystal structure of ChsH1-ChsH2N (PDB code: 4W78) was fitted into its SAXS envelope for comparison. The X and Y dimensions are calculated from the distance of the two most separated beads in the X and Y directions, respectively.
We used the SAXS data to generate ab initio bead models for the three protein complexes using the ATSAS suite of programs (Figure S3).22 Molmaps were generated from the bead models to 20 Å resolution in Chimera, and the map dimensions were calculated from the distance of the two most separated beads in the X and Y directions, respectively (Figure 2C, 2D, and 2E). Within expectation, the SAXS envelope of ChsH1-ChsH2N displayed a globular shape, and fitting the corresponding crystal structure into its envelope is straightforward (Figure 2C). The SAXS envelope of ChsH1-ChsH2 clearly showed two distinct regions on its two ends, which accounts for the inclusion of ChsH2C in the sample (Figure 2D). Upon the addition of Ltp2, the ChsH1-ChsH2-Ltp2 protein complex exhibited an even longer and wider SAXS map (Figure 2E). The three SAXS maps have similar widths in their X-dimensions, but their Y-dimensions are 60 Å, 150 Å, and 190 Å for ChsH1-ChsH2N, ChsH1-ChsH2, and ChsH1-ChsH2-Ltp2, respectively. The comparison revealed a clear trend of increased map dimensions for these three proteins in the solution state.
Negative stain EM analysis provides further structural information about the ChsH1-ChsH2-Ltp2 complex.
EM is a robust way to structurally characterize proteins that resist crystallization. Therefore, we carried out negative-stain EM studies on the ChsH1-ChsH2-Ltp2 complex. Single particle analysis showed homogeneously dispersed protein particles (Figure S4A), a key characteristic to facilitate three-dimensional reconstructions. After particle picking and extraction, all the extracted particles were used for initial two-dimensional (2D) analysis (Figure S4B). Particle stacks were cleaned by removing outliers from 2D classes. Three-dimensional (3D) classification revealed five major classes. After careful inspection, the best 3D class was selected based on its similarity with the initial model and the number of particles it contains. Refinement was performed with 3D autorefine in Relion (Figure S5) and the angular distribution of the refined model was calculated (Figure S6). Back-projections of the 3D map were calculated by Xmipp in Scipion,23 and the results agreed with the 2D class averages observed experimentally (Figure 3A). The refined model, determined to a resolution of ~24 Å (Figure 3B), exhibited a dumbbell shape from the front view and a wide C-shape from the side view. A groove feature was observed on the map at the back of the EM map (Figure 3C).
Figure 3. EM analysis of ChsH1-ChsH2-Ltp2 complex.

(A) Experimental reference-free 2D class averages, E, and the back-projections, P, calculated from the final 3D map. (B) Fourier shell correlation (FSC) curve of the map. (C) EM model of ChsH1-ChsH2-Ltp2 complex from single particle analysis of negative stain images. The density threshold was set at 0.05 in UCSF Chimera.
As an additional check, we calculated the theoretical X-ray scattering curve from the negative stain EM model and compared it to the experimental scattering data (Figure S7). A good fit was observed at small angles (q < 0.06 Å−1) but the curves diverged at larger angles which could result from distortion during dehydration of the EM samples.
AUC analysis revealed that the ChsH1-ChsH2-Ltp2 complex exists predominately as a diprotomer.
ChsH1-ChsH2 and Ltp2 form a stable complex according to the SAXS and EM experiments. To determine the actual oligomeric states, we performed AUC sedimentation velocity on the protein at three different concentrations (Figure S8 and Table S3). All three independent runs showed similar profiles with one major peak and two minor peaks (Figure 4A). The main peak matches the theoretical molecular weight of a protein complex composed of two copies of ChsH1, two copies of ChsH2 and two copies of Ltp2 (Figure 4B). We, thus, propose that ChsH1-ChsH2-Ltp2 complex predominately exists as a diprotomer (αβγ)2, where α, β, and γ represent a single copy of ChsH1, ChsH2, and Ltp2, respectively. This is consistent with our previous observation that ChsH1-ChsH2N exists as an α2β2 heterotetramer that is a dimer of a heterodimer. The two minor AUC peaks match the theoretical molecular weights of monoprotomer (αβγ), and tetraprotomer (αβγ)4, respectively (Figure 4B). These two oligomeric states, monoprotomer and tetraprotomer, may be in slow exchange with the dominant diprotomer form. At increasing concentrations of protein, the dispersity of states increased consistent with a shift toward more tetraprotomers at higher concentrations.
Figure 4. ChsH1-ChsH2-Ltp2 occupies three different oligomer states in solution and the dominant oligomer state is a diprotomer.

(A) Sedimentation velocity AUC experiments with ChsH1-ChsH2-Ltp2 at three protein concentrations (0.425, 0.85, and 1.7 mg/ml). (B) Estimated molecular weights from AUC analysis and theoretical molecular weights. Each protomer (αβγ) is a heterotrimer comprised of ChsH1 (α), ChsH2 (β), and Ltp2 (γ).
Rosetta protein-protein docking to build models of ChsH1-ChsH2-Ltp2.
Based on the EM and SAXS maps, we modeled the structure of the ChsH1-ChsH2-Ltp2 complex into the maps using the available crystal structures of ChsH1-ChsH2N from Mtb and DUF35-Ltp2 from T. curvata. Protein alignment of the ChsH2C and DUF35T. curvata sequences revealed that they are similar in their lengths and they share 39% identity in amino acid composition. Ltp2 and Ltp2T. curvata share 79% identity in amino acid composition.
As the EM map had a better resolution than SAXS, we used the EM map as a reference to model the structure of the ChsH1-ChsH2-Ltp2 complex. The AUC data identified the oligomeric states of ChsH1-ChsH2-Ltp2 in solution but the positions of the oligomer components relative to one another remained unknown. Thus, we constructed two possible models. In the first model, heterotetrameric ChsH1-ChsH2N is in the middle and two copies of heterodimeric DUF35 (ChsH2C)-Ltp2T. curvata are positioned at each end of the complex. Whereas in the second docked model, heterotetrameric DUF35 (ChsH2C)-Ltp2T. curvata is in the middle and two copies of heterodimeric ChsH1-ChsH2N are on the two ends (Figure S9).
We selected the second docked model as the final model to proceed based on the following observations (Figure 5A and 5B). First, the EM model has a distinct dent in the middle (Figure 3C and Figure S9D), which fits better with the saddle-like shape of DUF35 (ChsH2C)-Ltp2T. curvata αββα tetramer than with the heterotetrameric ChsH1-ChsH2N. Second, the DUF35 (ChsH2C)-Ltp2T. curvata αββα tetramer has a much stronger heterodimer-heterodimer interface than ChsH1-ChsH2N (Figure 6). The buried surface areas of their interfaces are 1769.6 Å2 and 679.8 Å2, respectively, and the corresponding desolvation energies gained from complex formation, are ~13 kcal/mol and ~1 kcal/mol. Furthermore, the second docked model has a more complete fit in the middle of the map (Figure S9D).
Figure 5. Full length ChsH1-ChsH2-Ltp2 complex model.

(A) Rosetta protein-protein docking model of ChsH1-ChsH2-Ltp2 complex. ChsH1 chains are colored in different shades of orange, ChsH2N chains are colored in different shades of pink, DUF35T. curvata (ChsH2C) chains are colored in different shades of green, and Ltp2T. curvata chains are colored in different shades of blue. (B) The ChsH1-ChsH2-Ltp2 model is fitted into its EM map. (C) The ChsH1-ChsH2-Ltp2 model is fitted into its SAXS molmap. (D) Full length ChsH1-ChsH2 was constructed and fitted into its SAXS molmap.
Figure 6. Molecular interface comparison between ChsH1-ChsH2 and DUF35-Ltp2T. curvata structures.

(A) In the ChsH1-ChsH2N structure, heterodimer-heterodimer interfacing are selected and shown in surface. The buried surface area is 679.811 Å2. Hot pink and light pink each represent a heterodimeric ChsH1-ChsH2N. (B) In the DUF35-Ltp2T. curvata structure, heterodimer-heterodimer interfacing residues are selected and shown in surface. The buried surface area is 1769.587 Å2. Dark blue and light blue each represent a heterodimeric DUF35-Ltp2T. curvata.
Subsequently, the selected model of the ChsH1-ChsH2-Ltp2 complex was fitted into its SAXS envelope with a high quality of superimposition (Figure 5C). The full length ChsH1-ChsH2 was computationally isolated from the ChsH1-ChsH2-Ltp2 model and a heterotetrameric full length ChsH1-ChsH2 was created based on the heterodimer interface in the ChsH1-ChsH2N structure. The structure was fitted into its SAXS envelope with good correlation, which further validated the ChsH1-ChsH2-Ltp2 docked model (Figure 5D).
The modeled ChsH1-ChsH2-Ltp2 structure clearly shows that each heterodimer of ChsH1-ChsH2 (αβ) binds with a monomer of Ltp2 (γ) to form a protomer (αβγ). The ChsH1-ChsH2-Ltp2 protein complex is comprised of two protomers to form a heterohexameric (αβγ)2 structure through the Ltp2 dimerization interface.
Since heterodimeric ChsH1-ChsH2 forms a di-heterodimeric structure through the above-mentioned heterodimer-heterodimer interface (Figure 6A), the oligomer may be elongated by adding another di-protomeric ChsH1-ChsH2-Ltp2 complex along the long axis through dimerization of the ChsH1-ChsH2 ends. This elongation model fits with the tetraprotomer peak observed in the AUC profile (Figure 4).
His344 plays an essential role in catalyzing the retro-aldol reaction.
The retro-aldol cleavage catalyzed by Ltp2 is similar to a β-ketoacyl-CoA cleavage reaction catalyzed by a thiolase since both reactions require formation of a CoA-ester enolate species. Protein sequence alignment reveals that among the canonical thiolase active site residues,24 a cysteine and a histidine are conserved in Ltp2. The conserved cysteine in Ltp2 aligns with the nucleophilic cysteine in thiolases (Figure 7A).
Figure 7. Ltp2 His344 is required for formation of propionyl-CoA from 3-OPDC-CoA.

(A) Sequence alignment of Ltp2 active site with thiolase active sites. Key catalytic residues are highlighted. The alignment was performed with ClustalW2. (B) Analysis of product formation catalyzed by 10 μM wild-type or mutant enzyme (a) ChsH1-ChsH2-Ltp2, (b) ChsH1-ChsH2-Ltp2C85A, (c) ChsH1-ChsH2-Ltp2H344A, (d) ChsH1-ChsH2-Ltp2C85A,H344A, and (e) ChsH1-ChsH2 with 3-OPDC-CoA (100 M) as substrate at 25 °C. Reactions were analyzed by MALDI-TOF mass spectrometry analysis in negative ionization mode, the peaks at i. 1089.2 m/z [M-H]−, ii: 1107.2 m/z [M-H]−, and iii: 822.4 m/z [M-H]-correspond to 3-OPDC-CoA, 17-HOPC-CoA, and propionyl-CoA, respectively.
To elucidate the degree of conservation of catalytic mechanism between thiolases and Ltp2, Cys85 and His344 were mutated to alanine in Ltp2. Three mutants were constructed: ChsH1-ChsH2-Ltp2C85A, ChsH1-ChsH2-Ltp2H344A, and ChsH1-ChsH2-Ltp2C85A,H344A. As observed by MALDI-TOF spectrometry, in the reactions catalyzed by wild-type ChsH1-ChsH2-Ltp2 and ChsH1-ChsH2-Ltp2C85A, the substrate 3-OPDC-CoA was completely converted to propionyl-CoA (Figure 7Ba and 7Bb). In contrast, in reactions catalyzed by ChsH1-ChsH2-Ltp2H344A and ChsH1-ChsH2-Ltp2C85A,H344A, 17-HOPC-CoA accumulated, as was observed in the reaction of ChsH1-ChsH2 alone, which is consistent with the abrogation of the retro-aldolase reaction (Figure 7Bc, 7Bd and 7Be). The results showed that the H344A mutation completely abolished the production of propionyl-CoA by Ltp2, whereas, the C85A mutation did not affect the retro-aldol reaction under these conditions.
Discussion
ChsH1-ChsH2-Ltp2 in the igr operon of Mtb is required for the complete degradation of the cholesterol side chain. In earlier work, two enzyme complexes, ChsH1-ChsH2 and ChsE1-ChsE2, encoded by genes from this operon have been shown to form unusual α2β2 heterotetramers.13,14 In addition, DUF35-Ltp2 from T. curvata forms an αββα heterotetramer.17 Herein, we provide structural information about a protein complex in this operon, full length ChsH1-ChsH2 and Ltp2. The protein complex consists of two protomers and each protomer contains three protein components: a chain of ChsH1, a chain of ChsH2, and a chain of Ltp2.
Two protomers dimerize at the interface of Ltp2 to form a heterohexameric structure. Based on the ChsH1-ChsH2-Ltp2 model, the formation of its diprotomer requires disruption of the ChsH1-ChsH2 heterotetramer interface. When we compare the dimer interface of ChsH1-ChsH2 and Ltp2T, curvata, we found that the buried surface area for ChsH1-ChsH2 is 679.8 Å2 and for Ltp2 is 1769.6 Å2 (calculated in Pymol). Therefore, energetically the interaction between two subunits of Ltp2T, curvata is stronger than the dimer-dimer interaction of ChsH1-ChsH2.
Interestingly, AUC experiments showed that besides diprotomers, there were also monoprotomer and tetraprotomer ChsH1-ChsH2-Ltp2 species present in solution. At the high sedimentation coefficient (s) region, the AUC peak became broad indicating coexistence of different large molecular weight species. Therefore, it is possible that the protein complex dynamically exchanges between ChsH1-ChsH2 heterotetramers and heterodimers at the ends of the protomer (Figure 8). It is worth noting that the tetraprotomer peak is more distinct at higher protein concentrations in AUC, indicating that the formation of large molecular weight species is concentration dependent. In vivo, localized cellular concentrations of ChsH1-ChsH2-Ltp2 could be sufficiently high for these dynamics to play a role.
Figure 8. The proposed tetraprotomer structure of ChsH1-ChsH2-Ltp2 complex.

Two diprotomers of ChsH1-ChsH2-Ltp2 interact through the dimer-dimer interface of ChsH1-ChsH2 and form a tetraprotomeric structure.
The shape of the EM model for ChsH1-ChsH2-Ltp2 is shorter than the SAXS model. However, the modeled structure fits well in both envelopes. We attribute the size difference to the conditions that were applied to the protein in EM analysis. As the protein was not in the solution state and a heavy metal was used for staining, the protein is in a dehydrated state, which may result in slight structure distortion.
We found that in the hydration step of 3-OPDC-CoA, the reaction reaches substrate-product equilibrium after the hydratase ChsH1-ChsH2, converts about 30% substrate. In contrast, the ChsH1-ChsH2-Ltp2 complex completely converts 3-OPDC-CoA to propionyl-CoA and androstenedione.15 We observed the same equilibrium constant in the reactions catalyzed by wild-type ChsH1-ChsH2-Ltp2 and mutants. When Ltp2 is absent or catalytically inactive, there is only about 30% substrate turnover; whereas when Ltp2 is present or catalytically active, the reaction substrate turnover is 100%.
Interestingly, in a methanogenic archaeon, Methanothermococcus thermolithotrophicus, an acetoacetyl-CoA thiolase, a 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase and a DUF35 protein form a protein complex.25 This protein complex contains two dimers of thiolase, two dimers of HMG-CoA synthase, and four monomers of the DUF35 protein. Importantly, the DUF35 protein acts as a bridge between the thiolase and the HMG-CoA synthase. The thiolase and the HMG-CoA synthase share one CoA binding pocket so that the product from the synthetic thiolase-catalyzed endergonic reaction is consumed immediately by the HMG-CoA synthase-catalyzed exergonic reaction. As a result, formation of the protein complex couples the thiolase reaction to the condensation reaction to ensure maximum production of 3-hydroxy-3methylglutaryl-CoA.
We hypothesize an analogous process occurs with formation of the ChsH1-ChsH2-Ltp2 complex. The CoA binding pocket in ChsH1-ChsH2N (PDB code: 4WNB) is oriented toward ChsH2C and Ltp2 in the modeled ChsH1-ChsH2-Ltp2 structure (Figure 9). Therefore, we propose that Ltp2 and ChsH1-ChsH2 share one fused CoA binding site to ensure the thermodynamically unfavored hydration reaction is coupled to the retro-aldolase lytic reaction (Figure 9).
Figure 9. The CoA binding site in ChsH1-ChsH2N is orientated toward ChsH2C and Ltp2 in the modeled ChsH1-ChsH2-Ltp2 structure.

The ChsH1 chain is colored in cyan, the ChsH2N chain in magenta, the DUF35T. curvata chain in blue, and Ltp2T. curvata chain in yellow.
The DUF35 domain is located at the interface between ChsH1-ChsH2 and Ltp2. There is buried surface area between DUF35 and both Ltp2 and ChsH1-ChsH2N. Although ChsH1-ChsH2N maintains enoyl-CoA hydratase activity in the absence of DUF35,14 the DUF35 motif likely helps to deliver the 17-HOPC-CoA molecule to Ltp2 for the second catalyzed reaction. This model is consistent with the proposed function of DUF35/DUF35_N motif in acyl-CoA-utilization processes.16
The production of propionyl-CoA is essential for Mtb’s central metabolism. Propionyl-CoA is mainly produced through cholesterol catabolism and odd chain fatty acid catabolism in Mtb. Propionyl-CoA is further connected to the methylmalonyl pathway, the methylcitrate cycle, and the biosynthesis of methyl branched polyketides. As it is related to mycobacterial key metabolic intermediates production and virulent lipids biosynthesis, thus, propionyl-CoA production and utilization are tied to mycobacterial central metabolism and virulent lipid biosynthesis and regulation of propionyl-CoA levels contributes to pathogenesis.
Additionally, Ltp2 shares high sequence identity with the thiolase family of enzymes. Thiolases catalyze cleavage of the Cα-Cβ bond in β-keto, acyl-CoA esters, whereas Ltp2 catalyzes cleavage of the Cα-Cβ bond in a β-hydroxy, acyl-CoA ester. In the degradative thiolase reaction, retro-Claisen cleavage reaction begins with nucleophilic attack of an active site cysteine on the β-keto carbonyl moiety of the acyl-CoA.26 Addition of cysteine generates an oxyanion intermediate that is stabilized by hydrogen bonds to backbone amides provided by the nucleophilic cysteine and a glycine (Figure 10A).24 Subsequent collapse of the oxyanion and Cα-Cβ bond cleavage provide a CoA ester enolate and an acyl-cysteine adduct. The CoA ester enolate is stabilized by hydrogen bonds with a histidine and water in a second oxyanion hole (Figure 10A).26 Nucleophilic addition of free CoA to the acyl-cysteine adduct followed by elimination of the cysteine generates a new acyl-CoA. The Ltp2 catalyzed retro-aldol reaction proceeds through a similar set of intermediates. However, nucleophilic attack of a cysteine is not required to form the oxyanion intermediate (Figure 10B).
Figure 10. Oxyanion holes in the Zoogloea ramigera biosynthetic acyl-CoA thiolase and the proposed catalytic mechanism of Ltp2.

A. In the Zoogloea ramigera biosynthetic acyl-CoA thiolase, oxyanion hole I, which consists of the acid-base histidine and a water molecule, stabilizes the enolate intermediate. Oxyanion hole II is formed by the nucleophilic cysteine and a glycine residue. It stabilizes the tetrahedral intermediate.24 B. For the retro-aldol reaction catalyzed by Ltp2, a base residue first deprotonates the hydroxyl group at C17 with the stabilization of the oxyanion in hole I. The steroid oxide anion then undergoes elimination to release one molecule of propionyl-CoA. The proton donor in this system is not defined. The proton could come from a water molecule or other residues in the substrate binding pocket.
All three oxyanion hole residues are conserved between thiolases and Ltp2: Cys89, Gly377 and His344. Therefore, we explored their role in retro-aldol reaction through mutagenesis. Mutation of His344 to Ala resulted in complete loss of Cα-Cβ bond cleavage, consistent with an essential role in stabilization of the CoA ester enolate. In contrast, mutation of Cys89 to alanine did not eliminate catalytic activity. Thus, the role of Cys89 in the Ltp2-catalyzed reaction appears to be limited to hydrogen bonding for oxyanion stabilization.
Interestingly, the non-nucleophilic cysteine that acts as a general base in the thiolase reaction is not conserved in Ltp2. Seah et al. identified two tyrosine residues in the active site of Ltp2 from T. curvata essential for catalysis of the retro-aldol reaction.17 Although a co-crystal structure of Ltp2 from T. curvata and its substrate is not available to directly determine the orientation, they proposed that Tyr294 is the general base that deprotonates the hydroxyl group at C17 and Tyr344 is the general acid that protonates the enolate.
Supplementary Material
Acknowledgment
The SAXS experiments used resources of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. Beamline [LiX] is primarily supported by the National Institute of Health, National Institute of General Medical Sciences (NIGMS) through a Biomedical Technology Research Resource P41 grant (P41GM111244), and by the DOE Office of Biological and Environmental Research (KP1605010). The negative stain EM data was collected from the McGill University Facility for Electron Microscopy Research (FEMR). We thank Xiaoxi Yu for ChsH1-ChsH2-Ltp2 SAXS data collection and Rayan Fakih for her help on AUC data analysis. We also thank Dr. Lindsay Eltis for providing the Rhodococcus jostii RHA1 strain and Dr. Tomohiro Tamura for pTipQC1 and pTipQC2 vectors. This research is funded by National Institutes of Health (NIH) Grant RO1AI134054 to N.S.S. Meng Yang is supported by Canadian Institute Health Research (CIHR) postdoc fellowship.
Footnotes
Conflict of interest: The authors declare they have no conflicts of interest with the contents of this article.
Supplemental Information
Additional information on the SAXS bead models, Guinier plots data, negative stain-EM images of ChsH1-ChsH2-Ltp2, EM data processing scheme, angular distribution of the EM model, comparison of SAXS and EM models, SDS-PAGE and AUC profiles and data points for ChsH1-ChsH2-Ltp2, and Rosetta protein-protein docking results are available in the Supplemental Information section.
Accession codes
EM reconstructions of ChsH1-ChsH2-Ltp2 complex have been deposited to the Electron Microscopy Data Bank (EMDB) under accession code EMD-20525.
Reference
- [1].WHO. (2018) Global Tuberculosis Report, World Health Organization, https://www.who.int/tb/publications/global_report/en/. [Google Scholar]
- [2].WHO. (2018) Tuberculosis, World Health Organization. [Google Scholar]
- [3].Yuan T, and Sampson NS (2018) Hit Generation in TB Drug Discovery: From Genome to Granuloma, Chem Rev 118, 1887–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Russell DG, Cardona PJ, Kim MJ, Allain S, and Altare F (2009) Foamy macrophages and the progression of the human tuberculosis granuloma, Nat Immunol 10, 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, Tsenova L, Kaplan G, and Russell DG (2010) Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism, EMBO Mol Med 2, 258–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pandey AK, and Sassetti CM (2008) Mycobacterial persistence requires the utilization of host cholesterol, Proc Natl Acad Sci U S A 105, 4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dubos RJ (1950) The effect of organic acids on mammalian tubercle bacilli, J Exp Med 92, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee W, VanderVen BC, Fahey RJ, and Russell DG (2013) Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress, J Biol Chem 288, 6788–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thomas ST, VanderVen BC, Sherman DR, Russell DG, and Sampson NS (2011) Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism, J Biol Chem 286, 43668–43678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chang JC, Harik NS, Liao RP, and Sherman DR (2007) Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice, J Infect Dis 196, 788–795. [DOI] [PubMed] [Google Scholar]
- [11].Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, and Sherman DR (2009) igr Genes and Mycobacterium tuberculosis cholesterol metabolism, J Bacteriol 191, 5232–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nesbitt NM, Yang X, Fontan P, Kolesnikova I, Smith I, Sampson NS, and Dubnau E (2010) A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol, Infect Immun 78, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thomas ST, and Sampson NS (2013) Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain, Biochemistry 52, 2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang M, Guja KE, Thomas ST, Garcia-Diaz M, and Sampson NS (2014) A distinct MaoC-like enoyl-CoA hydratase architecture mediates cholesterol catabolism in Mycobacterium tuberculosis, ACS Chem Biol 9, 2632–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gilbert S, Hood L, and Seah SYK (2018) Characterization of an Aldolase Involved in Cholesterol Side Chain Degradation in Mycobacterium tuberculosis, J Bacteriol 200, doi: 10.1128/JB.00512-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krishna SS, Aravind L, Bakolitsa C, Caruthers J, Carlton D, Miller MD, Abdubek P, Astakhova T, Axelrod HL, Chiu HJ, Clayton T, Deller MC, Duan L, Feuerhelm J, Grant JC, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, McMullan D, Morse AT, Nigoghossian E, Okach L, Reyes R, Rife CL, van den Bedem H, Weekes D, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, and Wilson IA (2010) The structure of SSO2064, the first representative of Pfam family PF01796, reveals a novel two-domain zinc-ribbon OB-fold architecture with a potential acyl-CoA-binding role, Acta Crystallogr Sect F Struct Biol Cryst Commun 66, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aggett R, Mallette E, Gilbert SE, Vachon MA, Schroeter KL, Kimber MS, and Seah SYK (2019) The steroid side chain-cleaving aldolase Ltp2-ChsH2DUF35 is a thiolase superfamily member with a radically repurposed active site, J Biol Chem doi: 10.1074/jbc.RA119.008889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang K (2016) Gctf: Real-time CTF determination and correction, J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling, Biophys J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Laue TM, Shah BD, Ridgeway TM & Pelletier SL Computer-aided interpretation of sedimentation data for proteins In Analytical ultracentrifugation in biochemistry and polymer science (eds Harding SE, Rowe AJ & Horton JC), pp. 90–125 (Royal Society of Chemistry, Cambridge, 1992) [Google Scholar]
- [21].Brautigam CA (2015) Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data, Methods Enzymol 562, 109–133. [DOI] [PubMed] [Google Scholar]
- [22].Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HD, Konarev PV, and Svergun DI (2012) New developments in the ATSAS program package for small-angle scattering data analysis, J Appl Crystallogr 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de la Rosa-Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, Vargas J, Oton J, Sharov G, Vilas JL, Navas J, Conesa P, Kazemi M, Marabini R, Sorzano CO, and Carazo JM (2016) Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy, J Struct Biol 195, 93–99. [DOI] [PubMed] [Google Scholar]
- [24].Kursula P, Ojala J, Lambeir AM, and Wierenga RK (2002) The catalytic cycle of biosynthetic thiolase: a conformational journey of an acetyl group through four binding modes and two oxyanion holes, Biochemistry 41, 15543–15556. [DOI] [PubMed] [Google Scholar]
- [25].Vogeli B, Engilberge S, Girard E, Riobe F, Maury O, Erb TJ, Shima S, and Wagner T (2018) Archaeal acetoacetyl-CoA thiolase/HMG-CoA synthase complex channels the intermediate via a fused CoA-binding site, Proc Natl Acad Sci U S A 115, 3380–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schaefer CM, Lu R, Nesbitt NM, Schiebel J, Sampson NS, and Kisker C (2015) FadA5 a thiolase from Mycobacterium tuberculosis: a steroid-binding pocket reveals the potential for drug development against tuberculosis, Structure 23, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.