

Abstract
Aims/hypothesis
Obesity during pregnancy increases offspring type 2 diabetes risk. Given that nearly half of women of child-bearing age in many populations are currently overweight/obese, it is key that we improve our understanding of the impact of the in utero/early life environment on offspring islet function. Whilst a number of experimental studies have examined the effect of maternal obesity on offspring islet architecture and/or function, it has not previously been delineated whether these changes are independent of other confounding risk factors such as obesity, postnatal high-fat-feeding and ageing. Thus, we aimed to study the impact of exposure to maternal obesity on offspring islets in young, glucose-tolerant male and female offspring.
Methods
Female C57BL/6J mice were fed ad libitum either chow or obesogenic diet prior to and throughout pregnancy and lactation. Offspring were weaned onto a chow diet and remained on this diet until the end of the study. An IPGTT was performed on male and female offspring at 7 weeks of age. At 8 weeks of age, pancreatic islets were isolated from offspring for measurement of insulin secretion and content, mitochondrial respiration, ATP content, reactive oxygen species levels, beta and alpha cell mass, granule and mitochondrial density (by transmission electron microscopy), and mRNA and protein expression by real-time RT-PCR and Western blotting, respectively.
Results
Glucose tolerance was similar irrespective of maternal diet and offspring sex. However, blood glucose was lower (p < 0.001) and plasma insulin higher (p < 0.05) in female offspring of obese dams 15 min after glucose administration. This was associated with higher glucose- (p < 0.01) and leucine/glutamine-stimulated (p < 0.05) insulin secretion in these offspring. Furthermore, there was increased mitochondrial respiration (p < 0.01) and density (p < 0.05) in female offspring of obese dams compared with same-sex controls. Expression of mitochondrial and nuclear-encoded components of the electron transport chain, L-type Ca2+ channel subtypes that play a key role in stimulus-secretion coupling [Cacna1d (p < 0.05)], and oestrogen receptor α (p < 0.05) was also increased in islets from these female offspring of obese dams. Moreover, cleaved caspase-3 expression and BAX:Bcl-2 were decreased (p < 0.05) reflecting reduced susceptibility to apoptosis. In contrast, in male offspring, glucose and leucine/glutamine-stimulated insulin secretion was comparable between treatment groups. There was, however, compromised mitochondrial respiration characterised by decreased ATP synthesis-driven respiration (p < 0.05) and increased uncoupled respiration (p < 0.01), reduced docked insulin granules (p < 0.001), decreased Cacna1c (p < 0.001) and Cacna1d (p < 0.001) and increased cleaved caspase-3 expression (p < 0.05).
Conclusions/interpretation
Maternal obesity programs sex differences in offspring islet function. Islets of female but not male offspring appear to be primed to cope with a nutritionally-rich postnatal environment, which may reflect differences in future type 2 diabetes risk.
Electronic supplementary material
The online version of this article (10.1007/s00125-019-05037-y) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Keywords: Beta cell, Islets, Maternal obesity, Type 2 diabetes

Introduction
Being overweight or obese is a major risk factor for type 2 diabetes. Importantly, overweight/obesity also has health implications for future generations since obesity during pregnancy increases offspring obesity and type 2 diabetes risk. This was recently highlighted in a study by Lahti-Pulkkinen et al showing that offspring of obese mothers have a 3.5-fold increased risk of type 2 diabetes compared with lean mothers [1]. Abnormalities in beta cell function are critical in delineating the risk of type 2 diabetes because the inability of beta cells to adapt and compensate for peripheral insulin resistance leads to type 2 diabetes pathogenesis [2]. In contrast, sustained beta cell adaptation is capable of preventing type 2 diabetes, even in the face of severe insulin resistance [3].
Given that, in many populations, nearly half of women of child-bearing age are overweight or obese [4], it is important that we improve our understanding of the impact of the in utero and early life environment on offspring islet function. A number of experimental studies have examined the effect of maternal obesity on offspring islet architecture and/or function (reviewed in [5]). Whilst findings of reduced beta cell mass and impaired glucose-stimulated insulin secretion (GSIS) suggest increased type 2 diabetes susceptibility, most previous studies have not delineated whether these changes in islet structure/function are independent of other confounding risk factors such as obesity/increased body weight in the offspring [6–9], postnatal high-fat-feeding [9, 10] and the effects of ageing [8, 10]. Importantly, research by Zambrano et al in 5-week-old rat offspring exposed to maternal obesity suggested that islet function may already be impacted in young life [11].
It is, therefore, key that we identify inherent changes in islet function, which are programmed by exposure to maternal obesity per se and are distinct from the response of the islets to an existing obesogenic, glucose intolerant milieu. This includes investigating the effects on both male and female offspring since sex affects glucose homeostasis and type 2 diabetes risk (reviewed in [12]). For example, using a mouse model of maternal diet-induced obesity, Samuelsson et al showed increased adiposity in both male and female offspring at 6 months of age; however, only male offspring developed type 2 diabetes. This was characterised by higher fasting glucose, lower fasting insulin and decreased pancreatic insulin content. In contrast, these variables were unchanged in female offspring, at the same age [13]. Thus, in the current study, using this same model we characterised pancreatic islet function in younger, metabolically healthy male and female offspring.
Methods
Animal model
This research was regulated under the UK Home Office Animals (Scientific Procedures) Act 1986 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Board. The model has been described in detail previously [14]. Briefly, female C57BL/6J mice were randomised to either ad libitum or a standard chow RM1 diet (∼7% simple sugars, 3% fat, 50% polysaccharide, 15% protein [wt/wt], 10.74 kJ/g) or a highly palatable energy-rich obesogenic diet (∼10% simple sugars, 20% animal lard, 28% polysaccharide, 23% protein [wt/wt], 28.43 kJ/g) and sweetened condensed milk (Nestle) (∼16% fat, 33% simple sugars, 15% protein, 13.7 kJ/g) fortified with mineral and vitamin mix (AIN93G), for 6 weeks before mating for first pregnancy. Diets were purchased from Special Dietary Services (Witham, UK). Mice were purchased from Charles River Laboratories (Wilmington, MA, USA). The first litter was culled post weaning. This first pregnancy ensured mice were proven breeders. Mice were then re-mated for a second pregnancy. Dams were maintained on their respective diets throughout both pregnancies and lactation periods. Dams fed an obesogenic diet have around 40% body fat on day one of pregnancy and are hyperinsulinaemic and have impaired glucose tolerance in late gestation [15]. Litters were standardised to six pups on postnatal day two. Offspring from ‘Control’ and ‘Obese’ groups were weaned onto RM1 on postnatal day 21 and remained on this diet until the end of the study. Mice were housed in ventilated cages, 2–3 mice per cage with same-sex littermates, and maintained in a humidity-controlled room with a 12 h light/dark cycle, with free access to food and water. An IPGTT was performed on 7-week-old offspring and the offspring were euthanised at 8 weeks of age. Blood glucose was measured between 07:00 and 08:00 hours on the day of euthanasia from tail blood sample using a blood glucose analyser (AlphaTRAK, Abbot Logistics, the Netherlands). Mice were euthanised by carbon dioxide inhalation. Blood was taken by cardiac puncture for serum insulin analysis. Insulin concentration was determined by ELISA (Mercodia, Sweden).
IPGTT
Following a 4 h fast, glucose (1 g/kg) was injected into the intraperitoneal cavity. Blood glucose measurements were made using a blood glucose analyser (AlphaTRAK) at 0, 15, 30, 45, 60, 90 and 120 min. Tail blood was also collected at 0, 15 and 30 min into glass micro-haematocrit capillary tubes with sodium heparin (Hirschmann-Laborgeräte, Germany) for plasma insulin analysis. The AUC for glucose was calculated using the trapezoidal rule.
Pancreatic islet isolation
Islets were isolated as described in [16]. Islets were hand-picked under a stereo microscope and incubated at 37°C overnight in RPMI-1640 medium supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin sulphate.
Islet insulin secretion
Groups of 20 islets were incubated in KRB [16] and 2.8 mmol/l glucose for 1 h at 37°C. Next, these groups of islets were incubated at 37°C for 1 h in KRB containing 2.8 mmol/l glucose, 16.7 mmol/l glucose or 10 mmol/l leucine/glutamine.
Islet insulin content
Islets were washed in PBS mixed with ethanol/hydrochloric acid and sonicated at 4°C. After centrifugation, the supernatant was stored at −20°C until assayed.
Oxygen consumption assay
Oxygen consumption was measured by the XF24 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA). Groups of 50 islets (in triplicate) were pre-incubated in KRB with 2 mg/ml BSA and 2.8 mmol/l glucose for 30 min at 37°C. Respiration was measured in the presence of 16.7 mmol/l glucose, oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) and rotenone as previously described [17]. Calculations of basal, glucose-stimulated, ATP-linked and proton leak-linked respiration and coupling efficiency were carried out as previously described [18].
Detection of reactive oxygen species
Reactive oxygen species (ROS) were detected as previously described [16]. After pre-incubation, islets were loaded with 10 μmol/l 2′,7′-dichlorofluorescin diacetate (DCFDA; Sigma) and incubated at 37°C for 1 h in KRB containing 2.8 mmol/l glucose or 16.7 mmol/l glucose. Fluorescence was measured using a microplate reader with excitation/emission at 495 nm/529 nm.
Quantification of ATP content
This was performed as previously described [16]. Islets were lysed and the content of ATP at 2.8 or 16.7 mmol/l glucose was assayed using a luciferase-based luminescent assay (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Quantification of mRNA expression using quantitative RT-PCR
RNA was extracted (Qiagen, Manchester, UK) and reverse transcribed into cDNA (Fermentas, Waltham, MA, USA). Quantitative RT-PCR (qRT-PCR) was performed using predesigned TaqMan Gene Expression assays or the SYBR Green system (Thermo Fisher Scientific, Carlsbad, CA, USA). mRNA expression was determined by the ΔΔCt method and normalised to Rplp0 and Hprt (females) and Actb and Hprt (males), expression of which was not influenced by maternal diet. Primer sequences are available in electronic supplementary material (ESM) Table 1.
Quantification of protein abundance using western blotting
Islets were lysed in RIPA buffer with protease inhibitor cocktail (Sigma, Gillingham, UK), subjected to SDS-PAGE and blotted using antibodies (ESM Table 2). Male and female samples were run on separate gels. Membranes were sectioned and some sections stripped (Restore Western Blot Stripping Buffer, Thermo Fischer Scientific) and re-probed to maximise the amount of data obtained from each western blot. Image Lab software version 5.2.1 (Bio-Rad, Hercules, CA, USA) was used to quantify the density of specific bands. Images of western blots showing statistically significant differences are included in ESM Fig. 1. All blots were made available to Diabetologia at the point of submission.
Mitochondrial DNA content
DNA was extracted (Qiagen) and quantified (Quant-iT PicoGreen dsDNA Reagent, Thermo Fisher Scientific). Equal quantities were amplified by qRT-PCR using primers for nuclear (Rplp0) and mitochondrial (mt-Nd5) DNA. Mitochondrial (mtDNA) content was determined as described in [19].
Transmission electron microscopy
Samples were prepared as previously described [16]. Electron micrographs of at least 40 different beta cells from 2–4 mice/sex were taken for each maternal diet group. Granules (large dense-core vesicles) were defined as docked when the centre of the granule was located within 190 nm (a half-length of the mean granule diameter) from the plasma membrane. The distance between the centre of the granule and the plasma membrane was calculated using an in-house software programmed in MatLab 7 (MathWorks, Natick, MA, USA) [20]. Granule density was estimated according to [21] and was normalised to granule diameter assuming spherical geometry [20]. Mitochondrial lesion in beta cells was determined by Photoshop Elements software (Adobe Systems, San Jose, CA, USA). The lesion area was outlined and the size was measured by ImageJ (National Institutes of Health, Bethesda, MD, USA) and expressed as the area density.
Immunofluorescence
Paraffin-embedded pancreas was exhaustively sectioned (5 μm thickness, 200 μm apart). Sections were incubated with primary antibody against insulin [Dako (Agilent Technologies] and glucagon (Bioss Antibodies, Woburn, MA, USA). The appropriate fluorescent-dye-conjugated secondary antibodies were used for identifying beta and alpha cells (Jackson ImmunoResearch, Ely, UK). Digital images were obtained using Axioscan Z1 Slide Scanner (Zeiss, Germany) and HALO image analysis platform (Indica Labs, Albuquerque, NM, USA) was used to calculate immune-positive and total tissue area. Beta and alpha cell mass was calculated as described in [22].
Statistical analyses
Investigators were not blind to group assignment and outcome assessment except for analyses of electron micrographs. Calculations were performed in IBM SPSS Statistics 23 (Armonk, NY, USA). ‘n’ represents the number of mice from separate litters. Data are presented as mean ± SEM of the indicated number of litters. mRNA expression, protein abundance, mtDNA content and AUCglucose were analysed using unpaired Student’s t test. Blood glucose and insulin levels during IPGTT were measured by two-way (repeated measures) ANOVA followed by Bonferroni’s multiple comparisons test (note: each sex was analysed separately). All other data were analysed by two-way ANOVA. A significant interaction indicated sex differences in the impact of maternal obesity. In this case, a simple main effect analysis was performed to isolate the effects of maternal diet on each sex separately. A probability level of 5% (p < 0.05) was taken to be significant.
Results
Body weight and glucose tolerance are comparable between offspring of control and obese dams at 8 weeks of age irrespective of sex
Body weight, (non-fasted) blood glucose and serum insulin levels were comparable in 8-week-old offspring between maternal diet groups (ESM Table 3). Male offspring, however, were heavier (p < 0.05) and had higher serum insulin (p < 0.01) (ESM Table 3).
Glucose tolerance in both sexes, as reflected by AUC, was also similar irrespective of maternal diet (Fig. 1a, b). We observed, however, that blood glucose was lower (p < 0.001) and plasma insulin higher (p < 0.05) in female offspring of obese dams 15 min after glucose administration compared with controls (Fig. 1b, c).
Fig. 1.
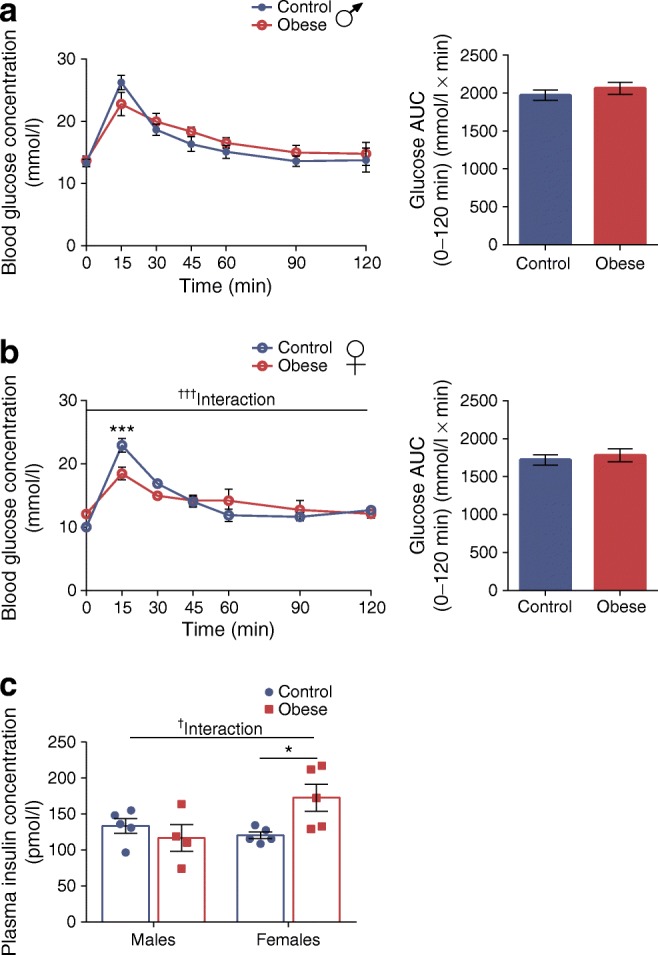
Body weight and glucose tolerance are comparable between offspring of control and obese dams at 8 weeks of age, irrespective of sex. (a, b) Glucose excursion curves and AUCs during an IPGTT performed on 7-week-old male (a) and female (b) offspring of control and obese dams after a 4 h fast. (c) Plasma insulin concentration at the 15 min time point following glucose administration during an IPGTT in male and female offspring of control and obese dams. (a, b) Data were analysed by two-way (repeated measures) ANOVA followed by Bonferroni’s multiple comparisons test. (c) Data were analysed by two-way ANOVA followed by a simple main effect analysis when a significant interaction was observed. †p < 0.05 and †††p < 0.001 for interaction; *p < 0.05 and ***p < 0.001 for obese vs control. (a, b) Males, n = 5 mice/group; females, n = 5 mice/group; (c) males, control: n = 5, obese: n = 4 mice; females, control: n = 5, obese: n = 5 mice. ‘n’ represents the number of mice from separate litters. All data are mean ± SEM
GSIS is higher in female but not male offspring that were previously exposed to maternal obesity
The amount of insulin secreted by beta cells depends on the mass and function of these cells. Beta cell mass was comparable between maternal diet groups (Fig. 2a). However, in line with having higher serum insulin, male offspring also had increased beta cell mass (p < 0.05). This was due to higher absolute pancreas weight (p < 0.05) and consequently greater total pancreatic tissue area (p < 0.05) with no difference in the relative number of insulin+ cells (ESM Fig. 2a–c). In contrast, alpha cell mass was comparable between sexes (Fig. 2b) owing to a lower number of glucagon+ cells (p < 0.01) in male compared with female offspring (ESM Fig. 2d).
Fig. 2.

GSIS is higher in female but not male offspring that were previously exposed to maternal obesity. (a, b) Beta cell (a) and alpha cell (b) mass in offspring from control and obese dams at 8 weeks of age. (c) Insulin secretion stimulated by low glucose (2.8 mmol/l glucose; LG), high glucose (16.7 mmol/l glucose; HG) and 10 mmol/l leucine/glutamine. (d) Islet insulin content. Experiments were performed on islets isolated from 8-week-old male and female offspring of control and obese dams. Data were analysed by two-way ANOVA followed by a simple main effect analysis when a significant interaction was observed. There was a significant interaction between maternal diet and offspring sex for data relating to basal (p < 0.05), glucose-stimulated (p < 0.01) and leucine/glutamine-stimulated (p < 0.05) insulin secretion, indicating a sex-specific effect on the outcome measured. *p < 0.05 and **p < 0.01 as shown. (a, b) Males, n = 4 mice/group; females, n = 6 mice/group; (c) LG: males, control: n = 8, obese: n = 6 mice; females, control: n = 6, obese: n = 7 mice; HG: males, control: n = 9, obese: n = 7 mice; females, control: n = 7, obese: n = 8 mice; Leu/Gln: males, control: n = 8, obese: n = 5 mice; females, control: n = 6, obese: n = 8 mice; (d) males and females, n = 5 mice/group. ‘n’ represents the number of mice from separate litters. All data are mean ± SEM
We found differences in beta cell function between male and female offspring of obese dams. Basal (p < 0.05), GSIS (p < 0.01) and amino acid-stimulated (p < 0.05) insulin secretion were increased only in female offspring of obese dams (Fig. 2c). Islet insulin content was not different between groups (Fig. 2d).
Sex differences in mitochondrial respiration in offspring of obese dams
To identify cellular components that may be contributing to enhanced GSIS in female offspring, we quantified GLUT2 protein abundance, the primary glucose transporter in rodent islets [23]. This was not altered by exposure to maternal obesity (Fig. 3a). Subsequent phosphorylation of glucose by glucokinase is the key step controlling glycolytic flux [24]. Glucokinase abundance was reduced in both male (p < 0.05) and female (p < 0.01) offspring of obese dams (Fig. 3b). This finding was unexpected given that the rate of glycolysis is an important determinant of GSIS from beta cells.
Fig. 3.
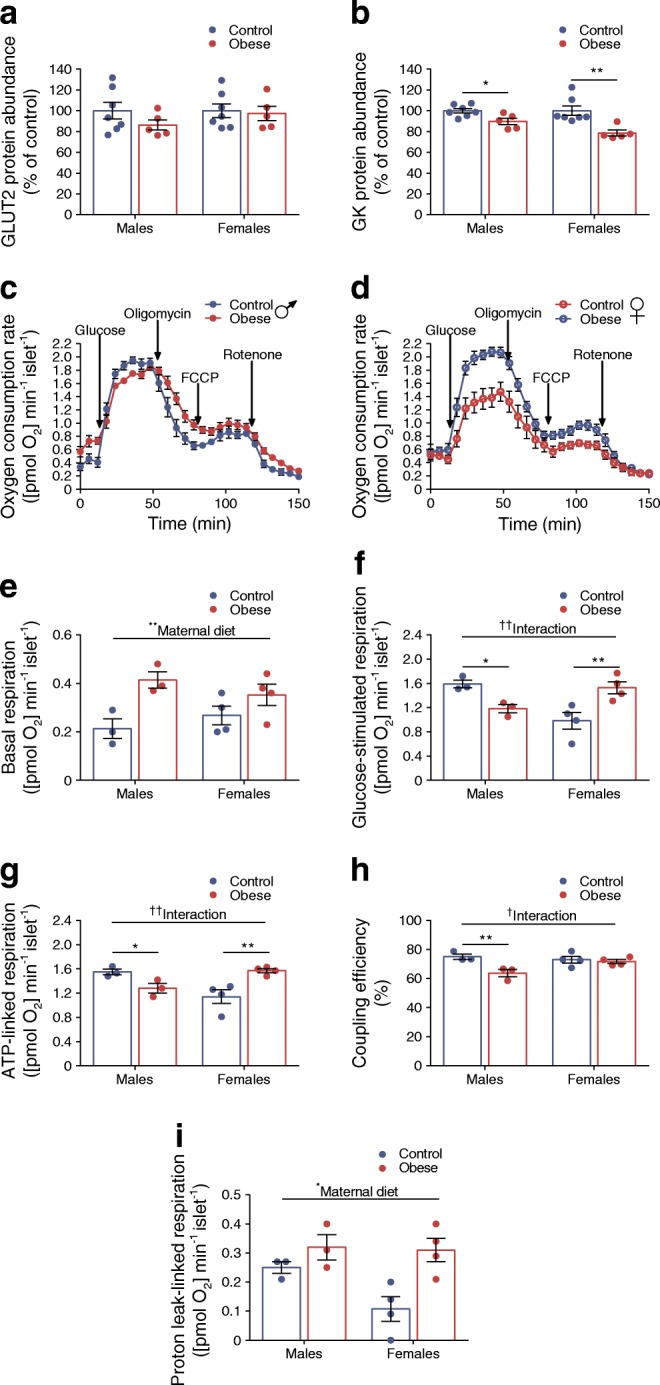
Sex differences in mitochondrial respiration in offspring of obese dams. (a, b) Western blot analysis of GLUT2 (a) and glucokinase (GK) (b); images of corresponding western blots are in ESM Fig. 1a, b, respectively. (c, d) Changes in islet oxygen consumption rate in male (c) and female (d) offspring of control and obese dams following treatment with 16.7 mmol/l glucose, 4 μg/ml oligomycin, 4 μmol/l FCCP and 5 μmol/l rotenone. (e–i) Basal (2.8 mmol/l glucose) respiration (e), glucose-simulated respiration (16.7 mmol/l glucose) (f), ATP-linked respiration (g), coupling efficiency (h) and proton leak-linked respiration (i). Experiments were performed on islets isolated from 8-week-old male and female offspring of control and obese dams. (a, b) Data were analysed by unpaired Student’s t test (control vs obese); (e–i) data were analysed by two-way ANOVA followed by a simple main effect analysis and a Sidak’s multiple comparisons test when a significant interaction was observed. †p < 0.05 and ††p < 0.01 for interaction; *p < 0.05 and **p < 0.01 as shown. (a, b) Males and females, control: n = 7, obese: n = 5 mice; (c–i) males, n = 3 mice/group; females, n = 4 mice/group. ‘n’ represents the number of mice from separate litters. All data are mean ± SEM
Mitochondrial metabolism couples glucose metabolism to insulin secretion. Since leucine/glutamine are mitochondrial fuels, this suggested that pathways downstream of glucokinase may be activated in these islets. We found sex differences in mitochondrial function in offspring of obese dams. Whilst exposure to maternal obesity resulted in higher basal respiration irrespective of offspring sex (p < 0.01) (Fig. 3c–e), glucose-stimulated (relative to basal) respiration was reduced in males (p < 0.05) but increased in female (p < 0.01) offspring of obese dams (Fig. 3c, d, f).
Respiration rate is mostly composed of ATP-linked and proton leak-linked respiration [25]. Similar to glucose-stimulated respiration, ATP-linked respiration was reduced in male offspring of obese dams (p < 0.05) (Fig. 3g). Moreover, coupling efficiency, which estimates the fraction of respiration used to drive ATP synthesis was also reduced in these islets (p < 0.01) (Fig. 3h). In contrast, ATP-linked respiration was increased in female offspring of obese dams (p < 0.01) (Fig. 3g). Finally, we also found that proton leak-linked respiration was higher in islets from maternal obesity-exposed offspring (p < 0.05) (Fig. 3i). This appears to be driven by a relatively large difference between female offspring groups due to lower proton leak-linked respiration in the control group.
Only female offspring of obese dams have increased expression of mitochondrial and nuclear-encoded components of the electron transport chain
Mitochondrial respiration/function is dependent on mtDNA, genes of the respiratory chain complexes that it encodes and its nuclear-encoded constituents. mRNA expression of some mitochondrial-encoded components of the respiratory complexes was significantly increased in maternal obesity-exposed female offspring (Fig. 4a). This was not due to higher levels of mitochondrial transcription factor A (TFAM) (Fig. 4b) but may be partly due to increased number of mitochondria as evidenced by higher mitochondrial area density (p < 0.05) (Fig. 4c). There was, however, no statistical difference in mtDNA content (p < 0.06; Fig. 4d). In addition to mitochondrial genes, mRNA expression of Sdha (Complex II) (p < 0.05) (Fig. 4a) and protein abundance of succinate dehydrogenase B (SDHB; Complex II) (p < 0.01), ubiquinol-cytochrome C reductase core protein 2 (UQCRC2; Complex III) (p < 0.05) and ATP5A (Complex V) (p < 0.01), which are nuclear-encoded, were also increased (Fig. 4e).
Fig. 4.
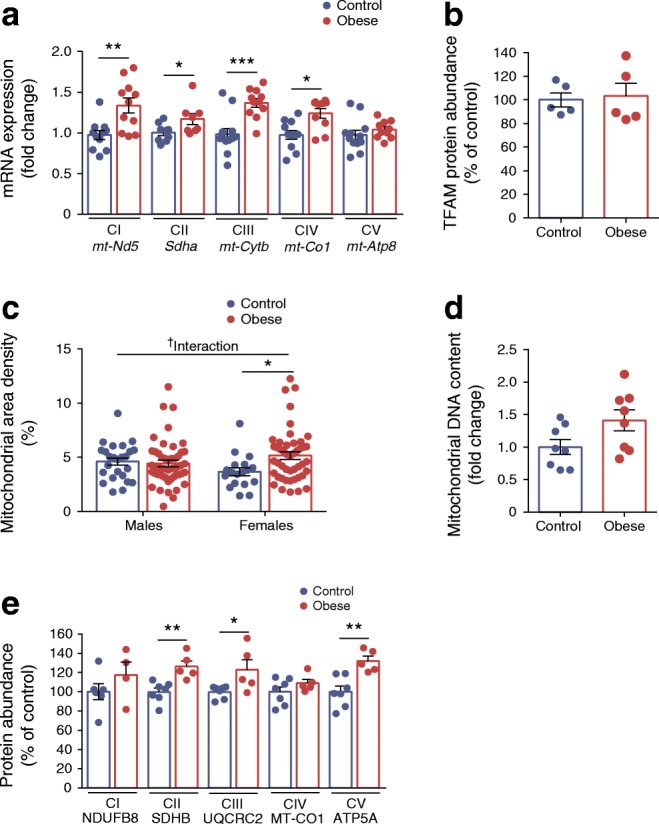
Among female offspring, only the offspring of obese dams have increased expression of mitochondrial and nuclear-encoded components of the electron transport chain. (a) qRT-PCR analysis of mRNA expression of mitochondrial (mt-Nd5, mt-Cytb, mt-Co1, mt-Atp8) and nuclear (Sdha) encoded components of the electron transport chain. (b) Western blot analysis of mitochondrial transcription factor A (TFAM); image of corresponding western blot is in ESM Fig. 1c. (c) Mitochondrial area density. (d) Mitochondrial DNA content. (e) Western blot analysis of mitochondrial (MT-CO1) and nuclear (NDUFB8, SDHB, UQCRC2 and ATP5A) encoded components of the electron transport chain; images of corresponding western blots are in ESM Fig. 1d. Experiments relating to (a, b, d, e) were performed on islets from female offspring and (c) was performed on islets isolated from 8-week-old male and female offspring of control and obese dams. (a, b, d, e) Data were analysed independently by unpaired Student’s t test (control vs obese). (c) Data were analysed by two-way ANOVA followed by a simple main effect analysis when a significant interaction was observed. †p < 0.05 for interaction; *p < 0.05, **p < 0.01 and ***p < 0.001 as shown. (a) mt-Nd5, control: n = 11, obese: n = 11; Sdha, control: n = 9, obese: n = 8; mt-Cytb, control: n = 12, obese: n = 11; mt-Co1, control: n = 11, obese: n = 10; Atp8, control: n = 12, obese: n = 10 mice; (b) n = 5 mice/group; (d) n = 8 mice/group; (e) NDUFB8, control: n = 6, obese: n = 4; SDHB, UQCRC2, MT-CO1 and ATP5A, control: n = 7, obese: n = 5 mice; ‘n’ represents the number of mice from separate litters; in (c) the data points shown correspond to the number of beta cells from: males, control: n = 2, obese: n = 4 mice; females, control: n = 2, obese: n = 3 mice. All data are mean ± SEM. For (a, b, d, e) the equivalent graphs for male offspring are in ESM Fig. 3. CI–V, Complex I–V
Our results suggest that higher mitochondrial respiration in female offspring of obese dams could be due to having more mitochondria and higher levels of respiratory complex proteins. The converse is not the case for male offspring (ESM Fig. 3a–d and Fig. 4c).
Increased ROS in islets from both male and female offspring of obese dams but increased expression of antioxidant enzyme only in female islets
Mitochondria are a major source of intracellular ROS [26]. Basal ROS levels were comparable between offspring groups (Fig. 5a). In response to glucose stimulation, ROS levels were higher in islets of maternal obesity-exposed offspring (p < 0.05) (Fig. 5b). Thus, in the case of male offspring of obese dams, ROS levels are elevated even with reduced mitochondrial respiration. Furthermore, female but not male offspring of obese dams displayed increased Sod2 (p < 0.01) (but not Cat) mRNA expression (Fig. 5c, d) suggesting that islets from these male offspring may be more vulnerable to oxidative stress.
Fig. 5.

ROS is increased in islets from both male and female offspring of obese dams but expression of antioxidant enzyme is increased only in female islets. (a, b) ROS levels measured by DCFDA fluorescence intensity following incubation with 2.8 mmol/l glucose (a) or 16.7 mmol/l glucose (b). (c, d) qRT-PCR analysis of Cat and Sod2 mRNA expression in male (c) and female (d) offspring. Experiments were performed on islets isolated from 8-week-old male and female offspring of control and obese dams. (a, b) Data were analysed by two-way ANOVA followed by a simple main effect analysis when a significant interaction was observed. (c, d) Data were analysed independently by unpaired Student’s t test (control vs obese). *p < 0.05 and **p < 0.01 as shown. (a, b) Males, control: n = 7, obese: n = 5 mice; females, control: n = 8, obese: n = 4 mice; (c, d) Cat: males, control: n = 7, obese: n = 8 mice; females, control: n = 7, obese: n = 7 mice; Sod2: males, control: n = 8, obese: n = 6 mice; females, control: n = 9, obese: n = 9 mice. ‘n’ represents the number of mice from separate litters. All data are mean ± SEM
Reduction in L-type Ca2+ channels and docked insulin granules in beta cells from male offspring that were previously exposed to maternal obesity
Activation of L-type voltage-gated Ca2+ (CaV1) channels are essential for insulin secretion [27, 28]. Exposure to maternal obesity resulted in sex-specific differences in expression of the two subtypes of CaV1 channels: Cacna1c and Cacna1d (which encode CaV1.2 and CaV1.3, respectively). Cacna1d expression was increased in females (p < 0.05) but both Cacna1c and Cacna1d expression was decreased in male (p < 0.001) offspring of obese dams (Fig. 6a, b). Downregulation of these genes has been observed in both human and rodent type 2 diabetic islets [29–31].
Fig. 6.

Reduction in L-type Ca2+ channels and docked insulin granules in beta cells from male offspring that were previously exposed to maternal obesity. (a, b) qRT-PCR analysis of Cacna1c and Cacna1d mRNA expression in male (a) and female (b) offspring. (c–f) Transmission electron microscopy analysis of insulin granule density (c) and docked granules estimated by the surface density (d). Relative distribution of granules at distance fractions from the plasma membrane (e). Granules were defined as docked if their distance from the plasma membrane was 0.19 μm, i.e. half the size of the mean granule diameter. Representative transmission electron microscopy micrographs of beta cells from male and female offspring of control and obese dams (f). The right-hand image is a magnification of the inset in the left-hand image; scale bars, 2 μm for the left-hand image in each set and 500 nm for the magnification. (g, h) Western blot analysis of STX1A (g) and STXBP1 (h); images of corresponding western blots are in ESM Fig. 1e, f, respectively. Experiments were performed on islets isolated from 8-week-old male and female offspring of control and obese dams. (c–e) Data were analysed by two-way ANOVA followed by a simple main effect analysis when a significant interaction was observed. Analyses based on individual animals (n = 2–4 mice/group) showed no statistically significant difference within the groups; thus, cell-based analyses (n = 40–59 cells/group) were performed. (a, b, g, h) Data were analysed independently by unpaired Student’s t test (control vs obese). ††p < 0.01 for interaction; *p < 0.05, **p < 0.01 and ***p < 0.001 as shown. In (b), p = 0.05 for Cacna1c mRNA expression in female offspring of control vs obese dams. (a, b) Cacna1c: males, control: n = 6, obese: n = 8 mice; females, control: n = 8, obese: n = 6 mice; Cacna1d: males, control: n = 6, obese: n = 8 mice; females, control: n = 7, obese: n = 6 mice; (g) males, control: n = 5, obese: n = 4 mice; females, control: n = 6, obese: n = 5 mice; (h) males, control: n = 6, obese: n = 4 mice; females, control: n = 6, obese: n = 6 mice; ‘n’ represents the number of mice from separate litters; in (c, d) the data points shown correspond to the number of beta cells from: males, control: n = 2, obese: n = 4 mice; females, control: n = 2, obese: n = 3 mice. All data are mean ± SEM
Docking of secretory vesicles is required for insulin exocytosis [20, 32]. Insulin granule density was reduced in beta cells of maternal obesity-exposed offspring of both sexes (p < 0.01) (Fig. 6c). We found, however, that the area of docked granules was decreased only in male offspring of obese dams (p < 0.001) (Fig. 6d). Furthermore, the number of granules in the first fraction beneath the plasma membrane (0.19 μm from the plasma membrane) was reduced (p < 0.01) whilst those in the second fraction (0.38 μm from the plasma membrane) were increased (p < 0.001) in these mice (Fig. 6e). These results suggest that granules may be stacking in the second fraction (which is considered to be the reserve pool) owing to dysfunctional docking in beta cells of these mice. Expression of syntaxin-1A (STX1A) and syntaxin binding protein 1 (STXBP1) proteins, which are required for docking [33] was unchanged in both male and female offspring (Fig. 6g, h).
Islets from female offspring of obese dams may be protected from dysfunction by increased oestrogen receptor α and reduced susceptibility to apoptosis
17β-oestradiol has been shown to protect females from beta cell death and hyperglycaemia via oestrogen receptor α (ERα) [34–36]. In our model, ERα protein abundance was higher in female offspring of obese dams (p < 0.05) (Fig. 7a). Furthermore, expression of cleaved (activated) caspase-3, the key mediator of the apoptotic cascade in mammalian cells [37] was decreased (p < 0.05) in these offspring (Fig. 7b). In contrast, cleaved caspase-3 abundance and Casp3 mRNA expression were increased in male offspring of obese dams (p < 0.05) (Fig. 7b, c).
Fig. 7.
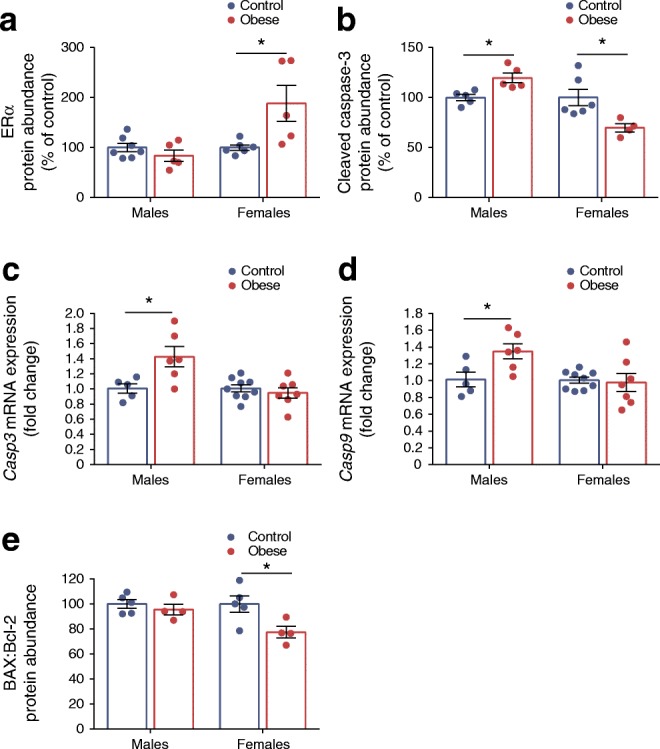
Islets from female offspring of obese dams may be protected from dysfunction by increased ERα and reduced susceptibility to apoptosis. (a, b) Western blot analysis of ERα (a) and cleaved caspase-3 (b) protein abundance; images of corresponding western blots are in ESM Fig. 1g, h, respectively. (c, d) qRT-PCR analysis of Casp3 (c) and Casp9 (d) mRNA expression in male and female offspring. (e) Western blot analysis of BAX:Bcl-2 ratio; images of corresponding western blots are in ESM Fig. 1i (BAX) and ESM Fig. 1j (Bcl-2). Experiments were performed on islets isolated from 8-week-old male and female offspring of control and obese dams. Males and females were analysed independently by unpaired Student’s t test (control vs obese). *p < 0.05, as shown. (a) Males, control: n = 7, obese: n = 5 mice; females, control: n = 6, obese: n = 5 mice; (b) males, n = 5 mice/group; females, control: n = 6, obese: n = 4 mice; (c, d) males, control: n = 5, obese: n = 6 mice; females, control: n = 9, obese: n = 7 mice; (e) BAX: males, n = 5 mice/group; females, control: n = 5, obese: n = 4 mice; Bcl-2: males and females, control: n = 5, obese: n = 4 mice. ‘n’ represents the number of mice from separate litters. All data are mean ± SEM
The mitochondrial phenotype in this study prompted us to investigate whether the mitochondrial intrinsic apoptosis pathway could be contributing to changes in caspase-3 activity. mRNA expression of Casp9, the initiator caspase involved in this pathway, was higher in male offspring of obese dams (p < 0.05) (Fig. 7d). Furthermore, the ratio of pro-apoptotic BAX to anti-apoptotic Bcl-2, which can profoundly influence the ability of a cell to respond to an apoptotic signal [38], was lower in female offspring of obese dams (p < 0.05) (Fig. 7e). This suggests a downregulation of the mitochondrial apoptosis pathway in these offspring. There were no differences in protein levels of BAX and Bcl-2 between offspring of maternal diet groups (ESM Fig. 4a, b).
Discussion
Whilst there are known genetic risk factors that may be transferred from mother to child, which explain future obesity and type 2 diabetes risk, the picture is far from complete. Studies in humans that have controlled for shared genetics found that individuals exposed to maternal obesity/gestational diabetes had a greater diabetes and obesity risk compared with unexposed individuals [39–41]. Furthermore, a recent record-linkage study involving 118,201 mothers and their offspring found that being overweight or obese in mothers is associated with increased incidence of offspring type 2 diabetes [1].
Given that in humans a mother’s genes and environment co-exist, rodent models of obesogenic diet-induced obesity across pregnancy and lactation, which resemble the human situation, have been key in our understanding of the non-genetic transfer of metabolic disease risk from mother to offspring. A number of studies have suggested that increased type 2 diabetes susceptibility in developmentally programmed offspring is due to altered pancreatic islet architecture and/or function (reviewed in [5]). The strength of this study is that it is the first to outline the sex-specific changes in islet function in offspring born to obese dams that are present before and are, therefore, independent of offspring obesity, glucose intolerance and ageing. This is important as it allows us to elucidate islet processes that are most vulnerable to dysfunction thus leading to higher type 2 diabetes risk in offspring exposed to maternal obesity.
A limitation of this study is that we did not measure adiposity in male and female offspring at 8 weeks of age. Previous studies in this model have shown, however, that fat mass is comparable in 8-week-old offspring from control and obese dams irrespective of sex [42, 43].
It has been shown previously, using the same model of maternal diet-induced obesity, that whilst both male and female offspring develop obesity and insulin resistance with age, female offspring of obese dams are less susceptible to type 2 diabetes at 6 months of age [13]. Our findings suggest that islets from female offspring are primed to handle a nutritionally-rich postnatal environment by upregulating mitochondrial respiration and CaV1 expression leading to higher GSIS. Therefore, despite being ‘overworked’ from a young age, these islets appear to have protective mechanisms in place to cope with the demands of increasing adiposity and insulin resistance (Fig. 8). For example, reduced glucokinase expression could act to temper glycolysis in the face of increased mitochondrial metabolism so as to maintain a healthy redox balance [44]. Increased proton leak-linked respiration may also not necessarily be damaging (especially since coupling efficiency is maintained). Mitochondrial superoxide production is steeply dependent on the protonmotive force across the inner mitochondrial membrane. Thus, increased proton leak may act to minimise oxidative damage by moderating the protonmotive force and, therefore, ROS production [45]. Furthermore, increased ERα levels could also have a positive influence on these islets by preserving mitochondrial function [46] and protecting beta cells from apoptosis [34–36]. Indeed, oestrogen-based replacement therapies have been shown to reduce type 2 diabetes incidence in post-menopausal women [47, 48]. As part of future studies, it will be important to determine if serum oestradiol levels are also different between offspring groups. Moreover, it will be key to elucidate islet function and type 2 diabetes risk in post-menopausal, maternal obesity-exposed mice.
Fig. 8.

Exposure to maternal obesity programs sex differences in pancreatic islets of the offspring in mice
In line with the observation that women have lowered risk for type 2 diabetes [49], females appear to be protected from beta cell death in most rodent models of type 2 diabetes [12, 50]. In the case of exposure to maternal diet-induced obesity/high-fat-feeding and future type 2 diabetes risk in the offspring, Yokomizo and colleagues showed that islets from female offspring were better able to compensate and adapt to a high-fat diet (HFD) in postnatal life compared with male offspring [9]. Interestingly, in a study of maternal obesity using the agouti viable yellow (Avy) mouse, (aged) female offspring developed glucose intolerance and had reduced GSIS following postnatal HFD compared with males [10]. This is in contrast to findings by Li and colleagues, which showed that the latent predisposition to metabolic disease in offspring of Avy dams, was more prominent in males who developed glucose intolerance and insulin resistance after only 3 weeks of HFD [51]. It should be noted that in contrast to most models of maternal diet-induced obesity, Avy mice are normoglycaemic during pregnancy.
In humans, men are diagnosed with type 2 diabetes at a younger age and at lower levels of adiposity than women [49]. The male sex is also acknowledged as a diabetes risk factor [52]. In this study, islets from maternal obesity-exposed male offspring appear not to fare as well as females (Fig. 8). This manifests as suboptimal mitochondrial respiration. Unlike in females, these changes could not be attributed to changes in mtDNA content or the expression of genes encoding components of the electron transport chain. The mitochondria in islets of male offspring seem particularly vulnerable to insults in utero/early life; male offspring that experienced intrauterine growth restriction followed by postnatal catch-up growth also showed impaired mitochondrial function and increased ROS production prior to the onset of diabetes [53]. There may, therefore, be common responses operating as a result of suboptimal nutrition in utero. There is a 50% reduction in the expression of Cacna1c and Cacna1d, which is key for GSIS, along with impairment of docking of insulin granules in male offspring of obese dams. The latter could be due to reduced ATP levels [54] owing to reduced ATP-linked respiration in these islets. Whilst not yet detrimental to GSIS at this age, these changes could mean that their islets are more vulnerable to metabolic insult/stress. This may partly explain why islets from maternal obesity-exposed male offspring that were subsequently exposed to a ‘second hit’ of high-fat feeding in postnatal life are more vulnerable to dysfunction compared with females [9]. Furthermore, our results highlight the role of pancreatic islets in conferring increased type 2 diabetes risk in male offspring exposed to maternal obesity.
Type 2 diabetes is associated with islet dysfunction and altered endocrine cell mass [55]. Whilst we found no difference in offspring beta/alpha cell mass, previous studies showed that maternal HFD impacted on offspring endocrine cell mass/number in neonatal and/or adult life (reviewed in [5]). Importantly, these studies also provided evidence for the dynamic nature of beta cell mass in these offspring and its ability to adapt to altered metabolic demand. Neonates exposed to maternal HFD throughout gestation have reduced beta cell number and volume at birth [56] but this difference disappears in adolescence [57]. Furthermore, exposure to maternal obesity led to reduced beta cell mass in 36-day-old offspring but this was higher at 110 days [11]. Thus, it is likely that differences between studies are due to differences in the age and metabolic status of the offspring at the time of investigation. Finally, it is also important to consider changes in beta cell mass in the context of its functional state since beta cells can occur under different phenotypes that vary with age and environmental conditions [58].
Looking to the future, it is important to improve our understanding of the possible maternal factors, e.g. hyperinsulinaemia, hyperglycaemia and hyperlipidaemia, underlying the programming of metabolic adversity across the life course of the offspring. Furthermore, it is also key that we identify the critical developmental period(s) during which suboptimal maternal nutrition programs altered islet function in the offspring.
In summary, the current findings suggest that sex differences in type 2 diabetes susceptibility in offspring as a consequence of exposure to maternal obesity could, at least in part, be driven by differences in islet function. Females, in response to nutritional cues from the mother signalling a nutrient-rich environment, prime islet development to thrive in this environment postnatally. In contrast, males may respond to these cues in a manner that minimises the risk of neonatal hypoglycaemia and maximises neonatal survival. However, this becomes maladaptive in later life following the onset of obesity and insulin resistance in these male offspring.
Electronic supplementary material
(PDF 1119 kb)
Acknowledgements
We would like to thank T. J. Ashmore and C. Custance (University of Cambridge Metabolic Research Laboratories and MRC Metabolic Diseases Unit, Wellcome Trust-MRC Institute of Metabolic Science, Addenbrooke’s Hospital, Cambridge, UK) for expert technical assistance. Parts of this study were presented in abstract form at the 53rd EASD Annual Meeting, Lisbon, 11–15 September 2017 and at the International Society for Developmental Origins of Health and Disease World Congress, Rotterdam, 15–18 October 2017. A non-peer-reviewed version of this article was submitted to the bioRxiv preprint server (10.1101/591586) on 27 March 2019.
Abbreviations
- ATP5A
ATP synthase F1 subunit α
- Avy
Agouti viable yellow
- BAX
Bcl-2-associated X protein
- Bcl-2
B cell lymphoma 2
- Cav1
L-type voltage-gated Ca2+ (channel)
- DCFDA
2′,7′-dichlorofluorescin diacetate
- ERα
Oestrogen receptor α
- FCCP
Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- GSIS
Glucose-stimulated insulin secretion
- HFD
High-fat diet
- mtDNA
Mitochondrial DNA
- ROS
Reactive oxygen species
- STX1A
Syntaxin-1A
- STXBP1
Syntaxin binding protein 1
- TFAM
Mitochondrial transcription factor A
- UQCRC2
Ubiquinol-cytochrome C reductase core protein 2
Author contributions
LMN conceived and performed experiments, analysed data and wrote the manuscript. MN performed experiments and analysed data. LCK and DSFT performed experiments. LE provided expertise and feedback. LMN, LE and SEO designed experiments and interpreted the data. LMN and SEO are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors read, commented on and approved the final version of the manuscript.
Funding
This work was supported by a fellowship from the National Health and Medical Research Council (GNT1092158) and grants from the Isaac Newton Trust [17.37(l)], Society for Endocrinology and the Diabetes Research and Wellness Foundation to LMN. LCK is funded by the Biotechnology and Biological Sciences Research Council Grant BB/M001636/1. SEO and DSFT are funded by the Medical Research Council (MC_UU_12012/4) and the British Heart Foundation (RG/17/12/33167). LE and MN used equipment funded by Swedish Foundation for Strategic Research (LUDC-IRC, IRC15-0067) and Swedish Research Council (EXODIAB, 2009-1039; LUDC 349-2006-237). LE acknowledges funding from Swedish Research Council (project grant, 2016-02124), Diabetes Wellness Sverige (25-505 PG) and Swedish Diabetes Foundation (DIA2016-130). MN is funded by the EFSD and the Japan Diabetes Society.
Data availability
The datasets generated during the current study are available from the corresponding author upon reasonable request.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lahti-Pulkkinen M, Bhattacharya S, Wild SH, et al. Consequences of being overweight or obese during pregnancy on diabetes in the offspring: a record linkage study in Aberdeen, Scotland. Diabetologia. 2019;62(8):1412–1419. doi: 10.1007/s00125-019-4891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 3.Wortham M, Sander M. Mechanisms of β-cell functional adaptation to changes in workload. Diabetes Obes Metab. 2016;18(Suppl 1):78–86. doi: 10.1111/dom.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branum AM, Kirmeyer SE, Gregory EC. Prepregnancy body mass index by maternal characteristics and state: data from the birth certificate, 2014. Natl Vital Stat Rep. 2016;65(6):1–11. [PubMed] [Google Scholar]
- 5.Elsakr JM, Gannon M. Developmental programming of the pancreatic islet by in utero overnutrition. Trends Dev Biol. 2017;10:79–95. [PMC free article] [PubMed] [Google Scholar]
- 6.Graus-Nunes F, Dalla Corte Frantz E, Lannes WR, da Silva Menezes MC, Mandarim-de-Lacerda CA, Souza-Mello V. Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition. 2015;31(2):380–387. doi: 10.1016/j.nut.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. 2006;291(4):E792–E799. doi: 10.1152/ajpendo.00078.2006. [DOI] [PubMed] [Google Scholar]
- 8.Taylor PD, McConnell J, Khan IY, et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol. 2005;288(1):R134–R139. doi: 10.1152/ajpregu.00355.2004. [DOI] [PubMed] [Google Scholar]
- 9.Yokomizo H, Inoguchi T, Sonoda N, et al. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic β-cell function in adult offspring with sex differences in mice. Am J Physiol Endocrinol Metab. 2014;306(10):E1163–E1175. doi: 10.1152/ajpendo.00688.2013. [DOI] [PubMed] [Google Scholar]
- 10.Han J, Xu J, Epstein PN, Liu YQ. Long-term effect of maternal obesity on pancreatic beta cells of offspring: reduced beta cell adaptation to high glucose and high-fat diet challenges in adult female mouse offspring. Diabetologia. 2005;48(9):1810–1818. doi: 10.1007/s00125-005-1854-8. [DOI] [PubMed] [Google Scholar]
- 11.Zambrano E, Sosa-Larios T, Calzada L, et al. Decreased basal insulin secretion from pancreatic islets of pups in a rat model of maternal obesity. J Endocrinol. 2016;231(1):49–57. doi: 10.1530/joe-16-0321. [DOI] [PubMed] [Google Scholar]
- 12.Gannon M, Kulkarni RN, Tse HM, Mauvais-Jarvis F. Sex differences underlying pancreatic islet biology and its dysfunction. Mol Metab. 2018;15:82–91. doi: 10.1016/j.molmet.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samuelsson AM, Matthews PA, Argenton M, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51(2):383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Gronert MS, Fernandez-Twinn DS, Poston L, Ozanne SE. Altered hepatic insulin signalling in male offspring of obese mice. J Dev Orig Health Dis. 2010;1(3):184–191. doi: 10.1017/s2040174410000231. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Twinn DS, Gascoin G, Musial B, et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci Rep. 2017;7:44650. doi: 10.1038/srep44650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholas LM, Valtat B, Medina A, et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol Metab. 2017;6(7):651–663. doi: 10.1016/j.molmet.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malmgren S, Nicholls DG, Taneera J, et al. Tight coupling between glucose and mitochondrial metabolism in clonal β-cells is required for robust insulin secretion. J Biol Chem. 2009;284(47):32395–32404. doi: 10.1074/jbc.M109.026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297–312. doi: 10.1042/bj20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rooney JP, Ryde IT, Sanders LH, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015;1241:23–38. doi: 10.1007/978-1-4939-1875-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olofsson CS, Gopel SO, Barg S, et al. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B cells. Pflugers Arch - Eur J Physiol. 2002;444(1–2):43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 21.Dehoff RT, Rhines FN. Determination of the number of particles per unit volume from measurements made on random plane sections: the general cylinder and the ellipsoid. Trans Metall Soc AIME. 1961;221:975–982. [Google Scholar]
- 22.Chintinne M, Stange G, Denys B, Ling Z, In ‘t Veld P, Pipeleers D. Beta cell count instead of beta cell mass to assess and localize growth in beta cell population following pancreatic duct ligation in mice. PLoS One. 2012;7(8):e43959. doi: 10.1371/journal.pone.0043959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and β-pancreatic islet cells. Cell. 1988;55(2):281–290. doi: 10.1016/0092-8674(88)90051-7. [DOI] [PubMed] [Google Scholar]
- 24.Meglasson MD, Matschinsky FM. New perspectives on pancreatic islet glucokinase. Am J Phys. 1984;246(1 Pt 1):E1–E13. doi: 10.1152/ajpendo.1984.246.1.E1. [DOI] [PubMed] [Google Scholar]
- 25.Divakaruni Ajit S., Paradyse Alexander, Ferrick David A., Murphy Anne N., Jastroch Martin. Methods in Enzymology. 2014. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data; pp. 309–354. [DOI] [PubMed] [Google Scholar]
- 26.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 27.Ammala C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B cells. J Physiol. 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davalli AM, Biancardi E, Pollo A, et al. Dihydropyridine-sensitive and -insensitive voltage-operated calcium channels participate in the control of glucose-induced insulin release from human pancreatic β cells. J Endocrinol. 1996;150(2):195–203. doi: 10.1677/joe.0.1500195. [DOI] [PubMed] [Google Scholar]
- 29.Dreja T, Jovanovic Z, Rasche A, et al. Diet-induced gene expression of isolated pancreatic islets from a polygenic mouse model of the metabolic syndrome. Diabetologia. 2009;53(2):309–320. doi: 10.1007/s00125-009-1576-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reinbothe TM, Alkayyali S, Ahlqvist E, et al. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia. 2013;56(2):340–349. doi: 10.1007/s00125-012-2758-z. [DOI] [PubMed] [Google Scholar]
- 31.Roe MW, Worley JF, 3rd, Tokuyama Y, et al. NIDDM is associated with loss of pancreatic beta-cell L-type Ca2+ channel activity. Am J Phys. 1996;270(1 Pt 1):E133–E140. doi: 10.1152/ajpendo.1996.270.1.E133. [DOI] [PubMed] [Google Scholar]
- 32.Daniel S, Noda M, Straub SG, Sharp GW. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes. 1999;48(9):1686–1690. doi: 10.2337/diabetes.48.9.1686. [DOI] [PubMed] [Google Scholar]
- 33.Gandasi NR, Barg S. Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat Commun. 2014;5:3914. doi: 10.1038/ncomms4914. [DOI] [PubMed] [Google Scholar]
- 34.Alonso-Magdalena P, Ropero AB, Carrera MP, et al. Pancreatic insulin content regulation by the estrogen receptor ERα. PLoS One. 2008;3(4):e2069. doi: 10.1371/journal.pone.0002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilic G, Alvarez-Mercado AI, Zarrouki B, et al. The islet estrogen receptor-α is induced by hyperglycemia and protects against oxidative stress-induced insulin-deficient diabetes. PLoS One. 2014;9(2):e87941. doi: 10.1371/journal.pone.0087941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le May C, Chu K, Hu M, et al. estrogens protect pancreatic β-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103(24):9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hui H, Dotta F, Di Mario U, Perfetti R. Role of caspases in the regulation of apoptotic pancreatic islet β-cells death. J Cell Physiol. 2004;200(2):177–200. doi: 10.1002/jcp.20021. [DOI] [PubMed] [Google Scholar]
- 38.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74(4):609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 39.Dabelea D, Hanson RL, Lindsay RS, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49(12):2208–2211. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- 40.Lawlor DA, Lichtenstein P, Langstrom N. Association of maternal diabetes mellitus in pregnancy with offspring adiposity into early adulthood: sibling study in a prospective cohort of 280,866 men from 248,293 families. Circulation. 2011;123(3):258–265. doi: 10.1161/circulationaha.110.980169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith J, Cianflone K, Biron S, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab. 2009;94(11):4275–4283. doi: 10.1210/jc.2009-0709. [DOI] [PubMed] [Google Scholar]
- 42.Alfaradhi MZ, Fernandez-Twinn DS, Martin-Gronert MS, Musial B, Fowden A, Ozanne SE. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol. 2014;307(1):R26–R34. doi: 10.1152/ajpregu.00049.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, et al. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab. 2014;3(3):325–333. doi: 10.1016/j.molmet.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu L, Nicholson W, Knobel SM, et al. Oxidative stress is a mediator of glucose toxicity in insulin-secreting pancreatic islet cell lines. J Biol Chem. 2004;279(13):12126–12134. doi: 10.1074/jbc.M307097200. [DOI] [PubMed] [Google Scholar]
- 45.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26(3):192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Z, Ribas V, Rajbhandari P, et al. Estrogen receptor α protects pancreatic β-cells from apoptosis by preserving mitochondrial function and suppressing endoplasmic reticulum stress. J Biol Chem. 2018;293(13):4735–4751. doi: 10.1074/jbc.M117.805069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanaya AM, Herrington D, Vittinghoff E, et al. Glycemic effects of postmenopausal hormone therapy: the Heart and Estrogen/progestin Replacement Study. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2003;138(1):1–9. doi: 10.7326/0003-4819-138-1-200301070-00005. [DOI] [PubMed] [Google Scholar]
- 48.Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47(7):1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- 49.Harreiter J, Kautzky-Willer A. Sex and gender differences in prevention of type 2 diabetes. Front Endocrinol. 2018;9:220. doi: 10.3389/fendo.2018.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Louet JF, LeMay C, Mauvais-Jarvis F. Antidiabetic actions of estrogen: insight from human and genetic mouse models. Curr Atheroscler Rep. 2004;6(3):180–185. doi: 10.1007/s11883-004-0030-9. [DOI] [PubMed] [Google Scholar]
- 51.Li CC, Young PE, Maloney CA, et al. Maternal obesity and diabetes induces latent metabolic defects and widespread epigenetic changes in isogenic mice. Epigenetics. 2013;8(6):602–611. doi: 10.4161/epi.24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.American Diabetes Association 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2018. Diabetes Care. 2018;41(Suppl 1):S13–S27. doi: 10.2337/dc18-S002. [DOI] [PubMed] [Google Scholar]
- 53.Simmons RA, Suponitsky-Kroyter I, Selak MA. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to β-cell failure. J Biol Chem. 2005;280(31):28785–28791. doi: 10.1074/jbc.M505695200. [DOI] [PubMed] [Google Scholar]
- 54.Eliasson L, Renstrom E, Ding WG, Proks P, Rorsman P. Rapid ATP-dependent priming of secretory granules precedes Ca2+-induced exocytosis in mouse pancreatic B cells. J Physiol. 1997;503(Pt 2):399–412. doi: 10.1111/j.1469-7793.1997.399bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meier JJ, Bonadonna RC. Role of reduced β-cell mass versus impaired β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36 Suppl 2:S113–S119. doi: 10.2337/dcS13-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cerf ME, Williams K, Nkomo XI, et al. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2005;288(5 57–5):R1122–R1128. doi: 10.1152/ajpregu.00335.2004. [DOI] [PubMed] [Google Scholar]
- 57.Cerf ME, Louw J (2014) Islet cell response to high fat programming in neonate, weanling and adolescent Wistar rats. JOP 15(3):228–236. 10.6092/1590-8577/1534 [DOI] [PubMed]
- 58.Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional β-cell mass in diabetes. Diabetes Obes Metab. 2008;10 Suppl 4:54–62. doi: 10.1111/j.1463-1326.2008.00941.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1119 kb)
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.