

Abstract
Tumor neovascularization/tumor angiogenesis is a pathophysiological process in which new blood vessels are formed from existing blood vessels in the primary tumors to supply adequate oxygen and nutrition to cancer cells for their proliferation and metastatic growth to the distant organs. Therefore, controlling tumor angiogenesis is an attractive target for cancer therapy. Structural abnormalities of the vasculature (i.e., leakiness due to the abnormal lining of pericytes on the microvessels) are one of the critical features of tumor angiogenesis that sensitizes vascular cells to cytokines and helps circulating tumor cells to metastasize to distant organs. Our goal is to repurpose the drugs that may prevent tumor angiogenesis or normalize the vessels by repairing leakiness via recruiting pericytes or both. In this study, we tested whether aspirin (ASA), which could block primary tumor growth, regulates tumor angiogenesis. We investigated the effects of low (1 mM) and high (2.5 mM) doses of ASA (direct effect), and ASA-treated or untreated triple negative breast cancer (TNBC) cells’ conditioned media (indirect effect) on endothelial cell physiology. These include in vitro migration using modified Boyden chamber assay, in vitro capillary-like structure formation on Matrigel, interactions of pericytes-endothelial cells and cell permeability using in vitro endothelial permeability assay. We also examined the effect of ASA on various molecular factors associated with tumor angiogenesis. Finally, we found the outcome of ASA treatment on in vivo tumor angiogenesis. We found that ASA-treatment (direct or indirect) significantly blocks in vitro migration and capillary-like structure formation by endothelial cells. Besides, we found that ASA recruits pericytes from multipotent stem cells and helps in binding with endothelial cells, which is a hallmark of normalization of blood vessels, and decreases in vitro permeability through endothelial cell layer. The antiangiogenic effect of ASA was also documented in vivo assays. Mechanistically, ASA treatment blocks several angiogenic factors that are associated with tumor angiogenesis, and suggesting ASA blocks paracrine-autocrine signaling network between tumor cells and endothelial cells. Collectively, these studies implicate aspirin with proper dose may provide potential therapeutic for breast cancer via blocking as well as normalizing tumor angiogenesis.
Electronic supplementary material
The online version of this article (10.1007/s12079-018-00499-y) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Aspirin, Hyperpermeability, Metastasis, Leaky blood vessels
Background
Metastasis is the most life-threatening event in various cancers including breast cancer. Despite advancement in breast cancer diagnosis and treatment, metastasis and recurrence of the disease are the common cause of cancer-related death worldwide. Metastatic cascade is a multistep process which includes invasion of cancer cell from primary tumor site to surrounding tissue, trans-endothelial migration into blood vessels (intravasation), survival in the blood and extravasation followed by colonization to distant organs. Therefore, tumor cell penetration through the vascular wall is a vital step in the metastatic process (Martin et al. 2009). Several studies showed that like other solid tumors, tumor angiogenesis plays a vital role in primary tumor growth and metastasis (Bielenberg and Zetter 2015; Longatto Filho et al. 2010).
Angiogenesis or neovascularization is a normal physiological process develops from the pre-existing blood vessels, and mostly occurs during embryonic development, wound-healing, pregnancy and essential for tumor growth and metastasis (Weis and Cheresh 2011). Blood vascular wall is composed of endothelial cells, pericytes, vascular smooth muscle cells, and the basal membrane that collectively support barrier functions. Disorganization of these layers’ results in leakiness and increased permeability, which is a common nominator of tumor angiogenesis (Bielenberg and Zetter 2015; Weis and Cheresh 2011). Endothelial barriers have a huge importance in vascular tissue homeostasis (Azzi et al. 2013). Under physiological condition, endothelial cell-cell junctional proteins (adherent junctional protein. Tight junctional protein) strictly regulate barrier function by restricting macromolecules and cells (Bazzoni and Dejana 2004; Dejana 2004). Any inefficiency of these junctional proteins leads to hampering vascular integrity (Zhou et al. 2014). Therefore, increased junctional proteins in endothelium lead to decrease in intravasation and extravasation. Moreover, the endothelial layer in tumor blood vessels shows abnormal morphology and the pericyte lining around the endothelial layer are less and loosely attached. This depletion and loosely connected pericyte also creates leaky vasculature and thereby facilitate tumor cell penetration and tumor metastasis. Multiple studies reported that pericyte depletion leads to increased vessel permeability and poor vessel integrity which favors tumor cells invasion and metastatic spread (Cooke et al. 2012; Sennino et al. 2007; Xian et al. 2006). All these abnormalities in tumor vasculature also impaired drug delivery to the solid tumors (Azzi et al. 2013). Several antiangiogenic therapies have been discovered not only for inhibiting tumor angiogenesis but also to normalize structural and functional abnormalities of tumor vessels (Carmeliet and Jain 2011), but still, their usage has not been widely accepted due to limitations of the widespread effects of these antiangiogenic drugs in different patients. Therefore, there is a need to develop an effective strategy to normalize leaky and hyperpermeable tumor vasculature to regulate tumor growth and metastasis (Jain 2005).
Aspirin (ASA) is a non-steroidal anti-inflammatory drug that is widely used for pain, arthritis, fever, and stroke. Studies showed that regular use of aspirin reduces the risk of different cancers (Akhmedkhanov et al. 2001; Ararat et al. 2011; Bardia et al. 2011; Din et al. 2004). Our study showed that ASA could effectively inhibit the primary growth of breast cancer cell and tumor-initiating cell in vitro and in vivo and delays the formation of a palpable tumor (Maity et al. 2015). We showed that the antitumor activity of aspirin was mediated by altering pathobiological changes and molecular signature (like migration, epithelial, mesenchymal and stem cell markers) in breast cancer (Maity et al. 2015). Based on our study and other findings (Bosetti et al. 2012; Huang et al. 2016; Maity et al. 2015; Mitra et al. 2012; Zhang et al. 2013), we can suggest that aspirin may have a role in tumor angiogenesis. To test the hypothesis, we examined the effect of ASA on tumor angiogenesis, pericyte recruitment and the permeability of the endothelial cell layer. Our studies found that ASA is a potent inhibitor of tumor angiogenesis. ASA helps in recruiting pericyte and shuttering vessel permeability.
Methods
Reagents
Aspirin (Acetylsalicylic Acid) was purchased from Sigma Aldrich (St. Louis, MO, USA). Matrigel was purchased from BD Biosciences (San Jose, CA). Penicillin–streptomycin, and trypsin–EDTA solution were purchased from Sigma (St Louis, MO, USA). Cell Tracker green CMFDA dye and Cell Tracker Red CMTPX dye were obtained from Life Technology (CA, USA). In vitro Vascular Permeability Imaging Assay kit was purchased from Millipore (MA, USA). Fetal bovine serum (FBS) was obtained from ATCC (Manassas, VA, USA). Antibodies like VE-cadherin and VEGF were purchased from Thermo-Fisher Scientific (Waltham, MA). ZO-1 antibody was purchased from Genetex (Irvine, CA).
Cell culture
Three different types of cell lines were used in angiogenesis experiments. These include human umbilical vein endothelial cells (HUVEC) and 10 T1/2 mouse pluripotent cells that transform to pericytes, and MDA-MB-231human triple negative breast cancer (TNBC) cells. These cell lines were bought from American Type Culture Collection (ATCC, Manassas, VA). HUVEC was grown in EGM2 (EBM2 media, growth factors, and 5% serum purchased from Lonza) media and supplemented with 100 units/ml penicillin and 100 units/ml streptomycin (Sigma, St. Louis, MO) at a 37 °C incubator in the presence of 5% CO2. These endothelial cells are the inner lining of the blood vessels and are used for in vitro angiogenesis. 10 T1/2 cells and TNBC cell lines (MDA-MB-231 cells and HCC-70 cell lines) were maintained and grown in Dulbecco’s Modified Eagle’s Medium (ATCC), supplemented with 10% fetal bovine serum (ATCC), 100 units/ml penicillin and 100 units/ml streptomycin (Sigma, St. Louis, MO) at 37 °C incubator in the presence of 5% CO2. Approximately, 60–70% confluent cells and not more than six passages of all cell lines were used for each experiment. Authentication of the cell lines was achieved using determining short tandem repeat (STR) profiles using the Promega PowerPlex 16 system. This was performed once and compared with external STR profiles of the cell lines (when available). Cell lines were also figured out to be mycoplasma-free before use.
Preparation of conditioned media (CM)
MDA-MB-231 and HCC-70 conditioned media (CM) were made to mimic the tumor microenvironment and the abnormalities on the blood vessel. CM was prepared according to our earlier method with some modifications (Maity et al. 2015). Briefly, the cells (60–70% confluent) were treated with ASA with the desired time (72 h) and dose (2.5 mM) or vehicle. In this study, we considered 2.5 mM dose of ASA because our earlier studies showed that the killing efficiency of ASA with 2.5 mM was significant but weak as compared to higher doses (Maity et al. 2015). Following treatment with ASA (2.5 mM) for 72 h, ~60% MDA-MB-231 cells were viable (Fig. S1). Thus, this concentration was used to generate conditioned media (CM) for further studies. The treated cells were washed to remove the drug, and then, HUVEC-specific growth media (EGM2) was added to the MDA-MB-231-ASA/HCC-70-ASA-treated or vehicle-treated MDA-MB-231 or HCC-70 cells for 24 h to make the CM. After 24 h, the debris-free CM was collected by centrifugation and used for further studies.
In vitro angiogenesis assay
In vitro angiogenesis assays were performed according to our earlier studies (Dhar et al. 2010; Maity et al. 2014). To detect the effect of CMs and ASA on angiogenesis, HUVEC and 10 T1/2 cells were stained with cell tracker red CMTPX dye (Fluorescent) and cell tracker green CMFDA dye (fluorescent) respectively, for an hour. After staining, the cells were washed with media thoroughly and used for the angiogenesis assay. For 3D cultures, HUVECs and 10 T1/2 cells (10,000 cells/well) were seeded on the Matrigel (200 μl) coated chambered slides (8-well) containing ASA-pre-treated or vehicle-treated MDA-MB-231-CMs or regular media (RM) and were incubated at 37 °C incubator for approximately 20 h which determined the binding efficiency of 10 T1/2 cells with capillary-like structure generated by HUVECs in RM or CM. Photographs were taken the next day using fluorescence imaging with a Nikon Ti-U photographic fluorescence microscope. The number of attached green fluorescence labeled 10 T1/2 cells were counted manually.
Cell viability
Cell viability was studied by trypan blue exclusion method using Cello-meter Auto T4 Bright Field Cell Counter (Nexcelom Bioscience LLC, Lawrence, MA, USA). Briefly, cells were exposed to CMs (vehicle or ASA-treated) for 72 h. Cells were then re-suspended in fresh medium and diluted in trypan blue (1:1) and cell viability was measured in cello-meter in triplicate. Each experimental condition was taken duplicate.
Migration assay
The in vitro migration was determined using modified Boyden chamber assay as described earlier (Maity et al. 2014). Briefly, the experimental HUVECs (10,000 cells/well) were trypsinized and added in the upper chambers of the modified Boyden chamber and were incubated at 37 °C in a humidified atmosphere with 5% CO2 overnight. After incubation overnight, the inner surface of the Boyden chamber insert was swabbed to remove excess cells and other debris that did not migrate and the outer membrane on the Boyden chamber insert was stained using crystal violet for 10 min. After staining, the membrane was washed with water, dried and photographed using Nikon Eclipse microscope. The membranes were placed in 10% glacial acetic acid and migration index was measured at 600 nm using V-Max Microplate Reader with current version of SoftMax Pro (Molecular Devices, Sunnyvale, CA).
Permeability assay
The permeability assay was performed using In Vitro Vascular Permeability Imaging Assay (Millipore) according to manufacturer’s instruction. In brief, each well of 8-well chamber slide glass was coated with poly-L Lysine, followed by glutaraldehyde and biotinylated gelatin coating to allow proteins to bind. After preparation of biotinylated gelatin coated surface, HUVEC (70–80% confluent) was plated into each well containing HUVEC growth media and was grown until uniform monolayer was formed. The regular growth media was removed and the endothelial cell monolayer were treated with MDA-MB-231-CM (with or without ASA pre-treated) as well as HUVECs specific RM for 48 h. The CM was removed and fluorescein-streptavidin was added to detect the permeable area of the HUVEC monolayer, as seen by green fluorescence.
Western blot
The Western blot was performed as described previously (Das et al. 2017). Briefly, fifty μg total proteins was separated by SDS-PAGE, and transferred to nitrocellulose membrane (Trans-blot transfer medium, Bio-Rad). The membranes were probed with primary antibodies for overnight. Blots were washed in TBS-T and incubated with secondary antibodies conjugated with horseradish peroxidase for 30 min at room temperature. Immunoreactions were detected with Super Signal Ultra Chemiluminescent substrate (Pierce, Rockford, IL, USA) using Carestream Imaging system.
Immunofluorescence
The immunofluorescence was carried out as described earlier (Sarkar et al. 2017). Briefly, the cells were seeded on coverslip, fixed in 100% methanol at -20 °C for 10 min. and were permeabilized with 0.1% Titron -X100 for 5 min. Then, the cells were blocked with a ready-to use blocking solution (Histostain kit, Invitrogen) and were incubated with specific mono- or polyclonal antibodies followed by FITC conjugated secondary antibody (Alexa Fluoro 488) at room temperature. Cells were washed with 1X PBS and nuclei were counter stained with DAPI solution and mounted in antifade mounting reagent (Molecular Probe). cells were visualized using a Nikon Eclipse TE-300 microscope and images were analyzed by software.
In vivo angiogenesis assay
To examine the effect of ASA on in vivo angiogenesis, a gel foam implantation angiogenesis assay was performed in 6–8-week-old immunocompetent FVB/N mice (Jackson Laboratories) as per our earlier method (Maity et al. 2014). Briefly, gel foam (8 X3 X8 mm; Gel foam H, Pharmacia & Upjohn Company, NY, USA) was presoaked either in regular medium (RM) or MDA-MB-231-CM. The foams were then implanted subcutaneously into the mice (N = 5) and kept them under the skin for 5-days for angiogenesis. In an added experiment, gel-foam was presoaked with either RM or MDA-MB-231-CM, and implanted into the mice. The gel-foam implanted mice were fed ASA (75 mg/Kg/day) with 10 ml of milk [Peptamen (Nestle)] or only milk (controls) for 5 days. The implanted gel foam was removed carefully from both experiments, and angiogenesis was monitored using a Nikon microscope. The number of blood vessels were counted in the gel-foam area. All studies performed on mice was approved by the Institutional Animal Care and Use Committee of Kansas City VA Medical Center.
Statistics
The quantifications of the signals were measured using ImageJ software, and GraphPad Prism6 was used for graphic representations and statistical analysis. Comparisons between groups were carried out with Student t-test and ANOVA analysis. All the analyses were two-sided and p values ≤0.05 were considered statistically significant.
Results
Aspirin (ASA) impairs the effect of breast cancer cell-conditioned media (CM) on in vitro capillary-like structure formation and recruitment of pericyte from multipotent stem cells
To investigate the effect of breast cancer cell-CM on in vitro capillary formation, and the recruitment of pericyte, we seeded HUVEC and pericyte forming multipotent stem cell 10 T1/2 on Matrigel with CM collected from MDA-MB-231 or HCC-70 cell cultures. We found that HUVEC (Red) formed tube like structure that was surrounded uniformly by 10 T1/2 (Green) in controls (Fig. 1a & b, left panel and 1C & D right panel). In the presence CM, the capillary-like structure forming ability of endothelial cells were increased significantly, but comparatively less and loosely attached 10 T1/2 was found around the HUVEC layer as compared to control (Fig. 1a–d). These studies, therefore, suggest that a leaky vasculature-like structure was formed by TNBC cell generated CM (Fig. 1a and b, Middle Panel). However, the effect of HCC-70-CM was significantly less as compared to MDA-MB-231-CM.
Fig. 1.

Normalization of tumor blood vessels by ASA. a–b Representative fluorescence image of capillary like structure. HUVEC (Red dye tracker labeled) and 10 T1/2 cells (Green dye tracker labeled) were seeded on Matrigel in presence of regular media (RM) or MDA-MB-231 or HCC-70 cell derived CM or ASA pretreated MDA-MB-231 or HCC-70 cell derived CM. RM served as control set. Scale bar is 500 μM. b–c The error-bar graphs show the mean numbers of capillary-like structures in different experimental conditions (left), and the quantification of attached 10 T1/2 cells with HUVEC in different experimental conditions (right). Data expressed as Mean ± SD of three sets of experiments
Next, we investigated whether ASA could reverse the effect of MDA-MB-231-CM or HCC-70-CM. To do so, cells were pretreated with ASA and ASA pre-treated CMs were used during tube formation assay. The studies showed that ASA suppresses significantly the CM-induced capillary-like structure forming ability of endothelial cells but simultaneously promotes recruitment of 10 T1/2 cell surrounding HUVEC (Right Panel; Fig. 1a–d).
Aspirin blocks tumor cell-CM-induced inhibition of 10 T1/2 cell viability
Next, we sought to figure out the effect of MDA-MB-231-CM and ASA pretreated MDA-MB-231-CM on 10 T1/2 cells. We found that cellular growth of 10 T1/2 cells were significantly decreased upon treatment with MDA-MB-231-CM, while ASA pretreatment significantly blocks MDA-MB-231-CM driven 10 T1/2 cell death as counted by increased survival cell numbers (Fig. 2).
Fig. 2.
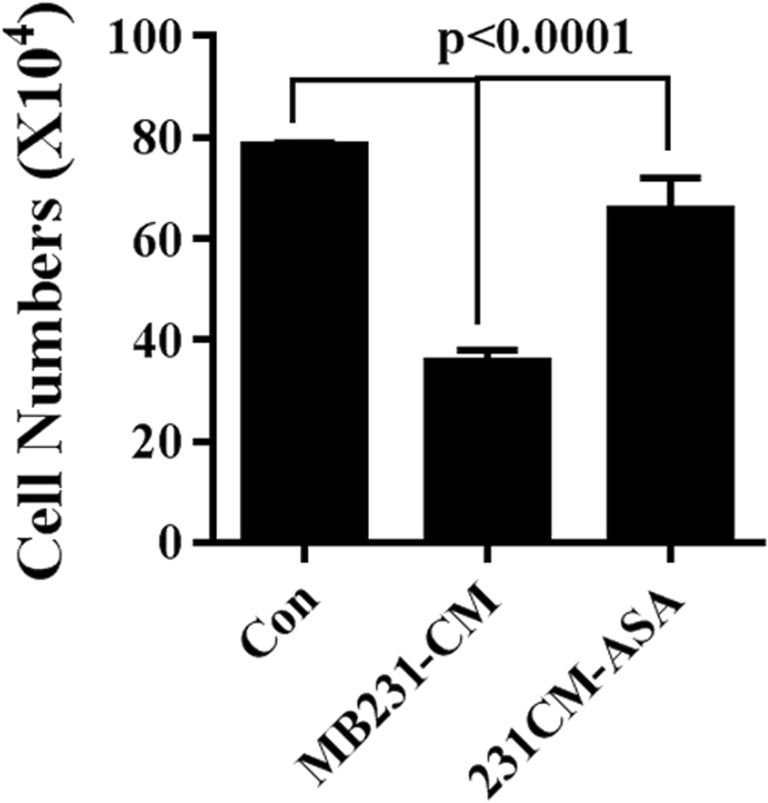
Effects of ASA on proliferation of 10 T1/2. Graphical representation of cellular count of 10 T1/2 in presence of MDA-MB-231 CM (with or without ASA pre-treatment). Regular medium (RM) was used as control. Data expressed as Mean ± SD of three sets of experiments. For statistical analysis ANOVA was done
Aspirin rescue loss of pericytes
Among diverse functions, pericytes have been associated mainly with permeabilization and stabilization of blood vessels (Ribeiro and Okamoto 2015). α-SMA is one of the few identified molecular markers for pericyte (Bergers and Song 2005). Therefore, in order to investigate the effect of breast cancer cells on multipotent stem cells-pericytes transition, we studied the effects of MDA-MB-231-CM on expression of α-SMA of 10 T1/2 cells. We found that majority of the 10 T1/2 multipotent stem cells were expressed α-SMA under normal cell culture condition suggesting 10 T1/2 cells to pericyte transition is a steady and continuous process in a normal tissue culture microenvironment (Fig. 3a). MDA-MB-231-CM treatment significantly downregulates α-SMA expression in 10 T1/2 cell cultures, and this effect of CM was impaired by ASA treatment (Fig. 3a and b). Therefore, the studies suggest that the stem cell-pericyte transition is blocked by breast cancer cell-generated signals, and these signaling pathways can be destroy by ASA treatment.
Fig. 3.

ASA rescue α-SMA expression. a Immunofluorescence of α-SMA in 10 T1/2 cells in presence of regular media (control) or MDA-MB-231 cell derived CM (MB-231-CM) or ASA pretreated MDA-MB-231 cell derived CM (MB-231-CM-ASA). White arrow indicates α-SMA positive cells and yellow arrow indicates α-SMA-negative cells, and (b). Immunoblots of α-SMA and β-Actin in treated and untreated 10 T1/2 cells. β-actin was used as internal loading control. Bar graph represents relative expression of α-SMA in treated and untreated 10 T1/2 cells. Error bars denote mean ± SD, n = 3. Scale, 50 μm. For statistical analysis two paired student’s t test and ANOVA were done
Aspirin prevents TNBC cell-induced endothelial cell migration
Endothelial cell migration is one of the notable events in tumor angiogenesis (Ausprunk and Folkman 1977). To investigate the direct effect of ASA on HUVEC migration, HUVECs were treated with 0.25, 0.5 and 1 mM ASA for 72 h, and was allowed to migrate through a modified Boyden chamber. Studies showed that ASA significantly decreased HUVEC migration at 0.5 and 1 mM dose, as compared to controls, but the migration was almost similar at 0.25 mM as compared to untreated cells (Fig. 4a). Next, we investigate the effect of ASA on breast cancer cell induced HUVEC migration. We have designed two experiments. In first set, MDA-MB-231-CM and ASA pretreated MDA-MB-231-CM were added on the upper Boyden chamber containing HUVECs (Fig. 4b, right), and in second set, MDA-MB-231 CM and ASA pretreated MDA-MB-231-CM in the lower chamber (Fig. 4b, left). We found that migration of HUVEC cell was increased significantly upon treatment with MDA-MB-231-CM, and this MDA-MB-231 CM driven HUVEC cell migration was inhibited when MDA-MB-231 was pretreated with ASA (Fig. 2b, right). Similarly, HUVEC migration was significantly enhanced when MDA-MB-231- CM was used in lower Boyden Chamber as chemoattractant and aspirin treatment reduces MDA-MB-231- CM driven chemotactic migration of HUVEC (Fig. 4b, left).
Fig. 4.

Effects of ASA on endothelial cell migration. a HUVEC was treated with different doses of ASA and in vitro migration was performed. The bar diagram shows the rate of HUVEC migration in different doses of ASA. The accompanying photograph (left) showing the view of migrated HUVEC. b In two set of experiments, HUVEC was allowed to migrate in the presence of MDA-MB-231-CM and ASA pretreated MDA-MB-231-CM either on upper Boyden chamber (Left bar diagram) or lower Boyden chamber (Right bar diagram). Regular medium (RM) was used in control set. The representative photograph of migrated cells in both experimental setup. Data shows mean±SD and represents at least 3 independent experiments. For statistical analysis two paired student’s t test was done
Aspirin inhibits TNBC cell induced endothelial hyper-permeability
In physiological condition, vascular permeability has a significant importance to supply materials properly. Increased vascular permeability allows extravasation of tumor cells leading to metastasis (Zhou et al. 2014). This barrier function is regulated by several factors including tight junction and gap junction proteins. We performed permeability assay to investigate whether ASA could recover TNBC cell-induced hyper-permeability. Permeability assay showed distinct high expression of adherent protein VE-cadherin with minimal FITC-fluorescence in the cell-cell junction of control cell monolayer (Fig. 5). But MDA-MB-231-CM significantly enhanced intercellular permeability as seen by enlarged areas of FITC–streptavidin staining with concomitant absent of VE-cadherin expression. Interestingly ASA treated MDA-MB-231-CM could recover endothelial hyper-permeability as reflected by regain of VE-Cadherin expression and decreased FITC fluorescence (Fig. 5a). Quantitative analysis showed ASA could able to reduce almost 50% breast cancer cell driven hyper-permeability as measured from FITC-positive area (Fig. 5b).
Fig. 5.

Effects of ASA treated or untreated MDA-MB-231-CM on vascular permeability in vitro. a HUVEC monolayer was formed on biotinylated gelatin on 8-well chamber slides as described in methods. Cell were treated with regular medium, untreated or ASA pretreated MDA-MB-231-CM or ASA alone for 48 h. Then Fluorescein-streptavidin was added as indicated in the protocol. Fixation and staining with anti-VE-cadherin (red) and DAPI (blue) were performed. Green fluorescence imaging between cell-cell junction was considered as permeable area and immunofluorescence staining of VE-cadherin (Red) detect cellular junctional integrity. b Permeabilization was quantified by of FITC coupled fluorescein-streptavidin using image J software. The histogram shows the positive staining of fluorescein-streptavidin and the bar diagram represents the quantitation of permeability. Photograph was taken under fluorescence microscope with magnification of X250. Data shows mean ± SD and represents at least 3 independent experiments. For statistical analysis two paired student’s t test was done
Tumor vessel’s hyper-permeability or barrier function is correlated with loss of junctional proteins and enhanced metastasis (Martin and Jiang 2009; Tornavaca et al. 2015; Zhou et al. 2014). Next, we performed western blot analysis to investigate the expression of junctional proteins (like VE-cadherin and ZO-1) under tumor microenvironment and to identify whether aspirin could regulate them. We showed that MDA-MB-231 derived CM significantly decreased expression of tight junction proteins like VE-cadherin and ZO-1 which has been rescued after ASA treatment (Fig. 6).
Fig. 6.

ASA revert the effects of breast cancer derived conditioned medium on expression of VE-cadherin, ZO-1 and VEGF. a–b Differential expression profile of VE-cadherin, ZO-1 and VEGF in different experimental conditions. GAPDH was used as the loading control. Data show mean ± SD and are representative of at least three independent experiments. For statistical analysis two paired student’s t test was done
Aspirin inhibits VEGF expression in HUVEC
VEGF not only induces endothelial cell proliferation, migration and angiogenesis but also it plays a crucial role in increasing vascular permeability by disrupting tight junction protein and thereby triggering tumor metastasis (Azzi et al. 2013). Therefore, we sought to determine whether ASA could regulate breast cancer cell induced VEGF expression. We found that VEGF expression in HUVEC was significantly increased in the presence of MDA-MB-231-CM. ASA pretreatment considerably reduced MDA-MB-231-CM-stimulated VEGF expression in HUVEC (Fig. 6).
Aspirin inhibits in vivo angiogenesis
To investigate whether ASA is equally effective in vivo, we performed an in vivo angiogenesis assay. To test this, we carried out two experiments. First, we presoaked gel foam with regular media (RM), MDA-MB-231-CM or ASA pretreated MDA-MB-231-CM. Respective presoaked gel foam was placed under the mouse skin for 5 days and examined the angiogenesis (Fig. 7a). The studies showed that both RM and CM promote angiogenesis in the pre-soaked gel-foam areas with significantly higher in CM (Fig. 7b–c). However, aberrant blood vessels were formed in the MDA-MB-231-CM pre-soaked gel foam areas, which was lacking in RM. The MDA-MB-231 CM-induced blood vessels were reasonably slugged and leaky as compared to RM-induced blood vessels (Fig. 7b, inset). Interestingly, we found that CM that collected from ASA-treated MDA-MB-231 cell culture was unable to induce aberrant vascularization in the gel-foam areas (Fig. 7c). Thus, these studies suggest that the TNBC cells can promote abnormal angiogenesis via a paracrine signaling pathway, and this effect of TNBC cells can be blocked by treating them with aspirin.
Fig. 7.

ASA prevents tumor cell induced angiogenesis in vivo. a Diagrammatic representation of in vivo gel-foam angiogenesis assay, b Illustrative image of blood vessel formation under mouse skin in the presence or absence of tumor cell derived conditioned media without (middle panel) or with (right panel) ASA treatment. Regular media (RM) was used as control. Note, CM-induced dilated blood vessels with high magnification are shown in the inset. Scale bar, 100 μm, (c–d). Error-bar show the mean numbers of capillary-like structures in different experimental conditions. Data show mean ± SD and are representative of at least five independent animals. Note, ASA was either directly used to treat MDA-MB-231 for CM collection (c) or fed mice through milk (d)
Next, we extended the experiments and explored whether treating mice with aspirin blocks RM or CM-induced blood vessel formation. To do so, RM or CM-presoaked gel foams were implanted under the skin of a mouse. Half of the mice (RM or CM) were fed ASA with the milk, and rest of them were given only milk (controls). After 5 days of ASA treatment, the gel foams were removed, and angiogenesis was measured. We found that ASA significantly impaired the blood vessels formation in ASA-fed mice (Fig. 7d), indicating both RM and CM induced angiogenesis can be regulated by ASA. However, the effect of ASA was less in RM (1.53-fold less) as compared to CM (2.23-fold less). The structural abnormalities associated with CM also minimized in ASA-fed mice (morphological data not included). Collectively, these studies suggest that ASA not only block the TNBC cells’-induced molecular signals required for tumor angiogenesis but it also blocks signaling in endothelial cells that interact with the paracrine signals generated from TNBC cells.
Discussion
We provide evidence that aspirin inhibits cancer cell-induced angiogenesis as well as normalizes tumor vasculature, and thereby we anticipate ASA treatment with appropriate dose could prevent metastasis in breast cancer.
Tumor angiogenesis is a pathophysiological process which not only triggers tumor growth by activating angiogenic factors (Nishida et al. 2006) from tumors cells but also is considered as a critical step in tumor metastasis (Bielenberg and Zetter 2015). There are several lines of thoughts and evidence that structural abnormalities in tumor vasculature play a key role in primary tumor growth and metastasis (Bielenberg and Zetter 2015; Folkman 2002; Mahadevan and Hart 1990). Tumor blood vessels exhibit different abnormal characteristics like immature, uneven vessel diameter, thin-walled, chaotic blood flow, leaky and sluggish with less pericyte coverage (Giverso and Ciarletta 2016; Laitakari et al. 2004). These abnormalities in tumor vasculature contribute to hypoxic conditions due to insufficient oxygen supply which does not allow drugs to pass through and reach the tumor, helping aggressive, uncontrolled tumor growth and metastasis (Liao and Johnson 2007). Therefore, normalization of structural and functional abnormalities of tumor vessels is important for delivering anti-cancer drugs efficiently (Carmeliet and Jain 2011; Jain 2005).
Pericyte plays a crucial role in maintaining blood vessels architecture (Bergers and Song 2005). Evidence showed that aberrant endothelial cells-pericyte network contributes leaky and sluggish blood vessel formation and tumor metastasis (Luk et al. 2012; Raza et al. 2010). Our study showed that ASA treatment promotes recruitment of pericytes from multipotent stem cells in an in vitro model of neovascularization (Fig. 1). Restoration of pericytes by ASA may be attributed by inhibiting tumor-induced cell death of pericytes (Figs. 2 and 3). We further showed that ASA could rescue MDA-MB-231-CM-mediated pericyte loss as seen from the reappearance of α-SMA expression (Fig. 3). Collectively, these studies suggest that ASA may have the potency to recover leaky blood vessels by recruiting pericytes. However further studies are warranted.
Endothelial cell migration is an essential step in normal and pathological angiogenesis including cancer (German et al. 2014; Lamalice et al. 2007). Our studies have shown that ASA inhibits endothelial cell migration in a dose-dependent manner. Moreover, ASA also blocks cancer cell-induced endothelial cell migration. This has been evident from our two independent experiments that ASA impedes MDA-MB-231-CM driven direct as well as the chemotactic migration of endothelial cells (Fig. 4). Thus, we suggest that the inhibition of neovascularization by ASA is mediated through suppressing the migration of endothelial cells.
Vascular permeability is a strictly regulated phenomenon which allows passing different molecules through the vessel. A rapid increase in permeability occurs in tumor vasculature due to loss of barrier function of the vascular wall like abnormal and weakly attached pericytes surrounding the endothelial layer, lack of smooth muscle and iregular basal lamina (Baluk et al. 2003). In addition to these structural features, vascular barrier also depends on functions of intercellular tight junction and adherent proteins. As a consequence of any impairment in these structural and functional barrier, tumor vasculature became leaky and hyperpermeable – leading to decreased blood flow and limits drug penetration and also facilitate distant metastasis by inducing invasion of tumor cells across the blood vessels (Claesson-Welsh 2015). Therefore, the therapeutic management of vascular hyperpermeability or leakiness is in demand to prevent tumor growth and progression. Our studies showed that MDA-MB-231-CM increased vascular permeability with the concomitant decrease of two junctional proteins such as VE-cadherin and ZO-1 expressions, and ASA treatment rescue blood vessel permeability significantly by triggering event causing upregulation of VE-cadherin and ZO-1 (Figs. 5 and 6). Therefore, ASA may be beneficial to regulate vascular barrier function and vascular homeostasis.
Vascular endothelial growth factor (VEGF) has a vital role in many functions in tumor development including vascular permeability (Senger et al. 1983). VEGF is secreted by various cells like the endothelial cells, smooth muscle cells, fibroblast, immune cell and tumor cells, and acts by autocrine-paracrine fashion. It has been reported that VEGF stimulates PAK-dependent VE-cadherin phosphorylation and internalization leading to destabilization of cell-cell contact and increased permeability (Azzi et al. 2013). Here, we show ASA-treatment causes a significant reduction in MDA-MB-231 stimulated VEGF upregulation but not the basal levels. Therefore, we propose that normalization of vascular hyperpermeability or barrier function by ASA may be mediated through regulation of VEGF expression.
To investigate whether ASA effectively regulates tumor angiogenesis in vivo, we performed in vivo experiments. Consistent with in vitro data, we found that ASA significantly blocks TNBC cell-induced aberrant vasculature in vivo model of angiogenesis (Fig. 7). The collective observations suggest that ASA destroys the molecular networking between tumor cells and endothelial cells, which is needed for promoting tumor angiogenesis. However, further studies are warranted.
In conclusion, we show the potency of aspirin as a regulator of tumor vasculature normalization by stabilizing barrier function of tumor blood vessels. Thus, the therapeutic value of aspirin has drawn great attention in breast cancer management not only for its efficacy as cytotoxic (Maity et al. 2015), also for anti-angiogenic or normalizing tumor blood vessels.
Electronic supplementary material
(DOCX 50 kb)
Acknowledgements
We thank the members of Kansas City VA Research Office and Midwest Biomedical Research Foundation Administrative and clerical supports.
Abbreviations
- ASA
Aspirin
- VEGF
Vascular Endothelial Growth Factor
- HUVEC
Human Umbilical Vein Endothelial Cell
- CM
Conditioned Media
Author contributions
Conception and design: S. K. Banerjee, S. Banerjee I. Haque and G. Maity; Development of Methodology: G. Maity and A. Ghosh, I. Haque and S. Banerjee; Acquisition of Data: G. Maity and J. Chakraborty; Analysis and Interpretation of Data: G. Maity, A. Ghosh, J. Chakraborty, S. Banerjee and S.K. Banerjee; Writing and review of the manuscript: G. Maity, J. Chakraborty, S. Banerjee and S. K. Banerjee; Administrative, technical or material support: G. Maity, A. Ghosh and S. K. Banerjee, and Study supervision: S. K. Banerjee.
Funding
The work is supported by Merit review grant from Department of Veterans Affairs (Sushanta K. Banerjee, 5I01BX001989–04 and Snigdha Banerjee, I01BX001002–05), KUMC Lied Basic Science Grant Program (SKB), and Grace Hortense Greenley Trust, directed by The Research Foundation in memory of Eva Lee Caldwell (SKB).
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Compliance with ethical standards
Ethics approval and consent to participate
Compliance with ethical standard of VA Medical Center.
Consent for publication
All the authors of this manuscript have agreed to publish this article.
Competing interests
No potential conflicts of interest were disclosed.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Akhmedkhanov A, Toniolo P, Zeleniuch-Jacquotte A, Kato I, Koenig KL, Shore RE. Aspirin and epithelial ovarian cancer. Prev Med. 2001;33:682–687. doi: 10.1006/pmed.2001.0945. [DOI] [PubMed] [Google Scholar]
- Ararat E, Sahin I, Altundag K. Aspirin intake may prevent metastasis in patients with triple-negative breast cancer. Med Oncol. 2011;28:1308–1310. doi: 10.1007/s12032-010-9636-7. [DOI] [PubMed] [Google Scholar]
- Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211. doi: 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–1815. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardia A, Olson JE, Vachon CM, Lazovich DA, Vierkant RA, Wang AH, Limburg PJ, Anderson KE, Cerhan JR. Effect of aspirin and other NSAIDs on postmenopausal breast cancer incidence by hormone receptor status: results from a prospective cohort study. Breast Cancer Res Treat. 2011;126:149–155. doi: 10.1007/s10549-010-1074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015;21:267–273. doi: 10.1097/PPO.0000000000000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosetti C, Rosato V, Gallus S, Cuzick J, la Vecchia C. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23:1403–1415. doi: 10.1093/annonc/mds113. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- Claesson-Welsh L. Vascular permeability--the essentials. Ups J Med Sci. 2015;120:135–143. doi: 10.3109/03009734.2015.1064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke VG, LeBleu VS, Keskin D, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. 2012;21:66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Dhar K, Maity G, Sarkar S, Ghosh A, Haque I, Dhar G, Banerjee S, Banerjee SK. Deficiency of CCN5/WISP-2-driven program in breast cancer promotes Cancer epithelial cells to mesenchymal stem cells and breast Cancer growth. Sci Rep. 2017;7:1220. doi: 10.1038/s41598-017-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Dhar K, Dhar G, Majumder M, Haque I, Mehta S, van Veldhuizen PJ, Banerjee SK, Banerjee S. Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1. Mol Cancer. 2010;9:209. doi: 10.1186/1476-4598-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Din FV, Dunlop MG, Stark LA. Evidence for colorectal cancer cell specificity of aspirin effects on NF kappa B signalling and apoptosis. Br J Cancer. 2004;91:381–388. doi: 10.1038/sj.bjc.6601913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- German AE, Mammoto T, Jiang E, Ingber DE, Mammoto A. Paxillin controls endothelial cell migration and tumor angiogenesis by altering neuropilin 2 expression. J Cell Sci. 2014;127:1672–1683. doi: 10.1242/jcs.132316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giverso C, Ciarletta P. Tumour angiogenesis as a chemo-mechanical surface instability. Sci Rep. 2016;6:22610. doi: 10.1038/srep22610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lichtenberger LM, Taylor M, Bottsford-Miller JN, Haemmerle M, Wagner MJ, Lyons Y, Pradeep S, Hu W, Previs RA, Hansen JM, Fang D, Dorniak PL, Filant J, Dial EJ, Shen F, Hatakeyama H, Sood AK. Antitumor and antiangiogenic effects of aspirin-PC in ovarian Cancer. Mol Cancer Ther. 2016;15:2894–2904. doi: 10.1158/1535-7163.MCT-16-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Laitakari J, Nayha V, Stenback F. Size, shape, structure, and direction of angiogenesis in laryngeal tumour development. J Clin Pathol. 2004;57:394–401. doi: 10.1136/jcp.2002.004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- Longatto Filho Adhemar, Lopes José Manuel, Schmitt Fernando C. Angiogenesis and Breast Cancer. Journal of Oncology. 2010;2010:1–7. doi: 10.1155/2010/576384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K, Boatman S, Johnson KN, et al. Influence of morphine on pericyte-endothelial interaction: implications for antiangiogenic therapy. J Oncol. 2012;2012:458385. doi: 10.1155/2012/458385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Hart IR. Metastasis and angiogenesis. Acta Oncol. 1990;29:97–103. doi: 10.3109/02841869009089997. [DOI] [PubMed] [Google Scholar]
- Maity G, Mehta S, Haque I, et al. Pancreatic tumor cell secreted CCN1/Cyr61 promotes endothelial cell migration and aberrant neovascularization. Sci Rep. 2014;4:4995. doi: 10.1038/srep04995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity G, De A, Das A, et al. Aspirin blocks growth of breast tumor cells and tumor-initiating cells and induces reprogramming factors of mesenchymal to epithelial transition. Lab Investig. 2015;95:702–717. doi: 10.1038/labinvest.2015.49. [DOI] [PubMed] [Google Scholar]
- Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788:872–891. doi: 10.1016/j.bbamem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Martin DN, Boersma BJ, Yi M, Reimers M, Howe TM, Yfantis HG, Tsai YC, Williams EH, Lee DH, Stephens RM, Weissman AM, Ambs S. Differences in the tumor microenvironment between African-American and European-American breast cancer patients. PLoS One. 2009;4:e4531. doi: 10.1371/journal.pone.0004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Wang X, Khaidakov M, Ding Z, Ayyadevera S, Hearnsberger E, Goyal T, Mehta JL. Aspirin downregulates angiotensin type 1 receptor transcription implications in capillary formation from endothelial cells. J Cardiovasc Pharmacol. 2012;60:187–192. doi: 10.1097/FJC.0b013e31825b61e2. [DOI] [PubMed] [Google Scholar]
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–219. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol. 2010;85:593–598. doi: 10.1002/ajh.21745. [DOI] [PubMed] [Google Scholar]
- Ribeiro AL, Okamoto OK. Combined effects of pericytes in the tumor microenvironment. Stem Cells Int. 2015;2015:868475. doi: 10.1155/2015/868475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ghosh A, Banerjee S, Maity G, Das A, Larson MA, Gupta V, Haque I, Tawfik O, Banerjee SK. CCN5/WISP-2 restores ER- proportional, variant in normal and neoplastic breast cells and sensitizes triple negative breast cancer cells to tamoxifen. Oncogenesis. 2017;6:e340. doi: 10.1038/oncsis.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- Sennino B, Falcon BL, McCauley D, le T, McCauley T, Kurz JC, Haskell A, Epstein DM, McDonald DM. Sequential loss of tumor vessel pericytes and endothelial cells after inhibition of platelet-derived growth factor B by selective aptamer AX102. Cancer Res. 2007;67:7358–7367. doi: 10.1158/0008-5472.CAN-07-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821–838. doi: 10.1083/jcb.201404140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- Xian X, Hakansson J, Stahlberg A, et al. Pericytes limit tumor cell metastasis. J Clin Invest. 2006;116:642–651. doi: 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang Z, Wang Z, et al. Impact of acetylsalicylic acid on tumor angiogenesis and lymphangiogenesis through inhibition of VEGF signaling in a murine sarcoma model. Oncol Rep. 2013;29:1907–1913. doi: 10.3892/or.2013.2339. [DOI] [PubMed] [Google Scholar]
- Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O'Connor ST, Chin AR, Yen Y, Wang Y, Marcusson EG, Chu P, Wu J, Wu X, Li AX, Li Z, Gao H, Ren X, Boldin MP, Lin PC, Wang SE. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 50 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article.