

Abstract
Background and purpose
Hypomyelinating leukodystrophies are a heterogeneous group of genetic disorders with a wide spectrum of phenotypes and a high rate of genetically unsolved cases. Bi‐allelic mutations in NKX6‐2 were recently linked to spastic ataxia 8 with hypomyelinating leukodystrophy.
Methods
Using a combination of homozygosity mapping, exome sequencing, and detailed clinical and neuroimaging assessment a series of new NKX6‐2 mutations in a multicentre setting is described. Then, all reported NKX6‐2 mutations and those identified in this study were combined and an in‐depth analysis of NKX6‐2‐related disease spectrum was provided.
Results
Eleven new cases from eight families of different ethnic backgrounds carrying compound heterozygous and homozygous pathogenic variants in NKX6‐2 were identified, evidencing a high NKX6‐2 mutation burden in the hypomyelinating leukodystrophy disease spectrum. Our data reveal a phenotype spectrum with neonatal onset, global psychomotor delay and worse prognosis at the severe end and a childhood onset with mainly motor phenotype at the milder end. The phenotypic and neuroimaging expression in NKX6‐2 is described and it is shown that phenotypes with epilepsy in the absence of overt hypomyelination and diffuse hypomyelination without seizures can occur.
Conclusions
NKX6‐2 mutations should be considered in patients with autosomal recessive, very early onset of nystagmus, cerebellar ataxia with hypotonia that rapidly progresses to spasticity, particularly when associated with neuroimaging signs of hypomyelination. Therefore, it is recommended that NXK6‐2 should be included in hypomyelinating leukodystrophy and spastic ataxia diagnostic panels.
Keywords: hypomyelination, leukodystrophy, NKX6‐2, spastic ataxia 8, SPAX8
Introduction
Hypomyelinating leukodystrophies are a heterogeneous group of genetic disorders with a wide spectrum of phenotypes. Given that myelination is a highly regulated process these disorders usually result from genetic abnormalities. However, the majority of individuals with hypomyelinating disorders have no genetic diagnosis 1.
Hypomyelination can result from dysfunctions in myelin generation or maintenance pathways including mutations in the myelin proteins (PLP1), protein translation (POLR3A, POLR3B, POLR1C) and gap junction proteins linking astrocytes and oligodendrocytes (GJC2).
A new phenotype associated with bi‐allelic mutations in NKX6‐2 leading to spastic ataxia 8 (SPAX8), autosomal recessive, with hypomyelinating leukodystrophy (OMIM: 617560) has recently been described by our group 2. The reported NKX6‐2 mutations were bi‐allelic truncating or located in the highly conserved homeobox domain. Clinically, they presented with early onset spastic ataxia or hypotonia progressing to severe spasticity within a few months and were associated with hypomyelination 2.
Here, a large, ethnically diverse cohort is presented, describing comprehensively an expanding clinical and neuroimaging syndrome and a large genotypic spectrum of NKX6‐2‐related disease.
Methods
The study included affected individuals with spastic ataxia and hypomyelination from unrelated families of different ethnic backgrounds. Families were recruited under Institutional Review Board/ethics‐approved research protocols (UCLH: 04/N034) with informed consent. For comprehensive genotype–phenotype analyses, all reported genetically diagnosed NKX6‐2 mutations were included. Extended methods are given in Appendix S1.
Results
Genotype spectrum in NKX6‐2‐related disease
In this study, 11 new cases from eight families (Fig. 1) carrying pathogenic variants in NKX6‐2 were identified. Eight distinct mutations were found including four novel variants (Fig. 2a). One was present in gnomAD with very low allele frequency in the heterozygous state (MAF 0.0001170) but absent as homozygous (c.541C>G) (Fig. 2b), and three were known pathogenic variants. The c.196delC identified in families IV and V was present in a shared homozygous region (Fig. 2c). All missense variants were located in conserved amino acid positions (Fig. 2d).
Figure 1.
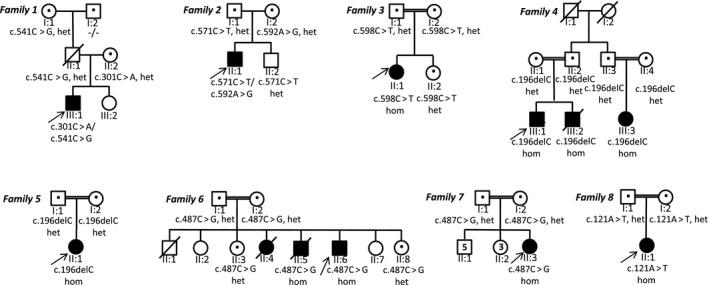
Family trees in all new families reported in this study. het, heterozygous; hom, homozygous; the individuals tested in this study are indicated with a dot.
Figure 2.
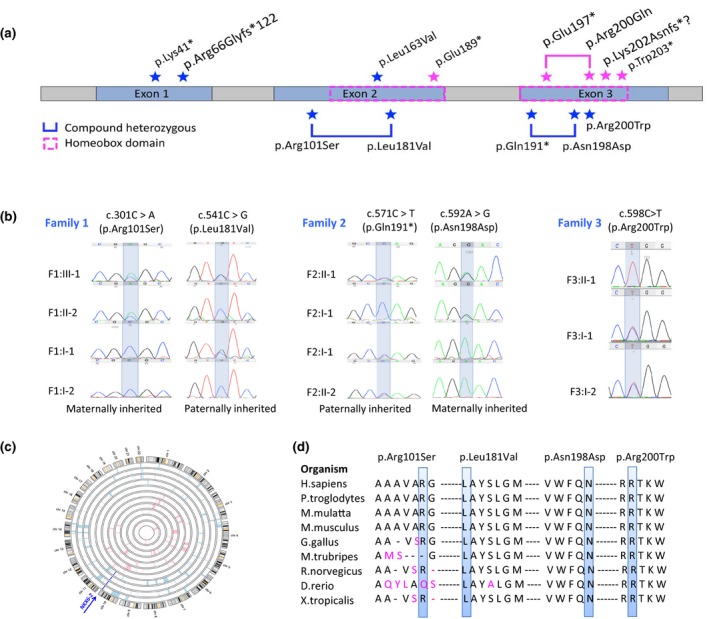
Mutation spectrum of NKX6‐2‐related disease. (a) NKX6‐2 gene with all the mutations identified. All known and novel mutations identified in our cohort are labelled with a blue star; all mutations previously reported are labelled with a magenta star and plotted on top of the gene. (b) Sanger sequencing confirmation with segregation analysis for novel NKX6‐2 variants reported in this study. (c) Homozygosity mapping in family IV and V identified a homozygous region on chromosome 10, shared by affected individuals and containing the pathogenic homozygous variant c.196delC in NKX6‐2. (d) Conservation across species of each novel missense mutation reported in this study.
Our analysis was extended to include 33 individuals from 21 families carrying NKX6‐2 mutations identified in this study and previously reported 2, 3, 4, 5 (Table 1, Appendices S2 and S3). So far, 13 distinct NKX6‐2 variants have been linked to SPAX8 disease. Several mutations (c.121A>T, c.487C>G, c.196delC) were reported in multiple families. The c.196delC and c.487C>G were identified in five families each, all originally from the Middle East. Haplotype analysis from three families confirmed that c.196delC was a founder mutation. However, the c.487C>G carriers did not share the same haplotype, the mutation arising recurrently 5. The c.121A>T was identified in three families of Indian origin. Haplotype analysis data from two of these families confirmed a founder effect 2.
Table 1.
Variant description of all NXK6‐2 mutations reported to date
| Zygosity | cDNA change | Amino acid change | Type of mutation | Novel/known | ACMG score | ACMG classification | Onset | Phenotype | Additional signs reported | Hypomyelination | Cerebellar atrophy | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound heterozygous | c.301C>A | p.Arg101Ser | Missense | Novel | PM2, PM3, PP1, PP3 | Likely pathogenic | Childhood | Predominantly motor delay | Seizures | Mild | Yes | This study |
| c.541C>G | p.Leu181Val | Missense | Known (rs369901030) (MAF = 0.0001170) | PM1, PM3, PP1, PP3 | Likely pathogenic | This study | ||||||
| Compound heterozygous | c.571C>T | p.Gln191* | Nonsense | Novel | PVS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | No | Diffuse, severe | No | This study |
| c.592A>G | p.Asn198Asp | Missense | Novel | PM1, PM2, PM3, PP1 | Likely pathogenic | This study | ||||||
| Homozygous | c.598C>T | p.Arg200Trp | Missense | Novel | PS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | No | Diffuse, severe | Yes | This study |
| Homozygous | c.121A>T | p.Lys41* | Nonsense | Known | PVS1, PS3, PM2, PM3, PP1 | Pathogenic | Childhood | Predominantly motor delay | Dystonia. Limitation of eye movements | Diffuse, severe | Yes | This study, Chelban et al. 2 |
| Homozygous | c.196delC | p.Arg66Glyfs*122 | Frameshift | Known | PVS1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | Limitation of eye movements, hearing impairment. Gastrostomy for severe dysphagia. Scoliosis | Diffuse, severe | No | This study, Anazi et al. 4, Baldi et al. 5 |
| Homozygous | c.487C>G | p.Leu163Val | Missense | Known | PM1, PM2, PM3, PP1 | Likely pathogenic | Neonatal | Severe global psychomotor delay | Seizures. Gastrostomy for severe dysphagia | Variable. 2 cases reported with no hypomyelination | Yes | This study, Chelban et al. 2, Baldi et al. 5 |
| Homozygous | c.565G>T | p.Glu189* | Nonsense | Known | PVS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | Severe dystonia | Diffuse, severe | Yes | Dorboz et al. 3 |
| Compound heterozygous | c.589C>T | p.Gln197* | Nonsense | Known | PVS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | Swallowing difficulties. Poor visual acuity | Diffuse, severe | No | Dorboz et al. 3 |
| c.599G>A | p.Arg200Gln | Missense | Known | PM1, PM2, PM3, PP1 | Likely pathogenic | |||||||
| Homozygous | c.606delinsTA | p.Lys202Asnfs?1 | Frameshift | Known | PVS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | Severe dystonia | Diffuse, severe | Yes | Dorboz et al. 3 |
| Homozygous | c.608G>A | p.Trp203* | Nonsense | Known | PVS1, PM1, PM2, PM3, PP1 | Pathogenic | Neonatal | Severe global psychomotor delay | Seizures | Diffuse, severe | No | Baldi et al. 5 |
ACMG, The American College of Medical Genetics and Genomics.
With one exception (p.Arg101Ser), all missense mutations affected the homeobox domain. To establish the deleterious effect of p.Arg101Ser variant, reverse transcription polymerase chain reaction (RT‐PCR) and western blot were performed. Control RT‐PCR for glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) confirmed the presence of cDNA in all samples. In the patient, NKX6‐2 cDNA was severely reduced compared to controls (Fig. 3a). Immunoblot analysis confirmed a significant reduction in NKX6‐2 protein levels in the patient compared to controls (Fig. 3b, c, Appendix S4).
Figure 3.
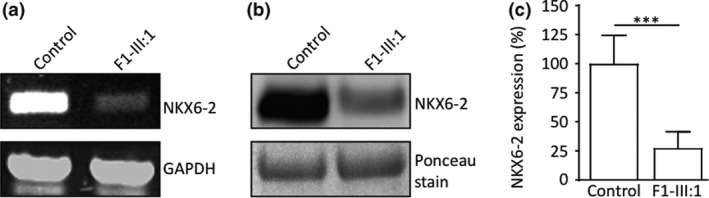
Functional analyses and pathogenicity of variants identified in family 1. (a) Reverse transcription polymerase chain reaction in compound heterozygous missense case F1‐III:1 showed absent or severely reduced NKX6‐2 compared to control; glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (b) Reduced NKX6‐2 protein levels confirmed by western blot in individual F1‐III:1. Total protein lysate extracted from human fibroblasts assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis and analysed by western blotting using anti‐NKX6‐2 antibody (left panel). (c) Densitometry analysis shows significant reduction in NKX6‐2 protein levels in fibroblasts harbouring the NKX6‐2 missense mutation compared to control cells. ***P < 0.01, replicate values, mean and SD are shown; one‐way anova with Bonferroni post hoc test.
The majority of reported cases (81.8%, 27/33) presented in the first year of life, half of these (14/27) as neonates. Whether the mutation type influenced the age of onset was analysed. Sixteen cases carried two truncating alleles, 13 had bi‐allelic missense mutations and four had compound heterozygous variants including one truncating mutation. Although an earlier mean onset age was observed in the group harbouring two truncating alleles versus the group with two missense alleles (7.3 months vs. 8.3 months), the difference was not statistically significant (P = 0.78).
Phenotype spectrum in NKX6‐2‐related disease
Assessment of the age of onset and disease severity revealed two ends of an expanding phenotype spectrum of NKX6‐2 mutations.
Neonatal and very early onset
Neonatal onset was associated with a higher rate of severe global psychomotor disability compared with onset after 1 month of age (P = 0.05) (Fig. 4a). Twenty cases with information on motor milestones showed very severe motor deficit, all children failing to achieve independent ambulation and 70% (14/20) failing to achieve head control. Furthermore, 90% of children with disease onset before 1 year never achieved verbal milestones/meaningful speech.
Figure 4.
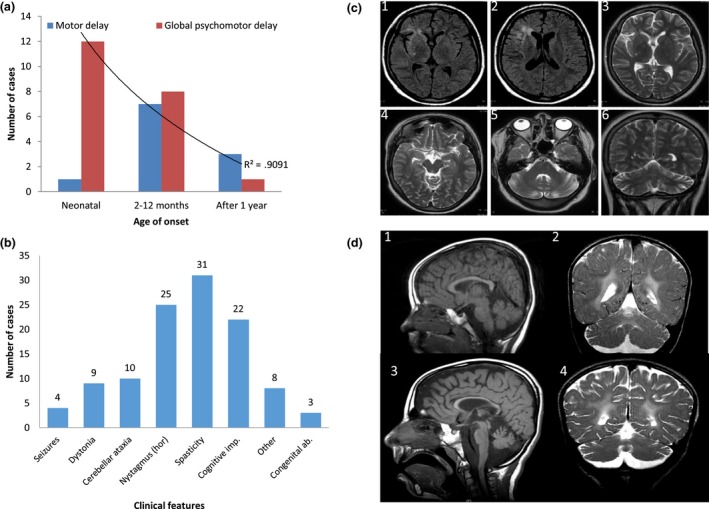
Genetype–phenotype correlation and neuroimaging spectrum of NKX6‐2 mutations. (a) The neonatal‐onset group has a statistically significant higher frequency of global psychomotor developmental delay (red) compared with the other two groups (onset from 2 months to 1 year and onset after 1 year). Childhood onset is associated with predominantly motor delay (blue). P = 0.05, r 2 = 0.9. (b) Clinical features associated with NKX6‐2 mutations (the horizontal axis) with the number of cases on the vertical axis (total n = 33). hor, horizontal gaze‐evoked nystagmus. (c) Fluid‐attenuated inversion recovery and T2‐weighted MRI acquisitions from case F1‐III:1 exhibiting T2 hyperintense signal change in periventricular WM surrounding the frontal horn of the right lateral ventricle, and frontal and temporal opercular and subinsular WM T2‐weighted hyperintense signal change associated with a degree of cortical volume loss. Note the normal signal intensity of the globi pallidi, thalami and external capsules, mesencephalon and pons. There is disproportionate cerebellar volume loss with mild T2‐weighted hyperintense signal change in the peri‐dentate WM. (d) Longitudinal MRI in case F7‐II:3 at ages of 4 years (D1, D2) and 8½ years (D3, D4) (D1, D3, mid‐sagittal T1‐weighted; D2, D4, coronal T2‐weighted) showing progressive thinning of the corpus callosum and cerebellar atrophy associated with WM abnormality sparing the U fibres (2, 4). There is progressive enlargement of the cortical sulci and extra axial cerebrospinal fluid spaces indicating underlying global brain atrophy.
Complex medical needs included severe dysphagia requiring gastrostomy (6/33 cases), congenital heart disease (1/33), respiratory failure (2/33) leading to death, undescended testicles (1/33), severe dental and/or gum abnormalities (6/33 cases) and inguinal hernia (1/33). Survival in this group was shorter with 12.1% (4/33) of children dying in their first 5 years of life.
Childhood onset
Childhood onset with predominantly motor delay and complex spastic ataxia was identified in seven individuals in this study and those previously reported 2. Case F1‐III:1 has the least severe motor phenotype in our series (Appendix S5). Case F8‐II:1 has a phenotype resembling the previously reported cases with the c.121A>T mutation – severe spastic ataxia but relatively preserved cognition 2. The seven cases all achieved ambulation, although they required walking aids (walking frame) 1–3 years into the disease and wheelchair 3–8 years later. Features including head titubation and severe dystonia in the previously reported patients who achieved adulthood suggest that they may be related to disease progression rather than genotype.
Complex spastic ataxia and developmental delay
The most common symptom at onset was nystagmus (25/33 cases) (Fig. 4b) described as horizontal gaze‐evoked in the majority of cases. Other ocular manifestations were square wave jerks, hypometric saccades, impaired smooth pursuit and reduced visual acuities (four cases). Limitation of bilateral, lateral gaze eye movements was seen later in three cases who reached adulthood.
Spasticity with brisk reflexes in the upper and lower limbs and upgoing plantar responses were present in all cases. Axial hypotonia was reported at presentation in nine patients, all with onset of disease before 1 year of age associated with upper and lower limb spasticity soon after presentation. Cervical and/or limb dystonia was present in around 40% of cases where information was available. Examination of the peripheral nervous system was normal in all cases and normal nerve conduction studies were reported in two cases who reached adulthood.
Seizures were present in four (12.5%) patients harbouring NKX6‐2 mutations. Case F1‐III:1 presented primary tonic progressing to secondarily generalized seizure at 6 years. Electroencephalography at the age of 13 showed loss of age‐based background activity and absent anterior–posterior gradient of background activity with focal tonic seizure presenting clinically as hemifacial seizures. There were intermittent frequency slowing (most pronounced at frontal electrodes), multifocal epileptic discharges (sharp waves and sharp‐slow waves) pronounced over the right hemisphere, and several focal tonic seizures. The seizures were multidrug‐resistant. No details regarding seizure phenomenology are available in the other three cases 3, 5.
Cognitive function varied greatly between patients. Interestingly, two cases reported here (F1‐III:1 and F8‐II:1) and four previously reported 2 (total 6/33) had normal cognitive development for their age. Case F8‐II:1 and the four previously reported cases with normal cognitive development carry the homozygous nonsense mutation p.Lys41* whilst F1‐III:1 carries two missense compound heterozygous variants (p.Arg101Ser, p.Leu181Val). However, severe cognitive impairment with arrested speech development was present in 66% of all children with bi‐allelic NKX6‐2 pathogenic variants and in all reported children with neonatal onset. It is acknowledged that, in most cases, an accurate cognitive function assessment was difficult due to severe motor impairment and/or developmental language delay.
Other features present in SPAX8 patients included strabismus (4/33 patients), scoliosis (6/33), neck or/and limb dystonia (9/33), contractures (4/33 patients), dysmorphism (2/33), hip dislocation (2/33) and single cases of hearing impairment and hirsutism.
Neuroimaging spectrum in NKX6‐2‐related disease
The key neuroimaging feature in the majority of cases was a hypomyelinating leukodystrophy. Magnetic resonance imaging (MRI) (Appendix S6) showed signal abnormality with atrophy noted supratentorially within the thalami and the globus pallidi. Infratentorially, there was notable involvement of the pons with signal abnormality involving the transverse pontine fibres with relative expansion to the entire pons. This contrasted with the orthogonal oriented fibres of the corticospinal tracts of relatively normal signal, providing a distinctive prominent appearance of the mid‐pons on the axial T2‐weighted sequences. Furthermore, signal change was noted in the cerebellar hemispheres, particularly involving the subcortical white matter (WM) and the dentate nuclei. Cerebellar volume was relatively increased in very young patients, probably related to the underlying WM changes, and demonstrated mild atrophic change over time. Not all children developed cerebellar atrophy (Appendix S7).
Interestingly, in case F1‐III:1 with two missense compound heterozygous mutations neuroimaging findings were milder compared to cases carrying homozygous truncation mutations (Fig. 4c). Marked cerebellar atrophy was a key finding in this case. Furthermore, two other paediatric NKX6‐2‐related cases were reported previously without overt hypomyelination 4. Thinning of the corpus callosum was present in 6/33 patients. A longitudinal MRI study in F7‐II:3 at 4 and 8 years old shows progressive thinning of the corpus callosum and cerebellar atrophy, associated with WM abnormality sparing the U fibres (Fig. 4d).
Discussion
Recently the first cases of NKX6‐2 mutations leading to the hypomyelination and spastic ataxia phenotype in humans were described 2. Given the rarity of hypomyelinating disorders and the absence of an unbiased cohort to screen, it is difficult to estimate the frequency of NKX6‐2 mutations. However, genetic analysis of the University College London (UCL) leukodystrophies cohort identified 10 cases of hypomyelination with pathogenic mutations in PLP1 (four families) and single cases of TUBB4A, POLR3A/B and CLCN2 6. Here are presented six additional cases (three families) of hypomyelination due to NKX6‐2 mutations from the same research group suggesting a significant burden of NKX6‐2 mutations.
In this study, the phenotypic spectrum was expanded and it was shown that NKX6‐2 mutations lead to a neonatal onset at the severe end and childhood onset at the milder end. Interestingly, the compound heterozygous missense variants c.301C>A (p.Arg101Ser) and c.541C>G (p.Leu181Val) led to a significant reduction (>70%) of the NKX6‐2 protein. It is possible that the small amount of NKX6‐2 protein identified by western blot provided a degree of myelination leading to a less severe clinical picture. This individual and the three cases previously published 5 presented with multidrug‐resistant epilepsy. In focal cortical dysplasia, a common cause of drug‐resistant epilepsy, hypomyelination abnormality was confirmed in numerous histopathological epilepsy surgery specimen studies 7. Focal dysplasia is currently linked to abnormalities in the differentiation of glial cells from their progenitors and their migration to the cortical place 8, a process regulated by transcription factors including NKX6‐2 9.
Additional clinical features associated with NKX6‐2 mutations – cervical and/or limb dystonia, congenital abnormalities (congenital heart disease, undescended testes), severe dental and/or gum abnormalities – reflect the developmental role of NKX6‐2 as a member of the homeobox gene family 10. These genes are involved in development, specifying geographical orientation of the body by directing the formation of limbs and organs 11. Some homeobox genes such as PAX6 12, OTX2 13 and MEOX1 14 are involved in a variety of developmental and neurological disorders with brain abnormalities.
Clinical and neuroimaging findings in NKX6‐2‐related disease reflect the involvement of WM and pyramidal tracts (spasticity, brisk reflexes, upgoing plantars), cerebellum (truncal and limb ataxia, nystagmus) and bulbar function (dysarthria, dysphagia). Complications related to these clinical manifestations led to significant impairment in vital functions including swallowing (although dysphagia was not routinely assessed in all cases, severe dysphagia requiring gastrostomy was reported in several cases), respiration and functionally disabling spasticity, similarly to other myelin‐related diseases 15. Early screening and recognition of these disease‐related complications are important aspects during the clinical assessments of these patients.
Furthermore, our study expands the MRI spectrum of NKX6‐2 mutations from diffuse hypomyelination to focal T2‐weighted hyperintensity and parenchymal volume loss. Recently, other hypomyelinating leukodystrophy genes such as TUBB4A and POLR3A have been shown to have distinct phenotypes and neuroimaging spectrum, ranging from spastic paraplegia to spastic ataxia without overt hypomyelination to hypomyelinating leukodystrophies resulting from different mutations 16, 17. A similar pattern was identified in NKX6‐2 with cases presenting clinically with spastic ataxia in the absence of hypomyelination and a reduction of NKX6‐2 protein levels (case F1‐III:1) compared to extended hypomyelination in cases with truncating mutations resulting in no expression of NKX6‐2 protein, as previously reported 2. Therefore, the importance of molecular diagnosis with additional functional work and confirmatory evidence is highlighted.
In conclusion, it is shown that the phenotypic and neuroimaging expression in NKX6‐2 mutations can range from a complex, neonatal onset at the severe end and a childhood onset at the milder end of the spectrum and that phenotypes with epilepsy in the absence of overt hypomyelination, and diffuse hypomyelination without seizures, can occur. It is recommended that NXK6‐2 should be included in hypomyelinating leukodystrophy and spastic ataxia diagnostic panels.
Acknowledgements
The authors would like to thank the patients and their families for their essential help with this work. The authors are grateful to the UK HSP Society. This study was supported by the Spastic Paraplegia Foundation, the Medical Research Council (MRC UK MR/J004758/1, G0802760, G1001253), the Wellcome Trust in equipment and strategic award (Synaptopathies) funding (WT093205MA and WT104033/Z/14/Z), Ataxia UK, the British Neurological Surveillance Unit (BNSU) and the National Institute for Health Research (NIHR). The Sequencing and Genotyping Core Facilities at KFSHRC are also thanked for their technical help. N.K. is supported by KACST Grant (14‐MED2007‐20) and KFSHRC seed grant (RAC#2120022). I.D. receives support from the NIHR UCL/UCLH Biomedical Research Centre. Supported in part by Doris Duke Clinical Scientist Development Award 2014112 MCK and NIH NINDS NS083739 (MCK). M.A.S. was supported by the Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia, through the research group project number RGP‐VPP‐301.
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
Supporting information
Appendix S1. Extended methods.
Appendix S2. Genotype–phenotype description of all NKX6‐2 mutations reported to date. Legend: NA‐not available, m‐months, y‐years, VEP‐visual evoked potentials, ERG‐electroretinogram, BAEP = brainstem auditory evoked potential, PDA = Patent Ductus Arteriosus.
Appendix S3. Pathogenicity and novelty assessment of all NKX6‐2 variants identified in this study.
Appendix S4. Western blot analysis in individual F1‐III:1. Experiments performed three times and three lysates from case and controls are shown on the blot.
Appendix S5. Genotype–phenotype description of the eight new families reported in this study.
Appendix S6. Hypomyelination in NKX6‐2‐related disease. From left to right: multiple T1‐weighted (column 1) and T2‐weighted (columns 2–5) MRI acquisitions through four cases (top to bottom rows: F4‐III:1, F3‐II:1, F6‐II:6, F2‐II:1). Normal to hyperintense T1 white matter (WM) signal (column 1) in areas corresponding to the T2‐weighted hyperintense signal (column 2) confirmed hypomyelination. Column 2 demonstrates diffuse T2‐weighted hyperintense signal change in subcortical, deep WM including external capsules, globi pallidi and thalami. Columns 3 and 4 demonstrate dorsal mesencephalic and diffuse pontine T2‐weighted hyperintense signal change. Column 5 demonstrates diffuse cerebellar WM T2‐weighted hyperintense signal change including the peri‐dentate WM with relative preservation of cerebellar volume.
Appendix S7. Neuroimaging spectrum of NKX6‐2‐related disease.
Contributor Information
V. Chelban, Email: v.chelban@ucl.ac.uk.
N. Kaya, Email: nkaya@kfshrc.edu.sa.
References
- 1. Vanderver A, Simons C, Helman G, et al Whole exome sequencing in patients with white matter abnormalities. Ann Neurol 2016; 79: 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chelban V, Patel N, Vandrovcova J, et al Mutations in NKX6‐2 cause progressive spastic ataxia and hypomyelination. Am J Hum Genet 2017; 100: 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dorboz I, Aiello C, Simons C, et al Biallelic mutations in the homeodomain of NKX6‐2 underlie a severe hypomyelinating leukodystrophy. Brain 2017; 140: 2550–2556. [DOI] [PubMed] [Google Scholar]
- 4. Anazi S, Maddirevula S, Salpietro V, et al Expanding the genetic heterogeneity of intellectual disability. Hum Genet 2017; 136: 1419–1429. [DOI] [PubMed] [Google Scholar]
- 5. Baldi C, Bertoli‐Avella AM, Al‐Sannaa N, et al Expanding the clinical and genetic spectra of NKX6‐2‐related disorder. Clin Genet 2018; 93: 1087–1092. [DOI] [PubMed] [Google Scholar]
- 6. Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, et al Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017; 140: 1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blumcke I, Thom M, Aronica E, et al The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011; 52: 158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cepeda C, André VM, Levine MS, et al Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav 2006; 9: 219–235. [DOI] [PubMed] [Google Scholar]
- 9. Briscoe J, Pierani A, Jessell TM, Ericson J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 2000; 101: 435–445. [DOI] [PubMed] [Google Scholar]
- 10. Zhong YF, Holland PW. The dynamics of vertebrate homeobox gene evolution: gain and loss of genes in mouse and human lineages. BMC Evol Biol 2011; 11: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat Rev Genet 2005; 6: 893–904. [DOI] [PubMed] [Google Scholar]
- 12. Azuma N, Yamaguchi Y, Handa H, et al Mutations of the PAX6 gene detected in patients with a variety of optic‐nerve malformations. Am J Hum Genet 2003; 72: 1565–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ragge NK, Brown AG, Poloschek CM, et al Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 2005; 76: 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, Alkuraya FS. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel–Feil anomaly. Am J Hum Genet 2013; 92: 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Haren K, Bonkowsky JL, Bernard G, et al Consensus statement on preventive and symptomatic care of leukodystrophy patients. Mol Genet Metab 2015; 114: 516–526. [DOI] [PubMed] [Google Scholar]
- 16. Curiel J, Rodríguez Bey G, Takanohashi A, et al TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum Mol Genet 2017; 26: 4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. La Piana R, Cayami FK, Tran LT, et al Diffuse hypomyelination is not obligate for POLR3‐related disorders. Neurology 2016; 86: 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Extended methods.
Appendix S2. Genotype–phenotype description of all NKX6‐2 mutations reported to date. Legend: NA‐not available, m‐months, y‐years, VEP‐visual evoked potentials, ERG‐electroretinogram, BAEP = brainstem auditory evoked potential, PDA = Patent Ductus Arteriosus.
Appendix S3. Pathogenicity and novelty assessment of all NKX6‐2 variants identified in this study.
Appendix S4. Western blot analysis in individual F1‐III:1. Experiments performed three times and three lysates from case and controls are shown on the blot.
Appendix S5. Genotype–phenotype description of the eight new families reported in this study.
Appendix S6. Hypomyelination in NKX6‐2‐related disease. From left to right: multiple T1‐weighted (column 1) and T2‐weighted (columns 2–5) MRI acquisitions through four cases (top to bottom rows: F4‐III:1, F3‐II:1, F6‐II:6, F2‐II:1). Normal to hyperintense T1 white matter (WM) signal (column 1) in areas corresponding to the T2‐weighted hyperintense signal (column 2) confirmed hypomyelination. Column 2 demonstrates diffuse T2‐weighted hyperintense signal change in subcortical, deep WM including external capsules, globi pallidi and thalami. Columns 3 and 4 demonstrate dorsal mesencephalic and diffuse pontine T2‐weighted hyperintense signal change. Column 5 demonstrates diffuse cerebellar WM T2‐weighted hyperintense signal change including the peri‐dentate WM with relative preservation of cerebellar volume.
Appendix S7. Neuroimaging spectrum of NKX6‐2‐related disease.