

Abstract
Cognitive dysfunction in individuals with schizophrenia is thought to reflect, at least in part, altered levels of excitatory and inhibitory neurotransmission in the dorsolateral prefrontal cortex (DLPFC). Studies of the postmortem human brain allow for interrogation of the disease-related alterations in markers of excitatory and inhibitory neurotransmission at different levels of anatomical resolution. Here, we re-analyzed six published datasets from postmortem studies of schizophrenia to assess molecular markers of glutamate and GABA neurotransmission in the DLPFC at three levels of anatomical resolution: 1) total cortical gray matter, 2) gray matter restricted to layer 3, and 3) a layer 3 local circuit composed of excitatory pyramidal cells and inhibitory, parvalbumin-containing, GABA neurons. We formulated composite measures of glutamate and GABA neurotransmission from z-scores of key transcripts that regulate these functions. Relative to unaffected comparison subjects, the composite glutamate measure was higher in schizophrenia subjects in total gray matter homogenates but lower in samples restricted to layer 3 or the layer 3 local circuit. The composite index of GABA neurotransmission did not differ between subject groups in total gray matter homogenates but was lower in schizophrenia subjects in layer 3 and lower still in the local layer 3 circuit. These findings suggest that the balance of excitation and inhibition in the DLPFC of schizophrenia subjects differs depending on the level of anatomical resolution studied, highlighting the importance of layer- and cell type-specific studies to understand disease-related alterations in cortical circuitry.
Keywords: schizophrenia, E/I balance, layer 3, prefrontal cortex, GABA, glutamate
1. Introduction
Cognitive dysfunction, such as working memory impairments, in individuals with schizophrenia are thought to reflect, at least in part, imbalances between excitatory and inhibitory elements of neural circuitry in the dorsolateral prefrontal cortex (DLPFC) (Foss-Feig et al., 2017; Hoftman et al., 2017; Krystal et al., 2017). For example, working memory performance and its neural correlate in the DLPFC, gamma oscillation power, are both lower in subjects with schizophrenia (Chen et al., 2014; Cho et al., 2006; Minzenberg et al., 2010; Senkowski and Gallinat, 2015). As gamma oscillations are generated by a local circuit composed of excitatory, glutamatergic pyramidal neurons and inhibitory, GABAergic neurons in DLPFC layer 3 (Bartos et al., 2007; Gonzalez-Burgos et al., 2015; Whittington et al., 2000), disturbances to either excitatory (Tatard-Leitman et al., 2014) or inhibitory (Cardin et al., 2009; Fuchs et al., 2007; Sohal et al., 2009) strength in this circuit can impair gamma oscillatory activity. Thus, altered levels of glutamate and/or GABA neurotransmission might contribute to lower gamma oscillation power and working memory deficits in schizophrenia (Lewis and Moghaddam, 2006).
Consistent with this notion, in vivo studies of subjects with schizophrenia found that glutamate levels in the frontal cortex were positively correlated with performance on verbal working memory tasks (Rowland et al., 2016) and other indices of global cognitive performance (Bustillo et al., 2011). Similarly, lower frontal GABA levels predicted poorer working memory performance (Marsman et al., 2014) and abnormal gamma oscillatory activity (Chen et al., 2014). In addition, a diminished capacity to increase extracellular GABA levels was associated with lower gamma oscillation power and poorer cognitive performance in schizophrenia subjects (Frankle et al., 2015).
Although not conclusive, these findings suggest a relationship between altered frontal glutamate and GABA levels and cognitive performance in schizophrenia subjects. Thus, lower levels of glutamate or GABA neurotransmission might contribute to cognitive impairments in schizophrenia. However, in vivo magnetic resonance spectroscopy (MRS) studies of the levels of these molecules in schizophrenia and comparison subjects have produced mixed results. Thus, many meta-analyses and reviews of in vivo MRS studies of frontal cortical glutamate in schizophrenia do not find evidence of a deficit in schizophrenia subjects (Marsman et al., 2013; Merritt et al., 2016; Poels et al., 2014, 2013) and one recent study using MRS at high strength 7T magnetic fields found no group differences in DLPFC glutamate levels between control and schizophrenia subjects (Wang et al., 2019). Similarly, meta-analyses and reviews of in vivo GABA levels in frontal cortex failed to find consistent evidence of differences between schizophrenia and comparison subjects (de Jonge et al., 2017; Egerton et al., 2017; Taylor and Tso, 2015; Wang et al., 2019), although effect sizes suggest modestly lower levels in schizophrenia (Egerton et al., 2017). In addition to the variability in findings across studies, which might be due to differences in methodology (i.e., MRS field strength, spectral editing, methods for normalization, etc.) or subject cohorts (i.e., duration of illness, antipsychotic exposure, etc.), two other key issues limit the interpretive value of these in vivo studies for understanding excitatory/inhibitory imbalance in the DLPFC of subjects with schizophrenia. First, most MRS imaging studies require large voxel sizes to quantify levels of the molecules of interest, and thus do not possess the anatomical resolution needed to assess disease effects at the level of cortical layers or local circuits. Second, these studies index total tissue levels of glutamate and GABA, and therefore cannot distinguish levels of these molecules involved in synaptic neurotransmission versus other metabolic processes.
Clearly, understanding abnormalities in cortical excitatory and inhibitory neurotransmission in schizophrenia requires the ability to quantify markers that directly regulate glutamate and GABA signaling at laminar and circuitry levels of resolution. One study, which assessed gene products involved in glutamate and GABA synthesis, vesicular packaging and terminal reuptake, as well as critical postsynaptic receptor subunits, found evidence of lower levels of both excitatory and inhibitory markers in layer 3 of the DLPFC of schizophrenia subjects (Hoftman et al., 2018). These findings support the idea that lower levels of both excitation and inhibition in this layer might contribute to impaired gamma oscillatory activity and working memory deficits in schizophrenia (Lewis et al., 2012). However, this study was limited to layer 3 tissue homogenates and did not determine if these same alterations are present across all cortical layers or in the layer 3 glutamatergic pyramidal cell/parvalbumin (PV) GABAergic neuron circuit that appears to be crucial for generating gamma oscillations and mediating working memory (Goldman-Rakic, 1995; Gonzalez-Burgos et al., 2015; Miller et al., 2018).
Here, we explored how the schizophrenia disease effect on composite measures of the gene products regulating glutamate and GABA neurotransmission is influenced by these different levels of anatomical resolution. Utilizing six previously published datasets in the DLPFC of schizophrenia subjects (Arion et al., 2015; Curley et al., 2011; Enwright et al., 2018; Fromer et al., 2016; Hoftman et al., 2018; Volk et al., 2016), we examined composite measures of glutamate and GABA neurotransmission in 1) total gray matter tissue homogenates containing all layers of the DLPFC, 2) isolated layer 3 tissue homogenates, and 3) the layer 3 circuit composed of pyramidal cells and PV neurons.
2. Methods
2.1. Datasets and Transcripts Selection
Gene expression data were re-analyzed from six previously published datasets of schizophrenia and unaffected comparison subjects; summary characteristics of the subjects used in each study are shown in Table 1. In all six studies, each schizophrenia subject was matched for sex, and as closely as possible for age, to one unaffected comparison subject. As shown in Fig 1, most subject pairs were common across studies. To compare findings across datasets, we constrained our study to the same key transcripts indexing glutamatergic and GABAergic neurotransmission studied by Hoftman and colleagues (Hoftman et al., 2018): glutamate and GABA neurotransmitter synthesis (glutamine synthetase, GLS; and 67 kDa isoform of glutamic acid decarboxylase, GAD67), vesicular transport (vesicular glutamate transporter 1, vGLUT1; and vesicular GABA transporter, vGAT), reuptake (solute carrier family 1 member 2, SLC1A2, also known as excitatory amino acid transporter 2, EAAT2; and GABA reuptake transporter 1, GAT1), and critical postsynaptic receptors (the obligatory subunit of the N-methyl-D-aspartate (NMDA) receptor, GRIN1; the calcium-impermeable receptor subunit of the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor, GRIA2; and the obligatory subunit of the GABAA receptor, γ2 (GABRG2)). In cases where a given transcript was represented by more than one probe, the highest expressing probe was selected for study.
Table 1.
Cohort characteristics and methods used for mRNA quantification.
| RNAseq |
Quantitative PCR |
Microarray |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study | (Fromer et al., 2016) | (Volk et al., 2016) | (Curley et al., 2011) | (Hoftman et al., 2018) | (Arion et al., 2015; Enwright et al., 2018) | |||||
| Sample | All Layers |
All Layers |
All Layers |
Layer 3 |
PCs/PVINs |
|||||
| Subject Group | UC | SZ | UC | SZ | UC | SZ | UC | SZ | UC | SZ |
| n | 57 | 57 | 62 | 62 | 42 | 42 | 20 | 20 | 36 | 36 |
| Sex | 44 M/13 F | 44 M/13 F | 47 M/15 F | 47 M/15 F | 31 M/11 F | 31 M/11 F | 14 M/6 F | 14 M/6 F | 27 M/9 F | 27 M/9 F |
| Race | 48 W/9 B | 41 W/16 B | 52 W/10 B | 46 W/16 B | 34 W/8 B | 29 W/13 B | 16 W/4 B | 13 W/7 B | 30 W/6 B | 24 W/12 B |
| Age (y) | 49.0 (14.1) | 48.1 (13.0) | 48.8 (13.8) | 47.6 (12.7) | 48.1 (13.3) | 47.0 (12.8) | 47.2 (9.9) | 45.6 (9.5) | 48.1 (13.0) | 46.9 (12.4) |
| PMI (h) | 19.1 (5.6) | 20.1 (8.4) | 18.8 (5.5) | 19.2 (8.5) | 17.8 (5.9) | 18.1 (8.7) | 15.4 (5.8) | 14.4 (6.2) | 17.6 (7.1) | 18.0 (8.8) |
| Brain pH | 6.7 (0.2) | 6.6 (0.3) | 6.7 (0.2) | 6.6 (0.3) | 6.8 (0.2) | 6.6 (0.4) | 6.7 (0.2) | 6.5 (0.3) | 6.7 (0.2) | 6.6 (0.4) |
| RIN | 8.1 (0.6) | 8.0 (0.6) | 8.1 (0.6) | 8.1 (0.6) | 8.3 (0.6) | 8.2 (0.7) | 8.3 (0.5) | 8.2 (0.6) | 8.3 (0.6) | 8.2 (0.6) |
Abbreviations: PCR, polymerase chain reaction; PCs, pyramidal cells; PVINs, parvalbumin interneurons; UC, unaffected comparison; SZ, schizophrenia; M, male; F, female; W, white, B, black, PMI, postmortem interval; RIN, RNA integrity number. Values are mean (s.d).
Figure 1.
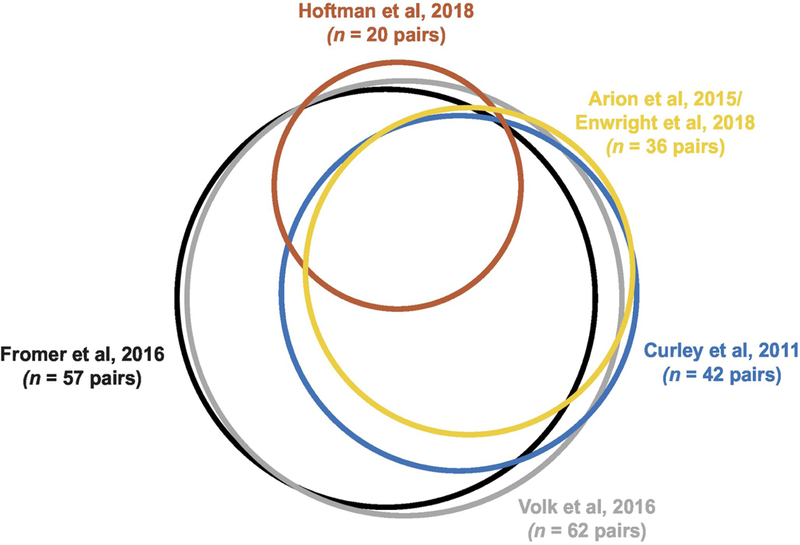
Euler diagram illustrating in proportionally-sized circles the number of subject pairs in each study and the overlap of identical subject pairs across studies. Between some studies, a different unaffected comparison subject was paired to the same schizophrenia subject, and these cases were omitted from the overlap in the diagrams. Plot was made in R using the eulerr package (Larsson, 2018).
2.2. Tissue samples and Methodology for Transcript Quantification
Transcript quantification was performed by RNA sequencing (RNAseq) in samples of total gray matter homogenates containing all DLPFC layers (Fig 2A; 2B) from 57 pairs of schizophrenia and unaffected comparison subjects (Fromer et al., 2016). Using quantitative PCR (qPCR), quantification of GAD67 was performed in comparable DLPFC total gray matter homogenates from 62 (Volk et al., 2016) or 42 (Curley et al., 2011) pairs of schizophrenia and unaffected comparison subjects. Similarly, qPCR was used to quantify all nine transcripts of interest in isolates of layer 3 tissue homogenates (Fig 2C; 2D), collected by laser-capture microdissection, from 20 matched pairs of schizophrenia and unaffected comparison subjects (Hoftman et al., 2018). Finally, microarray platforms were used to quantify transcripts in layer 3 pyramidal cells (Fig 2E; 2F) (Arion et al., 2015) or PV neurons, identified by the presence of perineuronal nets (Fig 2G) (Enwright et al., 2018), captured by laser-capture microdissection in 36 matched pairs of schizophrenia and unaffected comparison subjects.
Figure 2.
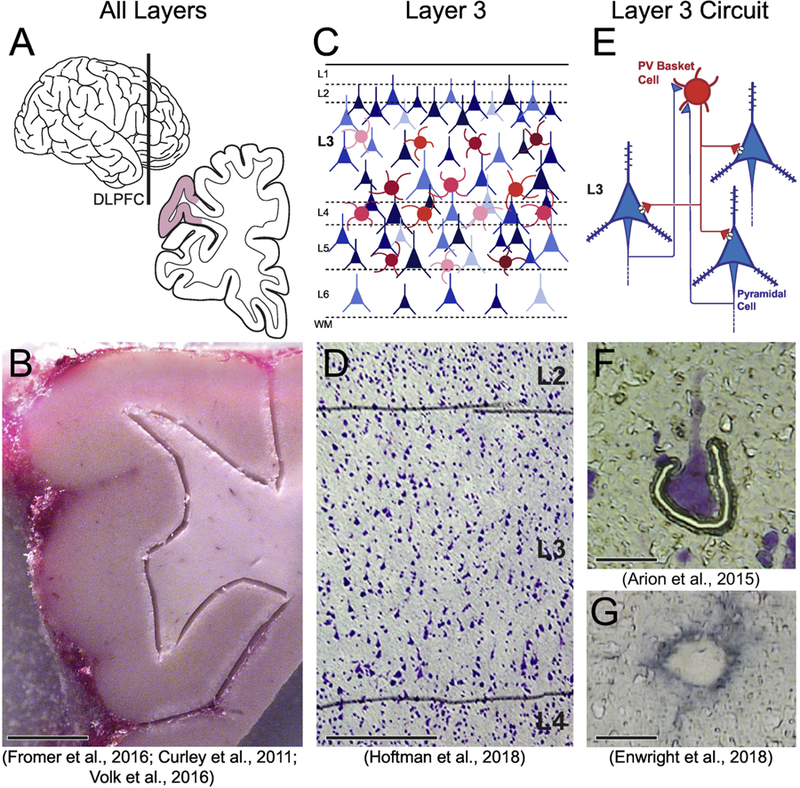
Approaches for studying postmortem tissue at multiple levels of anatomical resolution and techniques for capturing tissue at these resolutions for RNA quantification. (A) Schematic of the whole brain (left); the vertical line indicates the approximate location of the coronal tissue blocks (right) from which the DLPFC was sampled. Shaded area indicates the approximate location of the tissue block shown in (B). (B) Total gray matter tissue homogenates were dissected from the underlying white matter and collected for RNAseq or qPCR analyses (scale bar = 5mm). (C) Schematic of the laminar organization of the DLPFC. Objects of different colors and sizes represent the various cell types present in different cortical layers. (D) Under a laser microdissection 5x microscope objective (scale bar = 400 µm), the layer 2/3 and 3/4 borders were identified, and samples restricted to layer 3 tissue were collected for qPCR analysis. (E) Schematic drawing indicating the principal excitatory/inhibitory circuit within layer 3 between pyramidal cells (blue) and parvalbumin-containing interneurons (red). (F) Individual pyramidal cells were microdissected using a similar laser microdissection approach with a 40x microscope objective (scale bar = 25 µm). (G) Individual parvalbumin cells, identified by aggrecan which labels the perineuronal net surrounding parvalbumin cells, were collected using a laser microdissection approach with a 40x microscope objective (scale bar = 10 µm). Images D, F and G were adapted from Hoftman et al., 2018, Arion et al., 2015 and Enwright et al., 2018, respectively.
2.3. Calculation of Z-Scores, Composite Measures, and Effect Sizes
For each of the nine transcripts from four studies (Arion et al., 2015; Enwright et al., 2018; Fromer et al., 2016; Hoftman et al., 2018), expression levels were normalized by calculating a z-score for each transcript, using the mean and standard deviation of the given study sample. For each subject, glutamate and GABA composite measures were calculated by averaging the normalized value of excitatory (vGLUT1, GLS, EAAT2, GRIN1, GRIA2) and inhibitory (vGAT, GAD67, GAT1, and GABRG2) transcripts, thus providing equal weighting to each transcript in the composite measure. The mean and standard deviation of the z-scores across subjects in each group were then calculated, and 95% confidence intervals were plotted with the group means. Cohen’s D effect sizes were calculated for the group differences in composite glutamate and GABA measures between schizophrenia and unaffected comparison subjects and used to compare the disease effect on the composite measures across the three levels of anatomical resolution. Our interpretations of the effect sizes were as follows: 0.2 for small effects, 0.4 for medium effects, and 0.6 or greater for large effects. Because of the nature of the comparisons across studies, effect sizes and confidence intervals of the mean are reported, rather than p-values (Wasserstein et al., 2019). Pearson’s correlations and the associated p-values were used to compare methods for GAD67 mRNA quantification between studies.
3. Results
3.1. Glutamate and GABA Composite Measures in Total versus Layer 3 Gray Matter Homogenates
In gray matter homogenates containing all layers of the DLPFC (Fromer et al., 2016), the glutamate composite measure was higher in schizophrenia subjects with a medium effect size (D = +0.42, Fig 3A), whereas the GABA composite measure did not differ between subject groups (D = −0.05, Fig 3B). In contrast, in samples restricted to DLPFC layer 3 (Hoftman et al., 2018), both glutamate and GABA measures were lower in schizophrenia, with small-medium effect sizes (D = −0.33, Fig 3A, and D = −0.37, Fig 3B, respectively).
Figure 3.
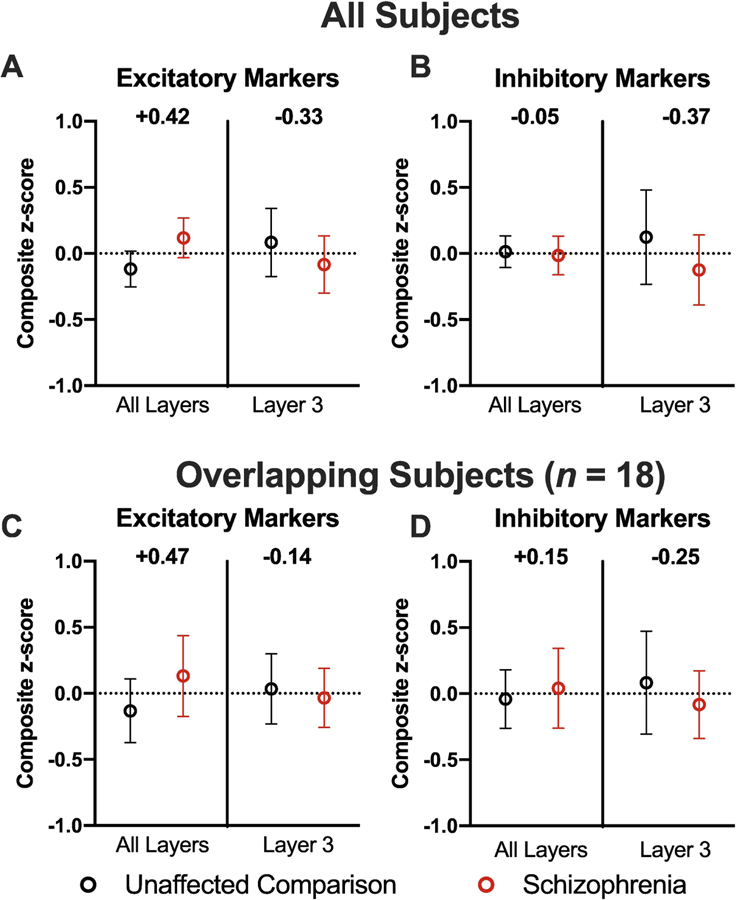
Comparison of composite measures for (A) excitatory and (B) inhibitory markers in all layers (i.e., total cortical gray matter homogenates) and in layer 3 tissue homogenates. Cohen’s D effect sizes are shown between the schizophrenia (red) and unaffected comparison (black) subject groups. Data in (A) and (B) are from all subjects in each study (n = 57 subject pairs for total gray matter, n = 20 subject pairs for layer 3). Data in (C) and (D) show composite measures for excitatory and inhibitory markers after these scores were re-calculated using only the subset of subjects (n = 18 subject pairs) common to both studies. Values are mean ± 95% confidence interval of the mean.
To determine if the different findings between these studies might be attributable to differences in the subject pairs sampled, we recalculated the z-scores based on the new sample means and standard deviations of the 18 subject pairs common to both studies (Fig 1). The differences between total gray matter and layer 3 gray matter for both glutamate (All layers: D = +0.47; Layer 3: D = −0.14, Fig 3C) and GABA (All layers: D = +0.15; Layer 3: D = −0.25, Fig 3D) composite measures were comparable to the findings from the full cohorts. In concert, these findings suggest that the disease effect of schizophrenia on the composite measure of glutamate neurotransmission is in the opposite direction between total gray matter and layer 3 samples. For GABA neurotransmission, the disease effect was not apparent in samples of total gray matter but was clearly seen in layer 3 samples.
These two studies also differed in the methods used for transcript quantification (RNAseq versus qPCR). Thus, to determine the potential influence of quantification method we compared within-subject levels of GAD67 mRNA, the only transcript that was quantified in the same subject pairs from 3 studies of total DLPFC gray matter (Fig 1). Between two studies using qPCR (Curley et al., 2011; Volk et al., 2016), GAD67 mRNA levels were highly positively correlated (r = 0.96, p < 0.0001, n = 82 subjects, Fig 4A). Similarly, GAD67 mRNA levels were highly positively correlated between the Curley et al. qPCR dataset and the Fromer et al. RNAseq dataset (r = 0.70, p < 0.0001, n = 72 subjects, Fig 4B) and between the Volk et al. qPCR dataset and the Fromer et al. RNAseq dataset (r = 0.73, p < 0.0001, n = 112 subjects, Fig 4C). These findings, in concert with prior reports of highly correlated mRNA levels between RNAseq and qPCR measures for many transcripts (Fromer et al., 2016), suggest that the differences in the disease effect between DLPFC total gray matter and layer 3 described above do not reflect methodological issues.
Figure 4.

Plots of individual subject data for GAD67 mRNA levels in total gray matter homogenates from three studies. (A) Correlation of GAD67 mRNA levels between Volk et al., 2016, and Curley et al., 2011 for separate qPCR analyses in the same 41 subject pairs. (B) Correlation of GAD67 mRNA levels as determined by RNAseq (Fromer et al 2016) or qPCR (Curley et al 2011) in the same 36 subject pairs. (C) Correlation of GAD67 mRNA levels as determined by RNAseq (Fromer et al 2016) or qPCR Volk et al 2016 (qPCR) in the same 56 subject pairs.
3.2. Comparisons to the Layer 3 Local Circuit
To determine if the observed differences in the disease effect between DLPFC total gray matter might be attributable to specific neural elements within layer 3, we compared the above findings to those obtained from layer 3 pyramidal cells and PV neurons. For this comparison, we excluded EAAT2 mRNA levels, given the relatively low expression of EAAT2 in neurons compared to astrocytes (Danbolt et al., 2016; Roberts et al., 2014; Rothstein et al., 1996), and re-calculated the glutamate composite measure in total gray matter and in layer 3 homogenates. The glutamate composite measure was elevated in schizophrenia subjects in total gray matter (D = +0.58) but lower in layer 3 (D = −0.53) (consistent with the previous analysis using all transcripts), and the lower glutamate measure found in layer 3 was also present in the cellular level analyses of the layer 3 local circuit (D = −0.25) (Fig 5A). The GABA composite measure did not differ between subject groups in total gray matter homogenates (D = −0.05) but was lower in schizophrenia subjects for layer 3 tissue homogenates (D = −0.37) and appeared to be more pronounced in the local layer 3 circuit (D = −0.59) (Fig 5B).
Figure 5.

Comparison of composite measures for (A) excitatory and (B) inhibitory markers in all layers (n = 57 subject pairs, left panels), layer 3 tissue homogenates (n = 20 subject pairs, middle panel), and layer 3 local circuit consisting of excitatory pyramidal cells and inhibitory PV cells (n = 36 subject pairs, right panels). Cohen’s D effect sizes are shown for the composite z-scores between schizophrenia (red) and unaffected comparison (black) subjects. Data for (A) and (B) show all subjects from each dataset, and (C) and (D) show the same composite excitatory and inhibitory measures, respectively, re-calculated using only the subset of subject pairs (n = 13) common to all three studies. Values are mean ± 95% confidence interval of the mean.
Because these findings might be influenced by differences in the cohorts used in each dataset, we re-calculated the composite measures for the same transcripts in the subset of 13 subject pairs common to all three datasets (Fig 1). The patterns of effect size differences across levels of anatomical resolution for both glutamate (Fig 5C: all layers, D = +0.72; layer 3, D = − 0.53; layer 3 circuit, D = −0.40) and GABA (Fig 5D: all layers, D = +0.26; layer 3, D = −0.34; layer 3 circuit, D = −0.92) composite measures were similar to those observed in the full datasets (Fig 5A and Fig 5B, respectively).
4. Discussion
In the present study, we utilized existing datasets to identify differences across anatomical levels of resolution in the disease effect of schizophrenia on composite measures of glutamate and GABA transcripts in the DLPFC. Relative to unaffected comparison subjects, the composite glutamate measure was higher in schizophrenia subjects at the level of total gray matter, but lower in samples restricted to layer 3 or the layer 3 local circuit. The composite GABA measure did not differ between subject groups in total gray matter but was lower in schizophrenia subjects in layer 3 and lower still in the layer 3 local circuit. These findings do not seem to be attributable to differences in the mRNA quantification methods or in the subject cohorts used in each study.
4.1. Differences in Disease Effect on DLPFC Excitatory Neurotransmission Markers Between Gray Matter and Layer 3
Our finding of different disease effects on the composite glutamate and GABA measures in total versus layer 3 gray matter highlights the importance of the anatomical level of resolution used in studies of schizophrenia. In total gray matter homogenates, we found evidence of higher levels of markers of glutamate neurotransmission, consistent with a model of cortical disinhibition in schizophrenia (Murray et al., 2014; Starc et al., 2017). This hypothesis was first proposed based on findings that antagonism of NMDA receptors by ketamine led to psychotic symptoms in psychiatrically-unaffected subjects (Krystal et al., 1994) and is supported by more recent studies reporting elevated global connectivity in schizophrenia subjects (Glahn et al., 2014). In addition, studies of schizophrenia subjects early in the disease course reported elevated glutamate levels in the prefrontal cortex by in vivo MRS (Poels et al., 2014, 2013) and greater prefrontal functional connectivity (Anticevic et al., 2015), although the latter appears to normalize over time. Together, these findings have been interpreted as evidence that the prefrontal cortex is disinhibited in schizophrenia due to reduced signaling from GABA neurons, as observed in an animal model of NMDA receptor antagonism (Homayoun and Moghaddam, 2007). Our finding in total DLPFC gray matter of a higher composite glutamate measure in schizophrenia is consistent with the hypothesis of a hyperactive DLPFC in the illness, although we did not find evidence of lower GABA neurotransmission in the same subjects at this level of anatomical resolution.
It is important to note, however, that layer 3 pyramidal cells in the DLPFC (and other cortical regions) in schizophrenia exhibit 1) a lower complement of dendritic spines (Garey et al., 1998; Glantz and Lewis, 2000; Konopaske et al., 2014), suggesting reduced excitatory input into these cells, 2) lower expression of activity-dependent transcripts (Kimoto et al., 2015), and 3) lower expression of gene products that index energy production (Arion et al., 2017, 2015). Together, these findings suggest that DLPFC layer 3 pyramidal cells are hypoactive, rather than hyperactive, in schizophrenia. Consistent with these findings, here we found that schizophrenia is associated with a lower composite measure of glutamate neurotransmission in layer 3, in contrast to the higher composite measure in total DLPFC gray matter. The higher composite measure in total gray matter might be due to elevated glutamate neurotransmission in other cortical layers, suggesting that studies comparable to that of Hoftman et. al., 2018 should be conducted in these layers. Thus, depending on the level of anatomical resolution studied, schizophrenia appears to be associated with either hyperactive or hypoactive excitatory signaling.
4.2. Differences in Disease Effect on DLPFC Inhibitory Neurotransmission Markers Between Gray Matter and Layer 3
Lower DLPFC inhibitory signaling is thought to contribute to (or to reflect a compensation for) neural network alterations underlying working memory impairments in schizophrenia (Dienel and Lewis, 2018). However, findings of alterations in GABA neurons are not uniform across all cell types; for example, PV-containing GABA neurons are clearly affected in schizophrenia, whereas calretinin-containing GABA neurons appear to be unaffected (Chung et al., 2016a, 2016b; Enwright et al., 2018; Fung et al., 2010; Hashimoto et al., 2003; Mellios et al., 2009). Because calretinin cells make up approximately 50% of the GABA neurons in the DLPFC (Condé et al., 1994; DeFelipe, 1993; Gabbott and Bacon, 1996), and are largely found outside of layer 3, these unaffected GABA neurons might mask a deficit in GABA neurotransmission when measured at the level of total gray matter, consistent with our finding that the composite GABA measure in total gray matter did not differ between subject groups.
In contrast, DLPFC layer 3 is enriched for the PV cells that are affected in schizophrenia; this enrichment might explain our finding of a lower composite measure of GABA neurotransmission in layer 3. Consistent with this interpretation, the effect size of the lower GABA composite measure in schizophrenia was even larger when the analysis was restricted to cellular level studies of the PV neuron-pyramidal cell circuit in layer 3. Thus, the contrasting findings, for both excitatory and inhibitory neurotransmission indices, between DLPFC total gray matter and layer 3 in schizophrenia subjects suggest that alterations in the excitatory/inhibitory balance in schizophrenia might not be a uniform elevation or reduction; instead, findings of alterations in this balance might depend on the level of anatomic resolution of the research methods employed.
4.3. Interpretative Considerations
The goal of the present study was to utilize composite measures that provide comparable indices of excitatory and inhibitory neurotransmission across multiple levels of anatomical resolution in the postmortem human brain (Lewis, 2002). At the protein level, synaptic strength is directly related to the protein levels of the synthesizing enzymes (Asada et al., 1997; Danbolt, 2001); vesicular transporters which regulate quantal size (Erickson et al., 2006); and the number and type of postsynaptic receptors (Henley and Wilkinson, 2016; Jacob et al., 2008; Traynelis et al., 2010). In addition, inhibition of EAAT2 or GAT1, the key reuptake transporters for glutamate and GABA, respectively, leads to a reduction in presynaptic release of the associated neurotransmitters (Jensen et al., 2006; Kalivas, 2009; Maki et al., 1994), likely through activation of presynaptic receptors by excess neurotransmitter in the synaptic cleft, suggesting that levels of EAAT2 and GAT1 also directly index the strength of glutamate and GABA neurotransmission, respectively. Thus, the composite measures employed in this study provide insight into alterations in the strength of glutamate and GABA signaling in schizophrenia. However, it is important to note that these composite measures are limited by 1) the inclusion of only some of the many factors that can influence synaptic strength and 2) measures of only mRNA levels which may not reflect the levels or functional state of their cognate proteins.
Many studies of schizophrenia are potentially confounded by exposure to antipsychotic medications. Three lines of evidence suggest the findings of the present study are not driven by antipsychotic use. First, antipsychotic use at time of death was not found to be a significant covariate of altered transcript expression levels in the published studies used in the present analysis (Arion et al., 2015; Enwright et al., 2018; Fromer et al., 2016; Hoftman et al., 2018). Second, findings from a large study of schizophrenia subjects suggest that the transcriptomic alterations observed in the disease were directionally discordant to transcript alterations observed in antipsychotic exposed monkeys (Gandal et al., 2018; Martin et al., 2015). Third, the key finding in the present study, that these measures of excitatory and inhibitory neurotransmission differ as a function of anatomical resolution, was true even for comparisons made within the same subject pairs, providing a within-subject control of antipsychotic use. Thus, the differences observed in these composite measures across different levels of anatomical resolution are likely not confounded by antipsychotic use.
4.4. Conclusions
Our findings, that alterations in indices of cortical excitatory and inhibitory neurotransmission in schizophrenia appear to differ depending on the level of anatomic resolution studied, illustrate that although studies at the level of total gray matter can reveal disease-related alterations, these findings require additional studies to determine the layers and cell types that contribute to such findings. Furthermore, gray matter studies might also conceal disease-related alterations that can only be resolved with more focused studies of cortical layers or circuits. Thus, the results of the present study highlight the importance of designing future studies to assess laminar- and cell type-specificity of cortical abnormalities in schizophrenia and other psychiatric illnesses. Indeed, understanding the disease process of schizophrenia requires study designs capable of interrogating disease-related differences in a manner that reflects the remarkable inherent diversity of cortical microcircuits.
Acknowledgements
We would like to thank Mary Brady for her excellent assistance in preparation of the figures for this manuscript and Dr. Jill Glausier for her scholarly input in the conception of the project. The work in the studies cited here were supported by the National Institutes of Health Grant MH043784 (D.A.L) and the National Institute of General Medical Sciences Training Grant (GM008208 to S.J.D).
Disclosures
Dr. Lewis currently receives research support from Pfizer and Merck.
David A. Lewis currently receives investigator-initiated research support from Pfizer and Merck.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
All other authors report no conflicts of interest.
References
- Anticevic A, Hu X, Xiao Y, Hu J, Li F, Bi F, Cole MW, Savic A, Yang GJ, Reprovs G, Murray JD, Wang X-J, Huang X, Lui S, Krystal JH, Gong Q, 2015. Early-Course Unmedicated Schizophrenia Patients Exhibit Elevated Prefrontal Connectivity Associated with Longitudinal Change. J. Neurosci 35, 267–286. 10.1523/jneurosci.2310-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, Cacace AM, Zaczek R, Albright CF, Tseng G, Lewis DA, 2015. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol. Psychiatry 20, 1397–1405. 10.1038/mp.2014.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, Huo Z, Enwright JF, Corradi JP, Tseng G, Lewis DA, 2017. Transcriptome Alterations in Prefrontal Pyramidal Cells Distinguish Schizophrenia From Bipolar and Major Depressive Disorders. Biol. Psychiatry 82, 594–600. 10.1016/j.biopsych.2017.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K, 1997. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94, 6496–6499. 10.1073/pnas.94.12.6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P, 2007. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci 8, 45–56. 10.1038/nrn2044 [DOI] [PubMed] [Google Scholar]
- Bustillo JR, Chen H, Gasparovic C, Mullins P, Caprihan A, Qualls C, Apfeldorf W, Lauriello J, Posse S, 2011. Glutamate as a marker of cognitive function in schizophrenia: A proton spectroscopic imaging study at 4 tesla. Biol. Psychiatry 69, 19–27. 10.1016/j.biopsych.2010.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai L-H, Moore CI, 2009. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. 10.1038/nature08002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-MA, Stanford AD, Mao X, Abi-Dargham A, Shungu DC, Lisanby SH, Schroeder CE, Kegeles LS, 2014. GABA level, gamma oscillation, and working memory performance in schizophrenia. NeuroImage Clin 4, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RY, Konecky RO, Carter CS, 2006. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc. Natl. Acad. Sci. U. S. A 103, 19878–83. 10.1073/pnas.0609440103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DW, Fish KN, Lewis DA, 2016a. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am. J. Psychiatry 173, 1131–1139. 10.1176/appi.ajp.2016.16010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA, 2016b. Dysregulated ErbB4 Splicing in Schizophrenia: Selective Effects on Parvalbumin Expression. Am. J. Psychiatry 173, 60–68. 10.1176/appi.ajp.2015.15020150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condé F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA, 1994. Local Circuit Neurons Immunoreactive for Calretinin, Calbindin D-28k or Parvalbumin in Monkey Prefrontal Cortex: Distribution and Morphology. J. Comp. Neurol 341, 95–116. [DOI] [PubMed] [Google Scholar]
- Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, Lewis DA, 2011. Cortical Deficits of Glutamic Acid Dexcarboxylase 67 Expression in Schizophrenia: Clinical, Protein, and Cell Type-Specific Features. Am. J. Psychiatry 168, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC, 2001. Glutamate uptake. Prog. Neurobiol 65, 1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Furness DN, Zhou Y, 2016. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int 98, 29–45. 10.1016/j.neuint.2016.05.009 [DOI] [PubMed] [Google Scholar]
- de Jonge JC, Vinkers CH, Hulshoff Pol HE, Marsman A, 2017. GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front. Psychiatry 8, 118 10.3389/fpsyt.2017.00118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, 1993. Neocortical Neuronal Diversity: Chemical Heterogeneity Revealed by Colocalization Studies of Classic Neurotransmitters, Neuropeptides, Calcium-binding Proteins, and Cell Surface Molecules. Cereb. Cortex 3, 273–289. [DOI] [PubMed] [Google Scholar]
- Dienel SJ, Lewis DA, 2018. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis in press. 10.1016/j.nbd.2018.06.020 [DOI] [PMC free article] [PubMed]
- Egerton A, Modinos G, Ferrera D, McGuire P, 2017. Neuroimaging studies of GABA in schizophrenia: a systematic review with meta-analysis. Transl. Psychiatry 7, e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enwright JF, Huo Z, Arion D, Corradi JP, Tseng G, Lewis DA, 2018. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol. Psychiatry 23, 1606–1613. 10.1038/mp.2017.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, De Gois S, Varoqui H, Schafer MKH, Weihe E, 2006. Activity-dependent regulation of vesicular glutamate and GABA transporters: A means to scale quantal size. Neurochem. Int 48, 643–649. 10.1016/j.neuint.2005.12.029 [DOI] [PubMed] [Google Scholar]
- Foss-Feig JH, Adkinson BD, Ji L, Yang G, Srihari VH, Mcpartland JC, Krystal JH, Murray JD, Anticevic A, 2017. Searching for Cross-Diagnostic Convergence: Neural Mechanisms Governing Excitation and Inhibition Balance in Schizophrenia and Autism Spectrum Disorders. Biol. Psychiatry 81, 848–861. 10.1016/j.biopsych.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, Walker C, Lewis DA, Narendran R, 2015. In Vivo Measurement of GABA Transmission in Healthy Subjects and Schizophrenia Patients. Am. J. Psychiatry 172, 1148–1159. 10.1176/appi.ajp.2015.14081031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, Klei LL, Kramer R, Pinto D, Gümüş ZH, Cicek AE, Dang KK, Browne A, Lu C, Xie L, Readhead B, Stahl EA, Xiao J, Parvizi M, Hamamsy T, Fullard JF, Wang YC, Mahajan MC, Derry JMJ, Dudley JT, Hemby SE, Logsdon BA, Talbot K, Raj T, Bennett DA, De Jager PL, Zhu J, Zhang B, Sullivan PF, Chess A, Purcell SM, Shinobu LA, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Schadt EE, Hirai K, Roeder K, Brennand KJ, Katsanis N, Domenici E, Devlin B, Sklar P, 2016. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci 19, 1442–1453. 10.1038/nn.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FEN, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JNP, Monyer H, 2007. Recruitment of Parvalbumin-Positive Interneurons Determines Hippocampal Function and Associated Behavior. Neuron 53, 591–604. 10.1016/j.neuron.2007.01.031 [DOI] [PubMed] [Google Scholar]
- Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS, 2010. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 167, 1479–1488. 10.1176/appi.ajp.2010.09060784 [DOI] [PubMed] [Google Scholar]
- Gabbott PLA, Bacon SJ, 1996. Local circuit neurons in the medial prefrontal cortex (areas 24a, b, c, 25 and 32) in the monkey: II. Quantitative Areal and Laminar Distributions. J. Comp. Neurol 364, 567–608. 10.1002/(SICI)1096-9861(19960122)364 [DOI] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, Liu C, White KP, Horvath S, Geschwind DH, 2018. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (80-. ) 359, 693–697. 10.1126/science.aad6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR, 1998. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 65, 446–453. 10.1136/jnnp.65.4.446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Savic A, Pearlson GD, Repovs G, Krystal JH, Wang X-J, Yang GJ, Glasser MF, Murray JD, Anticevic A, Cole MW, Pittenger C, 2014. Altered global brain signal in schizophrenia. Proc. Natl. Acad. Sci 111, 7438–7443. 10.1073/pnas.1405289111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA, 2000. Decreased Dendritic Spine Density on Prefrontal Cortical Pyramidal Neurons in Schizophrenia. Arch. Gen. Psychiatry 57, 65–73. 10.1001/archpsyc.57.1.65 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, 1995. Cellular Basis of Working Memory. Neuron 14, 477–485. 10.1016/0896-6273(95)90304-6 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Cho RY, Lewis DA, 2015. Alterations in Cortical Network Oscillations and Parvalbumin Neurons in Schizophrenia. Biol. Psychiatry 77, 1031–1040. 10.1016/j.biopsych.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA, 2003. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci 23, 6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA, 2016. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci 17, 337–350. [DOI] [PubMed] [Google Scholar]
- Hoftman GD, Datta D, Lewis DA, 2017. Layer 3 Excitatory and Inhibitory Circuitry in the Prefrontal Cortex: Developmental Trajectories and Alterations in Schizophrenia. Biol. Psychiatry 81, 862–873. 10.1016/j.biopsych.2016.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftman GD, Dienel SJ, Bazmi HH, Zhang Y, Chen K, Lewis DA, 2018. Altered Gradients of Glutamate and Gamma-Aminobutyric Acid Transcripts in the Cortical Visuospatial Working Memory Network in Schizophrenia. Biol. Psychiatry 83, 670–679. 10.1016/j.biopsych.2017.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B, 2007. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J. Neurosci 27, 11496–11500. 10.1523/JNEUROSCI.2213-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R, 2008. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci 9, 331–343. 10.1038/nrn2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Chiu C-S, Sokolova I, Lester HA, Mody I, 2006. GABA Transporter-1 (GAT1)-Deficient Mice: Differential Tonic Activation of GABA A Versus GABA B Receptors in the Hippocampus. J. Neurophysiol 90, 2690–2701. 10.1152/jn.00240.2003 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, 2009. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci 10, 561–572. 10.1038/nrn2515 [DOI] [PubMed] [Google Scholar]
- Kimoto S, Zaki MM, Bazmi HH, Lewis DA, 2015. Altered Markers of Cortical γ-Aminobutyric Acid Neuronal Activity in Schizophrenia: Role of the NARP Gene. JAMA Psychiatry 72, 747–756. 10.1001/jamapsychiatry.2015.0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, Benes FM, 2014. Prefrontal Cortical Dendritic Spine Pathology in Schizophrenia and Bipolar Disorder. JAMA Psychiatry 71, 1323–1331. 10.1001/jamapsychiatry.2014.1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Anticevic A, Yang GJ, Dragoi G, Driesen NR, Wang X-J, Murray JD, 2017. Impaired Tuning of Neural Ensembles and the Pathophysiology of Schizophrenia: A Translational and Computational Neuroscience Perspective. Biol. Psychiatry 81, 874–885. 10.1016/j.biopsych.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS, 1994. Subanesthetic Effects of the Noncompetitive NMDA Antagonist, Ketamine, in Humans: Psychomimetic, Perceptual, Cognitive, and Neuroendocrine Responses. Arch. Gen. Psychiatry 51, 199–214. [DOI] [PubMed] [Google Scholar]
- Larsson J, 2018. Eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses Lund University. [Google Scholar]
- Lewis DA, 2002. The human brain revisited: opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology 26, 143–54. 10.1016/S0893-133X(01)00393-1 [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW, 2012. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B, 2006. Cognitive Dysfunction in Schizophrenia: Convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol 63, 1372–1376. [DOI] [PubMed] [Google Scholar]
- Maki R, Robinson M, Dichter M, 1994. The Glutamate Uptake Inhibitor L-trans-pyrrolidine-2,4-dicarboxylate Depresses Excitatory Synaptic Transmission via a Presynaptic Mechanism in Cultured Hippocampal Neurons. J. Neurosci 14, 6754–6762. 10.1523/jneurosci.14-11-06754.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman A, Mandl RCW, Klomp DWJ, Bohlken MM, Boer VO, Andreychenko A, Cahn W, Kahn RS, Luijten PR, Hulshoff Pol HE, 2014. GABA and glutamate in schizophrenia: a 7T H-MRS study. NeuroImage Clin 6, 398–407. 10.1016/j.nicl.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DWJ, 2013. Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr. Bull 39, 120–129. 10.1093/schbul/sbr069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MV, Mirnics K, Nisenbaum LK, Vawter MP, 2015. Olanzapine Reversed Brain Gene Expression Changes Induced by Phencyclidine Treatment in Non-Human Primates. Mol. Neuropsychiatry 1, 82–93. 10.1159/000430786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Huang HS, Baker SP, Galdzicka M, Ginns E, Akbarian S, 2009. Molecular Determinants of Dysregulated GABAergic Gene Expression in the Prefrontal Cortex of Subjects with Schizophrenia. Biol. Psychiatry 65, 1006–1014. 10.1016/j.biopsych.2008.11.019 [DOI] [PubMed] [Google Scholar]
- Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK, 2016. Nature of Glutamate Alterations in Schizophrenia: A Meta-Analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry 73, 665 10.1001/jamapsychiatry.2016.0442 [DOI] [PubMed] [Google Scholar]
- Miller EK, Lundqvist M, Bastos AM, 2018. Working Memory 2.0. Neuron 463–475. 10.1016/j.neuron.2018.09.023 [DOI] [PMC free article] [PubMed]
- Minzenberg MJ, Firl AJ, Yoon JH, Gomes GC, Reinking C, Carter CS, 2010. Gamma Oscillatory Power is Impaired During Cognitive Control Independent of Medication Status in First-Episode Schizophrenia. Neuropsychopharmacology 35, 2590–2599. 10.1038/npp.2010.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH, Wang X-J, 2014. Linking Microcircuit Dysfunction to Cognitive Impairment: Effects of Disinhibition Associated with Schizophrenia in a Cortical Working Memory Model. Cereb. Cortex 24, 859–872. 10.1093/cercor/bhs370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poels EMP, Kegeles LS, Kantrowitz JT, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR, 2014. Glutamatergic abnormalities in schizophrenia: A review of proton MRS findings. Schizophr. Res 152, 325–332. 10.1016/j.schres.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poels EMP, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR, 2013. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol. Psychiatry 19, 20–29. 10.1038/mp.2013.136 [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, McCullumsmith RE, 2014. Localization of excitatory amino acid transporters EAAT1 and EAAT2 in human postmortem cortex: A light and electron microscopic study. Neuroscience 277, 522–540. 10.1016/j.neuroscience.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF, 1996. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. 10.1016/S0896-6273(00)80086-0 [DOI] [PubMed] [Google Scholar]
- Rowland LM, Summerfelt A, Wijtenburg A, Du X, Chiappelli JJ, Krishna N, West J, Muellerklein F, Kochunov P, Hong LE, 2016. Frontal Glutamate and gamma-Aminobutyric Acid Levels and Their Associations With Mismatch Negativity and Digit Sequencing Task Performance in Schizophrenia. JAMA Psychiatry 73, 166–174. 10.1001/jamapsychiatry.2015.2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkowski D, Gallinat J, 2015. Dysfunctional Prefrontal Gamma-Band Oscillations Reflect Working Memory and Other Cognitive Deficits in Schizophrenia. Biol. Psychiatry 77, 1010–1019. 10.1016/J.BIOPSYCH.2015.02.034 [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K, 2009. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. 10.1038/nature07991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starc M, Murray JD, Santamauro N, Savic A, Diehl C, Cho YT, Srihari V, Morgan PT, Krystal JH, Wang X-J, Repovs G, Anticevic A, 2017. Schizophrenia is associated with a pattern of spatial working memory deficits consistent with cortical disinhibition. Schizophr. Res 181, 107–116. 10.1016/j.schres.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, White R, Featherstone RE, Ray R, Ortinski PI, Banerjee A, Gandal MJ, Lin R, Alexandrescu A, Liang Y, Gur RE, Borgmann-Winter KE, Carlson GC, Hahn CG, Siegel SJ, 2014. Pyramidal Cell Selective Ablation of N-Methyl-D-Aspartate Receptor 1 Causes Increase in Cellular and Network Excitability. Biol. Psychiatry 77, 1–18. 10.1016/j.biopsych.2014.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SF, Tso IF, 2015. GABA abnormalities in schizophrenia: A methodological review of in vivo studies. Schizophr. Res 167, 84–90. 10.1016/j.schres.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, 2010. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk DW, Sampson AR, Zhang Y, Edelson JR, Lewis DA, 2016. Cortical GABA markers identify a molecular subtype of psychotic and bipolar disorders. Psychol. Med 46, 2501–2512. 10.1017/S0033291716001446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Pradhan S, Coughlin JM, Trivedi A, Dubois SL, Crawford JL, Sedlak TW, Nucifora FC, Nestadt G, Nucifora LG, Schretlen DJ, Sawa A, Barker PB, 2019. Assessing Brain Metabolism with 7-T Proton Magnetic Resonance Spectroscopy in Patients with First-Episode Psychosis. JAMA Psychiatry 76, 314–323. 10.1001/jamapsychiatry.2018.3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein RL, Schirm AL, Lazar NA, 2019. Moving to a World Beyond “p < 0.05.” Am. Stat 73, 1–19. 10.1080/00031305.2019.1583913 [DOI] [Google Scholar]
- Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH, 2000. Inhibition-based rhythms: Experimental and mathematical observations on network dynamics. Int. J. Psychophysiol 38, 315–336. 10.1016/S0167-8760(00)00173-2 [DOI] [PubMed] [Google Scholar]