

Abstract
Objective
Chronic obstructive pulmonary disease (COPD) is a progressive disease that causes significant mortality and morbidity worldwide and is primarily caused by the inhalation of cigarette smoke (CS). Lack of effective treatments for COPD means there is an urgent need to identify new therapeutic strategies for the underlying mechanisms of pathogenesis. Tristetraprolin (TTP) encoded by the Zfp36 gene is an anti‐inflammatory protein that induces mRNA decay, especially of transcripts encoding inflammatory cytokines, including those implicated in COPD.
Methods
Here, we identify a novel protective role for TTP in CS‐induced experimental COPD using Zfp36aa/aa mice, a genetically modified mouse strain in which endogenous TTP cannot be phosphorylated, rendering it constitutively active as an mRNA‐destabilising factor. TTP wild‐type (Zfp36 +/+) and Zfp36aa/aa active C57BL/6J mice were exposed to CS for four days or eight weeks, and the impact on acute inflammatory responses or chronic features of COPD, respectively, was assessed.
Results
After four days of CS exposure, Zfp36aa/aa mice had reduced numbers of airway neutrophils and lymphocytes and mRNA expression levels of cytokines compared to wild‐type controls. After eight weeks, Zfp36aa/aa mice had reduced pulmonary inflammation, airway remodelling and emphysema‐like alveolar enlargement, and lung function was improved. We then used pharmacological treatments in vivo (protein phosphatase 2A activator, AAL(S), and the proteasome inhibitor, bortezomib) to promote the activation and stabilisation of TTP and show that hallmark features of CS‐induced experimental COPD were ameliorated.
Conclusion
Collectively, our study provides the first evidence for the therapeutic potential of inducing TTP as a treatment for COPD.
Keywords: chronic obstructive pulmonary disease, inflammation, protein phosphatase 2A, tristetraprolin, ubiquitin–proteasome system
Cigarette smoke (CS) exposure increases the mRNA expression of inflammatory mediators resulting in pulmonary inflammation. Ongoing inflammation drives the development of airway remodelling and emphysema, subsequently leading to lung function decline. This study highlights the importance of having active TTP to control CS‐induced inflammation, pulmonary remodelling, emphysema and impaired lung function in mice.
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease that is characterised by chronic airway inflammation, airway remodelling, emphysema and impaired lung function.1 It is now the third leading cause of morbidity and death worldwide and imposes a substantial socioeconomic burden.2 The inhalation of cigarette smoke (CS) is a major risk factor for COPD.3 Other factors such as wood and cooking smoke, occupational exposure to dust and chemicals and air pollution have also been associated with the aetiology of COPD.4 Current mainstay treatments for COPD are bronchodilators and inhaled steroids, which are only partially effective in managing disease symptoms and do not treat the underlying causes or halt the progression of the disease.5 Hence, there is an urgent need to understand the mechanisms of disease pathogenesis and identify new potential therapeutic targets.
Tristetraprolin (TTP) encoded by the Zfp36 gene is an anti‐inflammatory protein that post‐transcriptionally suppresses many pro‐inflammatory factors, including cytokines that are known to drive the pathogenesis of COPD. TTP induces mRNA decay by binding to the adenylate and uridylate (AU)‐rich elements in the 3’‐untranslated regions of target cytokines.6, 7, 8 The mRNA‐destabilising function of TTP is regulated by the phosphorylation and dephosphorylation of two particular residues, serines 52 and 178 of murine TTP (serines 60 and 186 of the human orthologue).9 It is phosphorylated and inactivated by mitogen‐activated protein kinase (MAPK)‐activated protein kinase 2 (MK2), leading to the stabilisation of target mRNA.10 Protein phosphatase 2A (PP2A) can dephosphorylate TTP, enhancing its activity and promoting the de‐stabilisation of target mRNA.11, 12 However, active TTP is targeted by the ubiquitin–proteasome system for degradation.13 In these ways, TTP is controlled by phosphorylation at key serines that act as a molecular switch, phosphorylated – OFF and hypophosphorylated – ON. Thus, TTP is a tractable target for novel anti‐inflammatory treatments for COPD.
The role of TTP in lung inflammation and respiratory disease has not been studied. Given that TTP regulates the mRNA stability of multiple inflammatory genes, including those involved in COPD, such as IL‐6, TNF‐α and the chemokines CXCL1 and CXCL2 that are functional homologs of human IL‐8, we hypothesised that active TTP would be beneficial in COPD.14, 15, 16 To test this, we utilised a genetically modified constitutively TTP active mouse strain in which serine 52 and serine 178 of the endogenous TTP protein are replaced with non‐phosphorylatable alanine residues (Zfp36aa/aa).17 The substitution of two amino acids of the endogenous TTP protein renders it constitutively active as an mRNA‐destabilising factor. We examined the impact of active TTP in experimental CS‐induced COPD using Zfp36aa/aa mice. We show for the first time that active TTP protects mice from hallmark features of experimental COPD by reducing pulmonary inflammation, airway remodelling, alveolar enlargement and improving lung function. Hence, we identify a previously unrecognised role for active TTP in regulating CS‐induced inflammatory responses and the pathogenesis of experimental COPD. Thus, pharmacological strategies that promote TTP activity may be novel therapies for COPD. We then tested this by treating wild‐type C57BL/6J mice with a PP2A activator (AAL(S)) that activates TTP, alone and in combination with the proteasome inhibitor, bortezomib (BORT), to stabilise TTP, and assessed the impact on the induction of experimental CS‐induced COPD. Treatment ameliorated the hallmark features of disease paving the way for future drug development.
Results
Acute CS‐induced pulmonary inflammation is reduced in TTP active mice
To evaluate the impact of active TTP on acute CS‐induced inflammatory responses in the lung, wild‐type and TTP active (Zfp36aa/aa) mice were exposed to CS for four days (Figure 1a) and pulmonary inflammation was assessed in BAL fluid. CS exposure of wild‐type mice significantly increased the numbers of total leucocytes, neutrophils, macrophages and lymphocytes in BAL fluid compared to normal air‐exposed wild‐type controls (Figure 1b). CS exposure of Zfp36aa/aa mice significantly increased total neutrophils and leucocytes, but not macrophages or lymphocytes compared to their air‐exposed Zfp36aa/aa controls. Notably, CS‐exposed Zfp36aa/aa mice had reduced numbers of neutrophils and lymphocytes compared to CS‐exposed wild‐type mice.
Figure 1.
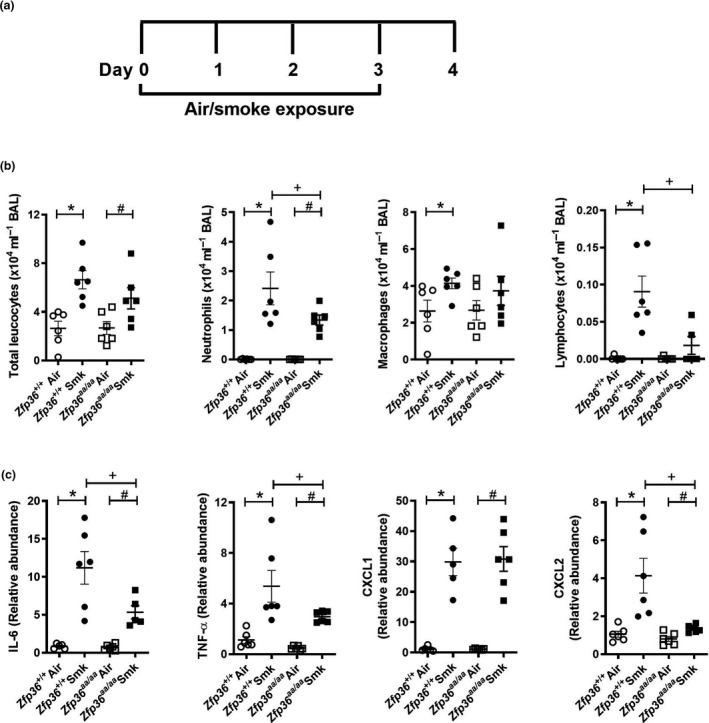
Acute cigarette smoke (CS)‐induced pulmonary inflammation is reduced in tristetraprolin (TTP) active mice. (a) Wild‐type (Zfp36 +/+) and TTP active (Zfp36aa/aa) C57BL/6J mice were exposed to CS or normal air for four days. (b) Total leucocytes, neutrophils, macrophages and lymphocytes in bronchoalveolar lavage (BAL) fluid. (c) Interleukin‐6 (Il6), tumor necrosis factor‐alpha (Tnfα), chemokine C‐X‐C motif ligand (Cxcl)1 and Cxcl2 mRNA expression levels in lung homogenates. Data (n = 5 or 6) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Zfp36 +/+ Air, # represents P ≤ 0.05 compared to Zfp36aa/aa Air, and + represents P ≤ 0.05 compared to Zfp36 +/+ Smoke (Smk).
Given that CS‐exposed Zfp36aa/aa mice had reduced pulmonary cellular inflammation, mRNA expression of associated pro‐inflammatory cytokines and chemokines that are also TTP targets and involved in COPD pathogenesis was assessed in lung homogenates. Acute CS exposure of both wild‐type and Zfp36aa/aa mice for 4 days increased the mRNA expression of the TTP targets Il6, Tnfα, Cxcl1 and Cxcl2 compared to air‐exposed wild‐type and Zfp36aa/aa controls (Figure 1c). There was a significant reduction in the mRNA expression of the pro‐inflammatory cytokines, Il6 and Tnfα, in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls (Figure 1c). There was no change in pro‐inflammatory chemokine Cxcl1 mRNA expression, but there was a significant reduction in Cxcl2 mRNA expression in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls.
Chronic CS‐induced pulmonary inflammation is reduced in TTP active mice
Since pulmonary inflammation is a key driver of chronic pathologies, we next assessed the impact of active TTP on chronic features of experimental CS‐induced COPD. Wild‐type and Zfp36aa/aa mice were exposed to CS for 8 weeks (Figure 2a), and chronic inflammatory responses were assessed. CS exposure of both strains significantly increased numbers of total leucocytes, neutrophils, macrophages and lymphocytes in BAL fluid compared to their respective air‐exposed controls (Figure 2b). Notably, CS‐exposed Zfp36aa/aa mice had reduced total leucocytes and neutrophils in BAL fluid, but not macrophages or lymphocytes compared to CS‐exposed wild‐type controls.
Figure 2.
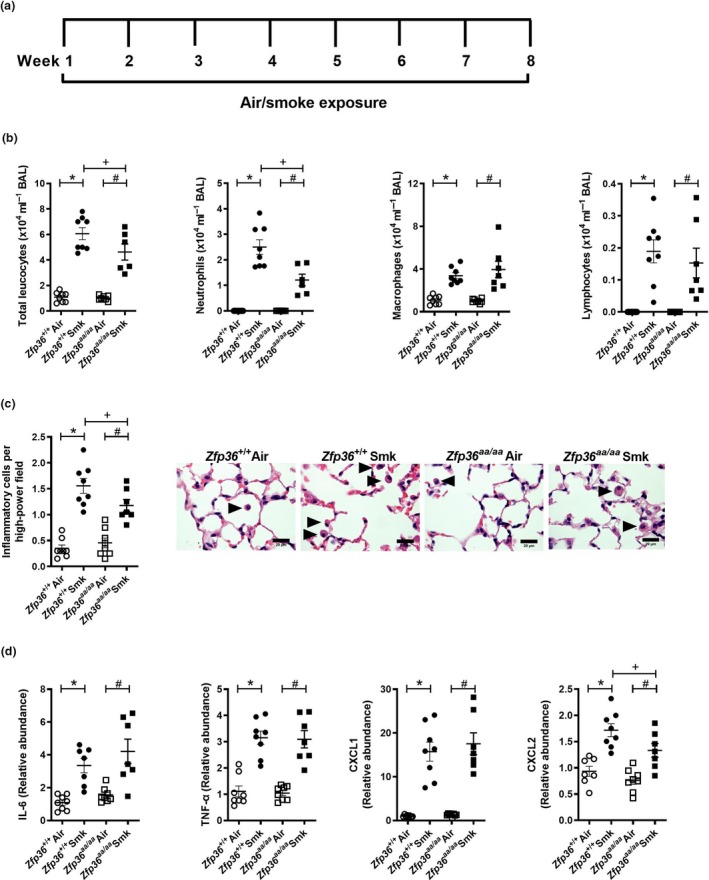
Chronic cigarette smoke (CS)‐induced pulmonary inflammation is reduced in tristetraprolin (TTP) active mice. (a) Wild‐type (Zfp36 +/+) and TTP active (Zfp36aa/aa) C57BL/6J mice were exposed to CS or normal air for eight weeks. (b) Total leucocytes, neutrophils, macrophages and lymphocytes in bronchoalveolar lavage (BAL) fluid. (c) Numbers of parenchymal inflammatory cells (arrowheads) were determined in haematoxylin and eosin‐stained lung sections. Scale bars = 20 μm. (d) Interleukin‐6 (Il6), tumor necrosis factor‐alpha (Tnfα), chemokine C‐X‐C motif ligand (Cxcl)1 and Cxcl2 mRNA expression were determined in lung homogenates. Data (n = 6‐8) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Zfp36 +/+ Air, # represents P ≤ 0.05 compared to Zfp36aa/aa Air, and + represents P ≤ 0.05 compared to Zfp36 +/+ Smoke (Smk).
Next, inflammatory cell numbers in the parenchyma were assessed. Eight weeks of CS exposure of both wild‐type and Zfp36aa/aa mice significantly increased the numbers of inflammatory cells in the parenchyma of both strains compared to their respective air‐exposed controls (Figure 2C). However, the numbers of parenchymal inflammatory cells in CS‐exposed Zfp36aa/aa were significantly reduced compared to CS‐exposed wild‐type controls.
Next, the mRNA expression of pro‐inflammatory cytokines and chemokines in the lungs after 8 weeks of CS exposure was assessed. CS exposure of both wild‐type and Zfp36aa/aa mice increased the mRNA expression of Il6, Tnfα, Cxcl1 and Cxcl2 compared to their respective air‐exposed controls (Figure 2d). There were no significant differences in the levels of mRNA expression of Il6, Tnfα or Cxcl1 between CS‐exposed wild‐type and Zfp36aa/aa mice, but there was a significant reduction in the expression of Cxcl2 in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls.
Chronic CS‐induced small airway remodelling is reduced in TTP active mice
We previously showed that mice develop small airway remodelling characterised by increases in collagen deposition, epithelial cell area and numbers of nuclei around and in the small airways in experimental COPD.18, 19, 20, 21, 22, 23 As expected, chronic CS exposure of wild‐type mice increased collagen deposition around the airways compared to normal air‐exposed wild‐type controls (Figure 3a). CS‐exposed Zfp36aa/aa mice were protected against collagen deposition with no differences compared to normal air‐exposed Zfp36aa/aa controls and had significantly reduced deposition compared to CS‐exposed wild‐type controls. In line with these data, CS exposure of wild‐type mice increased the mRNA expression of the pro‐fibrotic/remodelling protein Fn in the lungs compared to air‐exposed controls, but there was no increase in Zfp36aa/aa exposed to CS compared to their controls. Accordingly, CS‐exposed Zfp36aa/aa had significantly lower expression of Fn in the lungs compared to CS‐exposed wild‐type mice. CS exposure also significantly increased small airway epithelial cell area in wild‐type mice compared to air‐exposed wild‐type controls, but this did not occur in Zfp36aa/aa compared to their air‐exposed controls. Again, CS‐exposed Zfp36aa/aa mice had significantly reduced small airway epithelial cell area compared to CS‐exposed wild‐type controls. We then determined whether the increase in epithelial cell area was associated with an increase in the number of nuclei in the small airways, which is indicative of increased epithelial cell number. CS exposure increased nuclei numbers in the small airways of both wild‐type and Zfp36aa/aa mice. However, for this parameter there was no difference in nuclei numbers in the small airways between CS‐exposed wild‐type and Zfp36aa/aa mice.
Figure 3.
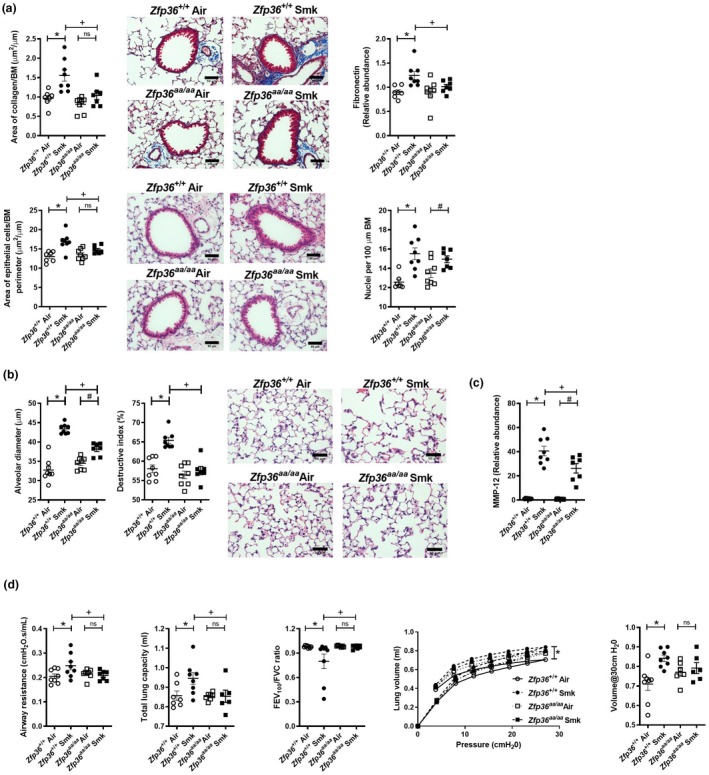
Chronic cigarette smoke (CS)‐induced airway remodelling, emphysema and impaired lung function are reduced in tristetraprolin (TTP) active mice. Wild‐type (Zfp36 +/+) and TTP active (Zfp36aa/aa) C57BL/6J mice were exposed to CS or normal air for eight weeks. (a) Area of collagen deposition (μm2) per basement membrane (BM) perimeter (μm) in Masson’s trichrome‐stained lung sections. Scale bars = 50 μm. Fibronectin mRNA expression was determined in lung homogenates. Small airway epithelial thickness in terms of epithelial cell area (μm2) per BM perimeter (μm) in haematoxylin and eosin (H&E)‐stained lung sections. Scale bars = 50 μm. Number of epithelial cells in H&E‐stained lung sections were assessed by enumerating the number of nuclei per 100 μm of BM perimeter. (b) Alveolar diameter (μm) was determined in haematoxylin and eosin‐stained lung sections using the mean linear intercept and destructive index techniques. Scale bars = 50 μm. (c) Matrix metalloproteinase (Mmp)12 mRNA expression was determined in lung homogenates. (d) Lung function was assessed in terms of airway resistance, total lung capacity, forced expiratory volume in 100 s/forced vital capacity (FEV100/FVC) ratio, pressure–volume loops and lung compliance at 30 cmH20. Data (n = 6–8) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Zfp36 +/+ Air, # represents P ≤ 0.05 compared to Zfp36aa/aa Air, + represents P ≤ 0.05 compared to Zfp36 +/+ Smoke (Smk), and ns represents not significant.
Chronic CS‐induced emphysema‐like alveolar enlargement and impaired lung function are reduced in TTP active mice
We previously showed that mice develop emphysema‐like alveolar enlargement and impaired lung function after eight weeks of CS exposure.18, 19, 20, 21, 22, 23, 24 In this study, chronic CS exposure of wild‐type mice for 8 weeks resulted in increased alveolar diameter and septal damage, which were assessed using the mean linear intercept and destructive index methods, respectively, compared to air‐exposed wild‐type controls (Figure 3b). In CS‐exposed Zfp36aa/aa groups, there were increases in alveolar diameter but not septal damage, compared to their air‐exposed controls. Notably, alveolar diameter and septal damage were also significantly reduced in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls.
We then assessed the impact of active TTP on matrix metalloproteinase‐12 (MMP‐12) as major protease associated with inducing emphysema. CS exposure of both wild‐type and Zfp36aa/aa mice increased the mRNA expression Mmp12 in whole lung tissue compared to their respective controls (Figure 3c). Notably, and concomitant with the reduction in emphysema, the expression of MMP‐12 was significantly reduced in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls.
Next, we assessed the potential effects of active TTP and suppressed inflammation, remodelling and emphysema on functional impacts and improved lung function in experimental COPD. CS exposure of wild‐type mice increased airway resistance and total lung capacity and reduced FEV100/FVC ratio (Figure 3d) compared to air‐exposed controls. Importantly, Zfp36aa/aa mice were protected against changes in airway resistance, total lung capacity and FEV100/FVC ratio, which were the same as in air‐exposed Zfp36aa/aa controls. Airway resistance and total lung capacity were significantly reduced, while FEV100/FVC ratio was significantly increased in CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls. CS exposure of wild‐type mice also increased compliance, determined during a pressure–volume loop manoeuvre compared to air‐exposed controls. CS‐exposed Zfp36aa/aa mice were protected against changes in pressure–volume loops and lung compliance, which were the same as in their air‐exposed controls.
BORT and BORT + AAL(S) treatment reduced acute CS‐induced pulmonary inflammation
Collectively, our data revealed that active TTP suppresses the hallmark features of human disease in CS‐induced experimental COPD. These data suggest that pharmacological strategies that promote the activation and stabilisation of TTP are novel anti‐inflammatory treatments with the potential to prevent irreversible damage and/or the progression of COPD. We tested this potential by examining the effects of treating mice with a PP2A activator (AAL(S)) to activate TTP, alone and in combination with the proteasome inhibitor, bortezomib (BORT), to stabilise TTP, on the induction of experimental CS‐induced COPD.
We first evaluated the impact of treatments on acute CS‐induced inflammation in the lungs (Figure 4a). Mice were treated daily with vehicle, BORT, AAL(S) or BORT + AAL(S), immediately before CS exposure for four days and controls were vehicle‐treated. Pulmonary cellular inflammation was increased in BAL fluid of vehicle‐treated CS‐exposed compared to air‐exposed mice (Figure 4b). Notably, BORT and BORT + AAL(S) treatment significantly reduced the numbers of neutrophils and lymphocytes in BAL fluid compared to vehicle‐treated CS‐exposed mice. Lymphocytes were significantly reduced with either treatment alone or in combination.
Figure 4.
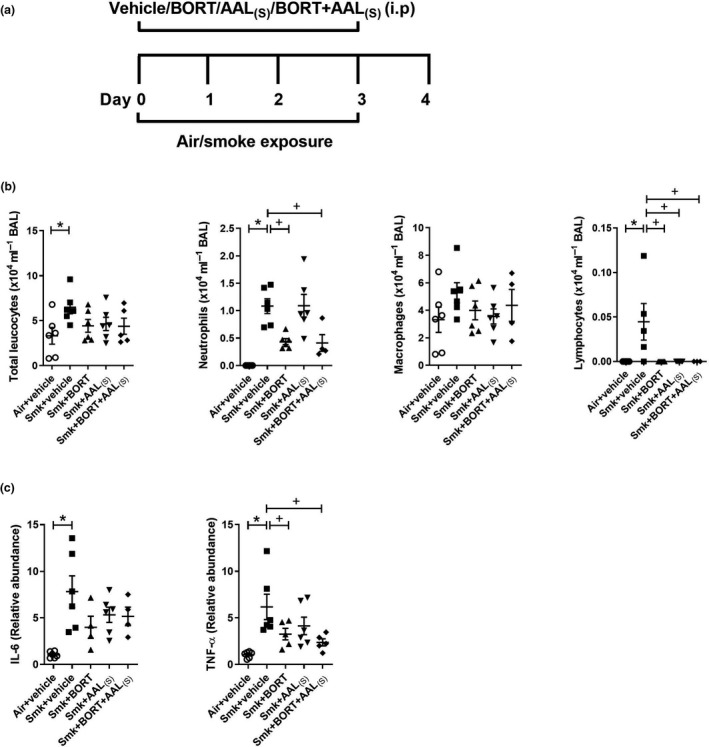
Acute cigarette smoke (CS)‐induced pulmonary inflammation is reduced in BORT‐ and BORT + AAL(S)‐treated mice. (a) Wild‐type C57BL/6J mice were treated daily (i.p.) with vehicle, BORT (0.2 mg kg−1), AAL(S) (0.8 mg kg−1) or BORT + AAL(S) and exposed to CS for four days. Controls were vehicle‐treated and exposed to normal air. (b) Total leucocytes, neutrophils, macrophages and lymphocytes in bronchoalveolar lavage (BAL) fluid. (c) Interleukin‐6 (Il6) and tumor necrosis factor‐alpha (TNF‐α) mRNA expression were determined in lung homogenates. Data (n = 6–8) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Air + vehicle and + represents P ≤ 0.05 compared to Smoke + vehicle.
We then assessed the impact of treatments on the expression of TTP targets Il6, Tnfα, Cxcl1 and Cxcl2. The mRNA expression of Il6, Tnfα (Figure 4c), Cxcl1 and Cxcl2 (not shown) was significantly increased in vehicle‐treated CS‐exposed compared to air‐exposed mice. Notably, there was a significant reduction in the mRNA expression of the pro‐inflammatory cytokine and archetypal TTP target TNF‐α in CS‐exposed mice treated with BORT or BORT + AAL(S) compared to vehicle‐treated CS‐exposed mice (Figure 4c). There was also a non‐significant trend to a reduction in IL‐6, but the chemokines were unaffected by treatment (not shown).
BORT and BORT + AAL(S) treatment reduced chronic CS‐induced pulmonary inflammation in experimental COPD
We next assessed the impact of treatments on chronic features of CS‐induced experimental COPD (Figure 5a). Vehicle‐treated chronically CS‐exposed mice had significantly increased total leucocytes, neutrophils, macrophages and lymphocytes in BAL fluid, compared to vehicle‐treated air‐exposed controls. Notably, BORT and BORT + AAL(S) treatment of CS‐exposed mice significantly reduced the numbers of neutrophils and lymphocytes in BAL fluid compared to vehicle‐treated CS‐exposed mice. Given that these inflammatory cells were also repressed with treatment in acute CS exposure model (Figure 4b), these data suggest that the effect of treatments on neutrophil and lymphocyte inflammation was sustained throughout chronic CS exposure. Although we measured pro‐inflammatory mRNA expression after chronic CS exposure (Figure 5c), there was no effect of treatments. This suggests that it is the early‐stage impact of TTP that is likely most important or that other factors are involved.
Figure 5.
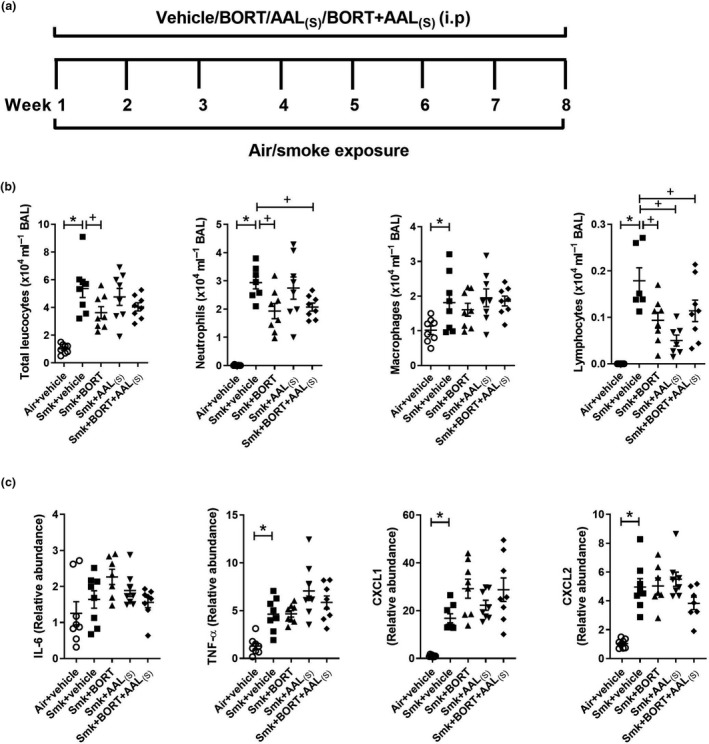
Chronic cigarette smoke (CS)‐induced pulmonary inflammation is reduced in BORT‐ and BORT + AAL(S)‐treated mice. (a) Wild‐type C57BL/6J mice were treated daily (i.p.) with vehicle, BORT (0.2 mg kg−1), AAL(S) (0.8 mg kg−1) or BORT + AAL(S) and exposed to CS for eight weeks. Controls were vehicle‐treated and exposed to normal air. (b) Total leucocytes, neutrophils, macrophages and lymphocytes in bronchoalveolar lavage (BAL) fluid. (c) Interleukin‐6 (Il6), tumor necrosis factor‐alpha (TNF‐α), chemokine C‐X‐C motif ligand (Cxcl)1 and Cxcl2 mRNA expression were determined in lung homogenates. Data (n = 6–8) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Air + vehicle and + represents P ≤ 0.05 compared to Smoke + vehicle.
BORT and BORT + AAL(S) treatment reduced chronic CS‐induced small airway remodelling but not emphysema‐like alveolar enlargement or lung function impairment in experimental COPD
We next examined the effect of the treatments on the chronic features of experimental COPD. Vehicle‐treated chronically CS‐exposed mice had increased collagen deposition (Figure 6a) and epithelial cell area around the small airways compared to vehicle‐treated air‐exposed controls. Importantly, both of these hallmark features of small airway remodelling were suppressed by treatment with BORT + AAL(S). Moreover, BORT alone was sufficient to significantly reduce CS‐induced collagen deposition around the small airways, and there was a trend towards lower epithelial cell area although the reduction was not significant (P = 0.0722).
Figure 6.
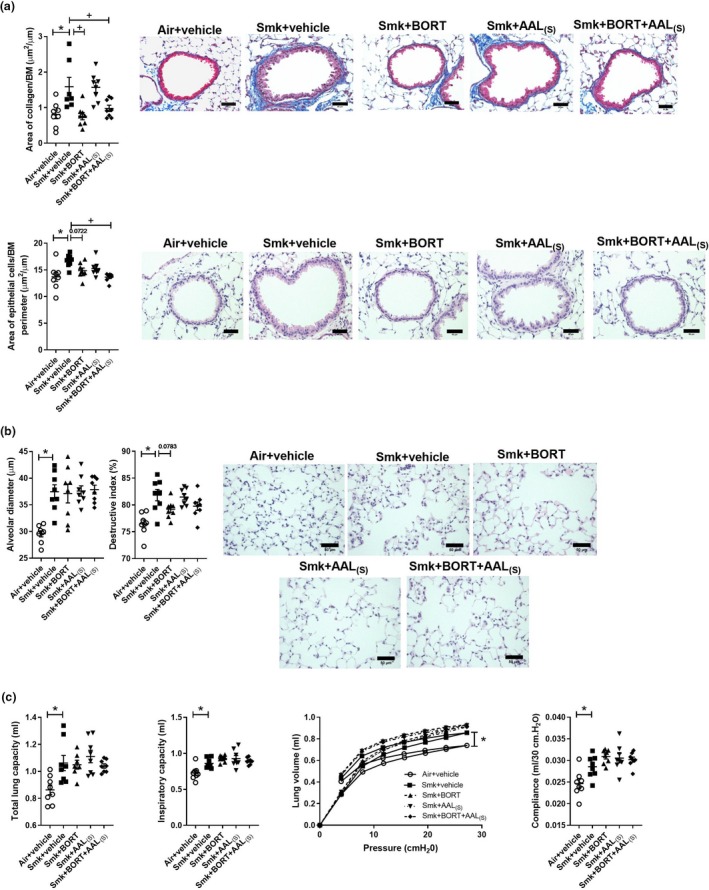
Chronic cigarette smoke (CS)‐induced airway remodelling but not emphysema or impaired lung function is reduced in BORT‐ and BORT + AAL(S)‐treated mice. Wild‐type C57BL/6J mice were treated daily (i.p.) with vehicle, BORT (0.2 mg kg−1), AAL(S) (0.8 mg kg−1) or BORT + AAL(S) and exposed to CS for eight weeks. Controls were vehicle‐treated mice exposed to normal air. (a) Area of collagen deposition (μm2) per basement membrane (BM) perimeter (μm) in Masson’s trichrome‐stained lung sections. Scale bars = 50 μm. Small airway epithelial thickness in terms of epithelial cell area (μm2) per BM perimeter (μm) in haematoxylin and eosin (H&E)‐stained lung sections. Scale bars = 50 μm. (b) Alveolar diameter (μm) was determined in haematoxylin and eosin‐stained lung sections using the mean linear intercept and destructive index technique. Scale bars = 50 μm. Lung function was assessed in terms of (c) total lung capacity, inspiratory capacity, pressure–volume loops and lung compliance at 30 cm H20. Data (n = 6–8) are presented as means ± sem from two independent experiments. *represents P ≤ 0.05 compared to Air + vehicle and + represents P ≤ 0.05 compared to Smoke + vehicle.
Vehicle‐treated chronically CS‐exposed mice also had emphysema‐like alveolar enlargement, compared to vehicle‐treated air‐exposed controls (Figure 6b). While there was a trend towards repression in destructive index in CS‐exposed mice treated with BORT and BORT + AAL(S), the reduction was not significant (P = 0.0783). There were no other changes in alveolar diameter or destructive index.
Finally, we assessed the potential of treatments to suppress CS‐induced impairment of lung function in experimental COPD. Vehicle‐treated chronically CS‐exposed mice had increased total lung capacity, inspiratory capacity and P‐V loop extracted lung compliance (Figure 6c), compared to vehicle‐treated air‐exposed controls. However, no treatments (alone or in combination) had significant effects on CS‐induced lung function impairment.
Discussion
In this study, a previously unrecognised role for TTP in suppressing CS‐induced pulmonary inflammation, airway remodelling, emphysema and impaired lung function was identified. Using established mouse models of CS‐induced experimental COPD and TTP constitutively active Zfp36aa/aa mice, we demonstrate that the presence of active TTP significantly reduced CS‐induced pulmonary cellular inflammation, characterised by reduced neutrophils in BAL fluid and numbers of inflammatory cells in the parenchyma. The reduced cellular inflammation was associated with concomitant reductions in the expression of pro‐inflammatory cytokines and chemokines that recruit neutrophils to the airway lumen and induce pulmonary inflammation in response to CS exposure. Mice expressing active TTP were also protected against chronic disease features including CS‐induced small airway remodelling, with no excess collagen deposition nor increases in small airway epithelial cell area. Importantly, these mice also had reduced emphysema‐like alveolar enlargement and improved lung function. Treatment of CS‐exposed wild‐type mice with the novel PP2A and TTP activator, AAL(S), and the clinically used proteasome inhibitor, BORT, also suppressed the important hallmark features of human disease of neutrophilic inflammation and airway remodelling in experimental COPD. Collectively, these data show for the first time that inducing TTP may have therapeutic potential in COPD and other chronic respiratory diseases, underscoring the need for drug discovery and development programmes that promote TTP.
CS exposure for four days enabled the evaluation of early smoke‐induced inflammatory responses in the lung,25, 26 while exposure for eight weeks induces the development of the hallmark features of COPD that are observed in humans.18, 21, 22, 27 These include pulmonary inflammation (chronic bronchitis), small airway remodelling, emphysema‐like alveolar enlargement and impaired lung function.28 The TTP active mouse (Zfp36aa/aa) is a valuable and novel tool to investigate roles in regulating inflammatory responses in the lung and in the pathogenesis of experimental COPD. Mice deficient in TTP develop severe inflammation and have a high mortality rate in the absence of exogenous inflammatory stimuli, and therefore, using these mice to study inflammatory diseases is not appropriate.15 Thus, we utilised TTP active mice to show that active TTP represses inflammation and protects mice against CS‐induced experimental COPD. Notably, as in humans, the hallmark features of COPD do not respond well to corticosteroid treatment, so the induction of TTP is an alternative approach.18
CS exposure for both four days and eight weeks resulted in increased pulmonary inflammation characterised by exaggerated levels of total leucocytes, neutrophils, macrophages and lymphocytes in the airway lumen. These cells have previously been shown to be increased in both experimental and human COPD.18, 21, 29, 30, 31 CS‐exposed Zfp36aa/aa mice had significantly reduced influx of neutrophils in the airway lumen after four days and eight weeks of CS exposure compared to CS‐exposed wild‐type controls. Neutrophils have been implicated in the development of alveolar destruction in COPD through the release of serine proteases.32, 33 Increased levels of sputum neutrophils have also been associated with rapid declines in lung function and increased COPD disease severity.29, 31, 34 We also quantified neutrophil numbers in the lung tissue by flow cytometry, but did not observe any difference between CS‐exposed Zfp36++/++ and Zfp36aa/aa mice (data not shown). In contrast, CS‐exposed Zfp36aa/aa mice did have reduced numbers of parenchymal‐associated inflammatory cells in lung tissue after eight weeks of CS exposure. These cells were predominantly macrophages, which were not reduced in the BAL fluid of CS‐exposed Zfp36aa/aa mice. Notably, the inflammatory cell profile from BAL fluid represents that from distal airways and alveoli while the inflammatory cells in lung tissue represent that from the parenchyma. These findings suggest that active TTP regulates inflammatory responses to CS in various parts of the airways and lungs. This likely results from active TTP inducing the degradation of mRNA of target cytokines such as Tnfα and Cxcl2 that recruit inflammatory cells.14, 15 Our findings are consistent with those of others that have previously shown that a lack of TTP leads to severe inflammation as a result of exaggerated TNF‐α expression.15 Notably, a recent study demonstrated that exposure to CS significantly reduced TTP levels in mouse alveolar macrophages and rat alveolar epithelial cells.35 This led to increased Tnfα mRNA stability and higher subsequent levels of TNF‐α in these cells.
In support of our inflammatory cell data, CS exposure also increased the mRNA expression of key pro‐inflammatory cytokines Il6 and Tnfα and chemokines Cxcl1 and Cxcl2, in both CS‐exposed wild‐type and Zfp36aa/aa mice. Levels of IL‐6 are increased in the sputum of COPD patients and are associated with reduced lung function.36, 37 Increased expression of TNF‐α occurs in mice and humans following CS exposure.18, 38, 39 This cytokine induces the expression of the neutrophil chemoattractants CXCL1 and CXCL2.40 Notably, the mRNA expression of Il6 and Tnfα was significantly lower in the lungs of Zfp36aa/aa mice compared to wild‐type controls after four days, but not after eight weeks of CS exposure. This may be attributed to differences in acute versus chronic phases of the disease, where the induction of pro‐inflammatory cytokines predominates in the acute phase, while this is less critical in the chronic phase. There was a significant reduction in the mRNA expression of Cxcl2, but not Cxcl1 in the lungs of CS‐exposed Zfp36aa/aa mice compared to CS‐exposed wild‐type controls after eight weeks of CS exposure. Both these chemokines are functional homologs of human IL‐8 and are increased in both mouse models and human COPD.18, 41, 42 The decreased Cxcl2 expression is consistent with the reduced neutrophil numbers in the BAL of CS‐exposed Zfp36aa/aa mice. Since CXCL2 is a known target of TTP,14 our findings suggest that the presence of active TTP may induce the decay of Cxcl2 mRNA in response to CS, and prevent the recruitment of neutrophils to the lung.
The continuous inflammatory response in COPD leads to the development of airway remodelling.43 Excessive production of extracellular matrix proteins such as fibronectin by airway epithelial and smooth muscle cells in response to CS is linked to the development of airway remodelling in COPD.44, 45 In our study, while CS exposure increased collagen deposition around the airways and increased airway epithelial cell thickness in wild‐type mice, these effects were not observed in CS‐exposed Zfp36aa/aa mice, highlighting a role for active TTP in suppressing airway remodelling. Increased collagen deposition occurs in emphysematous lung tissue and has been postulated to be associated with tissue destruction in human emphysema.46, 47, 48 Increased airway epithelial thickness has also been observed in COPD patients.49 The levels of the extracellular matrix protein fibronectin are also elevated in the airways of COPD patients, contribute to airway remodelling and have an inverse correlation with lung function in COPD patients.44, 45 The reduction in airway remodelling observed in Zfp36aa/aa mice was associated with reduced levels of fibronectin in the lung.
We previously showed that chronic CS exposure leads to emphysema‐like alveolar enlargement in experimental COPD.18, 19, 21, 23, 24, 50, 51 In our current study, Zfp36aa/aa mice were protected against CS‐induced alveolar enlargement. This was associated with reduced mRNA expression of Mmp12 in the lungs. MMP‐12 is a proteinase that is primarily produced by macrophages and plays a role in the destruction of alveolar walls and the development of emphysema.52, 53, 54 Mice deficient in MMP‐12 are protected against CS‐induced emphysema.32 Similarly, guinea pigs exposed to CS for 6 months and treated with the dual MMP‐9/MMP‐12 inhibitor were also protected against CS‐induced emphysema 55. Microarray data from our previous study showed reduced expression of MMP12 by ~ 50% in LPS‐treated Zfp36aa/aa compared to LPS‐treated Zfp36++/++ bone marrow‐derived macrophages, suggesting that MMP12 is highly likely a TTP target.56 Pulmonary MMPs degrade collagen and elastin that promote neutrophil and macrophage influx, and are also associated with gas trapping and small airways remodelling in COPD.57, 58 In our study, enhancing TTP activity reduced MMP12‐expressing macrophages that occur alongside reductions in remodelling and alveolar enlargement. Thus, we find that the reduction in MMP12 may contribute to amelioration of disease in Zfp36aa/a mice.
Impairment of lung function and breathing difficulties are the most important features in COPD patients. CS exposure of wild‐type mice for eight weeks resulted in impaired lung function with increased airway resistance and total lung capacity, reduced FEV100/FVC ratio and increased compliance. Importantly, CS‐exposed Zfp36aa/aa mice did not have impaired lung function. Protection against increased airway resistance in CS‐exposed Zfp36aa/aa is consistent with the decrease in collagen deposition around the airways, while no increases in total lung capacity or decreases in FEV100/FVC ratio are consistent with reduced alveolar enlargement. Taken together, our findings suggest that active TTP protects against a broad range of deleterious effects of CS exposure on lung function. This may be due to the ability of active TTP to induce the mRNA decay of pro‐inflammatory cytokines and chemokines, which then reduces pulmonary inflammation in response to CS and subsequently leads to reduced features of airway remodelling and emphysema and consequently improve lung function.
Our study highlights the importance of having active TTP to control CS‐induced inflammation, pulmonary remodelling, emphysema and impaired lung function in mice. Given that our CS‐induced experimental COPD mouse model is corticosteroid resistant, we have discovered that active TTP represses inflammation and the hallmark features of human COPD in vivo when corticosteroids cannot. As inflammation leads to progressive and irreversible tissue damage in COPD, our data also suggest that pharmacotherapeutic strategies targeting TTP represent novel anti‐inflammatory treatments that may not merely ameliorate the symptoms but may be viable interventions for preventing irreversible damage and/or the progression of COPD. We assessed this by systemically treating wild‐type mice with the novel PP2A and TTP activator, AAL(S), alone and in combination with the proteasome inhibitor, BORT. Notably, the combination of treatments exerted a sustained repression of pulmonary inflammation induced after 4 days and 8 weeks of CS exposure. The repression of neutrophils is particularly noteworthy, as neutrophilic inflammation in respiratory disease is refractory to corticosteroid treatment.18, 59, 60, 61, 62 While the impact of treatments on pro‐inflammatory cytokine and chemokine mRNA expression levels in the lungs after acute CS exposure was not as striking as the repression in TTP active mouse, BORT + AAL(S) significantly repressed Tnfα mRNA levels after four days, and features of CS‐induced airway remodelling at eight weeks were ameliorated. This is consistent with inflammation being a major driver of COPD pathogenesis and that developing therapies that restrain inflammation in conditions that are refractory to corticosteroids will be a clinical advance. Treatments did not alter CS‐induced emphysema‐like alveolar enlargement, nor lung function impairment. However, an important point is that the efficacy of treatments has not been optimised. They were administered intraperitoneally in standard dosing regimens, but these have not been refined to achieve maximum benefit in COPD. It is unknown whether the treatments reach the required tissues, whether exposure was for a sufficient period or whether administration directly to the lung would be more effective. The beneficial effects of these treatments would be maximised by performing drug development and formulation studies that optimise their efficacy that may see improve efficacy on suppressing chronic disease features. Further drug discovery is underway to develop these and other efficacious agents for pulmonary delivery. It is also important to note that we did not determine whether treatment activated and stabilised TTP expression. The amount of TTP protein and the mRNA‐destabilising activity of TTP are not directly related, and it is the phosphorylation status of TTP that determines its functional activity.9 Determining the amount of TTP in tissue will not indicate TTP activity. It is currently technically difficult to measure TTP activity (unphosphorylated form) and is beyond the scope of this study.
These data build upon our recent studies. We previously showed that PP2A activators (including AAL(S)) enhanced TTP anti‐inflammatory function and repressed cytokine production in airway epithelial cells in vitro.63, 64 In vivo, we also showed that targeting PP2A and proteasome activity ameliorates features of allergic airway disease; an effect speculated to be due to increased TTP activity.65 Our study also aligns with studies by the Clark group 66, where TTP active mice had an hypo‐inflamed phenotype and were protected against developing inflammatory arthritis. There, AAL(S) reduced inflammation and prevented bone erosion in vivo. We showed the effects of PP2A activators in vitro were mediated via TTP activation.66 Notably, others have shown that PP2A activity is significantly reduced in bronchial epithelial cells from COPD patients.67 While a direct link between TTP and PP2A activity was not investigated in that study, we propose that loss of PP2A activity may lead to loss of TTP function and that enhancing PP2A activity could be beneficial in COPD.
Collectively, these data suggest that TTP is a negative regulator of inflammation and confers protection from the development of CS‐induced emphysema, airway remodelling and lung function impairment (Figure 7). These studies support the compelling hypothesis that treating inflammation by targeting TTP with novel drug modalities (e.g. PP2A activators) is future pharmacotherapeutic strategies for treating corticosteroid‐insensitive respiratory diseases (including COPD) and, more broadly, may offer exciting potential for novel treatments for inflammatory diseases such as arthritis.
Figure 7.
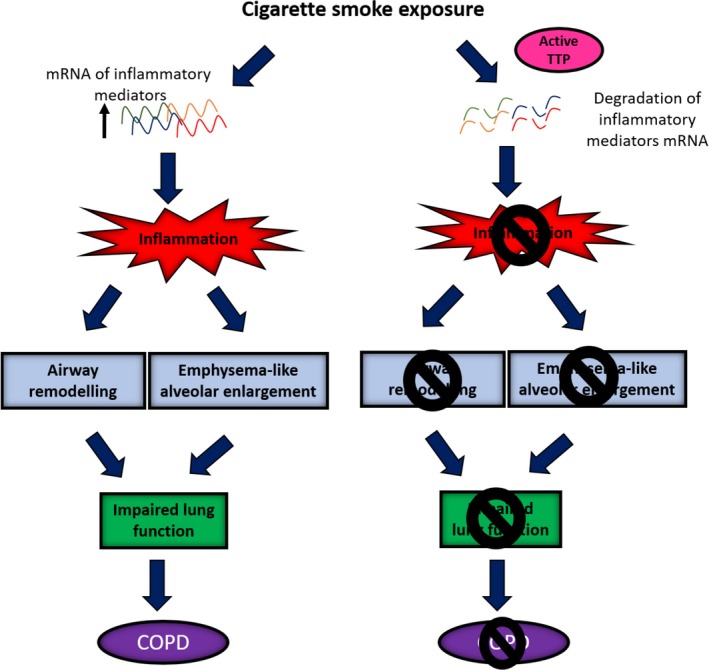
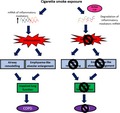
Proposed mechanisms of how active tristetraprolin (TTP) confers protection against the development of cigarette smoke (CS)‐induced experimental COPD. CS exposure increases the messenger ribonucleic acid (mRNA) expression of inflammatory mediators resulting in pulmonary inflammation. Ongoing inflammation drives the development of airway remodelling and emphysema. These features collectively result in lung function decline. Active TTP degrades the mRNA of inflammatory mediators, preventing the development of pulmonary inflammation, airway remodelling, emphysema and consequent lung function decline. TTP, tristetraprolin; mRNA, messenger ribonucleic acid; COPD, chronic obstructive pulmonary disease.
Methods
Ethics statement
This study was performed in strict accordance with the recommendations in the Australian code of practice for the care and use of animals for scientific purposes issued by the National Health and Medical Research Council of Australia. All protocols were approved by the Animal Ethics Committee of The University of Newcastle.
Experimental COPD
Female, 6–8 weeks old, wild‐type littermates (Zfp36++/++) or Zfp36aa/aa mice (generated on a C57BL/6J background)17 were exposed to normal air or CS through the nose only for four days (acute) or eight weeks (chronic) using well‐established methods.18, 19, 20, 21, 22, 23, 24, 30, 50, 51, 68, 69, 70 Exposures are equivalent to a pack‐a‐day smoker.28, 61 Females were used as women are more susceptible to developing COPD.71, 72
Drug treatments
Mice were treated daily by intraperitoneal (i.p.) injection with the PP2A and TTP activator, AAL(S) (0.8 mg kg−1 in 200 μL of PBS, synthesised in‐house); the proteasome inhibitor, bortezomib (BORT: 0.2 mg kg−1 in 200 μL of PBS, LC laboratories, Woburn, USA); or combined treatments (AAL(S) + BORT), daily before CS smoke exposure for four days (acute) or for five days per week for eight weeks (chronic). Doses used were based on previously published work.64, 65, 73 Controls were vehicle‐treated and exposed to normal air for the same time periods.
Airway and parenchymal inflammation
Bronchoalveolar lavage (BAL) was performed, cells were cytocentrifuged (300 g, 10 min) and stained with May–Grunwald–Giemsa, and differential leucocyte counts were determined according to morphological criteria from a total of 250 cells as previously described.74, 75, 76, 77, 78 Lungs were perfused, formalin‐fixed, embedded and sectioned. Longitudinal sections were stained with haematoxylin and eosin (H&E), and parenchymal inflammation was assessed by enumerating the numbers of inflammatory cells in 20 randomised high‐power fields as described previously.20, 21
mRNA expression
Whole lungs were collected and stored in RNA Stabilization Reagent, RNAlater (Qiagen, Chadstone Centre, Vic, Australia). RNA was extracted with guanidinium thiocyanate phenol chloroform (TRIzol). Extracted RNA was treated with DNAse I (Sigma, Castle Hill, NSW, Australia) and reverse‐transcribed using Bioscript (Bioline, Alexandria, NSW, Australia) and random hexamer primers (Invitrogen, Mount Waverley, Vic, Australia).21, 59, 79 The relative abundance of cytokine transcripts for IL‐6, TNF‐α, CXCL1, CXCL2, fibronectin (Fn) and matrix metalloproteinase‐12 (MMP‐12) was determined relative to the reference gene hypoxanthine phosphoribosyltransferase (HPRT) by real‐time qPCR using a ViiA 7 Real‐Time PCR System (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA). Custom‐designed primers (Integrated DNA Technologies, Coralville, Iowa, USA) were used (Supplementary table 1).
Airway remodelling
Longitudinal lung sections were stained with H&E or Masson’s trichrome. The area of collagen deposition (μm2), airway epithelial area (μm2) and cell (nuclei) number were assessed in a minimum of four small airways (basement membrane [BM] perimeter < 1000 μm) per section. Data were normalised to BM perimeter (μm) and quantified using ImageJ software (Version 1.49h; National Institutes of Health, New York City, NY, USA) 19, 21, 24, 80.
Emphysema‐like alveolar enlargement
H&E‐stained longitudinal lung sections were assessed for alveolar enlargement. Alveolar diameter was determined using the mean linear intercept method, and the percentage of alveolar destruction was determined using destructive index method.19, 20, 21, 22, 30
Lung function
Lung function parameters were assessed using forced oscillation (Flexivent, Scireq, Montreal, QC, Canada) and forced manoeuvre (Buxco electronics, New Brighton, MN, USA) techniques as previously described.20, 24, 50
Statistical analysis
Data are presented as means ± sem with 5–8 mice in each group and are representative of two independent experiments. Statistical significance for multiple comparisons was determined by one‐way ANOVA with Bonferroni post‐test, or non‐parametric equivalent, where appropriate using GraphPad Prism V.6 Software (San Diego, CA, USA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author contributions
PMN, MRS, ARC, AJA and PMH participated in the conception and design of the study. PMN performed all the experiments, analysed the data and wrote the draft of the manuscript. MRS, TJH and GL assisted with mouse experiments. JVM and NMV provided the AAL(S). All authors participated in the interpretation of data and editing of the manuscript for intellectual content. All authors read and approved the final manuscript.
Supporting information
Acknowledgments
This study was supported by grants (1099119, 1025637, 1141365) and fellowships (PMH 1079187) from the National Health and Medical Research Council, a Gladys Brawn Fellowship from the Faculty of Health and Medicine (PMH), University of Newcastle and a Cancer Institute NSW Fellowship. The authors thank Kristy Wheeldon, Nathalie Kiaos, Emma Broadfield and the staff from the Animal Service Unit of the University of Newcastle and Hunter Medical Research Institute for technical assistance.
References
- 1. Han MK, Agusti A, Calverley PM et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010; 182: 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lozano R, Naghavi M, Foreman K et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380: 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Willemse BW, Postma DS, Timens W et al. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J 2004; 23: 464–476. [DOI] [PubMed] [Google Scholar]
- 4. Driscoll T, Nelson DI, Steenland K et al. The global burden of non‐malignant respiratory disease due to occupational airborne exposures. Am J Ind Med 2005; 48: 432–445. [DOI] [PubMed] [Google Scholar]
- 5. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 2013; 131: 636–645. [DOI] [PubMed] [Google Scholar]
- 6. Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta 2013; 1829: 666–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackshear PJ. Tristetraprolin and other CCCH tandem zinc‐finger proteins in the regulation of mRNA turnover. Biochem Soc Trans 2002; 30: 945–952. [DOI] [PubMed] [Google Scholar]
- 8. O'Neil JD, Ammit AJ, Clark AR. MAPK p38 regulates inflammatory gene expression via tristetraprolin: Doing good by stealth. Int J Biochem Cell Biol 2018; 94: 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark AR, Dean JL. The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: a tale of two phosphatases. Biochem Soc Trans 2016; 44: 1321–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chrestensen CA, Schroeder MJ, Shabanowitz J et al. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14‐3‐3 binding. J Biol Chem 2004; 279: 10176–10184. [DOI] [PubMed] [Google Scholar]
- 11. Brook M, Tchen CR, Santalucia T et al. Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen‐activated protein kinase and extracellular signal‐regulated kinase pathways. Mol Cell Biol 2006; 26: 2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun L, Stoecklin G, Van Way S et al. Tristetraprolin (TTP)‐14‐3‐3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor‐α mRNA. J Biol Chem 2007; 282: 3766–3777. [DOI] [PubMed] [Google Scholar]
- 13. Hitti E, Iakovleva T, Brook M et al. Mitogen‐activated protein kinase‐activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine‐rich element. Mol Cell Biol 2006; 26: 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jalonen U, Nieminen R, Vuolteenaho K et al. Down‐regulation of tristetraprolin expression results in enhanced IL‐12 and MIP‐2 production and reduced MIP‐3α synthesis in activated macrophages. Mediators Inflamm 2006; 2006: 40691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor GA, Carballo E, Lee DM et al. A pathogenetic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996; 4: 445–454. [DOI] [PubMed] [Google Scholar]
- 16. Datta S, Biswas R, Novotny M et al. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J Immunol 2008; 180: 2545–2552. [DOI] [PubMed] [Google Scholar]
- 17. Ross EA, Smallie T, Ding Q et al. Dominant suppression of inflammation via targeted mutation of the mRNA destabilizing protein Tristetraprolin. J Immunol 2015; 195: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. EL Beckett SR, Jarnicki AG, Kim R et al. A new short‐term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol 2013; 131: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu G, Cooley MA, Jarnicki AG et al. Fibulin‐1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI insight 2016; 1: e86380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beckett EL, Phipps S, Starkey MR et al. TLR2, but not TLR4, is required for effective host defence against Chlamydia respiratory tract infection in early life. PLoS One 2012; 7: e39460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haw TJ, Starkey MR, Nair PM et al. A pathogenic role for tumor necrosis factor‐related apoptosis‐inducing ligand in chronic obstructive pulmonary disease. Mucosal Immunol 2016; 9: 859–872. [DOI] [PubMed] [Google Scholar]
- 22. Haw TJ, Starkey MR, Pavlidis S et al. Toll‐like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke‐induced chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 2018; 314: L298–L317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Donovan C, Starkey MR, Kim RY et al. Roles for T/B lymphocytes and ILC2s in experimental chronic obstructive pulmonary disease. J Leukoc Biol 2019; 105: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansbro PM, Hamilton MJ, Fricker M et al. Importance of mast cell Prss31/transmembrane tryptase/tryptase‐γ in lung function and experimental chronic obstructive pulmonary disease and colitis. J Biol Chem 2014; 289: 18214–18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vlahos R, Bozinovski S. Recent advances in pre‐clinical mouse models of COPD. Clin Sci (Lond) 2014; 126: 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vlahos R, Bozinovski S, Jones JE et al. Differential protease, innate immunity, and NF‐κB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol 2006; 290: L931–945. [DOI] [PubMed] [Google Scholar]
- 27. Starkey MR, Plank MW, Casolari P et al. IL‐22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur Respir J 2019; 54: 1800174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fricker M, Deane A, Hansbro PM et al. Animal models of chronic obstructive pulmonary disease. Expert Opin Drug Discov 2014; 9: 629–645. [DOI] [PubMed] [Google Scholar]
- 29. Keatings VM, Collins PD, Scott DM et al. Differences in interleukin‐8 and tumor necrosis factor‐alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153: 530–534. [DOI] [PubMed] [Google Scholar]
- 30. Jarnicki AG, Schilter H, Liu G et al. The inhibitor of semicarbazide‐sensitive amine oxidase, PXS‐4728A, ameliorates key features of chronic obstructive pulmonary disease in a mouse model. Br J Pharmacol 2016; 173: 3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Stefano A, Capelli A, Lusuardi M et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998; 158: 1277–1285. [DOI] [PubMed] [Google Scholar]
- 32. Shapiro SD, Goldstein NM, Houghton AM et al. Neutrophil elastase contributes to cigarette smoke‐induced emphysema in mice. Am J Pathol 2003; 163: 2329–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 2004; 56: 515–548. [DOI] [PubMed] [Google Scholar]
- 34. Stanescu D, Sanna A, Veriter C et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996; 51: 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao XK, Che P, Cheng ML et al. Tristetraprolin down‐regulation contributes to persistent TNF‐alpha expression induced by cigarette smoke extract through a post‐transcriptional mechanism. PLoS One 2016; 11: e0167451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donaldson GC, Seemungal TA, Patel IS et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 2005; 128: 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eickmeier O, Huebner M, Herrmann E et al. Sputum biomarker profiles in cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) and association between pulmonary function. Cytokine 2010; 50: 152–157. [DOI] [PubMed] [Google Scholar]
- 38. Petrescu F, Voican SC, Silosi I. Tumor necrosis factor‐α serum levels in healthy smokers and nonsmokers. Int J Chron Obstruct Pulmon Dis 2010; 5: 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vlahos R, Bozinovski S, Chan SP et al. Neutralizing granulocyte/macrophage colony‐stimulating factor inhibits cigarette smoke‐induced lung inflammation. Am J Respir Crit Care Med 2010; 182: 34–40. [DOI] [PubMed] [Google Scholar]
- 40. Vieira SM, Lemos HP, Grespan R et al. A crucial role for TNF‐α in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br J Pharmacol 2009; 158: 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Traves SL, Culpitt SV, Russell RE et al. Increased levels of the chemokines GROα and MCP‐1 in sputum samples from patients with COPD. Thorax 2002; 57: 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. John G, Kohse K, Orasche J et al. The composition of cigarette smoke determines inflammatory cell recruitment to the lung in COPD mouse models. Clin Sci (Lond) 2014; 126: 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chung KF. The role of airway smooth muscle in the pathogenesis of airway wall remodeling in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2: 347–354: discussion 371–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Annoni R, Lancas T, Yukimatsu Tanigawa R et al. Extracellular matrix composition in COPD. Eur Respir J 2012; 40: 1362–1373. [DOI] [PubMed] [Google Scholar]
- 45. Kranenburg AR, Willems‐Widyastuti A, Moori WJ et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol 2006; 126: 725–735. [DOI] [PubMed] [Google Scholar]
- 46. Cardoso WV, Sekhon HS, Hyde DM et al. Collagen and elastin in human pulmonary emphysema. Am Rev Respir Dis 1993; 147: 975–981. [DOI] [PubMed] [Google Scholar]
- 47. Lang MR, Fiaux GW, Gillooly M et al. Collagen content of alveolar wall tissue in emphysematous and non‐emphysematous lungs. Thorax 1994; 49: 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin‐Mosquero C, Peces‐Barba G, Rubio ML et al. Increased collagen deposition correlated with lung destruction in human emphysema. Histol Histopathol 2006; 21: 823–828. [DOI] [PubMed] [Google Scholar]
- 49. Hogg JC, Chu F, Utokaparch S et al. The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- 50. Hsu AC, Starkey MR, Hanish I et al. Targeting PI3K‐p110α Suppresses Influenza Virus Infection in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2015; 191: 1012–1023. [DOI] [PubMed] [Google Scholar]
- 51. Franklin BS, Bossaller L, De Nardo D et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol 2014; 15: 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hautamaki RD, Kobayashi DK, Senior RM et al. Requirement for macrophage elastase for cigarette smoke‐induced emphysema in mice. Science 1997; 277: 2002–2004. [DOI] [PubMed] [Google Scholar]
- 53. Zheng T, Zhu Z, Wang Z et al. Inducible targeting of IL‐13 to the adult lung causes matrix metalloproteinase‐ and cathepsin‐dependent emphysema. J Clin Invest 2000; 106: 1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morris DG, Huang X, Kaminski N et al. Loss of integrin αvβ6-mediated TGF-β activation causes Mmp12‐dependent emphysema. Nature 2003; 422: 169–173. [DOI] [PubMed] [Google Scholar]
- 55. Churg A, Wang R, Wang X et al. Effect of an MMP‐9/MMP‐12 inhibitor on smoke‐induced emphysema and airway remodelling in guinea pigs. Thorax 2007; 62: 706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smallie T, Ross EA, Ammit AJ et al. Dual‐specificity phosphatase 1 and Tristetraprolin cooperate to regulate macrophage responses to lipopolysaccharide. J Immunol 2015; 195: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Houghton AM. Matrix metalloproteinases in destructive lung disease. Matrix Biol 2015; 44–46: 167–174. [DOI] [PubMed] [Google Scholar]
- 58. Ostridge K, Williams N, Kim V et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax 2016; 71: 126–132. [DOI] [PubMed] [Google Scholar]
- 59. Kim RY, Horvat JC, Pinkerton JW et al. MicroRNA‐21 drives severe, steroid‐insensitive experimental asthma by amplifying phosphoinositide 3‐kinase‐mediated suppression of histone deacetylase 2. J Allergy Clin Immunol 2017; 139: 519–532. [DOI] [PubMed] [Google Scholar]
- 60. Simpson JL, Phipps S, Gibson PG. Inflammatory mechanisms and treatment of obstructive airway diseases with neutrophilic bronchitis. Pharmacol Ther 2009; 124: 86–95. [DOI] [PubMed] [Google Scholar]
- 61. Jones B, Donovan C, Liu G et al. Animal models of COPD: What do they tell us? Respirology 2017; 22: 21–32. [DOI] [PubMed] [Google Scholar]
- 62. Ngkelo A, Adcock IM. New treatments for COPD. Curr Opin Pharmacol 2013; 13: 362–369. [DOI] [PubMed] [Google Scholar]
- 63. Rahman MM, Rumzhum NN, Hansbro PM et al. Activating protein phosphatase 2A (PP2A) enhances tristetraprolin (TTP) anti‐inflammatory function in A549 lung epithelial cells. Cell Signal 2016; 28: 325–334. [DOI] [PubMed] [Google Scholar]
- 64. Collison A, Hatchwell L, Verrills N et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus‐induced asthma by inhibiting protein phosphatase 2A activity. Nat Med 2013; 19: 232–237. [DOI] [PubMed] [Google Scholar]
- 65. Nair PM, Starkey MR, Haw TJ et al. Targeting PP2A and proteasome activity ameliorates features of allergic airway disease in mice. Allergy 2017; 72: 1891–1903. [DOI] [PubMed] [Google Scholar]
- 66. Ross EA, Naylor AJ, O'Neil JD et al. Treatment of inflammatory arthritis via targeting of tristetraprolin, a master regulator of pro‐inflammatory gene expression. Ann Rheum Dis 2017; 76: 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nath S, Ohlmeyer M, Salathe MA et al. Chronic cigarette smoke exposure subdues PP2A activity by enhancing expression of the oncogene CIP2A. Am J Respir Cell Mol Biol 2018; 59: 695–705. [DOI] [PubMed] [Google Scholar]
- 68. Hsu AC, Dua K, Starkey MR et al. MicroRNA‐125a and ‐b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight 2017; 2: e90443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tay HL, Kaiko GE, Plank M et al. Correction: Antagonism of miR‐328 Increases the Antimicrobial Function of Macrophages and Neutrophils and Rapid Clearance of Non‐typeable Haemophilus Influenzae (NTHi) from Infected Lung. PLoS Pathog 2015; 11: e1004956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fricker M, Goggins BJ, Mateer S et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight 2018; 3: e94040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sorheim IC, Johannessen A, Gulsvik A et al. Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax 2010; 65: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Barnes PJ. Sex Differences in chronic obstructive pulmonary disease mechanisms. Am J Respir Crit Care Med 2016; 193: 813–814. [DOI] [PubMed] [Google Scholar]
- 73. Hatchwell L, Girkin J, Dun MD et al. Salmeterol attenuates chemotactic responses in rhinovirus‐induced exacerbation of allergic airways disease by modulating protein phosphatase 2A. J Allergy Clin Immunol 2014; 133: 1720–1727. [DOI] [PubMed] [Google Scholar]
- 74. Horvat JC, Starkey MR, Kim RY et al. Chlamydial respiratory infection during allergen sensitization drives neutrophilic allergic airways disease. J Immunol 2010; 184: 4159–4169. [DOI] [PubMed] [Google Scholar]
- 75. Essilfie AT, Horvat JC, Kim RY et al. Macrolide therapy suppresses key features of experimental steroid‐sensitive and steroid‐insensitive asthma. Thorax 2015; 70: 458–467. [DOI] [PubMed] [Google Scholar]
- 76. Thorburn AN, Foster PS, Gibson PG et al. Components of Streptococcus pneumoniae suppress allergic airways disease and NKT cells by inducing regulatory T cells. J Immunol 2012; 188: 4611–4620. [DOI] [PubMed] [Google Scholar]
- 77. Kim R, Horvat JC, Pinkerton JW et al. MicroRNA‐21 drives severe, steroid‐insensitive experimental asthma by amplifying phosphoinositide‐3‐kinase‐mediated suppression of histone deacetylase 2. J Allergy Clin Immunol 2017; 139: 519–532. [DOI] [PubMed] [Google Scholar]
- 78. Preston JA, Essilfie AT, Horvat JC et al. Inhibition of allergic airways disease by immunomodulatory therapy with whole killed Streptococcus pneumoniae . Vaccine 2007; 25: 8154–8162. [DOI] [PubMed] [Google Scholar]
- 79. Starkey MR, Nguyen DH, Brown AC et al. PD‐L1 promotes early‐life chlamydia respiratory infection‐induced severe allergic airway disease. Am J Respir Cell Mol Biol 2016; 54: 493–503. [DOI] [PubMed] [Google Scholar]
- 80. Liu G, Cooley MA, Nair PM et al. Airway remodeling and inflammation in asthma are dependent on the extracellular matrix protein fibulin‐1c. J Pathol 2017; 243: 510–523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials