

Abstract
Chalcogen‐bonding cascade switching was introduced recently to produce the chemistry tools needed to image physical forces in biological systems. In the original flipper probe, one methyl group appeared to possibly interfere with the cascade switch. In this report, this questionable methyl group is replaced by a hydrogen. The deletion of this methyl group in planarizable push‐pull probes was not trivial because it required the synthesis of dithienothiophenes with four different substituents on the four available carbons. The mechanosensitivity of the resulting demethylated flipper probe was nearly identical to that of the original. Thus methyl groups in the switching region are irrelevant for function, whereas those in the twisting region are essential. This result supports the chalcogen‐bonding cascade switching concept and, most importantly, removes significant synthetic demands from future probe development.
Keywords: fluorescent probes, chalcogen bonds, molecular switches, twisted fluorophores, push-pull fluorophores, lipid bilayer membranes
Not every atom matters: Chemistry tools made for the fluorescent imaging of physical forces in biological systems subjected to single methyl group deletions: Whereas methyl groups in the twisting region are essential, the pink methyl in the switching region does not interfere with function, a result that required significant multistep organic synthesis to obtain and will therefore be thoroughly appreciated in future probe design.
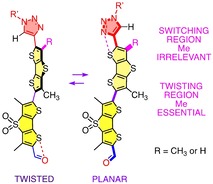
The fluorescence imaging of physical forces is one of the current challenges in biology that cannot be addressed without input from chemistry.1 To tackle this problem, planarizable push‐pull (PP) chromophores were introduced2 as a mechanosensitive fluorescent membrane probe.3 The first operational “flipper”4 probe consists of twisted dithienothiophene (DTT)5 dimers (Figure 1).6 Repulsion (
 ) between methyls (
) between methyls (
 ) and σ holes (
) and σ holes (
 )7 on the sulfurs next to the connecting bond is applied to twist the two flippers out of coplanarity (Figure 1a, 9).4, 6, 8 The primary “sulfide” donors in the DTT bridge and “sulfone” acceptors in the dithienothiophene S,S‐dioxide (DTTO2) bridge establish the primary push‐pull system (Figure 1a, 5, 6). Mechanical co‐planarization establishes conjugation between donor and acceptor DTT and shifts the excitation maximum to the red.4, 6, 8
)7 on the sulfurs next to the connecting bond is applied to twist the two flippers out of coplanarity (Figure 1a, 9).4, 6, 8 The primary “sulfide” donors in the DTT bridge and “sulfone” acceptors in the dithienothiophene S,S‐dioxide (DTTO2) bridge establish the primary push‐pull system (Figure 1a, 5, 6). Mechanical co‐planarization establishes conjugation between donor and acceptor DTT and shifts the excitation maximum to the red.4, 6, 8
Figure 1.
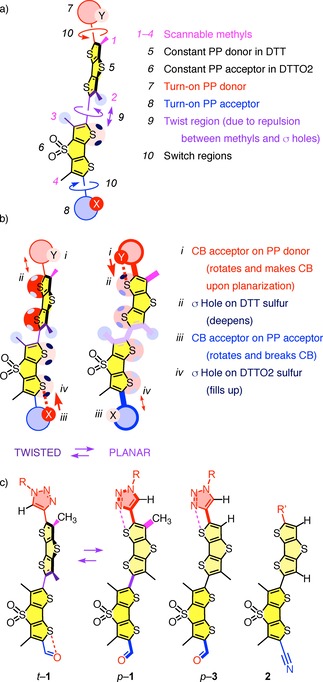
a) Design and b) mode of action of the chalcogen‐bonding cascade switch. c) Relevant structures for methyl scanning: Original cascade switch 1 in twisted (t) and planar (p) conformation, flipper 3 without the methyl (1) in the donor switching region and control 2 with methyls in neither switching (1) nor twisting (2) donor regions.
Additional exocyclic donors (D) and acceptors (A) are needed in planarized probes for high mechanosensitivity, i. e., large redshifts from strong PP systems. However, they are not tolerated in twisted probes because they promote oxidation and reduction of at least partially decoupled DTTs.9 To overcome this “flipper dilemma,” the bioinspired10 chalcogen‐bonding (CB)7 cascade switching concept was introduced.11 The exocyclic PP D and A (Figure 1a, 7, 8) are equipped with CB acceptors Y and X, respectively (Figure 1b, i, iii). In the twisted ground state, repulsion (↕) of Y (i) by the electron‐rich DTT (ii) turns off D (7), while a “back‐donating” (↑) CB (⋅⋅⋅) between X (iii) and the deep σ holes ()5, 7, 11, 12, 13 on the electron‐poor DTTO2 (iv) turns off A (8). Upon planarization by physical force (9, ), the deepened DTT σ holes (ii,
 →) produce a CB (⋅⋅⋅) to Y (i) and turn on (10,
→) produce a CB (⋅⋅⋅) to Y (i) and turn on (10,
 , ↑) D (7), and the filled DTTO2 σ holes (iv,
, ↑) D (7), and the filled DTTO2 σ holes (iv,
 →) repel (↕) X (iii) and turn on (10,
→) repel (↕) X (iii) and turn on (10,
 ) A (8).
) A (8).
Proof‐of‐concept for CB cascade switching was secured with flipper 1, containing a triazole as turn‐on donor (7) and an aldehyde as turn‐on acceptor (8, Figure 1).11 The results were chemically stable fluorescent force probes with redshifted absorption in planarized form and, probably more important, much chemical space opened up to explore. To venture into this space, we were concerned about the methyl group next to the triazole ring in flipper 1 (magenta, 1). This methyl could conceivably hinder the formation of the CB (⋅⋅⋅, ↑) between the nitrogen Y (i) in triazole PP donor (7) and DTT (ii, ) in the planar p‐1 conformation, i. e., weaken or even inactivate the turn‐on donor. Similar concerns apply to the methyl on the other end (4), possibly interfering with turn‐on acceptors. Although flipper 1 showed excellent mechanosensitivity,11 we wondered if the removal of this methyl (1) is necessary to unleash the full capacity of turn‐on donors. However, removal of both methyls (1, 2) on DTT donor (5) as in flipper probe 2 was shown previously to be detrimental to the mechanosensitivity because of insufficient twisting.14 The challenge then was to delete the methyl (1) in the switch region (10) and preserve the methyl (2) in the twist region (9). This design called for DTTs with four different substituents on the four available carbons, a synthetic challenge that has not been met so far with flipper probes. In the following, we report the synthesis of flipper 3 with one methyl group of original 1 cleanly deleted, and show that the mechanosensitivities of the two are nearly the same, i. e., validate the CB cascade switching concept as it stands.
Single methyl deletion in flipper 3 was initiated with tetrabromothiophene 4 (Scheme 1). DTTO2 5 was readily accessible in eight steps from 4 following previously reported procedures.4, 8, 11 A stepwise procedure was developed for the introduction of the two different β substituents in DTT 6. The first bromine‐lithium exchange of thiophene 4 was followed by the addition of acetaldehyde 7, and the second by the addition of 1‐formylpiperidine 8. The secondary alcohol in the resulting thiophene 9 was oxidized with Dess‐Martin periodinane, and product 10 was engaged in cascade cyclizations15 with thioacetate 11. Ester hydrolysis of DTT 12 followed by decarboxylation of diacid 13 afforded the previously reported16 DTT 14. Chemoselective bromination of 14 with NBS was achieved following the procedure in the literature.16 Iodination of the second α carbon in bromo‐DTT 15 afforded the first intermediate 6 with four different substituents on the four available DTT carbons. Sonogashira coupling with TIPS‐protected alkyne 16 prepared for direct coupling of the resulting alkynylated bromo‐DTT 17 with bromo‐DTTO2 5 in the presence of CuI and then Pd(PPh3)4. From the resulting product mixture, it was possible to isolate pure dimer 18. Deprotection of the alkyne and Cu‐mediated cycloaddition of alkyne 19 to azide 20 gave the desired target molecule 3. The yield of 14 % for the final two steps does not well reflect conversion because much material was sacrificed during purification of the final, poorly soluble amphiphile 3. Yields of the multistep synthesis of 3 were overall not vigorously optimized because the functional evaluation suggested that, fortunately, single methyl deletion will not be necessary for the future development of cascade switching (vide infra).
Scheme 1.
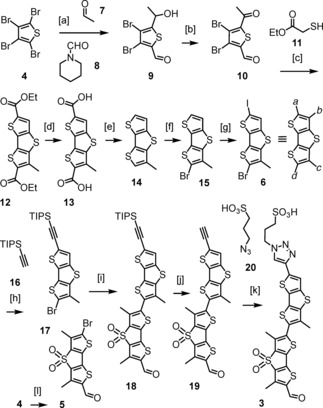
Synthesis of 3: [a] 1. 4, nBuLi, THF, −78 °C, 7, 30 min; 2. nBuLi, THF, −78 °C, 8, −78 °C to rt, 12 h, 49 %; [b] Dess‐Martin periodinane, CH2Cl2, rt, 1 h; [c] K2CO3, DMF, rt, 12 h; [d] KOH, EtOH, H2O, reflux, 4 h; [e] Ag2CO3, AcOH, DMSO, 120 °C, 48 h, 31 % (4 steps); [f] NBS, DMF, 95 %;16 [g] I2, PhI(OAc)2, CD2Cl2, rt, 12 h; [h] CuI, PdCl2(PPh3)2, TEA, N2, 55 °C, 12 h; [i] CuI, DMF, 80 °C, Pd(PPh3)4, 120 °C, 24 h, 8 % (3 steps); [j] TBAF, THF, 0 °C, 5 min; [k] CuSO4 ⋅ 5H2O, sodium ascorbate, TBTA, DMF/H2O 6 : 1, rt, 12 h, 14 % (2 steps); [l] 8 steps, as in [4, 8d].
The challenge to build demethylated flipper 3 is best appreciated by comparison with the previously reported, much less problematic and reasonably optimized synthesis of original 1 (Scheme 2).11 Here, the dimethylated DTT 21 is readily accessible from 4 by acetylation with 7 followed by cascade cyclization with 11. Importantly, dimer 22 is accessible with good yields from DTT 21 and DTTO2 23 by Stille coupling, and the terminal acetylene can be introduced on the dimer level with the TMS‐protected alkyne 24 rather than the TIPS‐protected alkyne 16, which in turn allows milder deprotection of dimer 25. Most of these conditions optimized for flipper 1 were not applicable to 3 because the deleted methyl not only asymmetrized but also affected the reactivity of DTT 6.
Scheme 2.
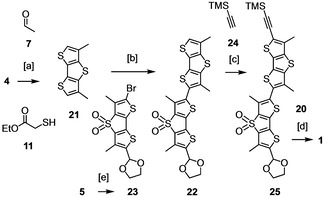
Synthesis of 1:11 [a] 1. nBuLi, 7, 2. Na2Cr2O7, 3. NaOEt, 11, 80 %; 4. LiOH, 5. Ag2CO3, 69 %; [b] 1. nBuLi, Bu3SnCl, 2. 23, Pd(PPh3)4, 54 %; [c] 1. NIS, 2. 24, PdCl2(PPh3)2, CuI, Et3N, 66 %; [d] 1. K2CO3, 2. 20, CuSO4 ⋅ 5H2O, sodium ascorbate, TBTA, 20 %; [e] ethylene glycol, 92 %.
Fluorescence spectra of the demethylated flipper 3 were recorded in large unilamellar vesicles (LUVs) composed of liquid‐disordered (Ld) DOPC (magenta), the liquid‐ordered (Lo) SM/CL 7 : 3 (green) and solid‐ordered (So) DPPC membranes (blue) at 25 °C, and Ld DPPC membranes at 55 °C (Figure 2a, red).11 In all membranes, increasing flipper concentration resulted in a roughly linear increase in fluorescence intensity until saturation was reached. Such behavior occurred at c>5 nM 3 in Lo SM/CL membranes and at c >32 nM in other membranes (Figure S6). The mechanosensitivity of flipper 3 was thus evaluated below these critical concentrations.
Figure 2.
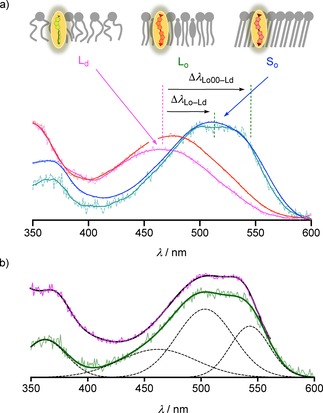
a) Normalized fluorescence excitation spectra (λ em=620 nm, original and smoothed) of 3 (32 or 4 nM) in Ld DOPC (magenta), Lo SM/CL 7 : 3 (green) and So DPPC (blue) at 25 °C and Ld DPPC LUVs (red) at 50 °C (75 μM lipid in 10 mM Tris, 100 mM NaCl, pH 7.4). b) Deconvolution (dashed) of fluorescence excitation spectra (λ em=620 nm) of 3 (4 nM) in Lo SM/CL 7 : 3 LUVs (original data and sum of the deconvoluted peaks, green) compared to that of 1 (original data, magenta; sum of the deconvoluted peaks, blue).
From Ld DOPC to So DPPC, the lowest energy maximum in the excitation spectrum shifted by Δλ So–Ld=+50 nm from λ Ld=470 nm to λ So=520 nm. At the same time, the fluorescence intensity increased by I So/I Ld=13, while the emission maxima λ em≈630 nm did not change at all (Figures S2, S4). These changes were in agreement with operational flipper probes, characterized by i) in‐equilibrium planarization and polarization in the ground state accounting for the redshift in excitation,4, 8, 11 ii) uniform emission from planarized and polarized lowest excited state for constant emission maxima,9 and iii) competition of viscosity‐dependent off‐equilibrium planarization and polarization in the excited state11, 14 with non‐radiative TICT states3 accounting presumably for the lower fluorescence intensity of twisted probes.11, 17
Demethylated flipper 3 perfectly reproduced the mechanosensitivity of the original cascade switch 1. For instance, the characteristic strongly shouldered peak appeared in more ordered membranes. Best developed in Lo membranes, these shoulders could be attributed to the vibrational fine structure upon in‐equilibrium planarization in the ground state.11, 14 Spectral deconvolution for 3 placed the formal 0–0 transition at λ Lo00=543 nm, followed by the most intense 0–1 transition at λ Lo01=503 nm and a broad band at λ LoE=462 nm which might contain the 0–2 transition but presumably also contributions from a misplaced subpopulation (Figure 2b, green, solid; black, dashed). The estimated Δν Lo12=1470 cm−1 appeared reasonable for vibrational transitions. The redshift of the excitation band of 3 from Ld to Lo membranes can be described as a maximal shift Δλ Lo00–Ld=+73 nm or an average redshift Δλ Lo–Ld≈+50 nm. This most revealing excitation spectrum of the demethylated probe 3 reproduced that of original cascade switch 1 in Lo membranes to perfection (Figure 2b, magenta).
With 3 in So membranes, the broad peak at average λ So=520 nm was as in Lo membranes, but the vibrational fine structure was less pronounced. However, deconvolution readily gave λ Lo00=547 nm, a dominant λ Lo01=506 nm and a broad λ LoE=444 nm (Figure 3a). As in Lo membranes, the deconvoluted spectra of 3 and 1 in So membranes were very similar (Figure 3b).
Figure 3.
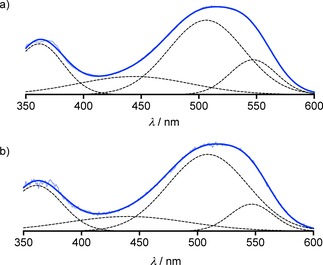
Deconvolution (dashed) of fluorescence excitation spectra (λ em=620 nm) of (a) 3 and (b) 1 in So DPPC LUVs at 25 °C (original data, light blue; sum of deconvoluted peaks, blue).
Chalcogen‐bonding cascade switching was introduced recently to overcome the flipper dilemma and open chemical space for fluorescent membrane tension probe development.11 Although validated experimentally and theoretically, there was the one methyl group that could have hindered rotation and chalcogen‐bond formation to turn on the triazole donor. The results disclosed in this report suggest that this concern is without foundation: Deletion of the single methyl in the switch region does not affect mechanosensitivity. In other words, whereas methyls in twist region, operating with chalcogen bonding, are essential,14 methyls in switch region are irrelevant. This result was delightful because it validates the chalcogen‐bonding cascade switching concept and thus opens much space for future probe development without the synthetic complications of single methyl deletion described in this study.
Conflict of interest
The original Flipper‐TR® is commercially available from Spirochrome, through the NCCR Store (https://nccr‐chembio.ch/technologies/nccr‐store/). The NCCR receives 15% of the revenues.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the NMR, the MS and the bioimaging platforms for services, and the University of Geneva, the Swiss National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.
X. Zhang, N. Sakai, S. Matile, ChemistryOpen 2020, 9, 18.
Dedicated to Jean‐Marie Lehn on the occasion of his 80th birthday
Contributor Information
Dr. Xiang Zhang, http://www.unige.ch/sciences/chiorg/matile/
Prof. Stefan Matile, Email: stefan.matile@unige.ch.
References
- 1.
- 1a. Krieg M., Fläschner G., Alsteens D., Gaub B. M., Roos W. H., Wuite G. J. L., Gaub H. E., Gerber C., Dufrêne Y. F., Müller D. J., Nat. Rev. Phys. 2019, 1, 41–57; [Google Scholar]
- 1b. Diz-Muñoz A., Weiner O. D., Fletcher D. A., Nat. Phys. 2018, 14, 648–652; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Pontes B., Monzo P., Gauthier N. C., Semin. Cell Dev. Biol. 2017, 71, 30–41; [DOI] [PubMed] [Google Scholar]
- 1d. Roca-Cusachs P., Conte V., Trepat X., Nat. Cell Biol. 2017, 19, 742–751; [DOI] [PubMed] [Google Scholar]
- 1e. Hamada T., Kishimoto Y., Nagasaki T., Takagi M., Soft Matter 2011, 7, 9061; [Google Scholar]
- 1f. Ho J. C. S., Rangamani P., Liedberg B., Parikh A. N., Langmuir 2016, 32, 2151–2163. [DOI] [PubMed] [Google Scholar]
- 2. Fin A., Vargas Jentzsch A., Sakai N., Matile S., Angew. Chem. Int. Ed. 2012, 51, 12736–12739; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12908–12911. [Google Scholar]
- 3.
- 3a. Klymchenko A. S., Acc. Chem. Res. 2017, 50, 366–375; [DOI] [PubMed] [Google Scholar]
- 3b. Kubánková M., López-Duarte I., Kiryushko D., Kuimova M. K., Soft Matter 2018, 14, 9466–9474; [DOI] [PubMed] [Google Scholar]
- 3c. Muraoka T., Umetsu K., Tabata K. V., Hamada T., Noji H., Yamashita T., Kinbara K., J. Am. Chem. Soc. 2017, 139, 18016–18023; [DOI] [PubMed] [Google Scholar]
- 3d. Chambers J. E., Kubánková M., Huber R. G., López-Duarte I., Avezov E., Bond P. J., Marciniak S. J., Kuimova M. K., ACS Nano 2018, 12, 4398–4407; [DOI] [PubMed] [Google Scholar]
- 3e. Guo R., Yin J., Ma Y., Wang Q., Lin W., J. Mater. Chem. B 2018, 6, 2894–2900; [DOI] [PubMed] [Google Scholar]
- 3f. Jiménez-Sánchez A., Lei E. K., Kelley S. O., Angew. Chem. Int. Ed. 2018, 57, 8891–8895; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9029–9033; [Google Scholar]
- 3g. Yang Z., He Y., Lee J.-H., Park N., Suh M., Chae W.-S., Cao J., Peng X., Jung H., Kang C., Kim J. S., J. Am. Chem. Soc. 2013, 135, 9181–9185; [DOI] [PubMed] [Google Scholar]
- 3h. Haidekker M. A., Theodorakis E. A., J. Mater. Chem. C 2016, 4, 2707–2718; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3i. Halabi E. A., Püntener S., Rivera-Fuentes P., Helv. Chim. Acta 2018, 101, e1800165; [Google Scholar]
- 3j. Xiong Y., Vargas Jentzsch A., Osterrieth J. W. M., Sezgin E., Sazanovich I. V., Reglinski K., Galiani S., Parker A. W., Eggeling C., Anderson H. L., Chem. Sci. 2018, 9, 3029–3040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3k. Grenier V., Daws B. R., Liu P., Miller E. W., J. Am. Chem. Soc. 2019, 141, 1349–1358; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3l. Kulkarni R. U., Vandenberghe M., Thunemann M., James F., Andreassen O. A., Djurovic S., Devor A., Miller E. W., ACS Cent. Sci. 2018, 4, 1371–1378; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3m. Collot M., Fam T. K., Ashokkumar P., Faklaris O., Galli T., Danglot L., Klymchenko A. S., J. Am. Chem. Soc. 2018, 140, 5401–5411; [DOI] [PubMed] [Google Scholar]
- 3n. Bauer C., Duwald R., Labrador G. M., Pascal S., Moneva Lorente P., Bosson J., Lacour J., Rochaix J.-D., Org. Biomol. Chem. 2018, 16, 919–923; [DOI] [PubMed] [Google Scholar]
- 3o. Leung C. W. T., Hong Y., Chen S., Zhao E., Lam J. W. Y., Tang B. Z., J. Am. Chem. Soc. 2013, 135, 62–65; [DOI] [PubMed] [Google Scholar]
- 3p. Baumgart T., Hunt G., Farkas E. R., Webb W. W., Feigenson G. W., Biochim. Biophys. Acta 2007, 1768, 2182–2194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3q. Yan P., Xie A., Wei M., Loew L. M., J. Org. Chem. 2008, 73, 6587–6594; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3r. Chen X.-M., Chen Y., Yu Q., Gu B.-H., Liu Y., Angew. Chem. Int. Ed. 2018, 57, 12519–12523. [DOI] [PubMed] [Google Scholar]
- 4. Dal Molin M., Verolet Q., Colom A., Letrun R., Derivery E., Gonzalez-Gaitan M., Vauthey E., Roux A., Sakai N., Matile S., J. Am. Chem. Soc. 2015, 137, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Strakova K., Assies L., Goujon A., Piazzolla F., Humeniuk H. V., Matile S., Chem. Rev. 2019, 119, 10977–11005; [DOI] [PubMed] [Google Scholar]
- 5b. Cinar M. E., Ozturk T., Chem. Rev. 2015, 115, 3036–3140; [DOI] [PubMed] [Google Scholar]
- 5c. Barbarella G., Di Maria F., Acc. Chem. Res. 2015, 48, 2230–2241; [DOI] [PubMed] [Google Scholar]
- 5d. Kim O.-K., Fort A., Barzoukas M., Blanchard-Desce M., Lehn J.-M., J. Mater. Chem. 1999, 9, 2227–2232; [Google Scholar]
- 5e. Kim O.-K., Lehn J.-M., Chem. Phys. Lett. 1996, 255, 147–150. [Google Scholar]
- 6.
- 6a. Colom A., Derivery E., Soleimanpour S., Tomba C., Molin M. D., Sakai N., González-Gaitán M., Matile S., Roux A., Nat. Chem. 2018, 10, 1118–1125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Goujon A., Colom A., Straková K., Mercier V., Mahecic D., Manley S., Sakai N., Roux A., Matile S., J. Am. Chem. Soc. 2019, 141, 3380–3384. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Bauzá A., Mooibroek T. J., Frontera A., ChemPhysChem 2015, 16, 2496–2517; [DOI] [PubMed] [Google Scholar]
- 7b. Lim J. Y. C., Beer P. D., Chem 2018, 4, 731–783; [Google Scholar]
- 7c. Scilabra P., Terraneo G., Resnati G., Acc. Chem. Res. 2019, 52, 1313–1324; [DOI] [PubMed] [Google Scholar]
- 7d. Pascoe D. J., Ling K. B., Cockroft S. L., J. Am. Chem. Soc. 2017, 139, 15160–15167; [DOI] [PubMed] [Google Scholar]
- 7e. Scheiner S., Chem. Eur. J. 2016, 22, 18850–18858; [DOI] [PubMed] [Google Scholar]
- 7f. Beno B. R., Yeung K.-S., Bartberger M. D., Pennington L. D., Meanwell N. A., J. Med. Chem. 2015, 58, 4383–4438; [DOI] [PubMed] [Google Scholar]
- 7g. Vogel L., Wonner P., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 1880–1891; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1896–1907. [Google Scholar]
- 8.
- 8a. Verolet Q., Soleimanpour S., Fujisawa K., Dal Molin M., Sakai N., Matile S., ChemistryOpen 2015, 4, 264–267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Soleimanpour S., Colom A., Derivery E., Gonzalez-Gaitan M., Roux A., Sakai N., Matile S., Chem. Commun. 2016, 52, 14450–14453; [DOI] [PubMed] [Google Scholar]
- 8c. Verolet Q., Molin M. D., Colom A., Roux A., Guénée L., Sakai N., Matile S., Helv. Chim. Acta 2017, 100, e1600328; [Google Scholar]
- 8d. Macchione M., Tsemperouli M., Goujon A., Mallia A. R., Sakai N., Sugihara K., Matile S., Helv. Chim. Acta 2018, 101, e1800014; [Google Scholar]
- 8e. Strakova K., Soleimanpour S., Diez-Castellnou M., Sakai N., Matile S., Helv. Chim. Acta 2018, 101, e1800019; [Google Scholar]
- 8f. Goujon A., Strakova K., Sakai N., Matile S., Chem. Sci. 2019, 10, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verolet Q., Rosspeintner A., Soleimanpour S., Sakai N., Vauthey E., Matile S., J. Am. Chem. Soc. 2015, 137, 15644–15647. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gamiz-Hernandez A. P., Angelova I. N., Send R., Sundholm D., Kaila V. R. I., Angew. Chem. Int. Ed. 2015, 54, 11564–11566; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11726–11729; [Google Scholar]
- 10b. Sheves M., Nakanishi K., Honig B., J. Am. Chem. Soc. 1979, 101, 7086–7088; [Google Scholar]
- 10c. Kiser P. D., Golczak M., Palczewski K., Chem. Rev. 2014, 114, 194–232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Baumeister B., Matile S., Chem. Eur. J. 2000, 6, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 11. Macchione M., Goujon A., Strakova K., Humeniuk H. V., Licari G., Tajkhorshid E., Sakai N., Matile S., Angew. Chem. Int. Ed. 2019, 58, 15752–15756; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15899–15903. [Google Scholar]
- 12. Benz S., Macchione M., Verolet Q., Mareda J., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 9093–9096. [DOI] [PubMed] [Google Scholar]
- 13. Fabbro C., Armani S., Carloni L.-E., Leo F. D., Wouters J., Bonifazi D., Eur. J. Org. Chem. 2014, 2014, 5487–5500. [Google Scholar]
- 14.K. Strakova, A. I. Poblador-Bahamonde, N. Sakai, S. Matile, Chem. Eur. J., DOI: 10.1002/chem.201903604. [DOI] [PubMed]
- 15.
- 15a. Frey J., Bond A. D., Holmes A. B., Chem. Commun. 2002, 2424–2425; [DOI] [PubMed] [Google Scholar]
- 15b. Hong W., Yuan H., Li H., Yang X., Gao X., Zhu D., Org. Lett. 2011, 13, 1410–1413. [DOI] [PubMed] [Google Scholar]
- 16. He M., Zhang F., J. Org. Chem. 2007, 72, 442–451. [DOI] [PubMed] [Google Scholar]
- 17. Macchione M., Chuard N., Sakai N., Matile S., ChemPlusChem 2017, 82, 1062–1066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary