

Abstract
The up-regulation of miR-92a in human cancer happens frequently, and is related to an increase of metastasis and decreased survival. However, its functions in colorectal cancer (CRC) are largely unknown. This study aimed to investigate the regulatory effect of miR-92a on cell invasion and migration in colorectal cancer (CRC). A total of 158 patients with CRC were included, and in situ hybridization was used to predict the expression of miR-92a in the paraffin sections from the patients. Quantitative reverse transcription PCR was used to detect the expression of miR-92a and its target gene. Protein levels were determined by western blotting. Luciferase assays confirmed the direct target of miR-92a. Furthermore, cell invasion and migration were detected using Transwell and wound healing assays. The expression level of miR-92a in tumor tissues was upregulated compared with that of paired normal tissues and negatively correlated with the RECK protein level. The 3-year disease-free survival (DFS) and overall survival (OS) rates of miR-92a expression in the high group were significantly lower than those in the miR-92a-low group. The RECK 3’-UTR reporter activity assay suggested that the RECK gene was a direct target of miR-92a. After transfection of the miR-92a-mimic, the miR-92a levels were increased in HCT116 and SW620 cell lines, while the protein expression of RECK was decreased instead of the mRNA level, along with downregulation of matrix metalloproteinase (MMP) protein expression. Conversely, after transfection with miR-92a-inhibitor, the opposite trend was achieved. In conclusion, miR-92a promotes the invasion and migration of CRC through the RECK-MMP signaling pathway, and the upregulation of miR-92a was associated with poor long-term prognosis in CRC.
Keywords: Colorectal cancer, miR-92a, invasion, migration, RECK
Introduction
Colorectal cancer (CRC) is one of the most common cancers and a leading cause of cancer-related death worldwide [1]. CRC treatment has shown great progress. Although lymph node metastasis or distant organ metastasis predicts poor survival outcomes, the cause of local invasion and distant metastasis of CRC is still not fully understood [2]. Previous studies have shown that the invasion and metastasis of tumors are related to the abnormal expression of microRNAs (miRNAs) in tissues [3]. However, the specific molecular mechanism is unknown.
MiRNAs are single-stranded noncoding RNAs containing 21-24 nucleotides, and they bind to the target mRNAs in the 3’-untranslated region (3’UTR) to induce translational repression or mRNA cleavage, attenuating protein expression [4]. More than 50% of these miRNA genes are located in cancer-associated genomic regions or fragile sites, suggesting that these miRNAs are deeply involved in the pathogenesis of cancer [5]. Indeed, genome-wide studies have revealed that miRNAs may be potential diagnostic or prognostic tools for cancer, and the identification of target mRNAs is a key step in assessing the role of aberrantly expressed miRNAs in human cancer. The miR-17-92 cluster is located at chromosomal locus 13q31.3 and encodes miR-17, miR-18a, miR-19a/b, miR-20a, and miR-92a [6-9]. Among them, miR-92a is dysregulated in many tumors, including lung cancer, ovarian cancer and gastric cancer [10-12]. Previous studies have also shown that miR-92a is involved in the development and progression of CRC. However, its related molecular mechanisms have not yet been fully elucidated [13].
The reversion-inducing cysteine-rich protein with Kazal motifs (RECK) gene was first identified as a tumor metastasis suppressor, which induced morphological reversion in v-Ki-Ras-transformed NIH/3T3 cells in 1998 [14]. RECK is a unique membrane-anchored protein and matrix metalloproteinase (MMP) regulator, and its expression is correlated with tumor cell aggression [15]. Previous studies have demonstrated that RECK can inhibit three MMP family members: MMP-2, MMP-9, and MT1-MMP [16]. In this study, we identified a potential link between miR-92a and the RECK gene through bioinformatic analysis. Consequently, we inferred that a noncoding RNA, miR-92a, acts as a local regulator of RECK by binding to the 3’-UTR of its mRNA, thereby modulating CRC metastasis. To verify this hypothesis, we investigated the regulatory effect of miR-92a on cell invasion and migration in CRC. We aimed to reveal a new regulatory mechanism of miR-92a in the metastasis of CRC and provide a new miRNA and target gene for clinical application.
Materials and methods
Patients
A total of 179 patients with pathologically confirmed CRC from the Nanfang Hospital Affiliated with Southern Medical University (Guangzhou, China) were enrolled. None of the patients received anticancer therapy before sampling. Postoperatively, patients with high-risk stage II and III CRC were treated with the mFOLFOX6 or Xelox regimen according to the National Comprehensive Cancer Network (NCCN) guidelines. Fresh specimens were taken from 21 patients in March 2017. Another 158 paraffin-embedded and formalin-fixed samples were obtained from patients who underwent their operation between 2013 and 2014 with complete follow-up data, and these patients were further enrolled for analyses of DFS and OS. We defined DFS as the time from the date of diagnosis to the date of confirmed tumor recurrence. OS was defined as the interval between the date of diagnosis and the date of death from any cause. All samples were coded for anonymity following local ethical guidelines (as stipulated by the Declaration of Helsinki). Written informed consent was obtained from all patients, and the protocol was approved by the Review Board of Southern Medical University. Demographic features and clinicopathologic data of the patients are shown in Table 1.
Table 1.
Expression of miR-92a in human CRC and its clinical significance
| Characteristics | Cases | Low miR-92a | High miR-92a | χ2 | P |
|---|---|---|---|---|---|
| Gender | 3.163 | 0.075 | |||
| Male | 93 | 52 (55.9%) | 41 (44.1%) | ||
| Female | 65 | 27 (41.5%) | 38 (58.5%) | ||
| Age (years) | 0.408 | 0.523 | |||
| <60 | 86 | 45 (52.3%) | 41 (47.2%) | ||
| ≥60 | 72 | 34 (47.7%) | 38 (52.8%) | ||
| BMI | 2.099 | 0.147 | |||
| <23 | 91 | 50 (54.9%) | 41 (45.1%) | ||
| ≥23 | 67 | 29 (43.3%) | 38 (56.7%) | ||
| Tumor size (cm) | 1.373 | 0.241 | |||
| <5 | 137 | 71 (51.8%) | 66 (48.2%) | ||
| ≥5 | 21 | 8 (38.1%) | 13 (61.9%) | ||
| Differentiation | 4.348 | 0.037 | |||
| Well/Moderate | 89 | 51 (57.3%) | 38 (42.7%) | ||
| Poor | 69 | 28 (40.6%) | 41 (59.4%) | ||
| T stage | 6.782 | 0.009 | |||
| T1/T2 | 31 | 22 (71.0%) | 9 (29.0%) | ||
| T3/T4 | 127 | 57 (44.9%) | 70 (55.1%) | ||
| N stage | 6.661 | 0.010 | |||
| N0 | 92 | 54 (58.7%) | 38 (41.3%) | ||
| N1/N2 | 66 | 25 (37.9%) | 41 (62.1%) | ||
| M stage | 5.725 | 0.017 | |||
| M0 | 138 | 74 (53.6%) | 64 (46.4%) | ||
| M1 | 20 | 5 (25.0%) | 15 (75.0%) | ||
| AJCC stage | 5.060 | 0.024 | |||
| I/II | 90 | 52 (57.8%) | 38 (42.2%) | ||
| III/IV | 68 | 27 (39.7%) | 41 (60.3%) |
In situ hybridization
The patient’s tumor tissue was used to generate 3-μm formalin-embedded sections, which were assessed with a digoxigenin-labeled probe (TaKaRa, Tokyo, Japan). The sequence of the digoxin-labeled probe of miR-92a was as follows: 5’-ACAGGCCGGGACAAGTGCAATA-3’. Positive controls and scrambled control RNAs were included for each hybridization procedure and analyzed using a Nikon 80i microscope with Nikon NISElements F 2.3 software (Nikon, Tokyo, Japan). Slides were examined independently by two investigators, and miR-92a expression was quantified using a visual grading system based on the extent of staining (the percentage of positive tumor cells on a scale of 0-4: 0, none; 1, 1%-25%; 2, 26%-50%; 3, 51%-75%; 4, >75%) and the intensity of staining (graded on a scale of 0-3: 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining). The product of the extent and intensity grades was used to define the miR-92a relative expression in tumor tissues.
Immunohistochemical staining
Paraffin-embedded and formalin-fixed samples were cut into 3-μm sections, which were then processed for immunohistochemistry. Following incubation with an antibody against human RECK (Invitrogen, Carlsbad, CA) overnight at 4°C, the secondary rabbit antibody (Invitrogen, Carlsbad, CA) labeled with horseradish peroxidase was incubated again, and immunohistochemistry results were visualized using a BAD coloring solution. At low power (200 ×), the tissue sections were screened using an inverted research microscope (Leica DM IRB, Germany), and the five most representative fields were selected. This analysis was performed by two independent observers who were blinded to the clinical outcome.
Cell culture and transfection
Human CRC-derived cell lines, including HCT116, Lovo, SW480, SW620, Caco2 and DLD1, were provided by the Shanghai Institutes for Biological Science (Shanghai, China). Cell lines were cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA) at 37°C in an atmosphere of 5% CO2. The culture medium was replaced every 3 days, and the cells were trypsinized using trypsin when they reached 85%-95% confluence. After suitable conditions were established, cells (1 × 105/well) were seeded in a 6-well plate, incubated for 24 h to ensure that they attached to the bottom of the well, and then transfected with 50 nmol/L negative control, miR-92a mimics or miR-92a inhibitor oligonucleotides (RiboBio, Guang Zhou, China). The sequences of the small RNAs used in this experiment were as follows: miR-92a mimics 5’-UAUUGCACUUGUCCCGGCCUGU-3’ and miR-92a inhibitor 5’-AUAACGUGAACAGGGCCGGACA-3’. Lipofectamine 3000 (Life Technologies, Gaithersburg, MD, USA) transfection was applied and operated in accordance with the manufacturer’s procedure.
Bioinformatic analysis
The analysis of miR-92a-predicted targets was performed using the TargetScan (http://targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), and miRanda (http://www.microrna.org/microrna/home.do) algorithms. The detailed bioinformatic analyses are described in the Supplementary Materials and Methods.
Luciferase activity assay
The blank reporter plasmid and the 3’-UTR reporter plasmid of the RECK gene were purchased from GeneCopoeia (Rockville, MD, USA). The CRC cells were evenly plated in 6-well plates and incubated overnight at 37°C in a cell incubator. Then, the CRC cells were divided into four groups, including negative control group, miR-92a mimics group, and miR-92a inhibitor group. Reporter plasmids and small RNAs were transfected into the cells, and 48 h later, firefly luciferase activity was measured and normalized with Renilla luciferase activity.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from CRC cells with TRIzol reagent (Invitrogen, Carlsbad, CA). Then, the extracted total RNA was dissolved in DEPC-treated water. A nucleic acid protein analyzer (TaKaRa, Tokyo, Japan) was used to determine the RNA concentration. To identify the purity and integrity of total RNA, we used two indicators: A260 nm/A280 nm≥1.8 and a band ratio of 28S to 18S RNA≥1.5 in formaldehyde denaturing gel electrophoresis. The expression levels of miR-92a and RECK mRNA were measured using the PrimeScriptTM RT reagent kit (TaKaRa, Tokyo, Japan). The bulge-loop miRNA RT-PCR primer sets (one reverse transcription primer and a pair of quantitative PCR primers for each set) were designed by RiboBio (RiboBio, Guang Zhou, China). U6 was utilized as an internal control for miRNAs, and GAPDH was used as an internal control for mRNA. The primers used in this study were as follows: RECK-forward 5’-TGTGAACTGGCTATTGCCTTG-3’ and reverse 5’-GCATAACTGCAACAAACCGAG-3’; GAPDH-forward 5’-GCACCGTCAAGGCTGAGAAC-3’ and reverse 5’-TGGTGAAGACGCCAGTGGA-3’. The relative quantitative and statistical analyses were carried out using the 2-ΔΔCt method.
Western blotting
The total protein was extracted 72 h after cell treatment. Then, the BCA method was used to determine the protein concentration. Equivalent amounts of protein were taken from each group, followed by SDS-PAGE. Proteins were transferred to nitrocellulose membranes, and the blots were probed with primary antibodies against RECK, MMP2, MMP9, and GAPDH (Cell Signaling Technology, CST, Danvers, MA, USA) at 4°C overnight and then incubated with horseradish-peroxidase conjugated secondary rabbit or mouse antibody (Invitrogen, Carlsbad, CA). The blots were developed using the ECL detection kit (Thermo Fisher, Waltham, ME, USA).
Transwell assay
Briefly, an 8 μm pore size polycarbonate membrane filter was inserted into each Transwell chamber (Corning, NY, USA) and coated with 50 ml of Matrigel (Sigma, St Louis, MO, USA) that had been previously diluted with serum-free medium to obtain a final concentration of 4 mg/ml. The transfected CRC cells (3.0 × 104) were then seeded into the upper chamber with 100 ml of serum-free medium; in the bottom chamber, 1 ml of growth medium containing 10% FBS was added as a chemoattractant. The cells were incubated at 37°C for 48 h, and then, the Matrigel-coated upper surface of the filter was thoroughly wiped with a cotton swab. The cells that invaded the lower surface of the filter were fixed with 4% paraformaldehyde and stained with crystal violet; cells from three random fields per filter were counted under a microscope at 100 magnification.
Wound healing assay
Each group of CRC cells was seeded in 6-well plates after treatment. After 48 h, the culture reached approximately 90% confluence, and an artificial homogeneous wound was created on the monolayer with a 200 ml plastic micropipette tip; cell debris was removed by washing with PBS twice. Closing of the wound was then observed at the indicated times, and images were captured under an inverted microscope with a 40 objective.
SiRNA experiments
We selected the CRC cell line HCT116 for siRNA interference experiments. The small-interfering (si)-RECK was provided by GeneCopoeia (Rockville, MD, USA). The sequences of si-RECK used in this experiment was as follow: 5’-CCAGAGAATGTGGAAAGCAA-3’. Cell lines were cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA) at 37°C in an atmosphere of 5% CO2. The culture medium was replaced every 3 days, and the cells were trypsinized when they reached 85-95% confluence. After suitable conditions were established, cells (1 × 105/well) were seeded in a 6-well plate, incubated for 24 h to ensure that they attached to the bottom of the well, and then transfected with 50 nmol/l negative control, si-RECK oligonucleotides. Lipofectamine 3000 (Life Technologies, Gaithersburg, MD, USA) transfection was applied in accordance with the manufacturer’s instructions. Then, the status of cell invasion and migration was determined by Transwell and wound-healing assays.
Statistical analysis
Results are displayed as the mean ± SEM. Data were analyzed with the SPSS statistical package for Windows version 20 (SPSS, Chi-cago, IL, USA) and GraphPad Prism 6 (Graph-Pad Software, Inc., San Diego, CA, USA). Means were compared using the Pearson chi-squared test, two-tailed Student’s t-tests, Kaplan-Meier plots, log-rank tests, or ANOVA as appropriate. Differences were considered significant at P<0.05.
Results
MiR-92a is aberrantly upregulated in CRC tissues and associated with the prognosis of CRC
To study the expression pattern of miR-92a in CRC, we performed in situ hybridization to detect miR-92a expression in twenty-one CRC samples and adjacent nontumorous tissue sections. All twenty-one cases exhibited miR-92a upregulation in CRC tissues compared with non-tumorous tissues (9.10 ± 2.23 vs 7.52 ± 2.63; n=21), and the difference was statistically significant (P=0.022; n=21, Figure 1A). Compared with non-tumor tissues, CRC tissues in 21 patients showed an upregulated mean expression of miR-92a (9.10 ± 2.23 vs 7.52 ± 2.63; n=21), and the difference was statistically significant (P=0.022; n=21, Figure 1A). We then investigated the correlation between the miR-92a expression profile and prognosis to determine whether miR-92a was a prognostic factor. We used samples from 158 patients who underwent an operation in our hospital and had their associated prognostic information available. A three-year disease outcome was known for all these patients; 36 patients showed recurrence, and 26 patients died of cancer within the follow-up period. The final follow-up date was November 1, 2016, and the median follow-up time was 36.2 months (range 34-38 months). Most patients were given anticancer drugs either orally or intravenously postoperatively as an adjuvant chemotherapy. After disease recurrence, these individuals were given other anticancer drugs. To generate survival curves, we converted continuous miR-92a expression levels measured by in situ hybridization to a dichotomous variable using the mean level of expression as a threshold (Figure 1B). This procedure enabled the division of samples into classes with high and low expression of miR-92a. Consequently, Kaplan-Meier survival curves were generated for miR-92a, and we compared survival curves by the log-rank test. As shown in Figure 1C, 1D, patients with high miR-92a expression had poorer DFS (P=0.001) and OS (P=0.001) than did patients with low expression of miR-92a. As shown in Table 1, positive expression was significantly associated with the degree of tumor differentiation (P=0.037), T stage (P=0.009), N stage (P=0.01), M stage (P=0.017), and AJCC stage (P=0.024). These results indicated that elevated expression of miR-9a is correlated with malignant clinicopathologic parameters of CRC. These results provide the first hint that the expression of miR-92a might be one of the mechanisms that promotes CRC development.
Figure 1.
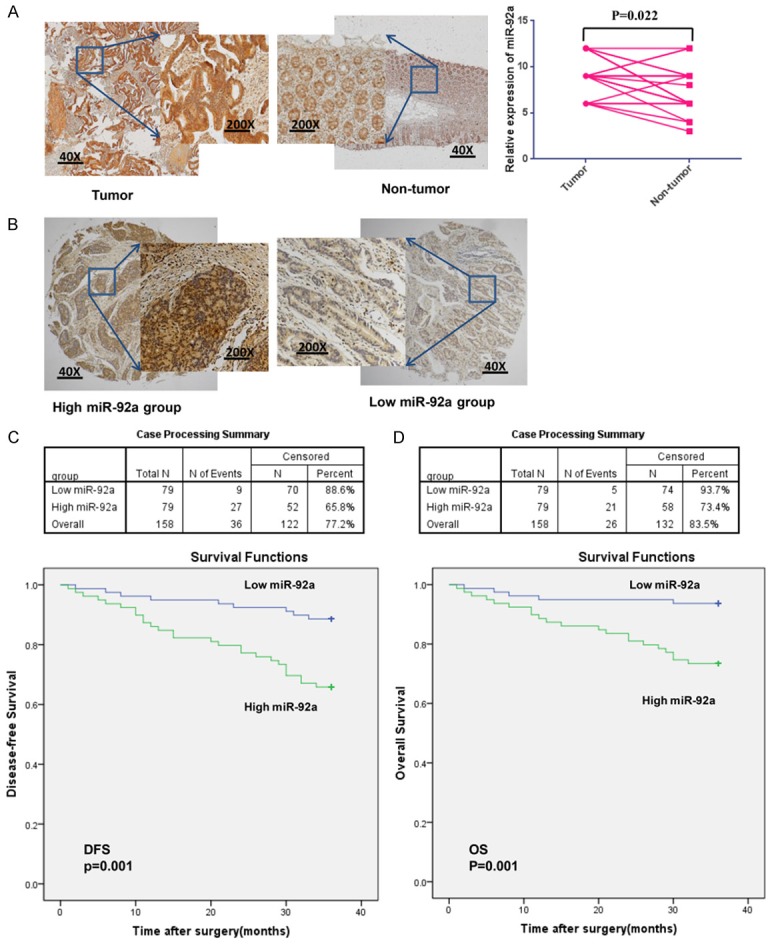
MiR-92a is aberrantly upregulated in CRC tissues and associated with the prognosis of CRC. (A) An in situ hybridization was used to detect the expression of miR-92a, and the miR-92a level was higher in CRC samples than in adjacent nontumorous tissues (*P=0.022; n=21). (B) An in situ hybridization was used to detect the expression of miR-92a in CRC samples (a selection of cases with relatively high or low miR-92a expression is shown). (C, D) The 158 CRC cases were divided into two groups according to the miR-92a relative expression level. The patients with high expression of miR-92a had poorer DFS (C; P=0.001) and OS (D; P=0.001) than did patients with low expression of miR-92a.
MiR-92a interacts with the 3’-UTR of RECK mRNA
Most miRNAs are thought to control gene expression by base-pairing with the miR-recognizing elements (miR-RE) found in their messenger target [17]. We then used all three currently available major prediction programs, including TargetScan, miRanda and PicTar, to analyze the potential interaction between miR-92a and RECK. RECK mRNA was predicted by all of the algorithms and has a potential miR-92a target site in its 3’-UTR (Figure 2A).
Figure 2.
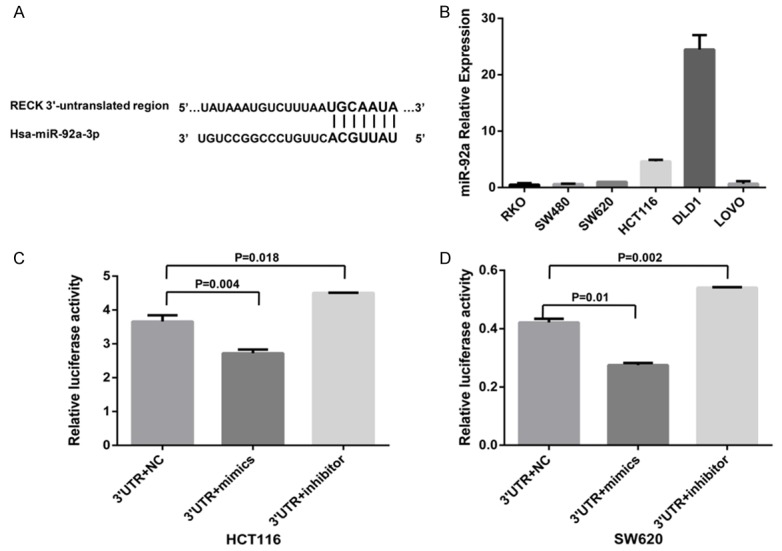
MiR-92a interacts with the 3’-UTR of RECK mRNA. (A) Bioinformatic analysis identified the miR-92a binding site in the 3’-UTR of RECK mRNA, which is located at the 1357-1363 region and is highly conserved in different species. (B) The level of miR-92a expression in six CRC cell lines. (C, D) HCT116 (C) and SW620 (D) cell lines were cotransfected with the RECK gene 3’-UTR reporter vector and miR-92a-mimic or miR-92a-inhibitor. Luciferase activity was determined and normalized to Renilla luciferase activity. Data represent the average of three independent experiments. Mean ± SD, P<0.05 was considered statistically significant. Abbreviations: RECK, reversion-inducing cysteine-rich protein with Kazal motifs; 3’-UTR, the 3’-UTR reporter plasmid of the RECK gene; mimics, miR-92a mimics; inhibitor, miR-92a inhibitor; NC, negative control.
We then selected six types of CRC cell lines, including SW480, SW620, HCT116, DLD1, RKO and Lovo, to detect the miR-92a expression levels. The HCT116 and SW620 cell lines were chosen for both miR-92a-mimic and miR-92a-inhibitor transfection in the successive experiment because of the intermediate miR-92a levels in these lines among the six cell lines tested (Figure 2B).
To demonstrate the direct interaction between miR-92a and RECK mRNA, we prepared the RECK gene 3’-UTR double luciferase vector plasmid and control plasmid. The double fluorescein carrier containing the RECK gene 3’-UTR was cotransfected into HCT116 cells with miR-92a-mimic or miR-92a-inhibitor. Compared with the activity of the corresponding negative pairs, the activity of the RECK 3’-UTR transcriptional vector and miR-92a-mimic-cotransfected HCT116 cells decreased significantly (P=0.004; Figure 2C). In contrast, compared with the activity of the negative control, the luciferase activity of HCT116 cells cotransfected with miR-92a-inhibitor and the 3’-UTR luciferase vector containing the RECK gene increased significantly, and the difference was statistically significant (P=0.018; Figure 2C). The results were verified in SW620 cells (P<0.05; Figure 2D). These results support the bioinformatic prediction of the RECK 3’-UTR as a target of miR-92a.
RECK is a target of miR-92a in CRC
To confirm the regulatory effect of miR-92a on RECK in CRC, we performed in situ hybridization to detect miR-92a expression (as shown in Figure 1) and immunohistochemical staining of RECK in twenty-one CRC samples and adjacent nontumorous tissue sections. Importantly, RECK protein expression was downregulated in CRC tissues compared with non-tumor tissues (5.35 ± 2.60 vs 8.40 ± 3.20; n=21; P<0.001; case selection for immunohistochemical staining is shown in Figure 3A). As shown in Figure 3B, when RECK protein levels were plotted against miR-92a expression, an inverse correlation was found (R2=0.737; P<0.01; Figure 3B). To determine whether miR-92a actually affects RECK expression in CRC cells, we analyzed the effects of ectopic expression of miR-92a. We transfected the miR-92a mimics or miR-92a inhibitor into CRC cells as described above, and we searched for changes in RECK protein levels by western blotting. As shown in Figure 3C and 3D, compared with the negative control, transfection of miR-92a mimics into CRC cells caused a significant increase in the miR-92a value and a decrease in the RECK protein level. Conversely, CRC cells transfected with miR-92a inhibitor exhibited a significant decrease in miR-92a levels and an increase in RECK protein levels. However, there was no significant change in the RECK mRNA level after transfection (P=0.931; P=0.476; Figure 3E and 3F). This result strongly validates the post-transcriptional regulation of the RECK protein by miR-92a.
Figure 3.
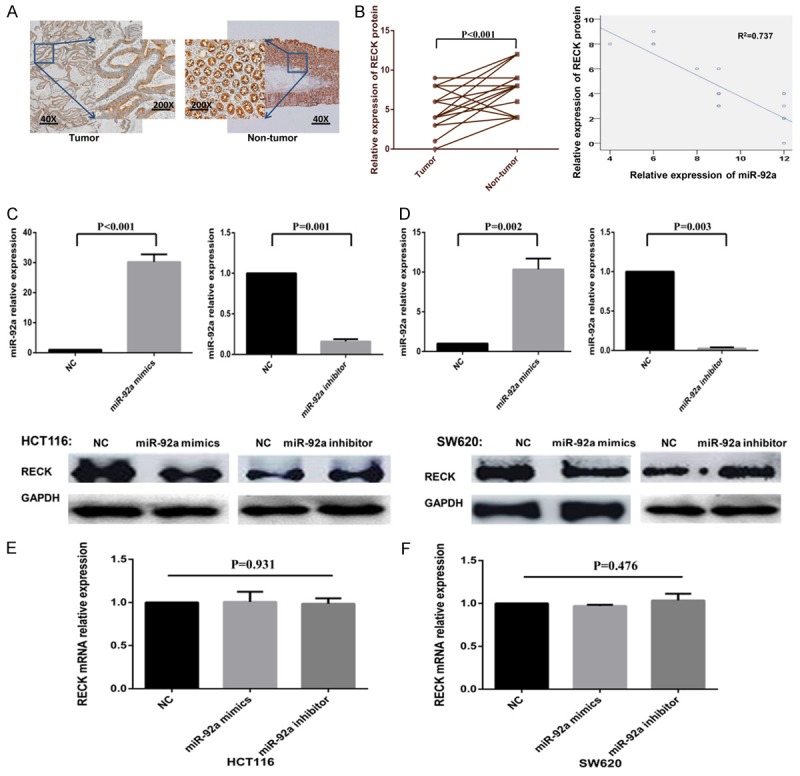
RECK is the direct target of miR-92a in CRC. (A) IHC staining of RECK protein in 21 pairs of CRC and nontumorous tissues. The expression of RECK protein was lower in CRC samples than in adjacent nontumorous tissues. (B) The expression of RECK protein was negatively correlated with the expression of miR-92a in CRC (R2=0.737; P<0.01). (C, D) After transfection with miR-92a mimics, the expression level of miR-92a increased in HCT116 (C) and SW620 (D) cell lines, while the expression of RECK protein decreased. Conversely, after transfection with miR-92a inhibitor, the opposite trend was achieved. (E, F) The expression of RECK mRNA was detected by qRT-PCR, and there was no significant change in RECK mRNA level after transfection. Abbreviations: RECK, reversion-inducing cysteine-rich protein with Kazal motifs; NC, negative control; IHC, immunohistochemistry.
MiR-92a promotes cell invasion and migration in CRC cells
MMPs can destroy the tissue barrier in tumor cell invasion, which is crucial in tumor invasion and metastasis [18,19]. Previous studies have shown that the membrane-anchored glycoprotein encoded by the RECK gene has a unique function of inhibiting the expression and activity of MMPs and is an MMP inhibitor [14]. Based on the above findings, we hypothesized that miR-92a might alter the MMP levels in CRC cells by regulating the expression of RECK, thus promoting the invasion and migration of tumor cells. To verify this hypothesis, we transfected miR-92a mimics or miR-92a inhibitor to overexpress or inhibit the expression of miR-92a in CRC cells. After CRC cells were transfected with miR-92a mimics, RECK protein expression was decreased, and the MMP2 and MMP9 protein levels were increased (Figure 4A). In contrast, after transfection with the miR-92a inhibitor, RECK protein expression was increased, and the MMP2 and MMP9 protein levels were decreased.
Figure 4.
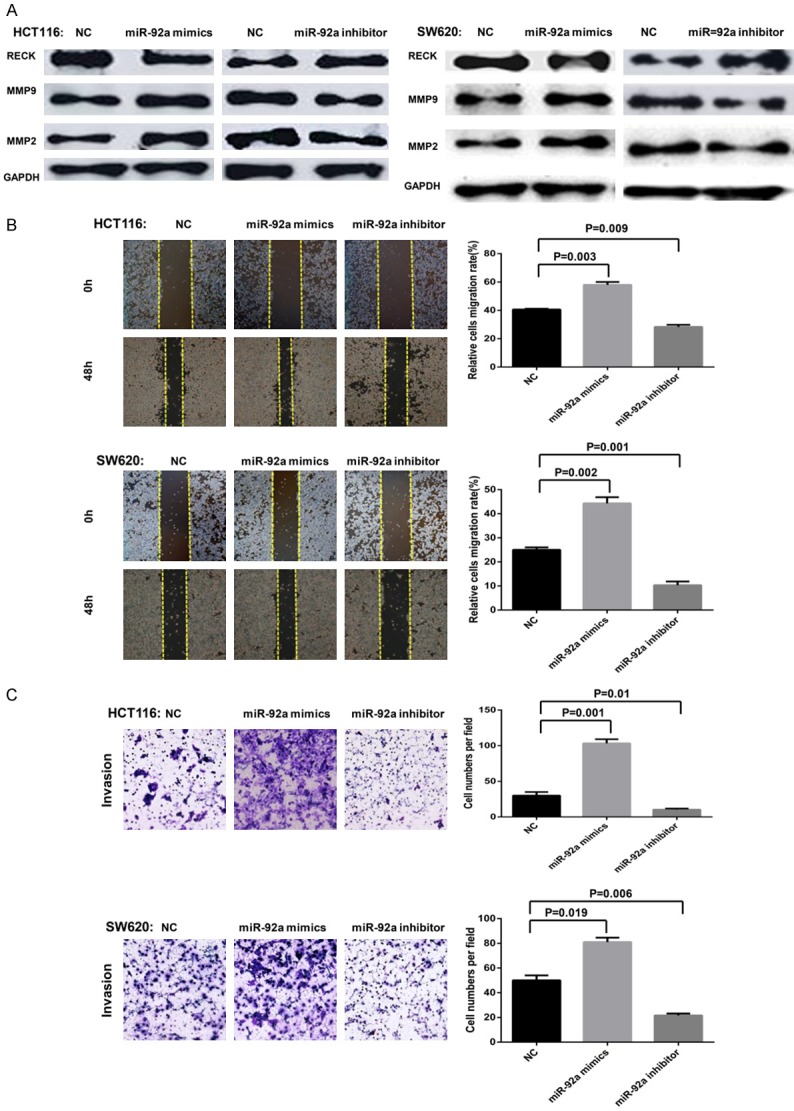
MiR-92a may promote the invasion and migration of CRC cells via the RECK-MMP signaling pathway. (A) HCT116 and SW620 cell lines were transfected with miR-92a mimics, and the expression of RECK protein was upregulated compared with that of the negative control and blank control groups, while the MMP2 and MMP9 protein levels were downregulated. Conversely, after transfection with miR-92a inhibitor, the opposite trend was achieved. (B, C) The status of cell invasion and migration was determined by transwell assays and wound healing assays. Cell invasion (B) and migration (C) were increased in cells treated with miR-92a mimics, whereas cell invasion (B) and migration (C) were decreased in cells treated with miR-92a inhibitor. Abbreviations: RECK, reversion-inducing cysteine-rich protein with Kazal motifs; NC, negative control.
To evaluate the effect of miR-92a on CRC cell invasion and migration, we transfected growing CRC cells with miR-92a mimics or miR-92a inhibitor for 48 h, and the status of cell invasion and migration was determined by Transwell assays and wound healing assays, respectively. Our data showed that miR-92a mimics transfection significantly promoted the invasion and migration of CRC cells, whereas knockdown of miR-92a expression decreased the invasion and migration of CRC cells (P<0.05; Figure 4B and 4C). These results confirmed the potential metastasis-promoting activity of miR-92a in CRC.
Suppression of CRC cell invasion and migration by miR-92a-inhibitor is mediated by RECK
If the suppression of CRC cell invasion and migration by miR-92a inhibitor is indeed mediated by RECK, we would expect the suppressive effect to be abolished by the RECK-specific and irreversible antagonist si-RECK. To test this hypothesis, we measured the invasion and migration variations induced by miR-92a inhibitor in CRC cells previously transfected with si-RECK. The aim of this experiment was to determine whether and how the RECK-depleted cellular environment responds to miR-92a inhibitor addition. CRC cells were pretreated with or without si-RECK for 24 h prior to the transfection of miR-92a inhibitor, and the status of cell invasion and migration was determined by transwell assays and wound healing assays. The data showed that when we pretransfected CRC cells with si-RECK that could reduce RECK mRNA (P=0.037) and protein expression (Figure 5A-C), the suppression of cell invasion and migration by the miR-92a inhibitor was largely abrogated (P=0.001; Figure 5D, 5E). These results indicated that the inhibitory effect of miR-92a inhibitor on CRC cell invasion and migration was largely mediated by RECK.
Figure 5.
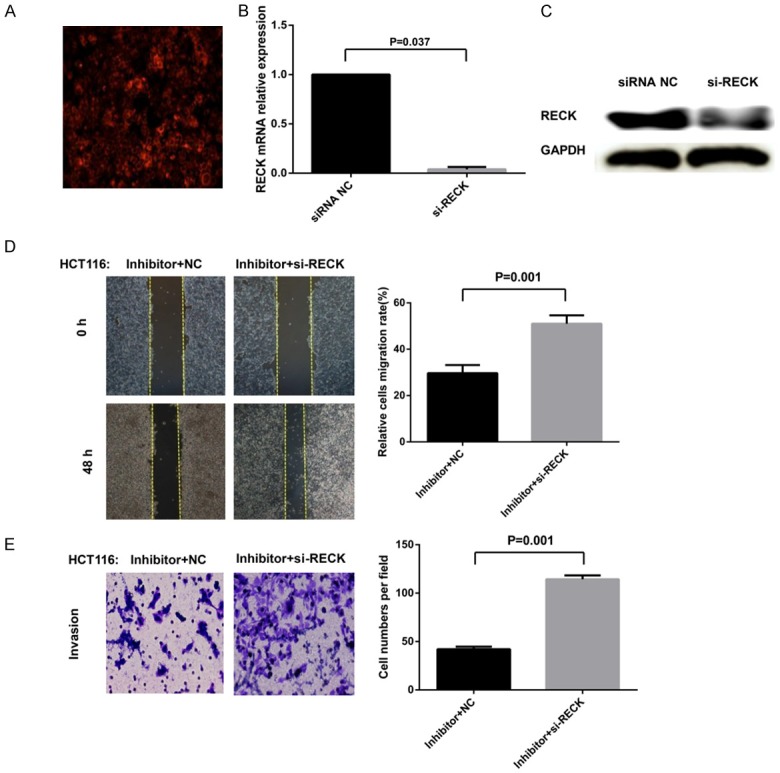
Ectopic expression of miR-92a affects the invasion and migration of CRC cells by targeting RECK. (A) si-RECK was transfected into the cells, which was verified by Cy3 fluorescently labeled transfection control siRNA. (B, C) qRT-PCR and western blotting analysis showing that RECK mRNA (P=0.037) and protein were markedly reduced after transfection with si-RECK. (D, E) The status of cell invasion and migration was determined by transwell assays and wound healing assays. The suppression of cell invasion (D) and migration (E) by miR-92a inhibitor was largely abrogated by si-RECK. Mean ± SD, P=0.001. Abbreviations: RECK, reversion-inducing cysteine-rich protein with Kazal motifs; NC, negative control; si-RECK, small-interfering (si)-RECK; Inhibitor, miR-92a inhibitor.
Discussion
With the advent of new chemotherapeutic agents, clarification of the molecular pathogenesis of CRC is crucial for developing effective therapy strategies to improve the outcome of patients with this disease. miRNAs are a new class of small noncoding RNAs that regulate the expression of target genes through translational repression or mRNA cleavage [3]. miRNAs play a key regulatory role in basic biological activities, such as the growth, development, and differentiation of organisms, and their unbalanced expression could also facilitate many human diseases, especially the occurrence and development of tumors [20]. Aberrant miRNA expression has been frequently reported in various tumors, including CRC, indicating that there is a close correlation between miRNAs and human malignancy [21].
The miR-17-92 cluster is located at chromosomal locus 13q31.3 and encodes miR-17, miR-18a, miR-19a/b, miR-20a, and miR-92a. Among them, miR-92a is dysregulated in many tumors. Several published reports have begun to elucidate the mechanisms that may link miR-92a upregulation to cancer. Ren et al. found that miR-92a was upregulated in lung cancer tissue and cell lines, and its expression level was significantly associated with tumor size, TNM stage, and lymph node metastasis [6]. Ren et al. reported that miR-92a expression was upregulated in gastric cancer, and high expression levels of miR-92a in tumors compared with neighboring normal tissues were associated with poor OS [11]. Chen et al. also found that miR-92a is required for the maintenance of ovarian cancer stemness in vitro as well as tumorigenicity in vivo and is critically involved in the downregulation of DKK1 by STAT3 [10]. However, the expression pattern and regulatory effect of miR-92a on CRC remain largely unknown.
In this study, we demonstrated that miR-92a expression was upregulated in CRC samples compared with nontumorous tissues, and the targets of miR-92a may be tumor suppressor genes or genes encoding proteins with potential tumor suppressor functions. Moreover, miR-92a was dramatically upregulated in human CRC-derived cell lines, which was in accordance with the results of clinical samples. Furthermore, we found that miR-92a was associated with an unfavorable DFS and OS in specimens from CRC patients treated by curative surgery and adjuvant chemotherapy. Although our findings should be validated in an independent trial, these results might help us screen individuals who are candidates for aggressive treatment because of their expresson status and who could become candidates for therapeutic treatment with miR-92a inhibitor sequences. However, the lack of knowledge about the targets of miR-92a has hampered a full understanding of the biologic functions deregulated by miR-92a aberrant expression.
The RECK gene is a tumor suppressor gene first studied by Takahashi in 1998 [14]. This gene encodes a membrane-anchored glycoprotein, the unique function of which is to inhibit the expression and activity of MMPs [22]. Previous studies have confirmed that the RECK gene is associated with the oncogenesis of various cancers. Xia et al. found that RECK protein levels were significantly lower in esophageal cancer cells than in normal cells. The overexpression of RECK could reverse malignant phenotypes of esophageal cancer cells [23]. Hong et al. reported that RECK impedes the DNA repair activity of cancer cells and induces hypersensitivity to chemotherapeutic drugs [24]. Du et al. also found that hypermethylation of the RECK promoter is a common event in gastric cancer patients. RECK methylation in peritoneal lavage fluid acts as a biomarker of peritoneal metastasis of gastric cancer. Recently, RECK was reported to suppress CRC invasion and metastasis by targeting MMPs [25]. With the application of bioinformatic prediction programs, such as TargetScan, PicTar and miRanda, we found that miR-92a and the 3’-UTR of RECK mRNA had complementary binding sites and inferred that RECK might be a new target of miR-92a in CRC; however, this relationship has not yet been reported. In this study, RECK proteins were downregulated in CRC tissues, which was confirmed by immunohistochemical analysis, and a significant inverse correlation between miR-92a and RECK was found in CRCs. Moreover, a direct interaction of miR-92a with a target site on the 3’-UTR of RECK mRNA was demonstrated by luciferase assays. Furthermore, downregulation of RECK occurred in response to miR-92a-mimic transfection into CRC cells, and a significant upregulation of RECK occurred in response to miR-92a-inhibitor transfection. miRNAs are known to regulate the expression of proteins associated with biological changes without producing a detectable change in the corresponding mRNA levels. In fact, numerous studies have reported that miRNAs can repress the production of hundreds of proteins without downregulating their mRNA levels [26-28]. Thus, it could be inferred that miR-92a binds to the 3’-UTR of RECK mRNA in an incomplete complementary pairing pattern. Therefore, this molecule could only inhibit further translation of RECK mRNA but could not ultimately degrade it, which was indicated by the finding that RECK mRNA had no significant change after transfection, while its protein level obviously decreased in CRCs. These results suggested that CRC cells probably gained an oncogenic capacity through miR-92a-mediated post-transcriptional silencing of RECK. Our data indicate the existence of more than one mechanism affecting RECK expression in CRC.
To extend our previous observations, we focused on the role of miR-92a in regulating the invasive and migrative capacity in CRC. In this study, we found that miR-92a upregulation promotes the invasion and migration of CRC cells accompanied by a decrease in RECK expression and an enhancement of MMP2 and MMP9. Conversely, miR-92a inhibition suppressed CRC cell invasion and migration accompanied by an increase in RECK expression and a decline in MMP2 and MMP9. Thus, we hypothesized that miR-92a promoted CRC cell invasion and migration likely through downregulating RECK. To confirm this, we performed RNA interference to knock down RECK in CRC cells before transfection with the miR-92a inhibitor. Our data showed that si-RECK could significantly reverse miR-92a-inhibitor-induced suppression of invasion and migration of CRC cells. Indeed, these results confirmed that miR-92a promoted CRC cell invasion and migration by downregulating RECK, which made it a novel potential strategy for CRC treatment.
In summary, our study demonstrates that miR-92a can promote the invasion and migration of CRC by directly binding to the 3’-UTR of RECK, its target. Although there is still much to learn about the role of miR-92a in CRC tumorigenesis, miR-92a provides us with a new potential target for CRC treatment.
Acknowledgements
This work was partly supported by Grants from the National Natural Science Foundation of China (81573731), the Natural Science Foundation of Guangdong Province (2015A030313248, 2015A030313275, 2016A030313538) and the Guangdong Science and Technology Project (2014A020212543).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Van Cutsem E, De Gramont A, Henning G, Rougier P, Bonnetain F, Seufferlein T. Improving outcomes in patients with CRC: the role of patient reported outcomes-an ESDO report. Cancers (Basel) 2017;9:59–67. doi: 10.3390/cancers9060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afifi R, Person B, Haddad R. The impact of surgeons: pathologists dialog on lymph node evaluation of colorectal cancer patients. Isr Med Assoc J. 2018;20:30–33. [PubMed] [Google Scholar]
- 3.Xu J, Xiao X, Yang D. In vitro methods for analyzing mirna roles in cancer cell proliferation, invasion, and metastasis. Methods Mol Biol. 2018;1733:159–171. doi: 10.1007/978-1-4939-7601-0_13. [DOI] [PubMed] [Google Scholar]
- 4.Sbarouni E, Georgiadou P. MicroRNAs in acute aortic dissection. J Thorac Dis. 2018;10:1256–1257. doi: 10.21037/jtd.2018.03.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vannini I, Fanini F, Fabbri M. Emerging roles of microRNAs in cancer. Curr Opin Genet Dev. 2018;48:128–133. doi: 10.1016/j.gde.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren P, Gong F, Zhang Y, Jiang J, Zhang H. MicroRNA-92a promotes growth, metastasis, and chemoresistance in non-small cell lung cancer cells by targeting PTEN. Tumour Biol. 2016;37:3215–3225. doi: 10.1007/s13277-015-4150-3. [DOI] [PubMed] [Google Scholar]
- 7.Tsuchida A, Ohno S, Wu W, Borjiqin N, Fujita K, Aoki T, Ueda S, Takanashi M, Kuroda M. miR-92 is a key oncogenic component of the miR-17-92 cluster in colon cancer. Cancer Sci. 2011;102:2264–2271. doi: 10.1111/j.1349-7006.2011.02081.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Zhang L, Zhang M, Zhang Y, Fan D, Jiang J, Ye L, Fang X, Chen X, Fan S, Chen X, Fan S, Chao M, Liang C. Prognostic value of high-expression of miR-17-92 cluster in various tumors: evidence from a meta-analysis. Sci Rep. 2017;7:8375–8384. doi: 10.1038/s41598-017-08349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conev NV, Donev IS, Konsoulova-Kirova AA, Chervenkov TG, Kashlov JK, Ivanov KD. Serum expression levels of miR-17, miR-21, and miR-92 as potential biomarkers for recurrence after adjuvant chemotherapy in colon cancer patients. Biosci Trends. 2015;9:393–401. doi: 10.5582/bst.2015.01170. [DOI] [PubMed] [Google Scholar]
- 10.Chen MW, Yang ST, Chien MH, Hua KT, Wu CJ, Hsiao SM, Lin H, Hsiao M, Su JL, Wei LH. The STAT3-miRNA-92-Wnt signaling pathway regulates spheroid formation and malignant progression in ovarian cancer. Cancer Res. 2017;77:1955–1967. doi: 10.1158/0008-5472.CAN-16-1115. [DOI] [PubMed] [Google Scholar]
- 11.Ren C, Wang W, Han C, Chen H, Fu D, Luo Y, Yao H, Wang D, Ma L, Zhou L, Han D, Shen M. Expression and prognostic value of miR-92a in patients with gastric cancer. Tumour Biol. 2016;37:9483–9491. doi: 10.1007/s13277-016-4865-9. [DOI] [PubMed] [Google Scholar]
- 12.Lin H, Chiang C, Hung W. STAT3 upregulates miR-92a to inhibit RECK expression and to promote invasiveness of lung cancer cells. Br J Cancer. 2013;109:731–738. doi: 10.1038/bjc.2013.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke T, Wei P, Yeh K, Chen WT, Cheng Y. MiR-92a promotes cell metastasis of colorectal cancer through PTEN-mediated PI3K/AKT pathway. Ann Surg Oncol. 2015;22:2649–2655. doi: 10.1245/s10434-014-4305-2. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, Noda M. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci U S A. 1998;95:13221–13226. doi: 10.1073/pnas.95.22.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu N, Zhou B, Zhu G. Potential role of reversion-inducing cysteine-rich protein with kazal motifs (RECK) in regulation of matrix metalloproteinases (MMPs) expression in periodontal diseases. Med Sci Monit. 2016;22:1936–1938. doi: 10.12659/MSM.896546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu M, Wang HF, Zhang HZ. Expression of RECK and MMPs in hepatoblastoma and neuroblastoma and comparative analysis on the tumor metastasis. Asian Pac J Cancer Prev. 2015;16:4007–4011. doi: 10.7314/apjcp.2015.16.9.4007. [DOI] [PubMed] [Google Scholar]
- 17.Lieberman J, Slack F, Pandolfi PP, Chinnaiyan A, Agami R, Mendell JT. Noncoding RNAs and cancer. Cell. 2013;153:9–10. doi: 10.1016/j.cell.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Ryu AR, Lee MY. Chlorin e6-mediated photodynamic therapy promotes collagen production and suppresses MMPs expression via modulating AP-1 signaling in P. acnes-stimulated HaCaT cells. Photodiagnosis Photodyn Ther. 2017;20:71–77. doi: 10.1016/j.pdpdt.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Naim A, Pan Q, Baig MS. Matrix metalloproteinases (MMPs) in liver diseases. J Clin Exp Hepatol. 2017;7:367–372. doi: 10.1016/j.jceh.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Ding J, Yang J, Guo X, Zheng Y. MicroRNA roles in the nuclear factor kappa B signaling pathway in cancer. Front Immunol. 2018;9:546–554. doi: 10.3389/fimmu.2018.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eslamizadeh S, Heidari M, Agah S, Faghihloo E, Ghazi H, Mirzaei A, Akbari A. The role of MicroRNA Signature as diagnostic biomarkers in different clinical stages of colorectal cancer. Cell J. 2018;20:220–230. doi: 10.22074/cellj.2018.5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu N, Zhou B, Zhu G. Potential role of reversion-inducing cysteine-rich protein with kazal motifs (RECK) in regulation of matrix metalloproteinases (MMPs) expression in periodontal diseases. Med Sci Monit. 2016;22:1936–1938. doi: 10.12659/MSM.896546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia H, Chen S, Chen K, Huang H, Ma H. MiR-96 promotes proliferation and chemo- or radioresistance by down-regulating RECK in esophageal cancer. Biomed Pharmacother. 2014;68:951–958. doi: 10.1016/j.biopha.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Hong KJ, Hsu MC, Hung WC. RECK impedes DNA repair by inhibiting the erbB/JAB1/Rad51 signaling axis and enhances chemosensitivity of breast cancer cells. Am J Cancer Res. 2015;5:2422–2430. [PMC free article] [PubMed] [Google Scholar]
- 25.Du YY, Dai DQ, Yang Z. Role of RECK methylation in gastric cancer and its clinical significance. World J Gastroenterol. 2010;16:904–908. doi: 10.3748/wjg.v16.i7.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razdan A, de Souza P, Roberts TL. Role of MicroRNAs in treatment response in prostate cancer. Curr Cancer Drug Targets. 2018;18:929–944. doi: 10.2174/1568009618666180315160125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rapado-González Ó, Majem B, Muinelo-Romay L, Álvarez-Castro A, Santamaría A, Gil-Moreno A, López-López R, Suárez-Cunqueiro MM. Human salivary microRNAs in cancer. J Cancer. 2018;9:638–649. doi: 10.7150/jca.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang P, Liu XM, Ding L, Zhang XJ, Ma ZL. mTOR signaling-related MicroRNAs and Cancer involvement. J Cancer. 2018;9:667–673. doi: 10.7150/jca.22119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.