

Abstract
Alcohol is harmful to the body, causing hepatic steatosis, alcoholic hepatitis and cirrhosis. The effects of alcohol on the liver can be offset using natural antioxidants. This study aimed to evaluate the effects of the administration of oral β-carotene on the morphoquantitative characteristics of mice livers exposed to ethanol consumption. Forty-eight male mice were used, divided into six groups: Control (C), Low-dose alcohol (LA), Moderate-dose alcohol (MA), β-carotene (B), Low-dose alcohol+β-carotene (LA+B) and Moderate-dose alcohol+β-carotene (MA+B). On day 28 the animals were euthanized and the organs were harvested. The morphoquantitative analysis, evaluation of the collagen fiber content and transmission electron microscopy were performed. A one-way ANOVA was used for statistical analysis. There were no differences between NVhep, VVhep, SVhep, VVbin, TVhep and TMhep in groups C and the MA+B (P < 0.001). The analysis of type I collagen fibers revealed that the MA+B group presented differences with groups C (P < 0.001), LA (P = 0.046) and LA+B (P = 0.009). The ultrastructural analysis for NAm, NVm, NTm, VVm, Vm, SVm and TSm did not reflect any significant differences between the groups. Our results suggest that the degree of hepatic steatosis produced by different doses of alcohol can be prevented. However, the following factors should be considered: amount of alcohol consumed, exposure time, regulatory mechanisms of alcoholic liver disease and signaling pathways involved in the ingestion of both ethanol and antioxidants.
Keywords: Ethanol intake, β-carotene, alcoholic fatty liver disease, morphoquantitative analysis
Introduction
Alcohol can cause toxicity and death if ingested in excessive amounts, producing a series of consequences detrimental to health including: alcoholic fatty liver, alcoholic hepatitis, cirrhosis [1] and an increased predisposition to several types of cancer [2], heart diseases and a variety of neurological, cognitive and behavioral deficits. Generally, alcohol is metabolized via oxidative pathways, where the enzymes alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) participate, or by non-oxidative pathways.
Ethanol metabolism can cause oxidative stress and tissue damage mediated by free radicals, causing morphological and functional alterations in the liver. Oxidative stress is produced through the activation of xanthine oxidase, cytochrome P450 2E1 proteins (CYP2E1) and the NADPH oxidase complex, which increases reactive oxygen species [3], promoting lipid peroxidation, increased catabolism of hepatic vitamin A and progression of liver damage [4].
ROS decrease mitochondrial-reduced glutathione (mGSH), making this organelle more susceptible to oxidative damage [5]. Also, ROS stimulate the release of TNF-α, which can contribute to tissue damage and scar tissue formation in the liver.
It has also been described that ROS can interact with lipids, proteins and DNA in a process called peroxidation, leading to the production of malondialdehyde (MDA) and 4-hydroxynonenal [6]. This peroxidation process increases the permeability of the inner mitochondrial membrane and deterioration of mitochondrial oxidative phosphorylation [6], which leads to ballooning degeneration and necrotic precipitates in the hepatocytes, while the release of cytochrome c, a result of the increase in mitochondrial permeability, induces apoptosis [7]. Moreover, the deterioration of hepatic antioxidant defenses and the alteration of iron homeostasis can contribute to the oxidative damage related to the hepatotoxicity of alcohol [8].
The processes that protect against the aggression can occur through antioxidant enzymes that exert their action fundamentally at intracellular level and interfere in the initiation phase of ROS synthesis, inactivating their precursory molecules. These enzymes include superoxide dismutase, catalase, glutathione and glutathione reductase [9]. Other protection processes are non-enzymatic mechanisms like reduced glutathione (GSH), which detoxifies the ROS produced in the mitochondrial electron transport chain; metal-binding proteins, which help iron and copper to remain in a non-reactive state and avoid the formation of hydroxyl radicals; and the action of vitamins and/or provitamins like vitamin C and E and carotenoids like β-carotene, which have antioxidant properties that aid in eliminating free radicals [10,11].
Although there are certainly different mechanisms that promote the antioxidant defense of hepatocytes, the therapeutic alternative for these patients has concentrated on obtaining high concentrations of GSH in the liver. However, neither GSH itself nor its precursor, the amino acid cysteine, can be used as supplements because they cannot enter the hepatic cell. Therefore, experimental studies have used supplementation with β-carotene, as this can boost antioxidant activity by increasing the concentration of GSH [12].
Nonetheless, tests on the effectiveness of β-carotene supplementation for the treatment of alcoholic hepatopathies are contradictory, and it has not been possible to confirm its therapeutic action [13]. Studies have revealed that ethanol interferes with its conversion to vitamin A, whereas others have reported that moderate alcohol ingestion may result in an increase in the β-carotene concentrations when this is administered in doses commonly used for supplementation [14].
In this context, there is still a paucity of scientific data that endorse the use of traditional natural products for the treatment of alcoholism and its effects on the liver. β-carotene administration offers an alternative therapy; however, it is necessary to continue with the studies that can clarify their use in chronic drinkers. Therefore, the aim of this study was to examine the effects of the oral administration of β-carotene on the morphoquantitative characteristics of the livers of C57BL/6 mice exposed to ethanol consumption.
Materials and methods
Animals
Forty-eight male C57BL/6 mice were used (Mus musculus), 50 days old, from the Chilean Public Health Institute. They were kept for 30 days in the Animal Facility of the Center of Excellence in Morphological and Surgical Studies (CEMyQ) at the Universidad de La Frontera under standardized conditions and a 12 h light/dark cycle (08:00 am-8 pm/8 pm-8:00 am), with a standard laboratory diet (AIN-93M) and water ad libitum for adaptation to their new surroundings. During the experimental period, all the mice were fed a standard laboratory diet (AIN-93M), following the guidelines established in the Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research [15]. This project was approved by the Scientific Ethics Committee of the Universidad de La Frontera (N°043/2016).
At the beginning of the experimental period (day 1), the mice were divided into six groups:
Control (C): mice not exposed to either alcohol or β-carotene.
Low-dose alcohol (LA): mice exposed to low-dose alcohol consumption (3% v/v ad libitum) [16] for 28 days.
Moderate-dose alcohol (MA): mice exposed to moderate-dose alcohol consumption (7% v/v ad libitum) [16] for 28 days.
β-carotene (B): mice exposed to the administration of 0.52 mg/kg body weight/day of β-carotene [17] for 28 days.
Low-dose alcohol+β-carotene (LA+B): mice exposed to low-dose alcohol consumption plus administration of 0.52 mg/kg body weight/day of β-carotene for 28 days.
Moderate-dose alcohol+β-carotene (MA+B): mice exposed to moderate-dose alcohol consumption plus administration of 0.52 mg/kg body weight/day of β-carotene for 28 days.
β-carotene was administered in a dose of 0.52 mg/kg body weight/day (Lucarotin 10 CWD F/O, Badische Anilin- und Soda-Fabrik, Ludwigshafen, Germany). β-carotene was diluted in water for groups C, LA, MA and B, whereas alcohol was used for the dissolution in groups LA+B and MA+B. In order to calculate the volume of the dilution, an average consumption of 5 mL per animal/day was considered.
Euthanasia
At the end of the experiment on day 28, the animals were fasted for 6 hours and euthanized with sodium pentobarbital.
Morphoquantitative analysis of the liver
For the stereological study and the evaluation of the collagen fiber content, five animals from each group were used. The liver mass was determined with an analytical balance and the volume with Scherle’s method [18]. Five random fragments from each liver were taken and fixed in 4% buffered formalin (1.27 mol/L of formaldehyde in phosphate buffer 0.1 M pH 7.2) for 48 hours, dehydrated, and embedded in Paraplast Plus (Sigma-Aldrich Co., St. Louis, MO, USA). Once the blocks were obtained, 5 μm thick sections were made (Leica® RM2255) and stained with H&E and Sirius Red.
For the stereological study, two fields per section were observed: altogether 50 fields per group [19]. The slides were visualized under a Leica® DM2000 LED stereological microscope and photographed with a Leica® MC170 HD digital camera. The 36-point test system designed by STEPanizer® was used and the following parameters were determined: number density (NVhep), volume density (VVhep) and surface density (SVhep) of the hepatocytes; VV of the binucleate hepatocytes (VVbi) and cytoplasmic vacuoles (VVcit). In addition, the total volume (TVhep) and total mass (TMhep) of the hepatocytes were determined. NVhep was quantified according to the following equation: NVhep = Q-/(AT × t), where Q- is the number of observations in a certain area considering the prohibited lines and the prohibited plane, AT is the total area of the test system and dissector thickness (t). VVhep was estimated using the formula: VVhep = PPhep/PT (100%), where PPhep is the number of points that touch the hepatocytes and PT the total number of points in the system. SVhep was determined according to the following equation: Svhep = (2 × I)/LT, where I is the number of intersections that touch the structure and LT is the total length of the lines of the 36-point test system. VVbi and VVcit were estimated as previously described. TVhep was obtained by multiplying VVhep by the volume of the liver. TMhep was obtained by multiplying VVhep by the liver mass.
For the evaluation of the collagen fiber content, one field per section was observed; in total 25 fields per group [19]. The histological sections were stained with Sirius Red F3BA 0.1% p/v (Sigma-Aldrich Co., St. Louis, MO, USA) for 1 hour in an aqueous solution saturated with picric acid (Merck, Darmstadt, Germany). The lower filter (Leica Microsystems, Wetzlar, Germany) was placed above the microscope’s field iris diaphragm ring, while the upper filter was constructed from a combination of quarter-wave plates (Leica Microsystems, Wetzlar, Germany) placed below a linear polarizer, aligned with the transmission axis at 45° to the fast axis of the wave plate [20]. These two filters were aligned to maximize darkness in the background (i.e., the filters were crossed). The histological images were obtained using a microscope (Leica® DM750, Wetzlar, Germany), a digital camera (Leica® ICC50 HD, Wetzlar, Germany) and projected onto a LCD View Sonic®. The total area (μm2) of collagen fibers, types I and III, was analyzed with the ImagePro Premier 9.1 software (Media Cybernetics, Warrendale, PA, USA).
Transmission electron microscopy
Three animals from each group were used for the evaluation of the livers through electron microscopy (in Nanoscale Biomedical Imaging Facility, The Hospital for Sick Children of the University of Toronto, Dr. Adeli). In order to evaluate the hepatic ultrastructure, focusing on mitochondrial remodeling due to alcohol intake and/or β-carotene supplementation, small fragments of liver were fixed with glutaraldehyde 1.5% in 0.1 mol of cacodylate buffer pH 7.4. Then, they were fixed with osmium tetroxide 1% in 0.1 mol of cacodylate buffer at 4°C, they were dehydrated through a graduated series of acetone solutions and embedded in epoxy resin (Sigma-Aldrich Co., St. Louis, MO, USA). Ultrathin sections were made using an ultramicrotome (Leica® EM UC7, Wetzlar, Germany). These sections were mounted on a 200-mesh grid (G200-Cu, Electron Microscopy Sciences, Hatfield, PA, USA), stained with uranyl acetate and lead citrate, examined and then photographed using a transmission electron microscope (Philips Tecnai F20, Massachusets, USA). The microphotographs were taken at low (x3500), intermediate (x11500) and high (x25000) magnification. Forty-five electronic micrographs were evaluated per group and they had the entire mitochondria within the testing area, including the prohibited lines [21]. Random test points were assigned on the low power micrographs (x3500), and the volume density (VVm) of the mitochondria was estimated in the cells [19]. The TVm was obtained by multiplying the VVm by the volume of the liver. The number of mitochondria per unit area (NAm) was obtained using random test points on the intermediate power micrographs (x11500). The numerical density (NVm) of particles from the NAm and their VVm have been calculated by:
NVm = k/β × (NAm 3/2/VVm 1/2)
The factor K (>1) is a dimensionless coefficient which depends on the mitochondrial size distribution, where a K = 1.05 has been ascribed. The shape of the object examined depends on β factor, which is therefore a variable and should be considered when calculating numerical density. Two β values have been employed, the one appropriate for spheres (β = 1.382) and the other for ellipsoids at an axial ratio 4:1 (β = 2.25). The total number of mitochondria (TNm) was obtained by multiplying the NVm by the volume of the liver. The surface density (SVm) of the mitochondrial outer membrane was estimated on the high-power micrographs (x25000). The surface area of the structure (TSm) was estimated using the formula:
TSm = 2 × I × V(organ)
Statistical analysis
The differences in the quantitative data were evaluated using the Kolmogorov-Smirnov test (analysis of normality of the data) and Levene’s test (homoscedasticity of the variances). The differences between the groups were analyzed with a one-way ANOVA, followed by Tukey’s post-hoc HSD test or Dunnett’s T3 test, as applicable. P < 0.05 was considered statistically significant (IBM SPSS Statistics, Version 21, IBM Corp., Armonk, NY, USA).
Results
Morphoquantitative analysis
In this regard, the LA group showed higher SVhep than the C group (P < 0.001), whereas the β-carotene supplementation rescued this finding in the LA+B group, which showed no difference to the C group. As for the hepatic steatosis quantification, the MA group showed higher Vvcit than the C and LA groups, and the β-carotene supplementation managed to reduce lipid accumulation within the hepatocytes in the MA+B group (P < 0.001) (Table 1).
Table 1.
Stereological analysis of mice livers exposed to ethanol consumption and oral supplementation of β-carotene
| Mean SD | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| C | LA | MA | B | LA+B | MA+B | P | |
| NVhep (mm-3) | 115322±43596 | 158285±65081a | 169026±52317a | 152632±35201a | 144153±49792a | 127382±42912c,d | <0.001 |
| VVhep (%) | 13±5 | 14±6 | 15±5 | 13±5 | 11±5c | 14±6 | 0.009 |
| SVhep (mm-1) | 25±11 | 33±12a | 32±13 | 26±13 | 23±11b,c | 28±14 | <0.001 |
| VVcit (%) | 42±13 | 48±14 | 70±14a,b | 63±15a,b | 60±13a,b,c | 60±14a,b,c | <0.001 |
| VVbin (%) | 1±1 | 2±1 | 2±2 | 2±2 | 2±2 | 2±2 | 0.056 |
| TVhep (%) | 0.20±0.07 | 0.18±0.07 | 0.20±0.06 | 0.19±0.07 | 0.15±0.07a,c | 0.21±0.09e | 0.002 |
| TMhep (g) | 0.22±0.07 | 0.19±0.08 | 0.22±0.07 | 0.21±0.07 | 0.17±0.08a,c | 0.23±0.09e | 0.003 |
Significant differences (P < 0.05) with the C group.
Significant differences (P < 0.05) with the LA group.
Significant differences (P < 0.05) with the MA group.
Significant differences (P < 0.05) with the B group.
Significant differences (P < 0.05) with the LA+B group.
The presence of types I and III collagen fibers in the liver can be observed (Figure 1), while the collagen fiber content is presented in Figure 2. The results showed that the type I collagen content of the liver was greater in the MA+B group than in the C group (P < 0.001), whereas the B group presented the greatest content of type III collagen. There were only significant differences between the B and LA+B groups (P = 0.047).
Figure 1.
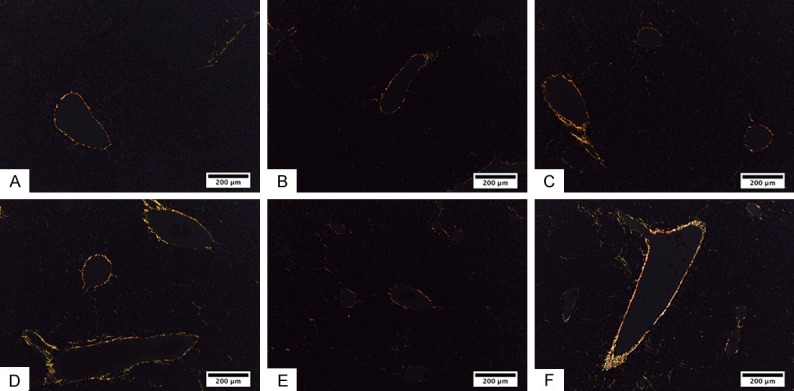
Presence of type I and III collagen fibers in hepatic tissue. Type I collagen fibers (red and yellow) and III (green) in mouse livers for each study group. A. Control group. B. Low-dose alcohol group. C. Moderate-dose alcohol group. D. β-carotene group. E. Low-dose alcohol with oral supplementation of β-carotene group. F. Moderate-dose alcohol group with oral supplementation of β-carotene. Sirius Red.
Figure 2.
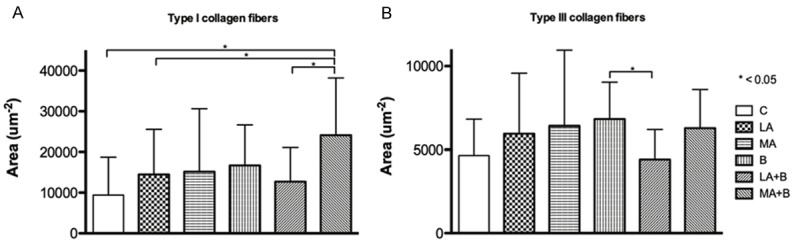
Quantification of collagen content in hepatic tissue. Evaluation of collagen content in mice liver exposed to ethanol consumption and oral supplementation of β-carotene using Image-Pro Premier. A. Type I collagen fibers. B. Type III collagen fibers.
Transmission electron microscopy
The hepatic ultrastructure evaluation (Figure 3) showed that the control group presented with well-preserved hepatocyte ultrastructure with little macrovesicular steatosis (asterisk, Figure 3A). Also, the control group showed numerous mitochondria (white arrowheads, Figure 3B). The low-alcohol dose damaged the hepatocyte ultrastructure, represented by widespread macrovesicular lipid droplets (asterisk, Figure 3C). Moreover, the LA group showed microvesicular steatosis (black arrowheads, Figure 3D), swollen mitochondria (white arrowheads, Figure 3D) coupled with enlarged cisterns of the endoplasmic reticulum (white arrows, Figure 3D).
Figure 3.
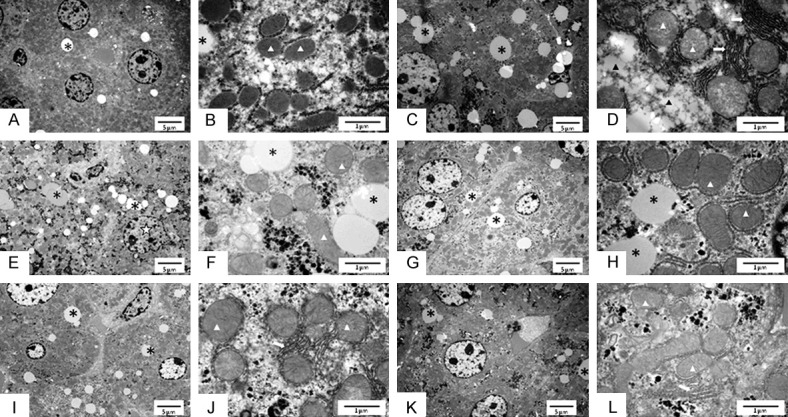
Ultrastructural analysis on liver of C57BL/6 mice. Ultrastructural analysis of the liver of male C57BL/6 mice exposed to ethanol consumption and oral supplementation of β-carotene. Transmission electron microscopy performed at low (x3500) and high magnification (x25000), respectively. A, B. Control group. C, D. Low-dose alcohol group. E, F. Moderate-dose alcohol group. G, H. β-carotene group. I, J. Low-dose alcohol with oral supplementation of β-carotene group. K, L. Moderate-dose alcohol group with oral supplementation of β-carotene. The symbols indicate mitochondrion (white arrowheads), macrovesicular steatosis (asterisk), microvesicular steatosis (black arrowheads), enlarged endoplasmic reticulum cistern (white arrows) and irregularly shaped nucleus (white star).
Moderate alcohol intake caused an impaired hepatocyte ultrastructure with an irregular nuclear shape (white star, Figure 3E), marked macrovesicular steatosis (asterisk, Figure 3E, 3F) and swollen mitochondria (white arrowheads, Figure 3F). The B group showed a well-preserved hepatocyte cytoarchitecture, albeit with noticeable macrovesicular steatosis (Figure 3G, 3H). Of note, the β-carotene supplementation did not damage the mitochondria, which showed visible cristae (arrowheads, Figure 3H), nor the endoplasmic reticulum, which was organized without fission or enlarged cistern (Figure 3H).
Although the β-carotene supplementation was able to reduce the macrovesicular steatosis in the LA+B group (asterisk, Figure 3I), it could not address the swollen mitochondria (white arrowheads, Figure 3J) and the endoplasmic reticulum alterations (white arrow, Figure 3J), resembling the untreated group. Of note, the β-carotene supplementation seemed to ameliorate the ultrastructure of hepatocytes from the MA+B group, which showed less macrovesicular steatosis than the MA group (asterisk, Figure 3K), in addition to mitochondrial and endoplasmic reticulum dispositions that resembled the B group (white arrowheads, Figure 3I).
The quantitative analysis of the mitochondria showed that there were no significant differences between the animals in the ethanol-exposed groups and those exposed to oral supplementation with β-carotene (Table 2).
Table 2.
Ultrastructural analysis of mice livers exposed to ethanol consumption and oral supplementation of β-carotene
| Mean SD | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| C | LA | MA | B | LA+B | MA+B | P | |
| NAm (μm-2) | 0.425±0.119 | 0.331±0.115 | 0.364±0.119 | 0.346±0.134 | 0.362±0.180 | 0.347±0.092 | 0.434 |
| NVm (μm-3) | 0.087±0.084 | 0.056±0.066 | 0.072±0.097 | 0.046±0.053 | 0.062±0.086 | 0.055±0.031 | 0.705 |
| TNm | 0.142±0.138 | 0.092±0.108 | 0.119±0.160 | 0.075±0.087 | 0.102±0.141 | 0.091±0.050 | 0.705 |
| VVm (μm3/μm3) | 0.581±0.150 | 0.550±0.224 | 0.530±0.185 | 0.600±0.090 | 0.572±0.187 | 0.435±0.164 | 0.127 |
| TVm (μm3) | 0.849±0.214 | 0.723±0.313 | 0.737±0.237 | 0.891±0.172 | 0.792±0.295 | 0.650±0.241 | 0.109 |
| SVm (μm-1) | 2.046±0.486 | 2.022±0.663 | 1.558±0.746 | 1.689±0.522 | 1.618±0.550 | 1.772±0.378 | 0.092 |
| TSm (μm2) | 33.687±8.478 | 29.432±9.453 | 24.939±13.690 | 28.432±10.923 | 25.027±9.236 | 29.588±5.956 | 0.158 |
Discussion
The doses of alcohol and β-carotene received, as well as the treatment periods, were established in previous studies [16,17]. In this sense, low doses of oral supplementation with β-carotene have demonstrated beneficial effects against liver damage induced by the antioxidant pathway [12]. However, Bast et al. [22] suggest that β-carotene could be effective as an antioxidant only at low O2 pressures, because when it is administered at high O2 pressures, it could even stimulate lipid peroxidation, producing ballooning degeneration and necrotic precipitates in the hepatocytes.
One of the mechanisms described to explain alcohol-induced liver disease is a decrease in sufficient oxygen supply so as to produce tissue hypoxia, particularly in perivenular hepatocytes due to an increased oxygen consumption induced by alcohol. It has been suggested that this causes hypoxia at the cellular level in pericentral regions of the liver [23] to the point of producing an anoxic injury of the perivenular hepatocytes [24] (Figure 1). From our results, we may infer that the oral administration of β-carotene reverses the reduction in partial oxygen pressure caused by chronic alcohol consumption. This would explain why there are no differences in the stereological analysis (NVhep, VVhep, SVhep, VVbin, TVhep and TMhep) in the C and MA+B groups (P = 0.926, P > 0.999, P = 0.937, P > 0.999, P > 0.999, P > 0.999 and P = 0.990; respectively) (Table 1). Other authors, however, did not observe any increases in oxygen consumption in rats fed a liquid ethanol diet [3] in a model that only develops fatty liver [25]. Thus, the reasons for these differences have yet to be clarified. Other studies have indicated that ethanol increases the binding of hypoxia markers prior to the onset of inflammation and necrosis [23]. It is possible that hypoxia has a key role in the progression of alcoholic liver disease [26]; this would explain the absence of damage in models that do not exhibit increases in hepatic oxygen extraction [3]. Nevertheless, ALD has been observed without tissue hypoxia [27].
Unlike other cells, most animal cells do not contain vacuoles. At the same time, the formation of giant vacuoles in animal cells in vivo and in culture occurs as a morphological phenomenon, called cytoplasmic vacuolization, which develops spontaneously or after exposure to bacterial or viral pathogens, as well as to several artificial compounds of low molecular weight [28,29]. The alcohol is a molecule of low molecular weight that contributes to anoxic injury in healthy hepatocytes [23]. Caraceni et al. [30] reported that chronic alcohol consumption reduces the hepatic glycogen content and produces moderate steatosis. In this sense, Lin et al. [12] and Peng et al. [17] showed that supplementation with β-carotene can prevent ethanol-induced liver damage and increase GSH concentrations in rats exposed to the chronic alcohol consumption. These findings agree with our results, where although there are no significant differences, greater VVcit was observed in the LA and MA groups exposed to chronic ethanol consumption compared to their analogs, LA+B and MA+B, supplemented with β-carotene (Table 1).
The products of lipid peroxidation stimulate the production of collagen and fibrosis. In turn, acetaldehyde forms protein adducts in hepatic stellate cells, promoting the production of these components by reducing the inhibition of collagen synthesis [31]. In this context, it has been reported that acetaldehyde is the key toxin in ALD, causing cell damage, inflammation, remodeling of the extracellular matrix and fibrogenesis [32]. It has been described that acetaldehyde increases the proportion of reduced nicotinamide adenine dinucleotide (NADH) to oxidized nicotinamide adenine dinucleotide (NAD+) in the hepatocytes to reduce fatty acid β-oxidation by the mitochondria, which produces a fatty liver [33]. Acetaldehyde can also form tubulin adducts that induce dysfunction of the microtubules, causing a decrease in the transport of liver lipoproteins [33]. Moreover, it has been described that the protein-acetaldehyde adducts promote collagen production and can act as neoantigens, inducing the immune response [34]. In our study, an increase was noted in the content of type I collagen (Figure 1), which although it is not significant, allows us to corroborate our findings with previous studies. Also, the analysis of type I collagen fibers revealed that only the MA+B group presented significant differences with the C (P < 0.001), LA (P = 0.046) and LA+B (P = 0.009) groups, which makes it possible to state that the effectiveness of the administration of oral β-carotene as a treatment will depend on the amount and type of alcohol consumed and consumption patterns (Figure 2).
Mitochondria are very important organelles of the eukaryotic cells. On the one hand, they produce the necessary energy, in the form of ATP, for the vital activity of these cells; on the other hand, they initiate the process of apoptosis through mitochondrial disintegration, which not only leads to the loss of their function, but also brings about the release of proapoptotic factors. Emerging evidence suggests that mitochondrial dysfunction caused by oxidative stress is an early event that plays an important role in the pathogenesis of ethanol-induced apoptosis [35]. It was recently shown that extrinsic and intrinsic pathways are related to apoptosis induced by the progression of ALD. As a result, the pan-caspase inhibitor could be a good approach to avoid ALD progression [36].
The first hepatic injury caused by alcohol abuse is the excessive accumulation of fat deposits [37]. From the first days, mitochondrial and cytoplasmic changes occur. In effect, alcoholic fatty liver or simple steatosis, which is usually macrovesicular, develops in ~90% of heavy drinkers and may be seen within 2 weeks of heavy and regular alcohol ingestion [38]. Ethanol effects have also been associated with mitochondrial, endoplasmic reticulum, ribosomal and vacuolar alterations. Steatosis is installed in the third week of ethylic intoxication, initially as the microvesicular, and subsequently as the macrovesicular kind [39]. In this sense, our results agree with Tutunaru et al. [39], where we found that the mitochondria of the ethanol groups were obviously damaged, such that the LA group showed widespread macrovesicular lipid droplets (Figure 3C) and microvesicular steatosis with enlarged cisterns of the endoplasmic reticulum (Figure 3D). Meanwhile, the MA group presented with an impaired hepatocyte ultrastructure with irregular nuclear shape (Figure 3E), compatible with proapoptotic stimuli, marked macrovesicular steatosis and swollen mitochondria (Figure 3F). Also, swollen mitochondria were described in the groups that received alcohol. These authors further described ethanol as causing increased liver regeneration activity, which could be observed through bi- or trinucleate hepatocytes. Our results establish that the administration of ethanol for a period of 4 weeks induces liver regeneration, with a greater VVbin being observed in the LA (2±1%; P = 0.110) and MA groups (2±2%; P = 0.235) compared to the C group (1±1%). In addition, this liver regeneration (VVbin) was greater in the MA+B group (2±2%; P = 0.045) than the C group (Table 1). These results also agree with the ultrastructural analysis, where it was found that the β-carotene supplementation seemed to significantly ameliorate the ultrastructure of hepatocytes from the MA+B group, which showed less macrovesicular steatosis than the MA group (Figure 3K), in addition to mitochondrial and endoplasmic reticulum dispositions that resemble the B group (Figure 3I).
Ethanol exposure of cells alters the NAD/NADH ratio that causes fatty liver through the inhibition of gluconeogenesis and fatty acid oxidation; CYP2E1 generates free radicals as a result of the oxidation of NADPH to NADP+(Stewart et al., 2001) and the activation of hepatic macrophages occurs to produce TNF-α [40], causing ROS production in the mitochondria. Also, ROS increase the permeability of the internal mitochondrial membrane and causes the mitochondrial oxidative phosphorylation process to deteriorate [6], instigating the release of cytochrome c, inducing cell apoptosis. Other histologic characteristics of ALD include glycogenated nuclei, megamitochondria, hemosiderin deposits, cholestasis and ductular reaction [41]. Although the ultrastructural analysis found macrovesicular lipid droplets, microvesicular steatosis and endoplasmic reticulum alterations, our ultrastructural analysis on the morphoquantitative characteristics of the mitochondria did not reflect significant differences in NAm, NVm, TNm, VVm, TVm, SVm or TSm in any of the groups, through which we can indicate that the effectiveness of the oral treatment with β-carotene will depend on the ethanol doses provided and/or on the period of ethanol exposure (Table 2).
Our study reveals, from a morphoquantitative point of view, the effects of alcohol on the hepatic cells and how these characteristics vary with the oral supplementation of β-carotene. From this perspective, we were able to observe that although it is possible to prevent the degree of hepatic steatosis produced by different doses of alcohol in order to avoid the progression to more serious injuries, other factors must be considered, such as: amount of alcohol consumed, exposure time, regulatory mechanisms of alcoholic liver disease and signaling pathways involved in the intake of both ethanol and antioxidants.
Acknowledgements
This work was supported by operating research grants from Universidad de La Frontera, DIUFRO Project DI18-0001; DIUFRO-FAPERJ BRASIL Nº FJP15-0013; CONICYT-PCHA/National Doctorate/2015-21150991 and the Canadian Institutes for Health Research awarded to Dr. Adeli.
Disclosure of conflict of interest
None.
References
- 1.Arias R. Reacciones fisiológicas y neuroquímicas del alcoholismo. Diversitas. 2005;1:138–47. [Google Scholar]
- 2.Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004;13:511–9. [PubMed] [Google Scholar]
- 3.Bredfeldt JE, Riley EM, Groszmann RJ. Compensatory mechanisms in response to an elevated hepatic oxygen consumption in chronically ethanol-fed rats. Am J Physiol. 1985;248:G507–11. doi: 10.1152/ajpgi.1985.248.5.G507. [DOI] [PubMed] [Google Scholar]
- 4.Albano E. Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Mol Aspects Med. 2008;29:9–16. doi: 10.1016/j.mam.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol. 2005;204:263–73. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245–54. [PMC free article] [PubMed] [Google Scholar]
- 7.Ronis MJ, Korourian S, Blackburn ML, Badeaux J, Badger TM. The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol. 2010;44:157–69. doi: 10.1016/j.alcohol.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 9.Chang P, Cheng E, Brooke S, Sapolsky R. Marked differences in the efficacy of post-insult gene therapy with catalase versus glutathione peroxidase. Brain Res. 2005;1063:27–31. doi: 10.1016/j.brainres.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 10.Nanji AA, Jokelainen K, Lau GK, Rahemtulla A, Tipoe GL, Polavarapu R, Lalani EN. Arginine reverses ethanol-induced inflammatory and fibrotic changes in liver despite continued ethanol administration. J Pharmacol Exp Ther. 2001;299:832–9. [PubMed] [Google Scholar]
- 11.Nieto N. Ethanol and fish oil induce NFkappaB transactivation of the collagen alpha2(I) promoter through lipid peroxidation-driven activation of the PKC-PI3K-Akt pathway. Hepatology. 2007;45:1433–45. doi: 10.1002/hep.21659. [DOI] [PubMed] [Google Scholar]
- 12.Lin WT, Huang CC, Lin TJ, Chen JR, Shieh MJ, Peng HC, Yang SC, Huang CY. Effects of beta-carotene on antioxidant status in rats with chronic alcohol consumption. Cell Biochem Funct. 2009;27:344–50. doi: 10.1002/cbf.1579. [DOI] [PubMed] [Google Scholar]
- 13.Bjelakovic G, Gluud LL, Nikolova D, Bjelakovic M, Nagorni A, Gluud C. Antioxidant supplements for liver diseases. Cochrane Database Syst Rev. 2011:CD007749. doi: 10.1002/14651858.CD007749.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leo MA, Lieber CS. Alcohol, vitamin A, and beta-carotene: adverse interactions, including hepatotoxicity and carcinogenicity. Am J Clin Nutr. 1999;69:1071–85. doi: 10.1093/ajcn/69.6.1071. [DOI] [PubMed] [Google Scholar]
- 15.Committee for the update of the guide for the care and use of laboratory animals, institute for laboratory animal research, division on earth and life studies, national research council. guide for the care and use of laboratory animals. Eighth Edi. Washington, DC: The National Academies Press; 2011. [Google Scholar]
- 16.Furuya D, Binsack R, Machado U. Low ethanol consumption increases insulin sensitivity in Wistar rats. Braz J Med Biol Res. 2003;36:125–30. doi: 10.1590/s0100-879x2003000100017. [DOI] [PubMed] [Google Scholar]
- 17.Peng H, Chen Y, Yang S, Ho P, Yang S, Hu J, Yang SC. The antiapoptotic effects of different doses of β-carotene in chronic ethanol-fed rats. Hepatobiliary Surg Nutr. 2013;2:132–41. doi: 10.3978/j.issn.2304-3881.2013.06.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie. 1970;26:57–60. [PubMed] [Google Scholar]
- 19.Mandarim-de-Lacerda CA, del Sol M. Tips for studies with quantitative morphology (morphometry and stereology) Int J Morphol. 2017;35:1482–94. [Google Scholar]
- 20.Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89:397–410. doi: 10.1007/BF00788278. [DOI] [PubMed] [Google Scholar]
- 21.Gundersen HJG. Notes on the estimation of the numerical density of arbitrary profiles: the edge effect. J Microsc. 1977;111:219–23. [Google Scholar]
- 22.Bast A, Haenen GR, van den Berg R, van den Berg H. Antioxidant effects of carotenoids. Int J Vitam Nutr Res. 1998;68:399–403. [PubMed] [Google Scholar]
- 23.Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–6. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- 24.Israel Y, Kalant H, Orrego H, Khanna JM, Videla L, Phillips JM. Experimental alcohol-induced hepatic necrosis: suppression by propylthiouracil. Proc Natl Acad Sci U S A. 1975;72:1137–41. doi: 10.1073/pnas.72.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieber CS, Decarli LM. Quantitative relationship between amount of dietary fat and severity of alcoholic fatty liver. Am J Clin Nutr. 1970;23:474–8. doi: 10.1093/ajcn/23.4.474. [DOI] [PubMed] [Google Scholar]
- 26.Albanes D, Heinonen OP, Taylor PR, Virtamo J, Edwards BK, Rautalahti M, Hartman AM, Palmgren J, Freedman LS, Haapakoski J, Barrett MJ, Pietinen P, Malila N, Tala E, Liippo K, Salomaa ER, Tangrea JA, Teppo L, Askin FB, Taskinen E, Erozan Y, Greenwald P, Huttunen JK. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–70. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 27.Lieber CS, Baraona E, Hernández-Muñoz R, Kubota S, Sato N, Kawano S, Matsumura T, Inatomi N. Impaired oxygen utilization. A new mechanism for the hepatotoxicity of ethanol in sub-human primates. J Clin Invest. 1989;83:1682–90. doi: 10.1172/JCI114068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aki T, Nara A, Uemura K. Cytoplasmic vacuolization during exposure to drugs and other substances. Cell Biol Toxicol. 2012;28:125–31. doi: 10.1007/s10565-012-9212-3. [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Duan CY, Chen SK, Zhang CY, He T, Li H, Liu YP, Dai RY. The suppressive role of p38 MAPK in cellular vacuole formation. J Cell Biochem. 2013;114:1789–99. doi: 10.1002/jcb.24522. [DOI] [PubMed] [Google Scholar]
- 30.Caraceni P, Ryu HS, Subbotin V, De Maria N, Colantoni A, Roberts L, Trevisani F, Bernardi M, Van Thiel DH. Rat hepatocytes isolated from alcohol-induced fatty liver have an increased sensitivity to anoxic injury. Hepatology. 1997;25:943–9. doi: 10.1002/hep.510250426. [DOI] [PubMed] [Google Scholar]
- 31.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Mello T, Ceni E, Surrenti C, Galli A. Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med. 2008;29:17–21. doi: 10.1016/j.mam.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Kane AB, Kumar V. Environmental and nutritional pathology. Philadephia: W. B. Saunders Company; 1999. [Google Scholar]
- 34.Stewart S, Jones D, Day CP. Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol Med. 2001;7:408–13. doi: 10.1016/s1471-4914(01)02096-2. [DOI] [PubMed] [Google Scholar]
- 35.Yan M, Zhu P, Liu HM, Zhang HT, Liu L. Ethanol induced mitochondria injury and permeability transition pore opening: role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2007;13:2352–6. doi: 10.3748/wjg.v13.i16.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao F, Cubero FJ, Ramadori P, Liao L, Haas U, Lambertz D, Sonntag R, Bangen JM, Gassler N, Hoss M, Streetz KL, Reissing J, Zimmermann HW, Trautwein C, Liedtke C, Nevzorova YA. Inhibition of Caspase-8 does not protect from alcohol-induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis. 2017;8:e3152. doi: 10.1038/cddis.2017.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arakawa M, Taketomi S, Furuno K, Matsuo T, Iwatsuka H. Metabolic studies on the development of ethanol-induced fatty liver in KK-Ay mice. J Nutr. 1975;105:1500–8. doi: 10.1093/jn/105.12.1500. [DOI] [PubMed] [Google Scholar]
- 38.Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol. 2018;113:175–94. doi: 10.1038/ajg.2017.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tutunaru D, Ciocoiu M, Coman M, Musat C, Nechita A, Bădescu M. Ethanol induced hepatic steatosis-morpho-functional aspects. Ann Rom Soc Cell Biol. 2010;15:362–7. [Google Scholar]
- 40.Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol. 2003;163:1137–46. doi: 10.1016/s0002-9440(10)63473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiniakos DG. Liver biopsy in alcoholic and non-alcoholic steatohepatitis patients. Gastroenterol Clin Biol. 2009;33:930–9. doi: 10.1016/j.gcb.2009.05.009. [DOI] [PubMed] [Google Scholar]