

Abstract
Background: p300, a transcription co-activator, plays an important role in multicellular organisms and inflammation. However, the mechanism of p300 in type 2 diabetes nephropathy (T2DN) remains largely unknown. Our aim is to explore the mechanism of p300 in T2DN. Methods: A T2DN mice model was induced by db/db transgenic mice or a high fat diet for 24 weeks. The levels of IL-6 and TNF-α were examined by real-time PCR (RT-PCR) in the renal cortex and by an enzyme linked immunosorbent assay (ELISA) in the serum of the T2DN mice. p300 siRNA was used to knockdown the expression of p300, and His-tagged-p300 plasmid was used to overexpress the p300 protein level in podocytes. Hematoxylin-eosin staining (H&E) and Masson trichrome analysis were used to detect the kidney pathology in T2DN. Results: The levels of IL-6 and TNF-α were significantly increased in T2DN. p300 was significantly increased in T2DN. Consistently, p300 silencing significantly suppressed the inflammatory response and the overexpression of p300 significantly promoted the production of IL-6 and TNF-α in T2DN. Conclusions: This study demonstrated that the production of IL-6 and TNF-α, and the expression of p300, were increased in T2DN. Furthermore, P300 significantly promoted the activation of the NF-κB subunit p65 through a direct association with p65 in T2DN, subsequently enhancing the production of IL-6 and TNF-α.
Keywords: Inflammation, transcription coactivator p300, type 2 diabetes nephropathy
Introduction
Type 2 diabetes is a long-term metabolic disorder characterized by hyperglycemia, insulin resistance, and inflammation [1-3]. Long-term hyperglycemia affects multiple tissues and organs of the body, leading to various chronic complications including nephropathy [4]. Diabetic nephropathy (DN), is one of the most common diabetic complications and occurs in both type 1 and type 2 diabetes and develops in approximately 30% of diabetic patients. Diabetic nephropathy can develop into nephritic syndrome, eventually leading to kidney failure and death [5]. DN is a leading cause of end-stage renal disease and has a high morbidity and mortality in diabetic patients [6,7]. The morbidity of DN has tremendously increased with the increasing incidence and prevalence of diabetes, and the pathogenesis of DN has not been fully understood [8,9]. Recent studies have shown that the inflammatory response is crucial in the development and progression of DN, even though DN has not been traditionally considered an inflammatory disease [2,10,11].
The nuclear factor-kappa B (NF-κB) family of transcription factors regulates multiple biological events including cell growth, survival, and the immune response. Structurally conserved members of the NF-κB/Rel family function as dimers in various combinations including p50, p52, p65 (Rel A), Rel B, and c-Rel. NF-κB exists in an inactive form in the cytoplasm because of its interaction with the inhibitory protein, IκBα [12,13]. Upon stimulation, the IκB kinase (IKK) phosphorylates IκB, causing ubiquitination and proteasomal degradation, which allows NF-κB to translocate into the nucleus and thus activates the expression of target genes [14,15]. NF-κB serves a major role in the inflammatory and innate immune responses by regulating genes that encode inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) [16,17]. The activation of NF-κB and the transcription of certain proinflammatory cytokines have been demonstrated as markers of progressive DN [18]. This finding strongly suggests that NF-κB-mediated inflammatory processes may represent a novel mechanism leading to DN. Therefore, reducing renal inflammation may be a novel therapeutic strategy for DN.
The most common active form of NF-κB is a heterodimer of two subunits, p65 (RelA) and p50 (NF-κB 1). The subunit p65 - but not p50 - contains an activation domain that binds to the co-activators for transcription initiation [19]. p300 and its paralog co-activator proteins, such as the CREB binding protein (CBP), are general transcriptional co-activators that help NF-κB bridge with the basal transcription machinery [19,20]. p300 possesses intrinsic histone acetylase (HAT) activity, which is necessary to open the chromatin structure through an acetylation-induced conformational change in histone protein [21]. Previous studies have shown that p300 aids in recruiting p65 NF-κB to the promoters of its target genes and may play an important role in the production of inflammatory cytokines. [22,23] However, the mechanism of the activation of NF-κB subunit p65 by p300 remains to be established in DN.
In this study, we tried to explore the role of p300 in the inflammation of T2DN and to find out whether the inhibition of p300 can suppress the production of inflammatory cytokines in T2DN.
Materials and methods
Animals
Db/wt (n = 12) and db/db (n = 12) mice with a C57BLKS/J background were purchased from the Institute of Model Animal Science (Nanjing University) and raised in SPF conditions. The mice were fed ad libitum with a standard diet and kept under a light-dark cycle of 12 h (n = 6 per group). The Db/wt and db/db mice were sacrificed at 18 weeks. At 8 weeks, the mice were randomly divided into two groups (n = 6 per group): the normal chow diet (ND) group and the high-fat diet group. All the mice were then fed for another 24 weeks. ND consists of 10% fat, 20% protein, and 70% carbohydrate (18.8 kJ/g). The HF (high fat) diet consists of 31.5% fat, 16% protein, and 52.5% carbohydrate (23.3 kJ/g). The T2DN mice were defined as high protein urea, high serum creatinine, and urea nitrogen. All study procedures and methods were approved and performed in accordance with the relevant guidelines and regulations by the Animal Care and Use Committee of Chengdu First People’s Hospital.
Blood and kidney examination
The mice were anesthetized using an intraperitoneal injection of pentobarbital, and blood samples were drawn from the tail vein after food and water fasting for a minimum of 10 h. The serum was collected after the blood samples were placed at room temperature for 30 minutes and spun at 3000 rpm for 15 minutes at 4°C, then subjected to an ELISA measurement of the inflammatory cytokines (IL-1β, IL-6 and TNF-α) according to the manufacturer’s instructions (R&D, Minnesota, USA). The mice were sacrificed by cervical dislocation and the two kidneys of each mouse were collected. One kidney was stored at -80°C for further experimental analysis, and the other one was fixed in formalin at 4°C overnight and then was subsequently dehydrated in a series of ethanol concentrations, then cleared in xylene and embedded in paraffin. The diabetic nephropathy mice were assessed as having a high serum level of blood urea nitrogen (> 50 mg/dl), creatinine (> 1 mg/dl), mesangial cell proliferation, and tubular injury.
Hematoxylin-eosin (H&E) and Masson trichrome staining
4 μm thick paraffin kidney sections were deparaffinized in xylene, then hydrated through a series of ethanol concentrations, and stained with hematoxylin and eosin (H&E) and Masson trichrome staining (MTS), following standard protocols. Histological observations were performed on a minimum of four sections of the different regions of the tissue. Images were acquired using an Olympus BX50 microscope connected to a digital camera. MTS was quantified using image pro-plus Software 6.0 (Media Cybernetics, Bethesda, MD, USA).
RNA extraction and real-time PCR (qRT-PCR)
Total RNA was extracted from the tissues using Trizol (Invitrogen) and subjected (3 μg) to reverse transcription (RT) with random hexanucleotide primers (Quanti-Tect RT kit; Qiagen, Tokyo, Japan) according to the manufacturer’s protocol. The resulting cDNA was then subjected to real-time PCR analysis with SYBR green PCR master mix (TaKaRa, Shiga, Japan) and 200 nM gene-specific primers. Assays were performed in triplicate with a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). The amplification protocol comprised an initial incubation at 60°C for 30 s and 95°C for 3 s, followed by 40 cycles. The sequences of the primers (sense and antisense) are shown in Supplementary Table 1. The expression levels of IL-1β, IL-6, and TNF-α were normalized to that of β-actin mRNA, which served as an endogenous control. The relative gene expression levels were calculated using the 2-ΔΔct method.
Podocyte cell culture and treatment
The conditional immortalized human podocyte line was obtained from ATCC. Cells were cultured in RPMI-1640 medium supplemented with 100 U/ml penicillin/streptomycin and 5% fetal bovine serum (FBS) at 37°C in a 5% CO2 humidified atmosphere. For the measurement of cytokine secretion from podocytes, the cells were cultivated in DMEM supplemented with 10% FBS and 25 mmol/L glucose for 48 h. The supernatants were harvested and centrifuged to get rid of the cell debris. The supernatants were then used for the determination of the inflammatory factors by ELISA (R&D, Minnesota, USA) following the manufacturer’s instructions. The levels of secreted inflammatory factors were normalized to the total protein determined by the BCA Protein Assay Kit (Thermo Fisher, Rockford, Illinois, USA). Three independent experiments were performed. For the p300 transfection, the cells were seeded at 4 × 105 cells per well in 6-well plates. After incubation for 12 h, the medium from the 6-well plate was changed with 1 ml, and then the cells were transfected with 3 μg of p300 plasmid (purchased and reconstructed from Zhen Shanghai and Shanghai Industrial Co., Ltd) using JetPEI Reagent (Life, MA, America) following the manufacturer’s instructions. After 8 h, the transfected cells were replenished with 2 ml of normal medium.
Western blotting
The proteins from the mouse kidney tissue and podocyte cells treated with high glucose for 48 h were extracted using the cell lysis buffer (CST, Danvers, America). Approximately 40 μg of total protein was loaded for each sample. The protein samples were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). After blocking, the membrane was probed with a primary antibody against the following proteins: p300 (1:500) (Merck Millipore, Massachusetts, USA), t-p65, p-p65 (1:1000) (CST, Danvers, America), t-IκBα, p-IκBα (1:1000) (CST, Danvers, America), GAPDH (1:1000) (CST, Danvers, America). After washing, the membrane was probed with appropriate horseradish peroxidase-conjugated secondary antibodies. The protein bands were revealed by the ECL detection reagent (Thermo Fisher, Rockford, Illinois, USA) on autoradiography films (Fuji Film, Tokyo, Japan). The western blot band density was quantified with Image J software (National Institutes of Health, Bronx, NY, USA). GAPDH was used as a loading control.
Statistical analysis
All data are shown as the mean value ± the standard error of the mean (SEM). All statistical analyses were carried out using SPSS 20 software. The results were subjected to a one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post-hoc test. A ‘p’ value less than 0.05 was considered statistically significant.
Results
The levels of IL-1β, IL-6, and TNF-α are significantly increased in T2DN
To explore the inflammation status in T2DN, we evaluated the production of inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, by using podocytes and db/db mice. First, we created a cell model by treating podocytes with high glucose (experimental group) or mannitol (control group). We found that the mRNA levels of IL-1β, IL-6, and TNF-α were significantly increased in the high glucose group compared to of the corresponding levels in the control group (Figure 1A). An ELISA assay reflected similar results in the supernatants (Figure 1B) Together, these data indicated that the inflammatory response was exaggerated in T2DN in vitro. Furthermore, the inflammation status in vivo was also observed. The mRNA levels of IL-1β, IL-6, and TNF-α in db/db mice kidney were increased compared to the levels of the db/wt mice kidneys (Figure 1C). The production of IL-1β, IL-6, and TNF-α in the serum was also more significantly elevated in the db/db mice than it was in the db/wt mice (Figure 1D). And the production of IL-1β, IL-6, and TNF-α was also increased in the serum of the HF diabetes mice compared to of the production in the normal mice (Figure 1E). The blood glucose level in the db/db mice was higher than it was in the control mice (Figure 1F), suggesting that the mice were hyperglycemic. Taken together, it can be inferred that the inflammatory response may be involved in T2DN.
Figure 1.
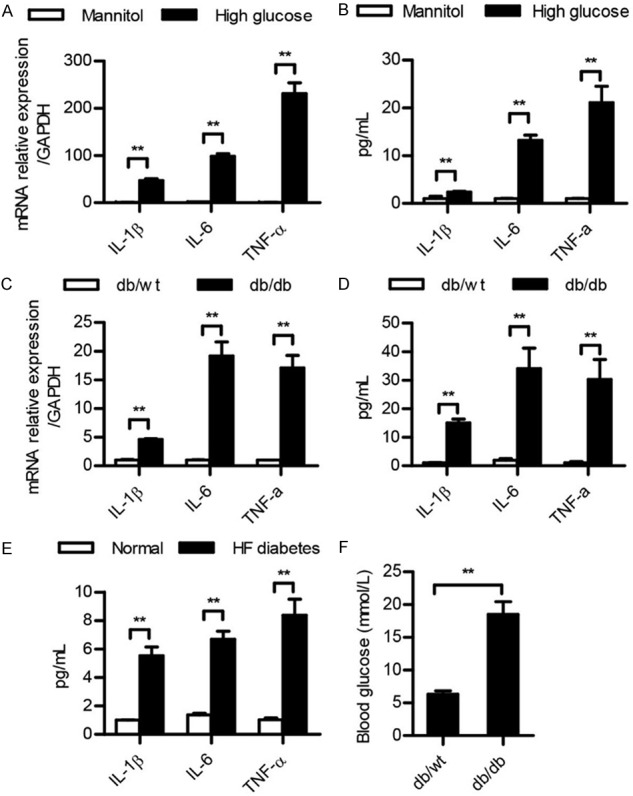
The levels of IL-1β, IL-6, and TNF-α were significantly increased in T2DN. (A-E) qRT-PCR and ELISA analysis of IL-1β, IL-6, and TNF-α in podocytes treated with mannitol or high glucose for 48 h (A and B), or in db/wt mice kidney (C and D) and serum of HF diabetes mice (E). The results were presented as a fold expression of IL-1β, IL-6, and TNF-α mRNA to that of β-actin. (F) Blood glucose levels were measured after db/wt and db/db mice were fed for 20 weeks. Data are shown as the means + SD. n = three independent experiments. **P < 0.01.
The expression of p300 was significantly unregulated both in vivo and in vitro
To investigate which factor was involved in the progression of inflammation, we evaluated the expression of co-activation factor p300. Interestingly, we found that both the mRNA and protein levels of p300 were elevated in the high glucose group compared to the levels of the mannitol group in the podocyte cells (Figure 2A, 2B). The mRNA level of p300 was significantly increased in db/db mice kidneys compared to the db/wt mice kidneys (Figure 2C). A Western blot analysis of the mice kidneys also showed similar results (Figure 2D). The expression of p300 was also more highly expressed in HF diabetes mice kidneys than it was in normal mice (Figure 2E).
Figure 2.

The expression of p300 was increased in T2DN. (A-E) qRT-PCR and western blot analysis of p300 expression in podocytes treated with mannitol or high glucose for 48 h (A and B), or in db/wt mice kidney (C and D) and HF diabetes mice kidneys (E). The results are presented as a fold expression of p300 mRNA to that of β-actin. Data are shown as the means + SD. n = three independent experiments (A and C). Similar results were obtained in three independent experiments (B, D and E). **P < 0.01.
p300 promotes the level of IL-1β, IL-6 and TNF-α after high glucose in podocyte cells
To further confirm the role of p300 in the inflammatory response in T2DN, we used specific siRNA to knockdown the expression of p300 in podocytes. The silencing of p300 significantly suppressed the mRNA and protein levels of IL-1β, IL-6, and TNF-α (Figure 3A). We used p300-His plasmid and showed that the overexpression of p300 promotes the mRNA and protein levels of IL-1β, IL-6, and TNF-α (Figure 3B). To further evaluate the inflammation in T2DN, H&E and Masson trichrome assays were performed on kidney sections to examine the morphology of the kidneys. We found that the inflammation of the db/db mice kidneys was more severe than that noted in the control mice kidneys (Figure 3C).
Figure 3.
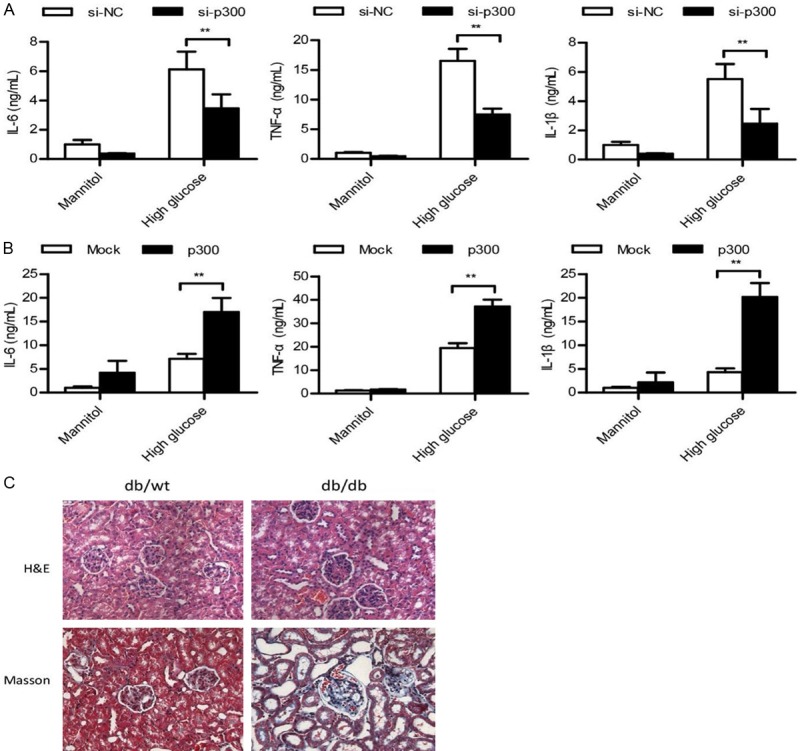
p300 significantly promoted the inflammatory response in T2DN. (A and B) ELISA analysis of IL-1β, IL-6, and TNF-α in podocytes transfected with si-NC or si-p300 for 36 h (A) or transfected with Mock or p300-His plasmid (B) stimulated with mannitol or high glucose for 48 h, or in db/wt mice kidneys. (C) H&E or Masson’s Trichrome analysis of kidney tissue in db/wt or db/db mice. Data are shown as the means + SD. n = three independent experiments (A and B). Similar results were obtained in three independent experiments (C). **P < 0.01.
p300 promotes the activation of NF-κB subunit p65 in T2DN
To elucidate the underlying mechanism for p300 in the elevation of inflammatory cytokines in T2DN, we next examined the effect of inflammation on the upstream signals, such as the NF-κB and MAPK signaling pathways, which are responsible for the production of IL-1β, IL-6, and TNF-α [24]. The results showed that p300 silencing significantly suppressed high glucose-induced phosphorylation of p65 (Figure 4A), while the overexpression of p300 robustly promoted the activation of p65 (Figure 4B). Likewise, the phosphorylation of p65 and IκBα was also found to be significantly increased in db/db mice kidneys, compared to the db/wt mice (Figure 4C). In HF diabetes mice kidneys, we also found that the activation of p65 and IκBα was higher than they were in the normal mice (Figure 4D). Taken together, these data suggest that the activation of NF-κB is responsible for the production of inflammatory cytokines in T2DN.
Figure 4.
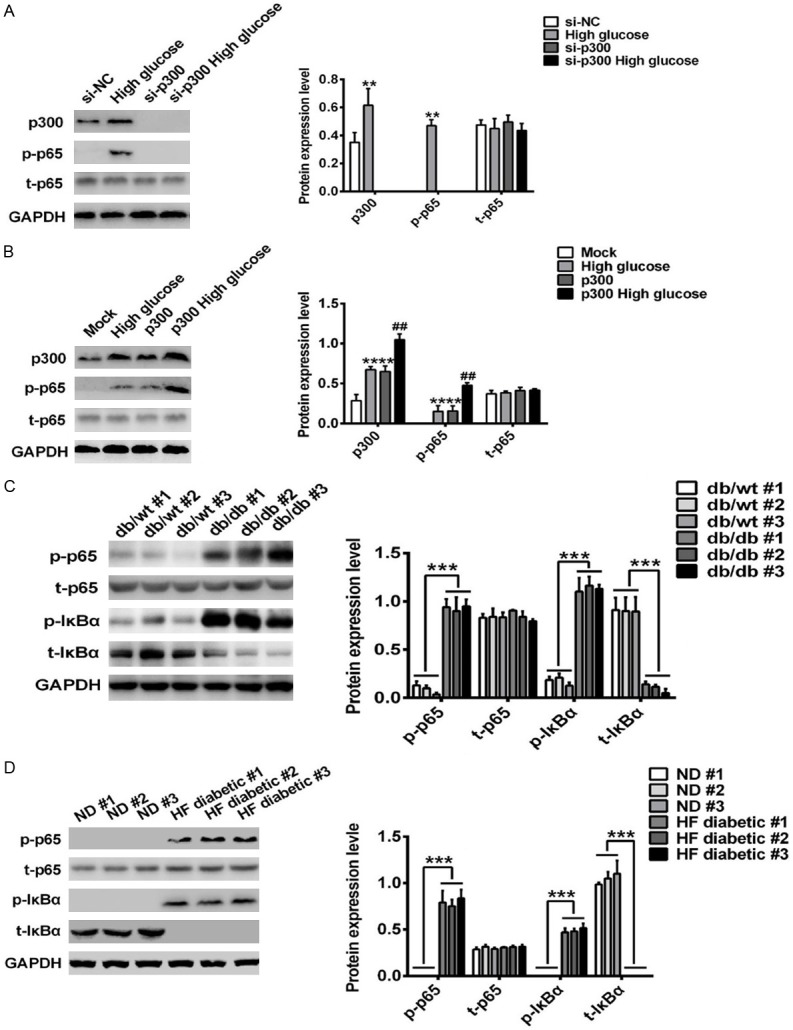
p300 promoted the activation of NF-κB subunit p65 in T2DN. (A-D) Western blot analysis in podocytes transfected with si-NC or si-p300 for 36 h (A) or transfected with Mock or p300-His plasmid stimulated with mannitol or high glucose for 48 h (B), or in db/wt mice kidneys (C) or in HF diabetes mice kidneys (D). Similar results were obtained in three independent experiments (A-D).
Discussion
p300 plays a critical role in the inflammatory response, which contributes to the progression of multiple diseases. However, it remains unclear whether p300 plays a role in T2DN, which we aimed to elucidate in the current study. We demonstrated that p300 enhanced the activation of NF-κB through a direct association with p65, which leads to the production of proinflammatory cytokines. Our findings identified a novel molecule that promotes the inflammatory response, and we provided a new insight into the mechanism that co-activation factor p300 uses to promote the activation of NF-κB in T2DN.
Our results showed that p300 expression is upregulated in db/db mice and podocytes after high glucose treatment. Previous studies have demonstrated that p300 is a prime factor leading to the development of hepatic insulin resistance by acetylating IRS1/2 in the early stages of obesity. In this study, we demonstrated that p300 could directly interact with p65, thus promoting the activation of the NF-κB signaling pathways. However, as an acetyltransferase, p300 also participates in the acetylation regulation in inflammation or fibrosis [25]. We have to point out that we did not investigate whether p300 could regulate the acetylation of p65. So further research is needed to address this issue.
Our results further demonstrated that p300 mediated its effects primarily in the nucleus by interacting with p65. CBP associates with p300 to form transcriptional co-activators that are essential for transcriptional regulation [26]. However, further investigations are needed to address whether CBP is involved in the p300-mediated activation of p65 in T2DN. NF-κB subunit p65 is an important modulator of the NF-κB signaling pathway and acts as a master regulator in inflammation and diseases. Thus, it is worthwhile to investigate the activity of p65.
The results showed that p300 was significantly increased in T2DN, which enhances the activation of NF-κB in association with p65, leading to the production of proinflammatory cytokines, and ultimately promoting the progression of T2DN. Therefore, our data indicate that p300 may act as a promoter in the progression of T2DN and may provide a potential target for the development of a novel therapeutic strategy for T2DN.
In this study, our results showed that p65 is activated in T2DN. The knockdown of p300 suppresses the activation of p65, but the overexpression of p300 significantly promotes the phosphorylation of p65 in T2DN. In addition, we demonstrated that p300 is directly associated with p65 and promotes the inflammation status. p300 contains five protein interaction domains, and each domain binds tightly to a sequence spanning both the transactivation domains 9aaTADs of transcription factor p53 [27]. In our study, we did not confirm which domain of p300 is responsible for the binding of p65. p300 can directly interact with p65, which ultimately can lead to the production of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α [17,28]. However, there are many other proinflammatory cytokines that may be involved in the progression of diabetes that remain to be detected in a future study. As is known, the NF-κB signaling pathway is responsible for the production of interferons such as IFN-α/β [29]. Interferon also play a critical role in the inflammatory response [30]. It will be interesting to explore whether there is interferon secretion in T2DN.
In conclusion, our data suggest that the inflammation status is increased in T2DN, and p300 may be involved in this process. Mechanistically, p300 could directly interact with p65 to promote the activation of the NF-κB signaling pathway. Therefore, targeting the p65/p300 signaling pathway could be a potentially promising strategy in the treatment of T2DN.
Acknowledgements
This work was supported by the National Natural Science Foundation of China, Young Scholars Program (81500645).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kohlgruber A, Lynch L. Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Curr Diab Rep. 2015;15:92. doi: 10.1007/s11892-015-0670-x. [DOI] [PubMed] [Google Scholar]
- 2.Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. Diabetes mellitus and inflammation. Curr Diab Rep. 2013;13:435–444. doi: 10.1007/s11892-013-0375-y. [DOI] [PubMed] [Google Scholar]
- 3.Drouin P, Classification A, Charbonnel B, Eschwege E, Guillausseau PJ, Plouin PF, Daninos JM, Balarac N, Sauvanet JP. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32:S62–S67. [PubMed] [Google Scholar]
- 4.Raptis AE, Viberti G. Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes. 2001;109(Suppl 2):S424–S437. doi: 10.1055/s-2001-18600. [DOI] [PubMed] [Google Scholar]
- 5.Al-Malki AL. Assessment of urinary osteopontin in association with podocyte for early predication of nephropathy in diabetic patients. Dis Markers. 2014;2014:493736. doi: 10.1155/2014/493736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab. 2008;4:444–52. doi: 10.1038/ncpendmet0894. [DOI] [PubMed] [Google Scholar]
- 7.Kanasaki K, Nagai T, Nitta K, Kitada M, Koya D. N-acetyl-seryl-aspartyl-lysyl-proline: a valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front Pharmacol. 2014;5:70. doi: 10.3389/fphar.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prasad N, Gupta P, Jain M, Bhadauria D, Gupta A, Sharma RK, Kaul A. Outcomes of de novo allograft diabetic nephropathy in renal allograft recipients. Exp Clin Transplant. 2013;11:215–221. doi: 10.6002/ect.2012.0193. [DOI] [PubMed] [Google Scholar]
- 9.Wu H, Kong L, Zhou S, Cui W, Xu F, Luo M, Li X, Tan Y, Miao L. The role of microRNAs in diabetic nephropathy. J Diabetes Res. 2014;2014:920134. doi: 10.1155/2014/920134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012;2012:146154. doi: 10.1155/2012/146154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 12.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa] B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 13.Trocha M, Merwid-Lad A, Piesniewska M, Kwiatkowska J, Fereniec-Golebiewska L, Kowalski P, Szelag A, Sozanski T. Age-related differences in function and structure of rat livers subjected to ischemia/reperfusion. Arch Med Sci. 2018;14:388–395. doi: 10.5114/aoms.2018.73470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 15.Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S49. doi: 10.1038/sj.ijo.0802501. [DOI] [PubMed] [Google Scholar]
- 16.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 17.Moreno-Eutimio MA, Espinosa-Monroy L, Orozco-Amaro T, Torres-Ramos Y, Montoya-Estrada A, Jose Hicks J, Rodriguez-Ayala E, Moral PD, Moreno J, Cueto-Garcia J. Enhanced healing and anti-inflammatory effects of a carbohydrate polymer with zinc oxide in patients with chronic venous leg ulcers: preliminary results. Arch Med Sci. 2018;14:336–344. doi: 10.5114/aoms.2016.59851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hajiaghaalipour F, Khalilpourfarshbafi M, Arya A. Modulation of glucose transporter protein by dietary flavonoids in type 2 diabetes mellitus. Int J Biol Sci. 2015;11:508–524. doi: 10.7150/ijbs.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, Schneider H, Ruizortega M, Egido J. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505–2512. doi: 10.1093/ndt/gfh207. [DOI] [PubMed] [Google Scholar]
- 20.Na SY, Lee SK, Han SJ, Choi HS, Im SY, Lee JW. Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor kappaB-mediated transactivations. J Biol Chem. 1998;273:10831–10834. doi: 10.1074/jbc.273.18.10831. [DOI] [PubMed] [Google Scholar]
- 21.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell. 1998;1:277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- 22.Gao Z, Chiao P, Zhang X, Zhang X, Lazar MA, Seto E, Young HA, Ye J. Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J Biol Chem. 2005;280:21091. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Ann Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 25.Cao J, Peng J, An H, He Q, Boronina T, Guo S, White MF, Cole PA, He L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat Commun. 2017;8:131. doi: 10.1038/s41467-017-00163-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stumpf C, Sheriff A, Zimmermann S, Schaefauer L, Schlundt C, Raaz D, Garlichs CD, Achenbach S. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch Med Sci. 2017;13:1086–1093. doi: 10.5114/aoms.2017.69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci U S A. 2007;104:7009. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Li C, Sun L, Chu G, Li J, Chen F, Li G, Zhao Y. Role of p300 in regulating neuronal nitric oxide synthase gene expression through nuclear factor-κB-mediated way in neuronal cells. Neuroscience. 2013;248:681–689. doi: 10.1016/j.neuroscience.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 29.Caglar FNT, Isiksacan N, Biyik I, Opan S, Cebe H, Akturk IF. Presepsin (sCD14-ST): could it be a novel marker for the diagnosis of ST elevation myocardial infarction? Arch Med Sci Atheroscler Dis. 2017;2:e3–e8. doi: 10.5114/amsad.2017.66827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.