

Abstract

The RNA-binding proteins TDP-43 and FUS are tied as the third leading known genetic cause for amyotrophic lateral sclerosis (ALS), and TDP-43 proteopathies are found in nearly all ALS patients. Both the natural function and contribution to pathology for TDP-43 remain unclear. The intersection of functions between TDP-43 and FUS can focus attention for those natural functions mostly likely to be relevant to disease. Here, we compare the role played by TDP-43 and FUS, maintaining chromatin stability for dividing HEK293T cells. We also determine and compare the interactomes of TDP-43 and FUS, quantitating changes in those before and after DNA damage. Finally, selected interactions with known importance to DNA damage repair were validated by co-immunoprecipitation assays. This study uncovered TDP-43 and FUS binding to several factors important to DNA repair mechanisms that can be replication-dependent, -independent, or both. These results provide further evidence that TDP-43 has an important role in DNA stability and provide new ways that TDP-43 can bind to the machinery that guards DNA integrity in cells.
Keywords: TDP-43, FUS, DNA damage repair, transcription, amyotrophic lateral sclerosis, frontal temporal dementia
Introduction
The 43 kDa RNA-binding protein TAR DNA-binding protein (TDP-43) has a role in regulating all levels of RNA metabolism, including transcription, splicing, trafficking, and translation.1−3 TDP-43 dysfunction is also closely associated with several neurodegenerative diseases.4−6 TDP-43 and another RNA-binding protein, fused in sarcoma (FUS), are tied as the third leading known genetic cause of amyotrophic lateral sclerosis (ALS).5,7 TDP-43 aggregates are also found in the central nervous system tissues of almost all patients with ALS.8 TDP-43 aggregation, the second most common pathology found in frontal temporal dementia (FTD), occurs in the brain in 40% of patients.4,5 TDP-43 pathology has also been associated with traumatic brain injury and a majority of Alzheimer’s patients, according to some studies.4,6,9
The role of TDP-43 in neurodegenerative disease places this RNA-binding protein in the company of several disease-associated and causative proteins associated with DNA damage repair.10−12 Most prominent among these is FUS, a key factor recruited to sites of DNA damage.13−15 Other genes that have functions relating to DNA damage repair may contain ALS-causing mutations such as p65/SQSTM1, VCP, OPTN, SETX, and SPG11.16−22 Indeed, recent studies have strengthened the connection between TDP-43 function and chromosome stability.10,11
Our original hypothesis was that one activity in which TDP-43 might have in common with FUS is in contributing to DNA damage repair. We performed siRNA knockdown of TDP-43, which resulted in unexpected levels of instability in chromosomal DNA. During the course of this study, two groups have reported these same findings following the loss of TDP-43.10,11 We then performed an interactome analysis of TDP-43 and FUS, with and without treatment of the DNA damaging agent etoposide. We found that TDP-43 and FUS both interact with numerous factors that have known ties to both replication-dependent and -independent pathways of DNA repair.
Materials and Methods
Cell Culture
HEK293T/17 cells were obtained from the ATCC (catalog# CRL-11268). HeLa-Kyoto cells stably expressing LAP-FUS, LAP-TDP-43, and LAP-SARS were obtained as gifts from the laboratory of A. Hyman (Max Planck Institute of Molecular Cell Biology and Genetics). HeLa reporter cells for negative controls were obtained from Cell Biolabs (catalog# AKR-213). Cells were treated with up to 5 μM etoposide (EMD Millipore) freshly diluted into phosphate-buffered saline from stock solutions in dimethyl sulfoxide (DMSO) before adding to cell media.
Antibodies
Western analyses were performed with the following antibodies: anti-TDP-43 polyclonal (Proteintech, catalog# 10782-2-AP), anti-FUS 4H11 (Santa Cruz, catalog# sc-47711), anti-β-tubulin (Sigma-Aldrich, catalog# T5201), anti-topoisomerase I (TOP1, Bethyl Laboratories, A302-589A), anti-KU80 C-terminal (Abcam, ab181435), anti-RFC3 (Abcam, ab182143), anti-NPM1 NA24 (Thermo Scientific, MA5-12508), and normal mouse IgG (EMD Millipore, catalog# 12-371).
siRNA Transfection
siRNA sequences from previously published works were used. Their forward strand sequences were as follows: siFUS (25 nM, CGGACAUGGCCUCAAACGAdTdT), siTDP1 (50 nM, GCGGGAAAAGUAAAAGAUGUU), and siTDP2 (50 nM, GGAUGAGACAGAUGCUUCAUU). siRNAs were transfected into HEK293T/17 cells as previously described, with the RNAiMax transfection reagent (Invitrogen, catalog# 13778150), and cells were harvested at 72 h. The cells were lysed in 50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 mM DTT, and 1× protease inhibitors (Gold Biotechnology, catalog# GB-108-5). Lysates were analyzed by western blotting.
Comet Tail Assays
Comet tail assays were performed with the OxiSelect Comet Assay Kit (Thermo Scientific) as per the manufacturer’s instructions. Briefly, after treatment with DMSO or etoposide, cells were harvested with trypsin and counted. The cells were resuspended in freshly prepared 1% low-melting agarose (Sigma-Aldrich, A4018) at a density of 1 × 104 per mL and applied to slides. After lysis, the slides were subjected to electrophoresis at 15 V for 15 min, dried, and stained with VISTA green (Cell BioLabs, Inc). Slides were imaged through confocal microscopy (Intelligent Imaging Innovations, Inc.). The number of cells displaying comet tails with lengths of more than three times the width of the head was assessed from 40 to 150 cells per biological replicate.
Affinity Enrichment Mass Spectrometry
Affinity enrichment was performed from HeLa-Kyoto cells harvested from confluent 150 mm diameter dishes and resuspended in lysis/IP buffer (1 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, and 0.5% NP-40). Lysis buffer was supplemented with protease inhibitors (Complete, Mini, EDTA-free, Roche) and 50 U benzonase (EMD Millipore). One milligram of the protein for each lysate was incubated for 1 h with 250 μL GFP-Trap beads (ChromoTek). The beads were washed three times with lysis/IP buffer without NP-40, and the protein was eluted with 200 mM glycine, TCA precipitated, resuspended in 8 M urea for reduction and alkylation with 10 mM TCEP and 40 mM chloroacetamide, respectively. Lysates were diluted to 4 M urea for LysC (Wako) digestion rocking at ambient for 4 h. Lysates were further diluted to ∼1 M urea for trypsin (Pierce) digestion rocking overnight at ambient conditions. Digested peptides were then desalted using an Oasis HLB cartridge (Waters) according to the manufacturer’s instructions.
Samples were suspended in 7 μL of 3% (v/v) acetonitrile/0.1% (v/v) trifluoroacetic acid, and 1 μL was directly injected on a C18 1.7 μm, 130 Å, 75 μm × 250 mm M-class column (Waters), using a Waters M-class UPLC. Tryptic peptides were eluted at 300 nL/min using a gradient from 3 to 20% acetonitrile over 100 min into an Orbitrap Fusion mass spectrometer (Thermo Scientific). Precursor mass spectra (MS1) were acquired at a resolution of 120 000 from 380 to 1500 m/z with an AGC target of 4.0 × 105 and a maximum injection time of 60 ms. Dynamics exclusion was set for 30 s with a mass tolerance of ±10 ppm. Precursor peptide ion isolation width for MS2 fragment scans was 1.6 Da using the quadrupole, and the most intense ions were sequenced using top speed with a 3 s cycle time. All MS2 sequencing procedures were performed using higher energy collision dissociation at 35% collision energy and scanning in the linear ion trap. An AGC target of 1.0 × 104 and 35 s maximum injection time was used. Rawfiles were searched against the Uniprot Human database using Maxquant version 1.5.2.8 with cysteine carbamidomethylation as a fixed modification.23−25 Methionine oxidation and protein N-terminal acetylation were searched as variable modifications. All peptides and proteins were thresholded at a 1% false discovery rate (FDR).
Quantification of Affinity Enrichment Mass Spectrometry Data and Gene Ontology
Potential interactors were identified from proteins detected whose adjusted quantified values for four biological replicates and two treatments summed to 15 or more for either FUS or TDP-43 enrichments. Interactors of TDP-43, FUS, or both were called if averaged label-free quantitation (LFQ) signals were greater than those for both LAP-SARS and GFP controls and with the p-value < 0.05 (Student’s t-test). The fold change was calculated for etoposide-treated samples over DMSO-treated samples. Ontology analysis was performed using Cytoscape v. 3.7.1 and the BiNGO tool set v. 3.0.3.26 Associated ontology terms were determined with the hypergeometric test with the Benjamini and Hochberg FDR correction.
Co-Immunoprecipitation Assays
Proteins interacting with LAP-TDP-43 and LAP-FUS were pulled down from HeLa-Kyoto cells as described in the above affinity enrichment mass spectrometry (AE-MS) protocol and then eluted with 0.1 M NaHCO3 and 1% sodium dodecyl sulfate at 90 °C for 10 min. The eluted proteins were detected by western analysis and quantified in ImageJ 1.50 g (http://imagej.nih.gov). The amounts of eluted protein detected were normalized to input samples, and the amounts of eluted LAP-TDP-43 or LAP-FUS were detected with anti-GFP antibodies for each biological replicate.
Results
TDP-43 is Required for the Stability of Chromosomal DNA
We hypothesized that TDP-43 and FUS might share a role in DNA damage repair. To test this possibility, we generated siRNAs to knock down TDP-43 (siTDP-1 and siTDP-2). By western analysis, we determined that siTDP-1 and siTDP-2 knocked down the TDP-43 protein in HEK293T cells, as compared with either a negative control-scrambled siRNA sequence (SCR) or our previously published siRNA for FUS knockdown (siFUS) (Figure 1A).
Figure 1.
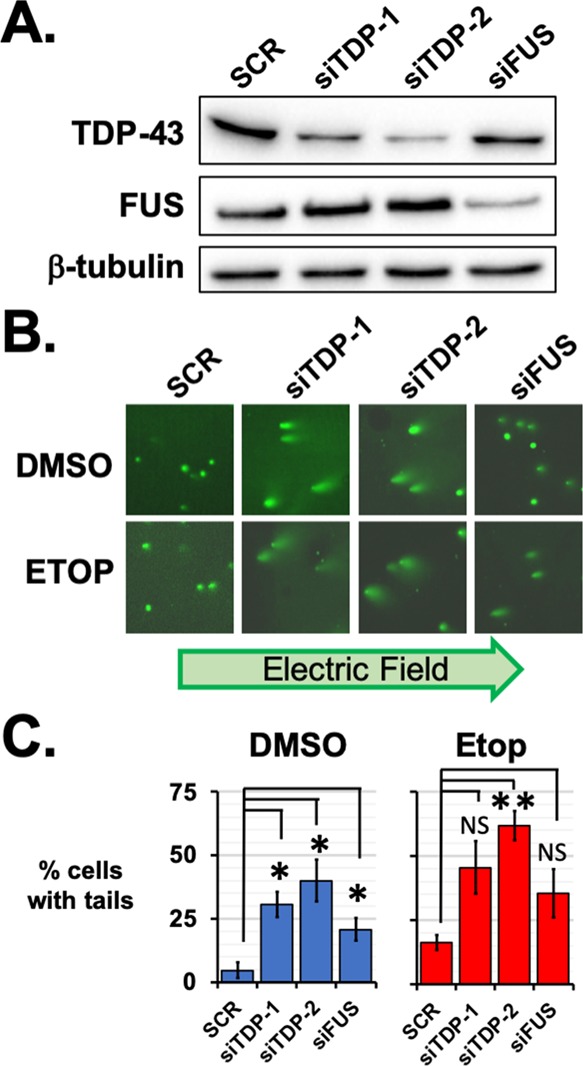
TDP-43 knockdown leads to chromatin instability. (A) By western analysis, TDP-43 protein levels were found to be lower in HEK293 cells after targeted by two siRNAs: siTDP-1 and siTDP-2. Knockdown of TDP-43 did not affect the FUS expression, nor did FUS knockdown with siFUS affect TDP-43 protein levels. Knockdown results were compared with those after transfection of an siRNA with a scrambled sequence, SCR. (B) Comet tail assays were performed for SCR, siTDP-1, siTDP-2, and siFUS-treated HEK293 cells after treatment with etoposide or DMSO as a vehicle control. Comet tails comprising fragmented DNA were observed for siTDP-1 and siTDP-2 knockdowns after DMSO or 5 μM etoposide treatment for 1 h. Comet tails were also observed after etoposide treatment of cells with FUS knocked down with siFUS. (C) For each treatment, the percentage of cells with comet tails was counted from 40 to 140 cells and three biological replicates. Error bars indicate the standard error about the mean of replicates. * indicates p < 0.05 and ** indicates p < 0.005 with Student’s t-test assuming equal variances and compared to the SCR-treated samples.
We tested whether the knockdown of TDP-43 or FUS in HEK293T cells might reduce the capacity to repair DNA after treatment with the topoisomerase poison etoposide. Recent studies have used comet tail assay to show DNA damage occurring after loss of TDP-43 by siRNA knockdown.10,11 To assess DNA stability, we used alkaline comet tail assays, which measure fragmented DNA as it is electrophoresed from cells harvested after 1 h of treatment with DMSO or 5 μM etoposide and embedded in an agarose gel. Alkaline comet tail assay can reveal damage from either single or double stranded breaks.27 Without addition of etoposide, both siTDP-1 and siTDP-2 produced a significant increase in the percentage of cells with fragmented DNA, as compared with SCR controls (p = 0.01 and 0.02 respectively, Student’s t-test, N = 3. Figure 1B). Treatment with etoposide increased the number of siTDP-treated cells with fragmented DNA (Figure 1C). Significantly more DNA fragmentation was found in cells treated with etoposide and siTDP-2 than in SCR-treated cells (p = 0.001, Student’s t-test, N = 3). Treatment with siTDP-1 and the knockdown of FUS produced more damage, but the results of comet tail assays were not significantly different from those of SCR controls (N = 3, p > 0.05, Student’s t-test, Figure 1C). Because etoposide treatment raised comet tails observed for SCR and TDP-43 or FUS knockdown, these results suggest that the effect of a loss of TDP-43 or FUS on chromatin stability is independent of the damage added by etoposide treatment.
TDP-43 and FUS Share Interactors before and after DNA Damage
We asked whether TDP-43 and FUS might share binding partners whose interactions are affected by DNA damage. We used HeLa-Kyoto cells stably expressing localization and affinity purification (LAP)-tagged TDP-43 (LAP-TDP43) or FUS (LAP-FUS) and then performed AE-MS.28−31 LAP-TDP43 and LAP-FUS were driven by a relatively weak phosphoglycerate kinase promoter. Fluorescence microscopy revealed the LAP-tagged proteins to be localized to the nucleus similar to their endogenous counterparts (Figure 1A). LFQ analysis revealed changes to protein abundance enriched by LAP-FUS or LAP-TDP43 pulldown. For negative controls, we used HeLa cells stably expressing either GFP, which makes up part of the LAP tag or a LAP-tagged seryl-tRNA synthetase, SARS (LAP-SARS). The SARS control protein was included to differentiate the effects of nonspecific interactors that might interact with RNA-binding proteins. SARS is a cytoplasmic protein, which we reasoned would be unlikely to have shared interactions with the nuclear proteins FUS and TDP-43.
We treated cells for 1 h with either DMSO or 5 μM etoposide. Cells were lysed and treated with benzonase to break apart interactions tethered by large nucleic acids rather than protein/protein interactions. Interacting proteins were affinity-enriched with anti-GFP nanobody beads (Figure 2A). Expression of tagged proteins, LAP-FUS and LAP-TDP43, was confirmed by western analysis and also that 1 h treatment of etoposide did not alter levels of the tagged or endogenous proteins (Figure 2B). Eluates from four biological replicates for each treatment were digested in solution and subjected to mass spectrometry analysis. By LFQ analysis, we determined enrichment over both GFP and LAP-SARS controls, as well as enrichment in etoposide-treated samples compared with DMSO controls.
Figure 2.

Interactions of FUS and TDP-43 are affected by DNA damage. (A) AE-MS was performed on HeLa-Kyoto cells stably expressing LAP-tagged TDP-43 or FUS and treated with either DMSO or 5 μM etoposide for 1 h. After enrichment with anti-GFP camelid antibodies, interactors were digested in solution for LC–MS/MS. LFQ analysis was used to determine enrichment above GFP or LAP-SARS control proteins (p < 0.05, Student’s t-test; N = 4 for each treatment and control). (B) Stable LAP-FUS (left) or LAP-TDP43 (right) cell lines were found by western analysis to express the tagged proteins below the endogenous levels and neither the tagged nor endogenous protein levels were affected by 1 h of etoposide treatment. (C) Heat maps show changes to interactions shared by both TDP-43 and FUS (left, N = 316), FUS only (center, N = 289), or TDP-43 only (right, N = 139). Fold changes are shown as increases (green) or decreases (red). Blue bars to the right of each heat map indicate significant changes (p ≤ 0.05, Student’s t-test). The Venn diagram summarizes the number of interactors shared by TDP-43 or FUS or unique to those enrichments. (D) Complex network analysis was performed for interactors with either TDP-43 (left, red) or FUS (blue, right). The log10 of the p-value for the significance of enrichment is plotted for each GO term. A list of interactors for each GO term can be found in Table 1.
We found 316 proteins significantly enriched over controls for both TDP-43 and FUS in either DMSO or etoposide-treated samples (Figure 2C). Because they are found in sizable and heterogeneous cellular granules, we expected TDP-43 and FUS to interact with a broad assortment of proteins.32−34 An additional 289 proteins were enriched only in FUS samples, and 139 were enriched only in TDP-43 samples. TDP-43 was significantly enriched in FUS pulldowns (p = 0.03, Student’s t-test), but TDP-43 enrichment of FUS did not reach significance.
We compared proteins identified by our analysis with those found in the previously published literature. Of the 262 previously published TDP-43 interactors, 180 were identified as significantly enriched in our AE-MS experiments.35,36 Of these, 55 were significantly enriched for FUS rather than TDP-43. Overall, 320 new interactions were found for TDP-43 and significantly enriched above controls, in addition to enrichment of 48% of previously published TDP-43 interactors. Many of the novel interactors provided a more complete list of members in protein complexes previously reported to bind TDP-43. The majority of published interactors not enriched in our study were recovered with TDP-43 but below significance compared to controls. Our results for FUS identified 89% of the 323 previously reported FUS interactors.37−39
FUS is Recruited to Interactions Shared with TDP-43 after DNA Damage
We quantified changes in the enrichment of TDP-43 and FUS interactors after DNA damage. We observed that those interactions that were most affected were those shared by FUS and TDP-43 (Figure 2C, left). Of these interactions, 38% of FUS interactions were significantly changed (N = 119, p ≤ 0.05), and 20% were highly significant (N = 63, p ≤ 0.01). In contrast, 6% of TDP-43 interactions were significantly changed (N = 19, p ≤ 0.05). Whereas DNA damage affected only a small number of TDP-43 interactions in common with FUS, FUS interactions generally showed greater enrichment after DNA damage (Figure 2C). Of these interactions, 12% showed ≥2-fold (N = 39) increase in enrichment, whereas 1% were decreased by 2-fold (N = 3). For TDP-43, the enrichment for 7% of interactors identified were increased and 3% were decreased by a fold change ≥2 (N = 20 and 10, respectively).
Of the 289 interactions significantly enriched with FUS but not TDP-43, 20% (N = 61) were significantly changed after DNA damage with etoposide (Figure 2C, center). These changes were fairly evenly distributed: 7% increased and 5% decreased with a fold change ≥2 (N = 23 and 17, respectively). As seen for factors binding both TDP-43 and FUS, few interactors that were significantly enriched for only TDP-43 were affected by DNA damage (N = 12, p ≤ 0.05) (Figure 2C, right).
In summary, the TDP-43 changes after DNA damage were fewer and smaller in magnitude. Most of the interactions affected involved FUS and tended to be strengthened among factors also bound to TDP-43. This finding led us to consider that interactions of TDP-43 relevant to DNA damage might already have formed in the absence of damage. In contrast, FUS was required to be mobilized and form or strengthen many of its interactions upon DNA damage.
Complex Network Analysis of TDP-43 and FUS Interactions
We performed a complex network analysis to group and classify TDP-43 and FUS interactors according to gene ontology (GO) terms. The largest group of interactors shared by FUS and TDP-43 was ribosomal proteins (Figure 2D). Unexpectedly, these interactors included proteins specific to the mitochondrial ribosome (Table 1). The second most significant group of interactors was spliceosome proteins. These interactions were consistent with previously published results. Additional classes of interactors that confirmed findings from previously published studies were protein components of the heterogeneous nuclear ribonucleoprotein (hnRNP) complex, nuclear speckles, and stress granules.
Table 1. Interactors Associated with Cellular Processes through Complex Network Analysis.
| class | category | GO-id | shared interactors | FUS unique interactors | TDP-43 unique interactors | changed in DNA damage |
|---|---|---|---|---|---|---|
| hnRNP proteins | heterogeneous nuclear ribonucleoprotein complex | 30530 | hnRN AC, hnRNPF, hnRNPM, hnRNPR, hnRNPU, hnRNPUL1, PTBP1 | hnRNPA1, hnRNPA2B1, hnRNPA3, hnRNPAB, hnRNPC, hnRNPD, hnRNPDL, hnRNPH1, hnRNPH3, hnRNPK, hnRNPL, hnRNPUL2, RBMX | NA | down |
| SMN proteins | NA | NA | Gemin7 | Gemin3, Gemin5, SMN1 | NA | dowm for FUS |
| snRNP complex | small nuclear ribonucleoprotien complex | 30532 | PRPF4, PRPF31, PPIH, SNRNPB2, SNRPF | LSM4, SNRPB, SF3B3, SNRPC, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPG, SNRNP200 | SNRPA1 | no change |
| nuclear RNA exosome | exosome (RNase complex) | 178 | EXOSC1, EXOSC2, EXOSC3, EXOSC4, EXOSC5, EXOSC10 | EXOSC7 | EXOSC9 | up |
| snoRNP complex | small nucleolar ribonucleoprotein complex | 5732 | FBL, GAR1, NHR2, MPHOSPH10, POP1 | NAF1, NOP10 | NOP56 | down |
| RNAi | gene silencing by RNA | 16246 | ADAR, CELF1, MOV10, PRKRA, SND1 | AGO2, DGCR8, DROSHA, NCBP1, NCBP2, RBM3, TNRC6A, TNRC6B | NA | no change |
| ribosome | ribosome, mitochondrial ribosome | 5840, 5761 | ABCF1, DAP3, DNAJC21, FAU, FXR2, MRPL11, MRPL17, MRPL19, MRPL23, MRPL47, MRPS25, MRPS9, NPM1, NUFIP2, RPL10, RPL10A, RPL11, RPL12, RPL13, RPL13A, RPL14, RPL15, RPL17, RPL18, RPL18A, RPL19, RPL21, RPL22, RPL23, RPL23A, RPL24, RPL26, RPL27, RPL27A, RPL28, RPL3, RPL30, RPL31, RPL32, RPL34, RPL35, RPL35A, RPL36, RPL36A, RPL37A, RPL4, RPL5, RPL6, RPL7A, RPL8, RPL9, RPLP0, RPLP1, RPLP2, RPS10, RPS11, RPS13, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS23, RPS24, RPS25, RPS26, RPS27A, RPS28, RPS3, RPS3A, RPS4X, RPS5, RPS6, RPS7, RPS8, RPS9, RRBP1, RSL1D1, SRP68, UBA52 | MRPL15, MRPL22, MRPL3, MRPL39, MRPL4, MRPL40, MRPL41, MRPL44, MRPL45, MRPL46, MRPL49, MRPL50, MRPL54, MRPL9, MRPS10, MRPS11, MRPS12, MRPS17, MRPS18A, MRPS18B, MRPS2, MRPS22, MRPS23, MRPS26, MRPS27, MRPS28, MRPS31, MRPS35, MRPS6, MRPS7, RPL22L1, RPS12, RPS20, SF1 | RPL7 | up |
| nuclear speckles | nuclear speck | 16607 | CDC5L, DDX3X, EIF4A3, NPM1, NXF1, PPIH, PRPF19, PRPF3, PRPF31, RBM39 | CASC3, DDX39B, FRG1, LUC7L3, PLRG1, PRPF4, PRPF8, RBM27, RBM8A, SART3, SF3B1, SMNDC1,SON, SRSF1, SRSF10, TOE1, YLPM1, ZNF638 | NA | no change |
| paraspeckles | paraspeckles | 42382 | hnRNPM | NONO, NUDT21, PSPC1, SFPQ | NA | no change |
| nucleolus | nucleolar part | 44452 | FBL, GAR1, NPM1, PES1, POLR1A, POP1, RPP30, RPP38, RPP40 | CD3EAP, NAF1, NOP14, POLR1C | NOP56 | up |
| signal recognition particle | signal recognition particle | 48500 | SRP72, SRP68, SRP14 | SRPR, SRP9 | NA | up |
| replication fork | replication fork | 5657 | PURA, RFC1, RFC2, RFC3, RFC4, RFC5 | NA | NA | up |
| nucleosome | nucleosome | 786 | HIST2H2AA4, HP1BP3, HIST1H1E, HGNC:4736, HIST1H1B, HIST1H1C | NA | NA | up |
| mitochondria | mitochondrial matrix | 5759 | LRRC59, ACADVL, MRPS25, RPL23, MRPL47, MRPL23, POLRMT, MRPL11, MRPS9, QARS, LONP1, RPL13, DAP3 | MRPS17, MRPS28, MRPS26, MRPS35, MRPS11, MRPS22, MRPS12, MRPS18B, MRPS18A, MRPL39, MRPL41, MRPL3, MRPL40, KARS, TFAM, MRPL9, RPS12 | POR, FH, ECHS1, SHMT2, MDH2, GOT2, CYCS, ETFB, LRPPRC | no change |
| proteosome | proteosome core complex | 5839 | PSME2 | PSMB5, PSMB2, PSMB3, PSMB1 | PSMDS | no change |
| cajal body | cajal body | 15030 | FBL, DKC1, PRPF3, GAR1, NGP2, PRPF31, GEMIN7 | PRPF4, SART1, FRG1, DDX20, GEMIN5, PHAX, U2AF1, SMN1, SNRPC, SMNDC1, NOP10 | NA | up for FUS |
| G-protein complex | heterotrimeric G-protein complex | 5834 | NA | NA | GNA13, GNB1, GNAS, GNG12, GNAI1 | no change |
| Membrane-anchored proteins | anchored to membrane | 31225 | NA | NA | CPM, NEGR1, CD109, PLAUR, LYPD3, RTN4RL2, MSLN, FOLR1, CD55 | no change |
| RNA polymerases | RNA polymerases | NA | POLR1A, POLR1E, POLRMT | POLR1C, POLR2C, POLR2E | NA | no change |
| nucleotide excision repair | nucleotide excision repair | NA | RFC1, RFC2, RFC3, RFC4, RFC5, POLR1A, PRPF19, RPS27A, UBA52, USP7, AQR, COPS3, COPS4 | COPS6 | NA | up |
Several novel interactions that we identified provided a more comprehensive list of the members within cellular complexes that had been reported or inferred by previous publications, enhancing the significance quantified of those findings.36,37,39 In addition to the RNP complexes described above, we identified interactions with nucleolar proteins, nearly every member of the nuclear RNA exosome and every member of the DNA replication fork complex C (RFC) (Figure 2D). Among the interactors for TDP-43 that were not found for FUS were five associated with G-protein-coupled receptor functions, as well as other membrane-anchored proteins (Table 1).
TDP-43 and FUS Interact with Transcription-Coupled Repair Proteins
We completed our analysis by determining quantitative changes among TDP-43 and FUS interactors either grouped by GO associations or established in the published literature. Among interactions shared between FUS and TDP-43, nuclear speckle proteins, small nucleolar RNA proteins, and hnRNP proteins showed little change after DNA damage (Table 1). Interactions shared with FUS and TDP-43 and affected by DNA damage included the nuclear RNA exosome, the ribosome, chromatin-associated proteins, and transcription-coupled DNA repair proteins. Members of the RNA exosome and transcription-coupled DNA repair showed significantly more enrichment with FUS, and, though trending in the same direction, few changes achieved significance for TDP-43 (Figure 3A,B). TDP-43 interactions with ribosomal protein and chromatin- and transcription-associated proteins did not reveal a significant change in either direction (Figure 3A). In contrast, FUS interactions became significantly more enriched for ribosomal and chromatin-associated proteins (Figure 3B).
Figure 3.
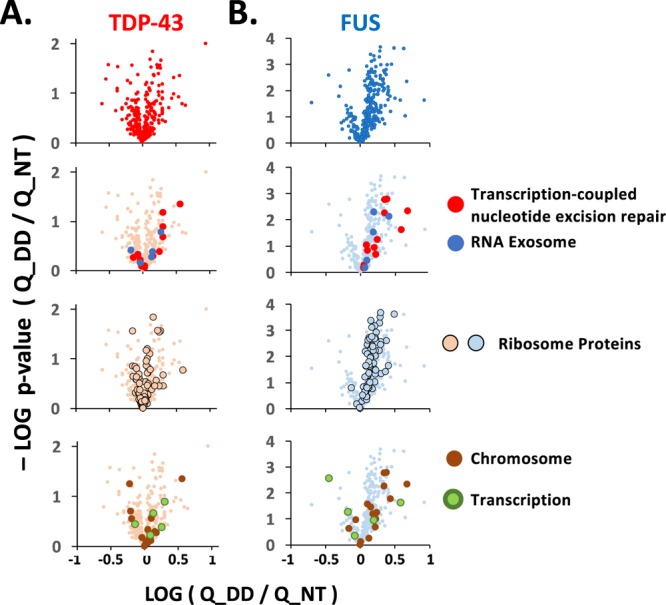
Interactions with DNA repair proteins are affected by DNA damage. Changes in enrichment determined by LFQ analysis are shown as volcano plots with the log10 of the fold change after etoposide treatment (x-axis) vs the log10 of p-values of the change for interactors shared by TDP-43 (A) and FUS (B). At the top are changes in all shared interactors detected at levels significantly above controls. Second, interactors are superimposed from the RNA exosome and transcription-coupled nucleotide repair machinery, including the RFC complex. Third, interactors that are members of the cytosolic and mitochondrial ribosomes are shown. Last are proteins classified as chromatin-bound and the few transcription-related proteins shared by FUS and TDP-43. The identity of the interactors plotted is found in Table 1. Interactors unique to TDP-43 or FUS are included in Figure 1B,C and Table 1.
Transcription machinery not included in the excision repair pathway and shared by TDP-43 and FUS were not substantially affected by etoposide treatment (Figure 3A,B). However, transcription machinery proteins bound only to FUS, as well as several hnRNP and small nuclear RNA proteins, were significantly decreased in their enrichment by DNA damage (Figure 1B,C and Table 1). Among the interactors uniquely associating with TDP-43, G-protein coupled receptors were increased in enrichment with TDP-43 after DNA damage by etoposide.
We selected a collection of interactors to validate with co-immunoprecipitation (co-IP) assays. The chosen interactors were also known to be associated with DNA damage repair mechanisms. We first targeted TOP1. A small but significant 1.4-fold increase was found in the LFQ analysis of AE-MS data for FUS (p = 0.03), and no change was found for TDP-43. Our co-IP assays confirmed an interaction for FUS and TDP-43 with TOP1, as well as more TOP1 in a FUS co-IP after DNA damage (Figure 4A). Another protein closely associated with DNA damage repair, Ku80, showed high AE-MS signals for FUS and TDP-43 samples, although the TDP-43 signals did not reach significance over the controls. Nevertheless, we did find that TDP-43 bound to Ku80 before and after DNA damage in our co-IP assays (Figure 4B). We also confirmed an average 1.6-fold decrease for Ku80 in the FUS co-IP, a result similar to the 1.8-fold decrease found in AE-MS.
Figure 4.

TDP-43 binds DNA damage repair proteins. co-IP was performed for LAP-tagged FUS (blue) and TDP-43 (orange). Western blots show inputs and eluted proteins for TOP1 (A), Ku80 (B), RFC3 (C), and NPM1 (D). Levels of protein in western analyses were quantified and then normalized to western blots for the input samples and for the eluted LAP-FUS or LAP-TDP43. Only changes for LAP-FUS interacting with Ku80 and NPM1 were significant (p < 0.05, Student’s t-test assuming equal variances). Each western blot includes samples treated with DMSO (D), 5 μM etoposide for 1 h (ET), and pulldown with a negative control nonspecific IgG antibody (IgG). Error bars show standard error from three or four biological replicates.
The RFC complex was the most affected among the interactors by DNA damage from AE-MS. All six known members of the RFC complex were significantly enriched among FUS and TDP-43 samples. The enrichment of all RFC members but RFC4 was increased for FUS, whereas no changes were found for TDP-43. One member, RFC3, was not detected before DNA damage. We tested whether the interactions for this member of the RFC complex were affected for both FUS and TDP-43. Our co-IP analysis revealed that RFC3 was bound by both FUS and TDP-43 (Figure 4C). The changes in RFC3 levels observed after DNA damage mirrored those for the rest of the complex members: a 2-fold increase in RFC3 was observed in the FUS co-IP, and no change was observed for TDP-43. Finally, the nucleolar protein NPM1 had especially high signals in AE-MS. We likewise observed strong recovery of NPM1 for FUS and TDP-43 (Figure 4D). Again, FUS co-IP recovered 1.4-fold more NPM1, as found in AE-MS, whereas TDP-43 showed no change.
Recent reports have suggested an unexpected role for TDP-43 during DNA damage repair. Here, in a direct comparison and in the same cell line, we found that TDP-43 appears to be more essential to genomic stability and DNA damage repair than FUS. FUS and TDP-43 are tied as the third leading causes of ALS. Moreover, TDP-43 disruption is ubiquitous in nearly all patients with ALS. This connection between TDP-43 and FUS suggests convergence toward a shared biological pathway.
Discussion
The RNA-binding proteins TDP-43 and FUS share roles in neurodegenerative disease, including ALS and FTD,40,41 thus potentially suggesting that their functions may overlap. Here, we have tested the ability of TDP-43 to affect DNA damage repair and found evidence of its importance in this vital cellular function. We observed significant DNA damage in HEK293 cells after a loss of TDP-43, even without the addition of the DNA damage agent etoposide (Figure 1C). Adding to recent reports also reporting TDP-43 involvement in DNA damage repair for dividing cells and nondividing neurons, we have provided a broader list of interactors and the changes to those interactions induced by DNA damage.10,11,22,27
The effects of FUS knockdown on DNA damage repair were used as a benchmark to assess the effects of loss of TDP-43. The effects of TPD-43 and FUS on transcription and RNA splicing have been widely reported, and those mechanisms are undergoing vigorous investigation.42−45 Interestingly, TDP-43 has been reported to affect DNA damage through prevention of RNA/DNA hybrids, referred to as R-loops.10,11 Indeed, nascent RNAs stripped of the protection provided by hnRNP proteins and splicing factors have a higher propensity to form R-loops during transcription.12,46
HEK293T cells used in this study are dividing and able to employ replication-associated DNA repair pathways.47,48 FUS has a role in DNA damage repair that spans both replication and nonreplication-associated repair pathways.49,50 Breaks induced by etoposide, as performed in our study, can be repaired by mechanisms either associated with cell cycle or independent of this.51,52 It is notable that DNA damage was already found in cells without the etoposide treatment. This can imply that these originate from damage occurring during cycle or other consequences of the loss of FUS or TDP-43, such as changes to transcription or chromatin (Figure 1C).5,7,45 While many factors bound to TDP-43 and FUS in our study have connections to transcription-coupled repair, which is used by nondividing cells such as neurons, some of those and additional interactors also have known roles in DNA repair mechanisms that occur during cell division.
Because of the importance of FUS and TDP-43 to transcription regulation, a connection with transcription-coupled DNA repair may seem natural or expected. Transcription-coupled DNA repair has a role in nucleotide excision repair.53,54 However, the influence of transcription on DNA repair has recently been suggested to also encompass base exchange repair, nonhomologous end joining (NHEJ), and homologous recombination repair.12,46,55−57 Our study revealed that the RNA-binding proteins TDP-43 and FUS to bind a number of factors with known connections to transcription-coupled DNA repair, including factors central to resolving RNA/DNA hybrids or R-loops, XRN2, DHX9, and AQR (Figure 5).54 Factors were found that are important to either nucleotide or base excision repair, such as DDB1, the COPS9 complex, and APEX1.58 Other factors were found that are important to repair broken DNA in mechanisms associated or independent of cell replication, including XRCC1, TOP1, PARP1, and the RFC complex. Additional factors were found that have yet undetermined connections to transcription during DNA repair: such as Ku70/Ku80 and the SMC3/SMC4 condensin complex (Figure 5).
Figure 5.
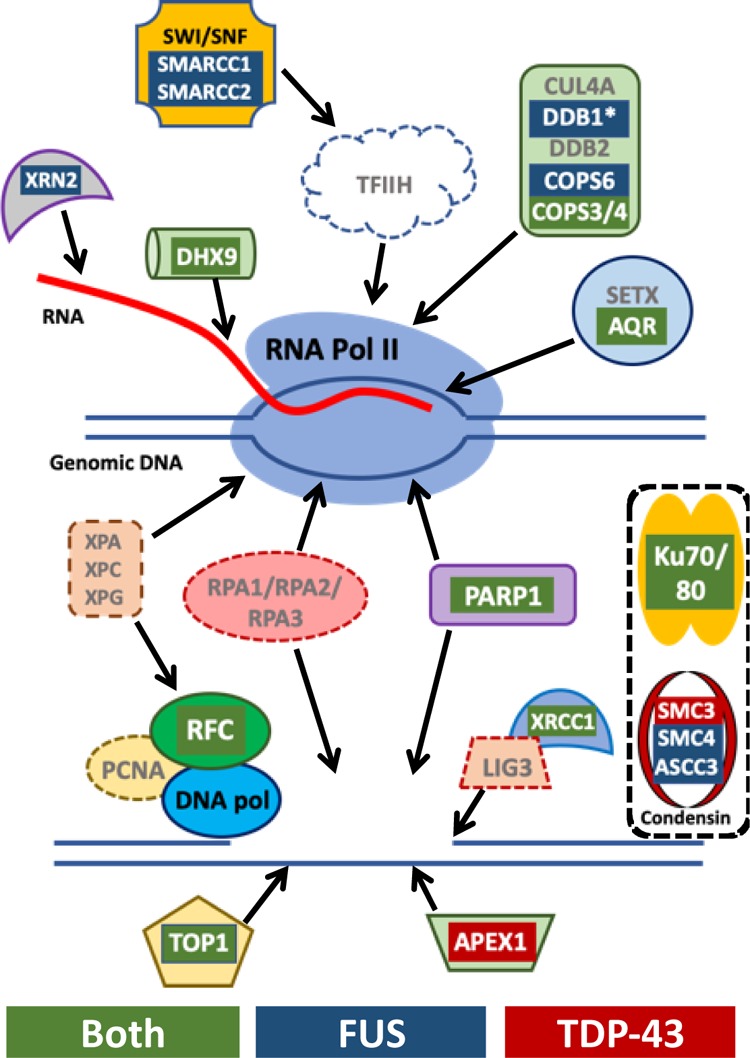
Summary of TDP-43 and FUS interactors associated in transcription-coupled DNA repair. Several of the key factors in the transcription-coupled repair pathway are depicted and interactors identified by AE-MS are highlighted as red for those bound to TDP-43, blue for those bound to FUS, and green for those bound to both. Additional key factors not found in AE-MS to be enriched above controls are indicated by the gray text. The top diagram indicates factors that interact with or recruited by the stalled polymerase at sites of DNA damage. Below that are factors subsequently recruited to repair the damaged DNA. Also shown are the bound complexes Ku70/Ku80 and condensin that are known to play important roles in several DNA repair pathways but their association with transcription-coupled repair remains unclear.
TOP1 is a key factor in repairing the DNA damage induced by R-loops. Through co-IP, we confirmed the interaction of TOP1 with TDP-43 and FUS that was revealed in our AE-MS analysis (Figure 4A).59 The RFC complex binds and helps to recruit the DNA polymerase during transcription-coupled DNA repair triggered by R-loops.60 We were able to identify every member of the RFC complex bound to TDP-43, FUS, or both (Table 1). We also confirmed the interaction of RFC3 by co-IP (Figure 4C). Both our results and those from previous reports find an interaction of FUS and TDP-43 with the Ku complex (Figure 4B). Although our observations concerning Ku70/Ku80 may differ from previous reports, there are important differences in experimental designs. First, previous reports used primary neurons, neuron progenitor cells, and differentiated neurons or SH-SY5Y cells, which depend heavily on NHEJ.10,49,61 Second, we treated with 5 μM etoposide for 1 h and previous reports used higher doses and longer treatments, such as 10 μM and 4 h treatment.10,49,62 The RFC complex is known to bind the DNA polymerase. However, we did not detect other factors in the NHEJ machinery that have been previously reported to bind TDP-43, such as XRCC4, LIG4, or Pol δ or γ.10
The DNA damage after TDP-43 knockdown was greater than that after FUS knockdown (Figure 1C), whereas DNA damage had greater effects on FUS interactions than TDP-43 interactions (Figure 2A). The first potential explanation for this result would be that more total interactors were identified for FUS and with higher LFQ values; consequently, some number of changes in TDP-43 interactions might fall below significance thresholds set by the negative controls in our experiments. If not a technical reason, other explanations can include that TDP-43 may need no more interactors to exert its effect on DNA damage repair than it would maintain constitutively in the absence of DNA damage. A third explanation would be that in comparison to FUS, TDP-43 may maintain relatively few interactions that are sufficient for TDP-43 to play its role in DNA repair, which make up a small subset of the total TDP-43 interactions that include functions not limited to the maintenance of chromatin integrity.
Numerous interactions found in this study are likely to be important in functions other than chromatin stability and DNA damage repair. First among these are interactions with the ribosome. The finding that interactions with the ribosome were significantly affected by DNA damage suggest that these associations may be biologically meaningful because they are subject to regulation during a cellular response (Figure 3A,B). Besides adding validity, these findings do not suggest an obvious connection between ribosome interactions and DNA damage repair. Multiple studies have reported effects on translation from the mutation or loss of TDP-43, which, as an RNA processing factor, is not required to directly interact with translating ribosomes or polysomes.63,64 Moreover, because TDP-43 and FUS are such robustly nuclear proteins, a direct mechanism is difficult to rationalize. We did observe FUS and TDP-43 interactions with nucleolar proteins, such as NPM1, thus potentially indicating that interactions may occur during ribosome assembly within the nucleus (Figure 4D).
An unexpected number of mitochondria-specific proteins were found among TDP-43 interactors. This finding supports reports of the TDP-43 function within and localization to mitochondria.65−67 Among the TDP-43 and FUS interactors, the mitochondrial polymerase POLRMT and the mitochondrial ribosome are notable as having no known function outside of mitochondria. Our interactors identified by AE-MS included 21 of the 51 members of the mitochondrial ribosome (Table 1). These include four of the seven mitochondrial ribosome proteins previously reported to bind TDP-43.
DNA damage is broadly associated with both neurodegeneration and aging.22,68 A relationship between TDP-43 and DNA damage has particular relevance to disease because of questions surrounding TDP-43 pathology, diseases of neurons, and aging. How does dysfunction of a ubiquitous protein such as TDP-43 or FUS, which are highly abundant in all cells and tissues, produce pathology late in life that is specific to neurons, such as motor neurons? DNA damage has an important place in the study of these questions because neurons do not divide and therefore lack several DNA repair pathways available to cells that are replenished over time. Because the chromosomes of a neuron must be maintained for a lifetime, a small deficit in their repair need not produce immediate catastrophe early in life but could hasten their eventual decay to produce the age association of neurodegeneration.22,68−70
In conclusion, we uncovered a role of TDP-43 in DNA damage repair with parallels to the role of FUS. First, these findings provide new insights into how transcription and RNA processing factors contribute to DNA damage repair. Second, the relationship that these proteins share with neurodegeneration may renew interest in links that connect DNA damage to diseases such as ALS, FTD, AD, and traumatic brain injury. Future research into the mechanism through which TDP-43 influences DNA damage repair can be expected to reveal aspects of this important cellular process that are both novel and provide new avenues to determine their contributions to disease.
Acknowledgments
This work was funded by the National Institute of Health [NS082376] to J.C.S. and by a DARPA cooperative agreement grant [13-34-RTA-FP-007] to W.M.O. Research was also supported by the Office of the Director, National Institutes of Health of the National Institutes of Health under award number S10OD013237. The authors thank Dr. Ivo Fierro-Monti for his technical assistance early in the preparation of samples for AE-MS. The authors thank Drs. Tony Hyman and Ina Poser for their assistance and providing the HeLa-Kyoto lines used in this study. The authors also thank Sarah Bolanos for her assistance in cell culture for AE-MS experiments. Imaging data were collected in the W. M. Keck Center for Nano-Scale Imaging in the Department of Chemistry and Biochemistry at the University of Arizona, which is partially supported by Arizona Technology and Research Initiative Fund (A.R.S. 15-1648).
Glossary
Abbreviations
- AE-MS
affinity enrichment mass spectrometry
- ALS
amyotrophic lateral sclerosis
- co-IP
co-immunoprecipitation
- DMSO
dimethyl sulfoxide
- DNA
deoxyribonucleic acid
- FTD
frontal temporal dementia
- FUS
fused in sarcoma protein
- GO
gene ontology
- hnRNP
heterogeneous nuclear ribonucleoprotein
- LAP tag
localization and affinity purification tag
- LFQ
label-free quantification
- RFC
DNA replication fork complex C
- RNA
ribonucleic acid
- SARS
seryl-tRNA synthetase
- TDP-43
TAR DNA-binding protein of 43 kDa
- TOP1
topoisomerase I
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00575.
Data of bound TDP, bound FUS, and both bound (XLSX)
IR profile (XLSX)
Fluorescence microscopy and LFQ analysis of AE-MS experiments, IR profile for AE-MS, changes to interactions after DNA damage, entire images for western in Figure 1A, entire images for Figure 2B westerns, entire images for Figure 4 westerns of LAP-FUS IP experiments, and entire images for Figure 4 westerns of LAP-TDP43 IP experiments (PDF)
Author Contributions
T.K. performed molecular studies of cellular responses to etoposide treatment and contributed to the conception and designs for the study. M.G.R., M.M., and A.A.M. performed cell culture, affinity enrichment, and co-IP experiments. M.M. and M.G.R. performed microscopy. C.C.E. and W.M.O. performed LC–MS/MS of AE-MS samples and analyzed data. J.C.S. contributed to conception and design of experiments, analyzed data for AE-MS and co-IP experiments, and wrote the manuscript. All co-authors contributed to writing, editing, and reviewing the manuscript.
The authors declare no competing financial interest.
Notes
All files associated with AE-MS experiments are deposited at http://massive.ucsd.edu: MSV000084486, doi: 10.25345/C5M953, or at http://proteomecentral.proteomexchange.org: PXD015972.
Supplementary Material
References
- Melnick M.; Gonzales P.; Cabral J.; Allen M. A.; Dowell R. D.; Link C. D. Heat shock in C. elegans induces downstream of gene transcription and accumulation of double-stranded RNA. PLoS One 2019, 14, e0206715. 10.1371/journal.pone.0206715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krach F.; Batra R.; Wheeler E. C.; Vu A. Q.; Wang R.; Hutt K.; Rabin S. J.; Baughn M. W.; Libby R. T.; Diaz-Garcia S.; Stauffer J.; Pirie E.; Saberi S.; Rodriguez M.; Madrigal A. A.; Kohl Z.; Winner B.; Yeo G. W.; Ravits J. Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. 2018, 136, 405–423. 10.1007/s00401-018-1870-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K.; Pratt G. A.; Vu A. Q.; Hutt K. R.; Martinez F. J.; Sundararaman B.; Batra R.; Freese P.; Lambert N. J.; Huelga S. C.; Chun S. J.; Liang T. Y.; Chang J.; Donohue J. P.; Shiue L.; Zhang J.; Zhu H.; Cambi F.; Kasarskis E.; Hoon S.; Ares M. Jr.; Burge C. B.; Ravits J.; Rigo F.; Yeo G. W. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun. 2016, 7, 12143. 10.1038/ncomms12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. N.; Gochenaur L.; Singh A.; Grant R.; Patel K.; Watkins S.; Wu J. Y.; Pandey U. B. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum. Mol. Genet. 2018, 27, 1366–1381. 10.1093/hmg/ddy047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purice M. D.; Taylor J. P. Linking hnRNP Function to ALS and FTD Pathology. Front. Neurosci. 2018, 12, 326. 10.3389/fnins.2018.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes N.; Eshleman N.; Buchan J. R. Stress Granules and ALS: A Case of Causation or Correlation?. Adv. Neurobiol. 2018, 20, 173–212. 10.1007/978-3-319-89689-2_7. [DOI] [PubMed] [Google Scholar]
- Schwartz J. C.; Cech T. R.; Parker R. R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355–379. 10.1146/annurev-biochem-060614-034325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne A. N.; Lorenzini I.; Chou C.-C.; Torvund M.; Rogers R. S.; Starr A.; Zaepfel B. L.; Levy J.; Johannesmeyer J.; Schwartz J. C.; Nishimune H.; Zinsmaier K.; Rossoll W.; Sattler R.; Zarnescu D. C. Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep. 2017, 21, 110–125. 10.1016/j.celrep.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs K. A.; Dickson D. W.; Tosakulwong N.; Weigand S. D.; Murray M. E.; Petrucelli L.; Liesinger A. M.; Senjem M. L.; Spychalla A. J.; Knopman D. S.; Parisi J. E.; Petersen R. C.; Jack C. R. Jr.; Whitwell J. L. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol. 2017, 16, 917–924. 10.1016/s1474-4422(17)30284-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra J.; Guerrero E. N.; Hegde P. M.; Liachko N. F.; Wang H.; Vasquez V.; Gao J.; Pandey A.; Taylor J. P.; Kraemer B. C.; Wu P.; Boldogh I.; Garruto R. M.; Mitra S.; Rao K. S.; Hegde M. L. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 4696–4705. 10.1073/pnas.1818415116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S. J.; Mordes D. A.; Cameron L. A.; Neuberg D. S.; Landini S.; Eggan K.; Livingston D. M. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E7701–E7709. 10.1073/pnas.1611673113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro C.; Bielli P.; Pagliarini V.; Sette C. The interplay between DNA damage response and RNA processing: the unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. 10.3389/fgene.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Macias M. I.; Moore D. A.; Green R. L.; Gomez-Herreros F.; Naumann M.; Hermann A.; Van Damme P.; Hafezparast M.; Caldecott K. W. FUS (fused in sarcoma) is a component of the cellular response to topoisomerase I-induced DNA breakage and transcriptional stress. Life Sci. Alliance 2019, 2, e201800222. 10.26508/lsa.201800222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.; Lee H. O.; Jawerth L.; Maharana S.; Jahnel M.; Hein M. Y.; Stoynov S.; Mahamid J.; Saha S.; Franzmann T. M.; Pozniakovski A.; Poser I.; Maghelli N.; Royer L. A.; Weigert M.; Myers E. W.; Grill S.; Drechsel D.; Hyman A. A.; Alberti S. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Sama R. R. K.; Ward C. L.; Bosco D. A. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 2014, 6, 175909141454447. 10.1177/1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi A. H.; Raviprakash N.; Mokhamatam R. B.; Gupta P.; Manna S. K. Profilin potentiates chemotherapeutic agents mediated cell death via suppression of NF-kappaB and upregulation of p53. Apoptosis 2016, 21, 502–513. 10.1007/s10495-016-1222-9. [DOI] [PubMed] [Google Scholar]
- Czarny P.; Pawlowska E.; Bialkowska-Warzecha J.; Kaarniranta K.; Blasiak J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. 10.3390/ijms16022641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Chen P.; Gao H.; Gu Y.; Yang J.; Peng H.; Xu X.; Wang H.; Yang M.; Liu X.; Fan L.; Chen S.; Zhou J.; Sun Y.; Ruan K.; Cheng S.; Komatsu M.; White E.; Li L.; Ji H.; Finley D.; Hu R. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014, 26, 106–120. 10.1016/j.ccr.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B.; Halder S.; Ramadan K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 60. 10.3389/fgene.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orebaugh C. D.; Fye J. M.; Harvey S.; Hollis T.; Wilkinson J. C.; Perrino F. W. The TREX1 C-terminal region controls cellular localization through ubiquitination. J. Biol. Chem. 2013, 288, 28881–28892. 10.1074/jbc.m113.503391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel O. J.; Yeo A. J.; Stellati A.; Heng E. Y. H.; Luff J.; Suraweera A. M.; Woods R.; Fleming J.; Carrie D.; McKinney K.; Xu X.; Deng C.; Lavin M. F. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. 2013, 9, e1003435. 10.1371/journal.pgen.1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero E. N.; Wang H.; Mitra J.; Hegde P. M.; Stowell S. E.; Liachko N. F.; Kraemer B. C.; Garruto R. M.; Rao K. S.; Hegde M. L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145–146, 78–97. 10.1016/j.pneurobio.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old W. M.; Meyer-Arendt K.; Aveline-Wolf L.; Pierce K. G.; Mendoza A.; Sevinsky J. R.; Resing K. A.; Ahn N. G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 2005, 4, 1487–1502. 10.1074/mcp.m500084-mcp200. [DOI] [PubMed] [Google Scholar]
- UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Carlson A.; Sinitcyn P.; Mann M.; Cox J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. 10.1002/pmic.201400449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R.; Smoot M. E.; Ono K.; Ruscheinski J.; Wang P.-L.; Lotia S.; Pico A. R.; Bader G. D.; Ideker T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero E. N.; Mitra J.; Wang H.; Rangaswamy S.; Hegde P. M.; Basu P.; Rao K. S.; Hegde M. L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 28, 2459–2476. 10.1093/hmg/ddz062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein M. Y.; Hubner N. C.; Poser I.; Cox J.; Nagaraj N.; Toyoda Y.; Gak I. A.; Weisswange I.; Mansfeld J.; Buchholz F.; Hyman A. A.; Mann M. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Hubner N. C.; Bird A. W.; Cox J.; Splettstoesser B.; Bandilla P.; Poser I.; Hyman A.; Mann M. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 2010, 189, 739–754. 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I.; Sarov M.; Hutchins J. R. A.; Hériché J.-K.; Toyoda Y.; Pozniakovsky A.; Weigl D.; Nitzsche A.; Hegemann B.; Bird A. W.; Pelletier L.; Kittler R.; Hua S.; Naumann R.; Augsburg M.; Sykora M. M.; Hofemeister H.; Zhang Y.; Nasmyth K.; White K. P.; Dietzel S.; Mechtler K.; Durbin R.; Stewart A. F.; Peters J.-M.; Buchholz F.; Hyman A. A. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods 2008, 5, 409–415. 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhauer E. C.; Hein M. Y.; Mann M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol. Cell. Proteomics 2015, 14, 120–135. 10.1074/mcp.m114.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S. F.; Lee H. O.; Hyman A. A.; Rosen M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S.; Wheeler J. R.; Walters R. W.; Agrawal A.; Barsic A.; Parker R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S. F.; Rice A. M.; Peeples W. B.; Lin Y.; Jain S.; Parker R.; Rosen M. K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. 10.1016/j.cell.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S.-C.; Albuquerque C. P.; Han J. S.; Lagier-Tourenne C.; Tokunaga S.; Zhou H.; Cleveland D. W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13318–13323. 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum B. D.; Chitta R. K.; High A. A.; Taylor J. P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Ling S.-C.; Qiu J.; Albuquerque C. P.; Zhou Y.; Tokunaga S.; Li H.; Qiu H.; Bui A.; Yeo G. W.; Huang E. J.; Eggan K.; Zhou H.; Fu X.-D.; Lagier-Tourenne C.; Cleveland D. W. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 6171. 10.1038/ncomms7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B.; O’Connell J. D.; Yamazaki T.; Gangopadhyay J.; Gygi S. P.; Reed R. Interactome analyses revealed that the U1 snRNP machinery overlaps extensively with the RNAP II machinery and contains multiple ALS/SMA-causative proteins. Sci. Rep. 2018, 8, 8755. 10.1038/s41598-018-27136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T.; Chen S.; Yu Y.; Yan B.; Haertlein T. C.; Carrasco M. A.; Tapia J. C.; Zhai B.; Das R.; Lalancette-Hebert M.; Sharma A.; Chandran S.; Sullivan G.; Nishimura A. L.; Shaw C. E.; Gygi S. P.; Shneider N. A.; Maniatis T.; Reed R. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012, 2, 799–806. 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. P.; Brown R. H. Jr.; Cleveland D. W. Decoding ALS: from genes to mechanism. Nature 2016, 539, 197–206. 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E.; Chiò A.; Traynor B. J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C.; Polymenidou M.; Hutt K. R.; Vu A. Q.; Baughn M.; Huelga S. C.; Clutario K. M.; Ling S.-C.; Liang T. Y.; Mazur C.; Wancewicz E.; Kim A. S.; Watt A.; Freier S.; Hicks G. G.; Donohue J. P.; Shiue L.; Bennett C. F.; Ravits J.; Cleveland D. W.; Yeo G. W. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P. J.; Daujotyte D.; Tollervey J. R.; Ule J.; Stuani C.; Buratti E.; Baralle F. E.; Damberger F. F.; Allain F. H.-T. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. 10.1038/nsmb.2698. [DOI] [PubMed] [Google Scholar]
- Avendano-Vazquez S. E.; Dhir A.; Bembich S.; Buratti E.; Proudfoot N.; Baralle F. E. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684. 10.1101/gad.194829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E. Y.; Russ J.; Cali C. P.; Phan J. M.; Amlie-Wolf A.; Lee E. B. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019, 27, 1409–1421. 10.1016/j.celrep.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H. Role of genome guardian proteins in transcriptional elongation. FEBS Lett. 2016, 590, 1064–1075. 10.1002/1873-3468.12152. [DOI] [PubMed] [Google Scholar]
- Walker C.; El-Khamisy S. F. Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain 2018, 141, 1247–1262. 10.1093/brain/awy076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon P. J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017, 31, 1180–1194. 10.1101/gad.301325.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.-Y.; Pan L.; Su S. C.; Quinn E. J.; Sasaki M.; Jimenez J. C.; Mackenzie I. R. A.; Huang E. J.; Tsai L.-H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola A. S.; Kim S. H.; Trinh A. T.; Rodenkirch L. A.; Tibbetts R. S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. 10.1074/jbc.m113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. W.; Chernikova S. B.; Kachnic L. A.; Banath J. P.; Sordet O.; Delahoussaye Y. M.; Treszezamsky A.; Chon B. H.; Feng Z.; Gu Y.; Wilson W. R.; Pommier Y.; Olive P. L.; Powell S. N.; Brown J. M. Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res. 2008, 68, 257–265. 10.1158/0008-5472.can-06-4497. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Suzuki H.; Iiizumi S.; Koyama H. Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: implications for the repair of topoisomerase II-mediated DNA damage. J. Biol. Chem. 2003, 278, 35897–35902. 10.1074/jbc.m306500200. [DOI] [PubMed] [Google Scholar]
- Massey T. H.; Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Models Mech. 2018, 11, dmm031930. 10.1242/dmm.031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J.; Stork C. T.; García-Rubio M. L.; Paulsen R. D.; Aguilera A.; Cimprich K. A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara T.; Kato R.; Hagiwara Y.; Shiotani B.; Yamauchi M.; Nakada S.; Shibata A.; Miyagawa K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570. 10.1016/j.cell.2018.08.056. [DOI] [PubMed] [Google Scholar]
- Marnef A.; Cohen S.; Legube G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017, 429, 1277–1288. 10.1016/j.jmb.2017.03.024. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe V. O.; Venkitaraman A. R. RNA Processing and Genome Stability: Cause and Consequence. Mol. Cell 2016, 61, 496–505. 10.1016/j.molcel.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt P. C.; Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Shen W.; Sun H.; De Hoyos C. L.; Bailey J. K.; Liang X.-h.; Crooke S. T. Dynamic nucleoplasmic and nucleolar localization of mammalian RNase H1 in response to RNAP I transcriptional R-loops. Nucleic Acids Res. 2017, 45, 10672–10692. 10.1093/nar/gkx710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J. L.; Chon H.; Cerritelli S. M.; Kunkel T. A.; Johansson E.; Crouch R. J.; Burgers P. M. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 2012, 47, 980–986. 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H.; Lee S.; Shang Y.; Wang W.-Y.; Au K. F.; Kamiya S.; Barmada S. J.; Finkbeiner S.; Lui H.; Carlton C. E.; Tang A. A.; Oldham M. C.; Wang H.; Shorter J.; Filiano A. J.; Roberson E. D.; Tourtellotte W. G.; Chen B.; Tsai L.-H.; Huang E. J. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 2014, 124, 981–999. 10.1172/jci72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q.; Holler C. J.; Taylor G.; Hudson K. F.; Watkins W.; Gearing M.; Ito D.; Murray M. E.; Dickson D. W.; Seyfried N. T.; Kukar T. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. 10.1523/jneurosci.0172-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Collins M.; An J.; Geiser R.; Tegeler T.; Tsantilas K.; Garcia K.; Pirrotte P.; Bowser R. Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein-protein interaction network. Brain Res. 2016, 1647, 79–93. 10.1016/j.brainres.2016.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbacher J. K.; Tabet R.; Yeo G. W.; Lagier-Tourenne C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. 10.1016/j.neuron.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S. A.; Itaman S.; Khalid-Janney C. M.; Sherard J. A.; Dowell J. A.; Cairns N. J.; Gitcho M. A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. 10.1016/j.neulet.2018.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Wang L.; Lu J.; Siedlak S. L.; Fujioka H.; Liang J.; Jiang S.; Ma X.; Jiang Z.; da Rocha E. L.; Sheng M.; Choi H.; Lerou P. H.; Li H.; Wang X. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. 10.1038/nm.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.-F.; Gendron T. F.; Zhang Y.-J.; Lin W.-L.; D’Alton S.; Sheng H.; Casey M. C.; Tong J.; Knight J.; Yu X.; Rademakers R.; Boylan K.; Hutton M.; McGowan E.; Dickson D. W.; Lewis J.; Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. 10.1523/jneurosci.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R.; Pan L.; Tsai L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen D. K.; Bohr V. A.; Stevnsner T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.; Wilson D. M. 3rd The involvement of DNA-damage and -repair defects in neurological dysfunction. Am. J. Hum. Genet. 2008, 82, 539–566. 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.