

Abstract
Secretory proteins are key modulators of host–pathogen interaction. The human opportunistic fungal pathogen Candida glabrata lacks secreted proteolytic activity but possesses 11 glycosylphosphatidylinositol-anchored aspartyl proteases, also referred to as Yapsins (CgYps1–11), that are essential for its virulence. To delineate the role of CgYapsins in interaction with host cells, we have profiled, through liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach, the total secretome of wild-type and Cgyps1-11Δ mutant. The wild-type secretome consisted of 119 proteins which were primarily involved in cell wall organization, carbohydrate metabolism, proteolysis, and translation processes. Of eight CgYapsins identified in the secretome, the release of two major CgYapsins, CgYps1 and CgYps7, to the medium was confirmed by Western analysis. Further, comparative analysis revealed 20 common proteins, probably signifying the core fungal secretome, among C. glabrata, Saccharomyces cerevisiae, and Candida albicans secretomes. Strikingly, the Cgyps1-11Δ secretome was 4.6-fold larger, and contained 65 differentially abundant proteins, as revealed by label-free quantitative profiling, with 49 and 16 being high- and low-abundant proteins, respectively, compared to the wild-type secretome. Importantly, the CgMsb2 mucin, a putative CgYapsins’ substrate, was six-fold underrepresented in the mutant secretome. Altogether, we demonstrate for the first time that CgYapsins are both bona fide constituents and key modulators of the C. glabrata secretome.
Keywords: Yapsins, secretory signal peptide, carbohydrate metabolism, cell wall organization, GPI proteins, core fungal secretome, pathogenic fungi
Introduction
Nosocomial bloodstream infections (BSIs) caused by fungal species are emerging as a major health problem.1Candida spp. are the leading cause of opportunistic fungal BSIs with C. albicans being the prime causal agent.1−3 A significant increase in the prevalence of BSIs due to non-albicans Candida spp., primarily represented by C. glabrata, C. tropicalis, C. parapsilosis, and C. krusei, has been observed in the last two decades.2,4,5 Based on the geographical distribution, C. glabrata can be the second to fourth most prevalent Candida bloodstream pathogen and accounts for up to ∼ 30% cases of BSIs caused by Candida spp.3,5−7
C. glabrata is a haploid budding yeast which belongs to the Nakesomyces clade.8 Compared to other Candida species, C. glabrata phylogenetically is more closely related to the nonpathogenic yeast Saccharomyces cerevisiae.8−10 Owing to this unique position in the phylogenetic tree, pathogenic traits of C. glabrata appear to have evolved independently of other Candida spp.8,11 In agreement, C. glabrata neither forms hyphae nor contains secretory aspartyl proteases.10,11 However, it is fully capable of establishing superficial mucosal and life-threatening BSIs in the human host.2,12 It also possesses the ability to survive and replicate in mouse and human macrophages.13−15 A family of 11 putative glycosylphosphatidylinositol (GPI)-anchored aspartyl proteases, also referred as CgYapsins (CgYps1-11), has been shown to be essential for the intracellular survival and virulence of C. glabrata.(13) Recently, CgYps1-11 proteases have been implicated in suppression of the proinflammatory immune response of the host.15 Human THP-1 macrophages displayed an increased activation of the spleen tyrosine kinase (Syk) signaling pathway and secretion of the interleukin-1β (IL-1β) upon infection with the C. glabrata mutant lacking 11 CgYapsins (Cgyps1-11Δ).15 Inhibition of the Syk signaling pathway rescued the intracellular survival defect of the Cgyps1-11Δ mutant, thereby underscoring the role of CgYapsins in facilitating the survival of C. glabrata in human macrophages.15
A strategy widely adopted by successful pathogens is to secrete virulence factors to facilitate invasion of, and survival inside, the host.16,17 Consistent with this, secretory aspartyl proteases are key virulence factor of many pathogenic fungi, including C. albicans, Cryptococcus neoformans, and Aspergillus fumigatus.17−19 The genome of C. glabrata does not code for any secretory aspartyl protease.10,11 The family of 11 CgYapsins represents the cell surface-associated aspartyl proteases in C. glabrata.13 Despite the predicted cell wall localization, CgYapsins have been reported to regulate the stationary-phase stress survival, pH and vacuole homeostasis, and energy production under in vitro conditions.13,19−22 Like their orthologs in S. cerevisiae, CgYapsins are required to survive the cell wall stress.13,23 Further, the Cgyps1-11Δ mutant has been reported to possess sunken cell walls, altered cell wall composition, enlarged vacuole, and diminished ATP levels.15,22 CgYapsins have also been implicated in shedding the major adhesin Epa1 off the cell wall, as processing of Epa1 from the cell wall was found to be reduced in the Cgyps1-11Δ mutant.13 Additionally, CgYapsins are required for proper trafficking of the vacuolar hydrolase carboxypeptidase Y, as it was missorted to the external environment in the Cgyps1-11Δ mutant.22 Owing to these multiple phenotypic traits of the Cgyps1-11Δ mutant, CgYapsins are considered as key players in the biology and pathogenesis of C. glabrata.(13,15,21,22)
The mass spectrometry-based approaches have been used to identify the proteome and secretome of pathogenic microbes that has advanced our understanding of infectious diseases.24−26 The secretome of a pathogen is modulated by environmental cues, including cell growth phase and external conditions, and regulates its pathogenesis.27−29 Consequently, the key secretome constituents, including candidalysin, gliotoxin, secretory hydrolases, and iron scavenger siderophores, are pivotal to the virulence of the pathogenic fungi.17,18,30 As mentioned earlier, CgYapsins are essential for the virulence of C. glabrata.(13,15) Hence, the goal of the current study was to identify and characterize the secretome of C. glabratawild-type and Cgyps1-11Δ mutant and to gain insights into the CgYapsin-mediated regulation of basic cellular processes. We report here for the first time that the secretome of C. glabratawild-type and Cgyps1-11Δ mutant contains 119 and 548 proteins, respectively. Although Cgyps1-11Δ mutant, compared to wild-type, secreted out 4.6-fold higher number of proteins, only 12% proteins carried the classical secretory signal peptide sequence underscoring the importance of the nonconventional secretion pathway in the mutant. Contrarily, 50% secretory proteins in the wild-type strain contained the signal peptide. Further, our quantitative secretome analysis revealed differential abundance of 65 proteins in the Cgyps1-11Δ mutant with 49 and 16 being high- and low-abundant proteins, respectively, compared to wild-type cells. Finally, we demonstrate unequivocally for the first time that two of putative GPI-anchored CgYapsins, CgYps1 and CgYps7, are present in the secretome of C. glabratawild-type cells. Overall, our study paves the path to a better understanding of the role of secretory proteins in the virulence of C. glabrata.
Materials and Methods
Strains, Media, and Growth Conditions
C. glabratawild-type (Cg 559), Cgyps7Δ (YRK1003), Cgyps2ΔypsCΔ (YRK1005), and Cgyps1-11Δ (YRK 85) strains were maintained on the rich yeast extract peptone dextrose (YPD) medium at 30 °C. All strains are derivatives of the C. glabrata vaginal isolate BG2.13
Secretome Collection
The secretome of wild-type (wt), Cgyps7Δ, Cgyps2ΔypsCΔ, and Cgyps1-11Δ cells was collected, as described previously.31 Briefly, C. glabrata cells were grown overnight in YPD medium at 30 °C. The cultures were inoculated in the minimal yeast nitrogen base (YNB) medium at a very low density (OD600 = 0.0005) and grown for 16–20 h at 30 °C. Once the OD600 reached 1.5, cultures were centrifuged and the supernatants were collected. The supernatants were passed through 0.4 μm membrane or syringe filters to remove the residual cells, if any. The resultant filtrates were concentrated using Amicon Ultra-15 and Ultra-0.5 (10 kDa cutoff) centrifugal filter units. The concentrated secretome fractions (200 μg) were resolved on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized with the Coomassie Brilliant Blue stain. The protein concentration in collected secretome samples was measured using the Bio-Rad Protein Assay Dye Reagent Concentrate.
Global Secretome Analysis
The global secretome analysis involved protein identification via the microcapillary LC-MS/MS (liquid chromatography-tandem mass spectrometry) method. For this, the secretome samples (200 μg) were run a 10% SDS-PAGE gel, until the bromophenol blue dye front entered 3 cm into the resolving gel, and the gel was stained with Coomassie Brilliant Blue. The gel lane containing all proteins was sliced into three 1 cm × 1 cm sections, with each section containing a different size range (<50 kDa, 50–120 kDa, and >120 kDa) of proteins. Each section was treated as an individual sample for mass spectrometry analysis. The secretomes of wt and Cgyps1-11Δ cells, collected in duplicates (total 12 gel slices), were sent to the Taplin Biological Mass Spectrometry Facility, Harvard Medical School, Boston, for analysis using the Orbitrap mass spectrometer. The overall secretome protein yield for wt and Cgyps1-11Δ strains was 1.60 and 2.23 mg per gram dry cell weight, respectively. Similarly, Cgyps7Δ and Cgyps2ΔypsCΔ samples were collected in duplicates (total 12 gel slices) and sent for analysis.
At the Taplin Facility, samples were digested overnight with trypsin in-gel at 37 °C, followed by washes and dehydration with acetonitrile for 10 min. After complete removal of acetonitrile and drying in a speed vac, samples were reconstituted in the high-performance liquid chromatography (HPLC) solvent-A (2.5% acetonitrile, 0.1% formic acid) and loaded onto a nanoscale reverse-phase HPLC capillary column (100 μm inner diameter × ∼30 cm length) containing Accucore C18-2.6 μm spherical silica beads (Thermo Fisher Scientific). The peptides were eluted with a gradient of increasing concentrations of the solvent B (97.5% acetonitrile and 0.1% formic acid) for 80 min. Eluted peptides were ionized by electrospray and analyzed using the LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific). The tandem mass spectrum of specific fragment ions for each peptide was generated by isolating and fragmenting the detected peptide.
The acquired fragmentation pattern for each peptide was analyzed using the Sequest software, and searches were run against the UniProt C. glabrata reference proteome database containing 5200 entries. The identified peptides were filtered to 1% false discovery rate. Peptides identified from the three individual gel pieces of each sample were combined using the Taplin core software, GFY Core Version 3.7—Module Search Version 3.3, and a selection criterion, of a minimum of 2 total peptides for each protein in both replicate samples, was applied to identify proteins present in the culture media of wt and mutant cells. The mass spectromtery parameters used for global secretome analysis are listed in Table S1.
Growth Curve and Viability Analysis
For time course analysis, C. glabrata strains were grown overnight in the YPD medium and inoculated in fresh YNB medium at a cell density corresponding to 0.0002 OD600. Cultures were incubated at 30 °C with constant shaking; an aliquot was taken out at regular intervals; and once the OD600 reached 0.1, absorbance at 600 nm was recorded. The absorbance values were plotted against time to obtain growth profiles. To assess the cell viability, a culture aliquot was taken out at select time intervals and diluted in phosphate-buffered saline (PBS). Appropriate culture dilutions were plated on YPD medium, and the number of colonies that appeared after 1–2 days’ incubation at 30 °C were counted. This number was multiplied by appropriate dilution factors to obtain the total number of colony-forming units (CFUs) per milliliter of culture. The cell viability at the point of secretome collection was also measured using methylene blue staining. The 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay was performed, as described previously.32 YPD-grown C. glabrata cultures were inoculated at 0.0002 OD600 and grown for 18 h. Cells corresponding to 0.5 OD600 were incubated with XTT (HiMedia #TC239) and menadione at 37 °C for 5 h. Following centrifugation at 6000 g for 15 min, absorbance of the clear supernatant was measured at 492 nm and data were plotted as absorbance units.
Quantitative Secretome Analysis
The quantitative secretome analysis involved label-free relative protein quantification, following LC-MS, using the Minora Feature Detector Node of the Proteome Discoverer 2.2. For this, secretomes (100 μg), prepared in duplicates, of log-phase wt and Cgyps1-11Δ cells, were sent to the Valerian Chem Private Limited (Vproteomics), New Delhi, India, on dry ice.
At the Vproteomics, protein samples (25 μg) were first reduced with TCEP solution [5 mM Tris(2-carboxyethyl) phosphine-HCl] followed by alkylation with iodoacetamide (50 mM) and 16 h digestion with trypsin at 37 °C. Digests were cleaned up using the C18 silica cartridge and dried using a speed vac. The dried pellet was suspended in Buffer-A (5% acetonitrile and 0.1% formic acid). All analyses were performed using the EASY-nLC 1000 system (Thermo Scientific) coupled to the QExactive mass spectrometer (Thermo Scientific) equipped with nanoelectrospray ion source.
The trypsin-digested sample (1 μg) was resolved on a 50 cm long PicoFrit column (360 μm outer diameter, 75 μm inner diameter, and 10 μm tip) filled with 1.8 μm-C18 resin. The peptides were eluted with a 5–15% gradient of the Buffer-B (95% acetonitrile, 0.1% formic acid) for 85 min, 15–40% gradient for 80 min, followed by 95% gradient for 6 min at a flow rate of 250 nL/min for the total run time of 180 min. The MS data were acquired using a data-dependent top10 method dynamically choosing the most abundant precursor ions from the survey scan.
The raw data files for all four samples were analyzed using the Proteome Discoverer 2.2 software against the UniProt C. glabrata reference proteome database (containing 8078 entries). For Sequest HT and MS Amanda 2.0 search, the precursor and fragment mass tolerances were set at 10 ppm and 0.5 Da, respectively. The enzyme specificity for trypsin/P was set as cleavage at the C terminus of “K/R”, unless followed by “P”, along with two allowed missed cleavage sites. Carbamidomethyl on cysteine as fixed modification, and methionine oxidation and N-terminal acetylation were considered as variable modifications for database search. Both the peptide spectrum match and the protein false discovery rate were set to 0.01 and determined using the percolator node. Relative protein quantification was performed using the Minora Feature Detector Node of the Proteome Discoverer 2.2 with default settings. The peptide spectrum matches with high confidence were only considered. The mass spectromtery parameters used for quantitative secretome analysis are listed in Table S2.
Antibody Generation
CgYPS1 and CgYPS7 genes without signal peptide- and pro-peptide-encoding sequences were cloned in the Escherichia coli expression plasmid pET28a+. N-terminally 6X-His-tagged CgYps1 and CgYps7 were expressed using IPTG (isopropyl β-d-1-thiogalactopyranoside) and purified with the TALON metal affinity resin. Purified CgYps1 was injected into Balb/C mice for polyclonal antibody generation at CDFD animal house, while purified CgYps7 protein was sent to the Bioklone Biotech Private Limited, Chennai, for generation of polyclonal antibody in New Zealand White rabbits. The specificity of anti-CgYps1 and anti-CgYps7 sera was checked using appropriate CgYPS-deleted strains.
Western Blot Analysis
Total cell lysates were prepared from log-phase grown cultures. Briefly, C. glabrata cells were pelleted down, washed, and suspended in the protein extraction buffer [50 mM Tris (pH 7.5), 2 mM EDTA] containing 1 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM sodium orthovanadate (NaOVa), 10 mM sodium fluoride (NaF), and 1X protease inhibitor cocktail. To this cell suspension, 50–100 μg of 0.5 mm glass beads were added, and cells were lysed on the Fastprep-24 bead-beater. Cell lysates were centrifuged to remove unlysed cells and debris.
For isolation of a total membrane fraction from log-phase-grown C. glabrata cultures, the protocol of Fernandes et al.33 was adopted. In short, cells were pelleted down, washed, and suspended in the protein extraction buffer [100 mM Tris (pH 10.7), 5 mM EDTA, 2 mM dithiothreitol (DTT)] containing 1X protease inhibitor cocktail. The cell suspension was rapidly frozen by keeping at −80 °C and left for overnight. Next day, 50–100 μg of 0.5 mm glass beads were added to this cell suspension and cells were lysed on Fastprep-24. The lysed homogenate was diluted five times in buffer containing 0.1 M Tris-HCl (pH 8.0), 0.33 M sucrose, 5 mM EDTA, and 2 mM DTT and centrifuged at 1000g for 3 min at 4 °C. The supernatant was collected in a new tube and centrifuged again at 3000 g for 5 min at 4 °C to remove the unbroken cells and cell debris. Next, the supernatant was centrifuged at 19 000g for 45 min at 4 °C to obtain the total membrane fraction pellet. After one wash with the buffer [0.1 M Tris-HCl (pH 8.0), 0.33 M sucrose, 5 mM EDTA and 2 mM DTT], the pellet was suspended in the membrane suspension buffer containing 20% glycerol (v/v), 0.1 mM EDTA, 0.1 mM DTT, and 10 mM Tris-HCl (pH 7.5) and stored at −80 °C till further use.
The protein concentration in all samples was measured using the Pierce BCA protein assay kit unless stated otherwise. Total cell lysate (100 μg), membrane fraction (100 μg), and secretome (50 μL) samples were resolved on a 10% SDS-PAGE gel. Secreted CgYps1 and CgYps7 were detected using polyclonal antibodies (1:500 dilution) raised against CgYps1 and CgYps7 proteins.
Measurement of Interleukin (IL) 1-β Levels
The human monocytic cell line THP-1 (ATCC TIB-202) was cultured in the RPMI medium containing 10% fetal bovine serum (FBS) and differentiated into macrophages with phorbol myristyl acetate (16 nM) treatment. After 12 h of incubation, the spent medium was removed and cells were grown, for recovery, in the fresh RPMI medium for 12 h. For measurement of the secreted IL-1β cytokine, THP-1 macrophages were either left as such or incubated with 50 μL of wt and Cgyps1-11Δ secretome. After 24 h incubation at 37 °C with 5% CO2, the supernatants of control and secretome-co-incubated THP-1 macrophages were collected and spun at 1000g for 10 min to remove any particulate material. Levels of secreted IL-1β in the supernatant were measured using the BD OptEIA ELISA kit. The concentration of the cytokine IL-1β was calculated from the curve prepared with the standard protein.
Bioinformatic Analyses
The CAGL IDs were used to obtain the protein sequences from the Candida Genome Database (http://www.candidagenome.org/) using the “Batch Download” option. The SignalP5.0 (http://www.cbs.dtu.dk/services/SignalP/), TargetP1.1 (http://www.cbs.dtu.dk/services/TargetP/), and DeepLoc1.0 (http://www.cbs.dtu.dk/services/DeepLoc-1.0/index.php) servers with default settings were used to determine the presence of secretory signal peptide sequence, secretory signal peptide, and intracellular localization, of identified proteins, respectively. All Venn diagrams were prepared using the Lucid Chart (www.lucidchart.com) server. The Excel tool of Microsoft Office 2019 was used to make the bar graph and line curves.
Functional Analysis
To identify the nature of proteins secreted by C. glabrata strains, we performed the functional enrichment analysis using three tools, gene ontology (GO) Slim Mapper (http://www.candidagenome.org/cgi-bin/GO/goTermMapper) and Term Finder (http://www.candidagenome.org/cgi-bin/GO/goTermFinder) of the Candida Genome Database (CGD), and FungiFun (https://elbe.hki-jena.de/fungifun/). For all analyses, default settings of tools were used. For FungiFun analysis, C. glabrata CBS138 was used as the reference strain. For the representation purpose, all categories showing a corrected p-value <0.05 are shown.
Data Availability
The global and quantitative secretome mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE34 partner repository with data set identifiers PXD015131, PXD015132, and PXD015147.
Results and Discussion
Global Proteomic Analysis of the Secretome of C. glabrataWild-Type and Cgyps1-11Δ Strains
For secretome analysis, we employed the global proteomics strategy to identify all proteins secreted by C. glabratawild-type (wt) and Cgyps1-11Δ mutant strains into the medium. Among all identified proteins, we selected proteins, which were represented by a minimum of two total peptides in the MS data of both biological replicate samples. We found a total of 119 and 548 proteins in the secretome of wt and Cgyps1-11Δ mutant, respectively, that fulfilled the minimum peptide number criterion (Figure 1). A set of 102 proteins were common to both strains, while 17 and 446 proteins were present uniquely in the wt and Cgyps1-11Δ mutant secretome, respectively (Figure 1). Unexpectedly, among 17 proteins present uniquely in the wt secretome, eight were CgYapsins, CgYps1, CgYps3, CgYps5, CgYps6, CgYps7, CgYps9, CgYps10, and CgYps11 (Table S3), indicating that putative GPI-anchored cell wall aspartyl proteases are secreted into the medium. All proteins identified in the wt and Cgyps1-11Δ secretome along with the total number of peptides identified are listed in Supplementary Tables S3 and S4, respectively.
Figure 1.

Global secretome analysis of C. glabrata. Venn diagram illustrating overlap in proteins identified in the secretomes of wild-type and Cgyps1-11Δ strains.
Intriguingly, the Cgyps1-11Δ mutant secretome was found to be 4.6-fold larger than the wt secretome. Hence, we next analyzed 119 and 548 secretory proteins constituting the wt and mutant secretome, respectively, for their putative cellular localization. First, the presence of the secretory signal peptide in identified proteins was checked using the SignalP 5.0 program (http://www.cbs.dtu.dk/services/SignalP/).35 This analysis revealed 60 and 64 proteins, in the wt and mutant secretome, respectively, to be secretory (Tables S3 and S4). An independent analysis using the TargetP1.1 software (http://www.cbs.dtu.dk/services/TargetP/)36 showed 61 proteins in wt and 84 proteins in mutant samples to be secretory in nature (Tables S3 and S4). A set of 47 proteins common to the wt and mutant secretome was predicted to be secretory by both tools (Tables S3 and S4). Further, of 119 proteins representing the C. glabrata secretome, 59 were predicted to have the canonical ER secretory signal by these two widely used web servers, Signal P and TargetP1.1 (Table S3).
The putative GPI-anchored proteins in the C. glabrata proteome have previously been identified, through in silico analysis, by Weig et al.37 Since several cell wall proteins have been reported to be shed off during cell growth,28,38,39 we, therefore, next compared proteins identified in our global secretome analysis with the published data set of predicted GPI-anchored proteins.37 We found that only 25 and 20 proteins present in the wt and Cgyps1-11Δ mutant secretome, respectively, have been predicted to contain the C-terminal, fungal-specific, consensus sequence for GPI modification (Tables S3 and S4). The functional classification of these putative GPI proteins is listed in Table S5.
Next, we used the DeepLoc-1.0 algorithm (http://www.cbs.dtu.dk/services/DeepLoc-1.0/index.php)40 to predict the subcellular localization of identified proteins. We found 10 (8%), 35 (29%), 6 (5%), and 6 (5%) proteins in the wt secretome with predicted nuclear, cytoplasmic, mitochondrial, and endoplasmic reticulum localizations, respectively (Tables 1 and S6). Contrarily, the Cgyps1-11Δ secretome contained 71 (13%), 244 (44%), 66 (12%), and 34 (6%) proteins with predicted nuclear, cytoplasmic, mitochondrial, and endoplasmic reticulum localizations, respectively, indicating a 1.5- to 2.5-fold increased secretion of cytoplasmic and mitochondrial proteins, upon deletion of the CgYPS1-11 genes (Tables S6 and S7).
Table 1. Summary of DeepLoc 1.0 Server-Based Subcellular Localization Analysis of Proteins Identified in Secretomes of Wild-Type and Cgyps1-11Δ Strainsa.
| Strain | Total proteins | NLS | CP | ExC | Mito | CM | ER | Plast | Golgi | Lysosome/Vacuole | Per |
|---|---|---|---|---|---|---|---|---|---|---|---|
| wild-type | 119 | 10 | 35 | 28 | 6 | 30 | 6 | - | 1 | 3 | - |
| Cgyps1-11Δ | 548 | 71 | 244 | 41 | 66 | 55 | 34 | 2 | 8 | 17 | 10 |
NLS = Nucleus; CP = Cytoplasm; ExC = extracellular; Mito = Mitochondrion; CM = Cell membrane; ER = Endoplasmic reticulum; Plast = Plastids; Golgi = Golgi apparatus; Per = Peroxisome.
Altogether, these data indicate that the C. glabrata secretome contains 119 proteins, of which 59 (50%), 24 (20%), 35 (29%), and 25 (21%) are predicted to be secretory, membrane, cytoplasmic, and GPI-anchored proteins, respectively. Further, the family of 11 cell surface-associated aspartyl proteases regulate the secretome of C. glabrata, as secretome of the mutant lacking these proteases contained 548 proteins, of which 64 (12%), 87 (16%), 244 (44%), and 20 (4%) are predicted to be secretory, membrane, cytoplasmic, and GPI-anchored proteins, respectively. This increased protein secretion in the mutant could either be through extracellular vesicles or the nonclassical secretion pathway. Since the substantially higher number of proteins in the Cgyps1-11Δ secretome could also arise from cell lysis, we performed four experiments to rule out this possibility. First, we performed growth curve analysis and showed that both wt and Cgyps1-11Δ mutant displayed increase in growth after reaching cell density corresponding to 1.5 OD600 (Figure S1A), the density at which secretomes were collected. Second, we measured CFUs at different time points and found no decrease in the cell number between 16 and 24 h growth period, the period during which secretomes were collected (Figure S1B). Third, we stained wt and Cgyps1-11Δ cultures with methylene blue and found no appreciable cell death in either culture (Figure S1C). Finally, the XTT-based assay also revealed no differences in cell viability of wt and Cgyps1-11Δ cultures at the secretome collection time point (Figure S1D). Collectively, these data highlight the differences in the secretomes of late-log phase cultures of wt and Cgyps1-11Δ strains and raise the possibility that the absence of CgYapsins leads to a protein missorting defect in C. glabrata.
Functional Analysis of the C. glabrata Secretome
Next, we functionally annotated the set of identified secretory proteins to GO terms for biological process, cellular component, and molecular functions using the Candida Genome Database (http://www.candidagenome.org/cgi-bin/GO/goTermFinder)41,42 and FungiFun (https://elbe.hki-jena.de/fungifun/fungifun.php)43 tools. The FungiFun analysis for Biological Process revealed fungal-type cell wall organization, carbohydrate metabolic process, translation, and proteolysis terms to be predominant in the wt secretome (Figure 2A). The Cgyps1-11Δ secretome analysis showed the enrichment of amino acid transmembrane transport, pentose-phosphate shunt, de novo cotranslational protein folding, and removal of superoxide radicals biological processes (Figure 2B).
Figure 2.
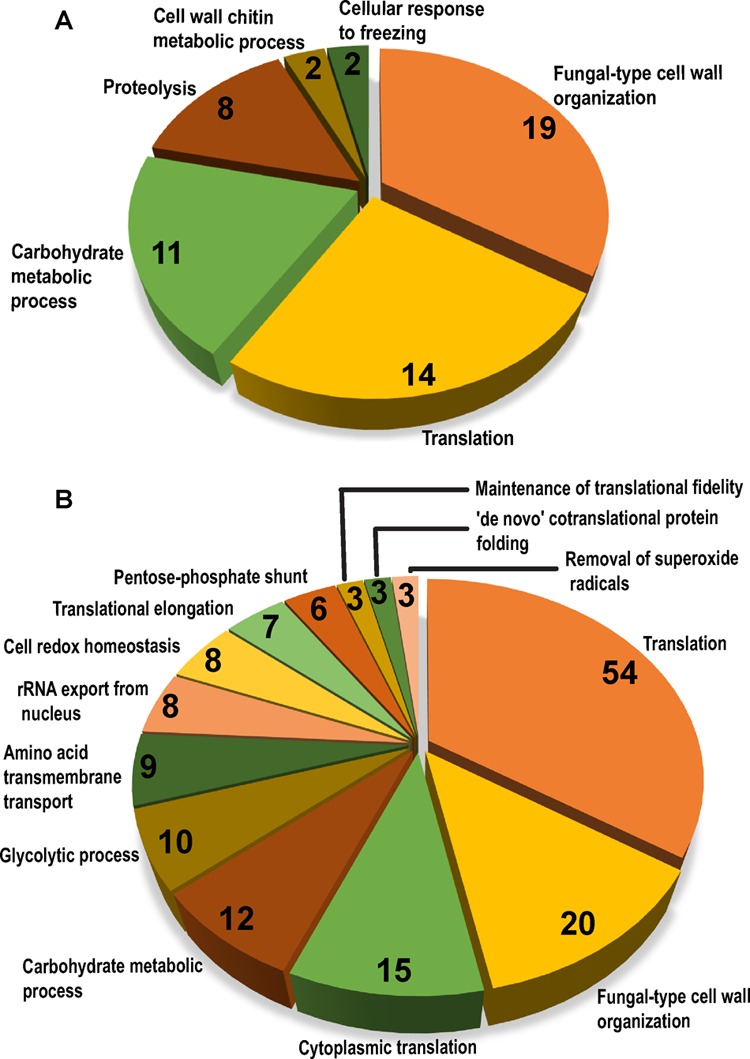
Functional enrichment analysis of secretomes. Pie chart illustrating enriched GO terms for biological process category in secretomes of wild-type (A) and Cgyps1-11Δ (B), as determined by the FungiFun tool. The number of proteins classified in each category are depicted inside pie slices.
The Cellular Component ontology analysis revealed that proteins in the wt secretome primarily belonged to the cell wall, extracellular region, plasma membrane, cytosolic large ribosomal subunit, and endoplasmic reticulum terms (Table S8). Comparison of the wt and Cgyps1-11Δ secretome revealed that besides cell periphery, a large number of proteins in the mutant secretome belonged to the cytosol category (Tables S8 and S9), which is consistent with our predicted subcellular localization of proteins in the Cgyps1-11Δ secretome (Tables S6, S7, and S9). Similarly, aspartic-type endopeptidase activity, O-glycosyl hydrolase activity, and glyceraldehyde-3-phosphate dehydrogenase (NAD+) (phosphorylating) activity function categories were exclusive to the wt secretome, while xenobiotic-transporting ATPase activity, unfolded protein binding, NAD binding, and proton-transporting ATP synthase activity categories were represented only in the Cgyps1-11Δ mutant secretome (Tables S8 and S9).
Next, we constructed, using the STRING software, the protein–protein interaction network for the wild-type secretome proteins involved in the fungal-type cell wall organization and carbohydrate metabolic process (Figure 3). We found the β-1,3-glucanosyltransferase Gas1 and endo-β-1,3-glucanase Bgl2 to be the key proteins linking cell wall organization and carbohydrate metabolism protein network, respectively (Figure 3).
Figure 3.

Protein–protein interaction network analysis. Networks, constructed by the STRING Protein–Protein Interaction Network (Ver 11.0) tool, depict interactions, among proteins identified in the wild-type secretome, that belonged to fungal-type cell wall organization (A) and carbohydrate metabolic process (B) categories. Since majority of C. glabrata proteins are uncharacterized, networks were constructed using the S. cerevisiae orthologs of identified C. glabrata proteins and S. cerevisiae as the reference strain. The line thickness reflects strength of the interaction.
Collectively, the GO analysis revealed that the C. glabrata secretome is primarily composed of proteins involved in cell wall organization, proteolysis, translation, and carbohydrate metabolism; however, lack of CgYapsins resulted in the secretion of several membrane transporters and proteins involved in the pentose-phosphate shunt pathway, superoxide detoxification, and cotranslational protein folding, to the extracellular environment.
Comparative Analysis of the Identified Secretome with the Published Glyco-Secretome and Secretome of C. glabrata
Two previous studies have reported 29 and 33 proteins to be present in the culture supernatant and filtrate of C. glabrata cells, respectively.44,45 However, despite these two reports, the C. glabrata secretome remains to be characterized fully. Our discovery of 119 proteins as secretome constituents in the current study typifies the largest catalogue of secretory proteins in C. glabrata. Previously, Stead et al. had identified all glycosylated proteins present in the secretome of wt and Cgace2Δ mutant strains.44 The Cgace2Δ mutant, which lacks the zinc finger motif-containing transcription factor, formed large cell clumps and was hypervirulent in mice.46 Both wt and Cgace2Δ strains (derivatives of the ATCC 2001 C. glabrata strain) contained 29 glycosylated proteins with two proteins being unique to the secretome of each strain.44 Of these 29 wt secretory proteins, 28 were found in our global secretome analysis (Table S3 and Figure 4). One protein not recovered in our secreteome analysis was the 60S acidic ribosomal protein, Cagl0a03168p (CgRpp2b; #Q6FYB0).
Figure 4.
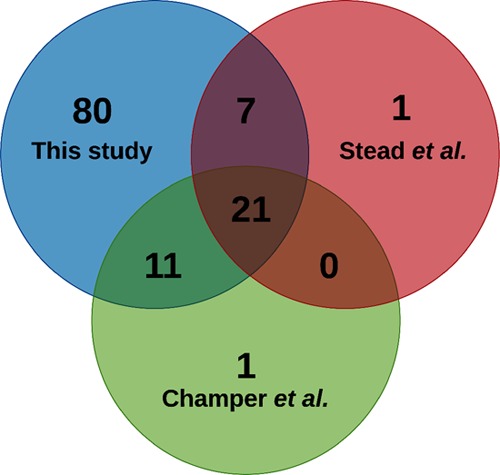
Comparative analysis of the identified wild-type secretome with published global secretome and glycosylated protein secretome of C. glabrata. Venn diagram illustrating overlap among proteins identified in our wild-type secretome with those identified in Champer et al.’s secretome (Champer et al. 2016) and Stead et al.’s glycosylated protein secretome (Stead et al., 2010).
Further, comparison of our secretome data with those of Champer et al.45 revealed 32 proteins to be common between the two analyses (Table S3 and Figure 4). One protein not recovered in our secreteome analysis was the putative adhesin Cagl0k00110p (CgAwp2; #Q6FNG1). Notably, Champer et al. had identified proteins, through LC/MSE analysis, of the cytosol, cell wall, and secretome of the strains of 13 fungal species, including one C. glabrata clinical isolate.45 Despite different genetic background of C. glabrata strains, 21 proteins were found to be common among three secretome analyses, viz., secretome reported herein, and by Stead et al. and Champer et al. (Table S3 and Figure 4).44,45 Of this set of 21 proteins, eight and five were involved in cell wall organization and carbohydrate metabolism, respectively (Table S3). Nine of these 21 proteins have previously been predicted to be GPI-anchored37 (Table S3). The presence of GPI-anchored proteins in fungal secretomes during normal growth is not unprecedented and has been implicated in modulation of cell adhesion.28,38,47,48 Similarly, the significant presence of carbohydrate metabolism proteins in the secretome of C. glabrata may hint toward their moonlighting functions, as reported for Paracoccidioides species.49 Finally, though the precise reason for the absence of CgRpp2b and CgAwp2 in our secretome data is yet to be determined, these comparative analyses together support our inference that 119 proteins identified in our study are the largest catalogue of the secretome constituents of C. glabrata.
Comparative Analysis of Identified Secretory Proteins with the Predicted Secretome of C. glabrata
Two previous studies have predicted, through in silico analysis, C. glabrata proteins that are secretory in nature.50,51 The Lum and Min group predicted 2.3% (121 proteins) of the entire proteome (5192 proteins) to be secretome constituents which were represented by 48 GPI-anchored and 73 soluble secretory proteins.51 Contrarily, using the three-layer hierarchical identification rule, Choi et al. considered an entry to be a secreted protein, if it was predicted to be so, by any one of the nine software tools (SignalP 3.0, SigCleave, SigPred, RPSP, TMHMM 2.0c, TargetP 1.1b, PSort II, SecretomeP 1.0f, and predictNLS).50 Accordingly, the secretory proteins were divided into four classes, with SP, SP3, SL, and NS classes containing 231, 290, 49, and 1767 proteins, respectively.50
To compare our identified secretome with the predicted secretome information, we combined the list of C. glabrata proteins predicted to be secretory by both Choi et al. and Lum and Min (Table S10).50,51 Comparison of our secretome with the predicted secretome data revealed that 76% of proteins (90 proteins) detected in our wt secretome were predicted to be secretome constituents (Table 2). In contrast, only half of the Cgyps1-11Δ secretome was predicted to be secretory (Table 2). The set of 29 proteins in the wt secretome, which were predicted to be secretory by neither study, included the plasma membrane proton pump, CgPma1, catalytic subunit of the phosphoinositide 3-kinase CgVps34, putative cell surface-associated aspartyl protease CgYps11, glycoside hydrolase CgGas1, and cytochrome c oxidase Cox12 (Table S11). Importantly, our in silico analysis predicted the presence of the secretory signal in three of these 29 proteins. Further, of these 29 proteins, 26 were detected in our all four samples (two replicate each for wt and Cgyps1-11Δ mutant) (Tables S3, S4, and S11), indicating that these are likely to be bona fide components of the C. glabrata secretome. It is possible that these 26 proteins are secreted out through unknown export signals and/or extracellular vesicles. These results also highlight the importance of experimental validation of predicted subcellular localizations.
Table 2. Summary of Comparative Analysis of Identified and Predicted Secretome of C. glabrata.
| Predicted by Choi et al. 2010 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Classical pathway | Nonclassical pathway |
|||||||
| Strain | Number of total proteins identified | Predicted by Lum & Min, 2011 | SP | NS | SP3 | SL | Number of total predicted proteins | % of Predicted secretory proteins |
| wild-type | 119 | 38 | 50 | 30 | 9 | 1 | 90 | 75.63 |
| Cgyps1-11Δ | 548 | 44 | 57 | 194 | 27 | 3 | 281 | 51.28 |
Comparative Analysis of the Secretomes of C. glabrata, C. albicans, and S. cerevisiae
The published secretome of C. albicans(52) and S. cerevisiae(53) consists of 61 and 180 proteins, respectively. These 180 proteins in the secretome of S. cerevisiae were identified in at least two replicate experiments.53 To determine the secretome similarity and difference, we compared the secretomes of C. glabrata, C. albicans, and S. cerevisiae. As mentioned earlier, we have identified a set of 119 proteins in the secretome of C. glabrata. Of these, 20 proteins were found to be common to all three yeast species (Figure 5 and Table S12), which may represent the core fungal secretome. The GO Slim Mapper analysis for biological process term revealed that of these 20 proteins, 11 and 8 are involved in cell wall organization and carbohydrate metabolism, respectively.
Figure 5.
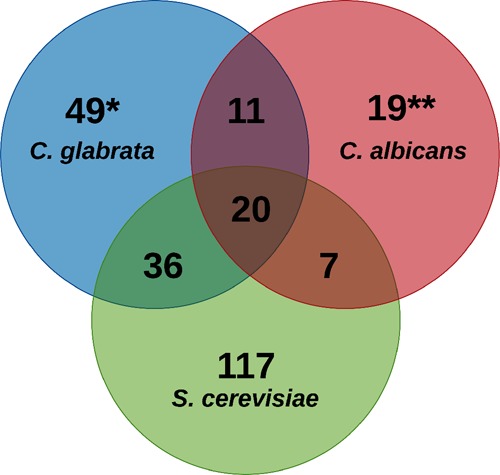
Comparative analysis of secretomes of C. glabrata, C. albicans, and S. cerevisiae. Venn diagram illustrating overlap among proteins identified in the C. glabratawild-type secretome with those identified in the secretomes of C. albicans (Gil-Bona et al. 2015) and S. cerevisiae (Smeekens et al. 2017). Of note, each of the three S. cerevisiae proteins, Scw11, Cwp1, and Gas1, corresponded to two different ORFs in C. glabrata, as indicated by *. Similarly, four of the S. cerevisiae proteins, Mkc7, Yps3, Cts1, and Plb3, corresponded to two, three, three, and two ORFs in C. albicans, respectively, as denoted by **.
Further, the pairwise comparison revealed that the C. glabrata secretome showed a higher overlap (56 common proteins) with the S. cerevisiae secretome compared to the C. albicans secretome (31 common proteins). This could be due to the closer phylogenetic relationship between C. glabrata and S. cerevisiae.(8−10) The proteins common specifically between S. cerevisiae and C. glabrata primarily belonged to cell wall organization, carbohydrate metabolism, and cell redox homeostasis, while the 11 protein set common specifically between C. albicans and C. glabrata contained phospholipase and aspartyl proteases, indicating that these secretory proteins may play a role in virulence. Of note, the C. albicans and S. cerevisiae secretome shared only 27 proteins, probably due to their discrete natural habitat and high evolutionary distance between these two fungal species.8,10 Overall, besides the common secretory proteins involved in cell wall organization and carbohydrate metabolism, these data also underscore fungal species-specific composition of the secretome.
Notably, in the C. albicans secretome study, authors also reported a set of 35 proteins present exclusively in the extracellular vesicle fraction.52 Of this set, seven proteins (Pma1, Kar2, Fet3, Ykt6, Ssa4, Tdh3, and Pgk1) were present in the secretome of both wt and Cgyps1-11Δ mutants of C. glabrata, while 17 proteins (Hxt6/7, Eft2, Pdi1, Tal1, Sah1, Cpr1, Sso2, Gpm1, Eno1, Ssa2, Rho3, Met6, Rho1, Gsc2, Sur7, Pep4, and Pdc1) were unique to the Cgyps1-11Δ mutant secretome, thereby raising the possibility that CgYapsin disruption may result in impaired extracellular vesicle formation and/or release of the extracellular vesicular proteins to the medium in C. glabrata. Of note, fungal extracellular vesicles are the major carriers of unconventionally secreted proteins, including heat shock proteins, glycolytic enzymes, virulence factors, and oxidative stress-counteracting proteins.54 Due to the vast nature of their cargo, extracellular vesicles could play an important role in infection establishment and progression. However, mechanisms underlying their biogenesis and selective cargo and transport across the cell wall are poorly understood. Extracellular vesicles may arise from plasma membrane, multivesicular bodies, and/or reverse micropinocytosis.54 A large presence of nonconventionally secreted proteins in the Cgyps1-11Δ secretome may reflect a requirement for CgYapsins in controlling extracellular vesicle cargo.
Further, a total of 49 proteins were found to be unique to the secretome of C. glabrata (Table S13.1). The GO Slim Mapper analysis found “structural molecule activity” (31%), “hydrolase activity” (14%), and “protein binding” (12%) function terms to be enriched in this set of 49 proteins (Table S13.2). A subset of these 49 unique proteins may hold promise as potential diagnostic biomarkers, and further studies will be designed to delve into this possibility.
Finally, while the C. albicans secretome contained seven secretory aspartyl proteases and two phospholipases,52 the S. cerevisiae secretome had three GPI-anchored aspartyl proteases and two phospholipases.53 Similarly, the identified C. glabrata secretome contained eight CgYapsins (CgYps1, CgYps3, CgYps5, CgYps6, CgYps7, CgYps9, CgYps10, and CgYps11) and two phospholipases (CgPlb1 and CgPlb2) (Tables S3 and S12). Altogether, these data indicate that though the release of both aspartyl proteases and phospholipases to the external environment appears to be a feature conserved among these three yeast species, the mechanism underlying the release of GPI-anchored yapsins from the cell wall in C. glabrata and S. cerevisiae needs to be elucidated.
Label-Free Quantitative Secretome Analysis of Wild-Type and Cgyps1-11Δ Cells
Since the secretome of Cgyps1-11Δ cells contained 4.6-fold higher number of proteins compared to the wt secretome, we next performed quantitative proteomics analysis, through the label-free quantitation approach, to identify proteins, which were present differentially in the secretome of the Cgyps1-11Δ mutant. The quantitative secretome profiling analysis identified a total of 85 and 193 proteins in the secretome of wt and Cgyps1-11Δ cells, respectively, with 79 secreted proteins being common to both strains (Figure 6A, Tables S14 and S15). Of these 79 proteins, 65 showed differential abundance (≥1.5-fold change), with 49 and 16 displaying increased and decreased abundance, respectively, in the secretome of the Cgyps1-11Δ mutant (Figure 6B,C). Interestingly, the five most abundant proteins in the secretome of the Cgyps1-11Δ mutant, CgTma7, CgHtb2, CgHtb1, CgMet6, and CgMbf1, were represented in the set of 49 proteins displaying higher levels than those in wt cells (Figure 6B; Table S15). A similar pattern was also observed for the least abundant protein set, CgMsb2, CgCrh1, CgCwp2, CgNca3, and CgScw11 (Figure 6C, Table S15). Of note, 6 and 114 proteins were unique to the secretome of wt and Cgyps1-11Δ mutant, respectively (Figure 6A and Tables S14 and S15). As observed in the global secretome analysis, CgYapsins CgYps1, CgYps6, CgYps7, CgYps9, CgYps10, and CgYps11, represented this unique six-protein set in the wt secretome (Table S14). Importantly, CgYps3, CgYps6, and CgYps9 have previously been reported in the glyco-secretome of C. glabratawt cells.44 Further, of 114 proteins, that were present uniquely in the Cgyps1-11Δ secretome, 21, 6, 6, and 4 were involved in translation, glycolysis, maturation of SSU-rRNA from tricistronic rRNA transcript, and cellular response to oxidative stress processes, respectively (Table S16). Of note, proteins involved in the detoxification of superoxide radicals were earlier found to be exclusively present in the global secretome of the Cgyps1-11Δ mutant (Figure 2B). Since the Cgyps1-11Δ mutant is known to have high reactive oxygen species (ROS) levels,22 it will be interesting to determine whether increased secretion/missorting of ROS detoxification proteins could account for the elevated ROS levels in the mutant.
Figure 6.

Quantitative secretome analysis of C. glabrata. (A) Venn diagram illustrating overlap in proteins identified in the quantitative secretomes of wild-type and Cgyps1-11Δ strains. (B) Bar graph depicting 49 proteins with higher abundance in the Cgyps1-11Δ secretome compared to the wild-type secretome. (C) Bar graph depicting 16 proteins with lower abundance in the Cgyps1-11Δ secretome compared to the wild-type secretome. The fold-difference in levels are presented as Log2 values.
Notably, the least abundant protein in the Cgyps1-11Δ secretome, compared to wt secretome, was CgMsb2 that was six-fold underrepresented (Figure 6C). CgMsb2 orthologs are present in many fungi, including S. cerevisiae(55) and C. albicans.(56) Importantly, Msb2, a signaling mucin, has previously been shown to be cleaved by Yps1 in S. cerevisiae(55) and Saps in C. albicans.(56) This aspartyl protease-mediated Msb2 shedding in S. cerevisiae and C. albicans is required for activation of MAPK signaling.55,56 To check if the reduced abundance of CgMsb2 in Cgyps1-11Δ secretome could be due to transcriptional downregulation, we measured transcript levels of the CgMSB2 gene by quantitative polymerase chain reaction (qPCR). However, we found a modest 1.6-fold upregulation in Cgyps1-11Δ cells, compared to wt cells (Figure S2). This result is consistent with 1.6-fold higher CgMSB2 transcripts observed in the RNA-Sequencing analysis of the Cgyps1-11Δ mutant.15 Together, these data suggest that the posttranscriptional regulation of CgMsb2 is likely to account for its underrepresentation in the Cgyps1-11Δ secretome and raise the possibility of CgMsb2 being a CgYapsin substrate, with aspartyl protease-dependent Msb2 cleavage as a common feature among C. glabrata, C. albicans, and S. cerevisiae.
Comparative Analysis of the Secretome Identified via Global and Quantitative Proteomic Approaches
Comparison of the global and quantitative secretomes of wt and Cgyps1-11Δ mutant revealed an overlap of 70–80% with 61 and 155 proteins being common to the secretome of wt and Cgyps1-11Δ, respectively, as identified by two methods (Figure 7). This set of 61 proteins in the wt secretome primarily belonged to fungal-type cell wall organization, carbohydrate metabolic process and proteolysis, with the latter process being represented by six CgYapsins, CgYps1, CgYps6, CgYps7, CgYps9, CgYps10, and CgYps11 (Tables S8 and S17). The set of 155 proteins identified, by both global and quantitative analyses, in the Cgyps1-11Δ secretome primarily belonged to translation, fungal-type cell wall organization, carbohydrate metabolic process, and glycolysis (Tables S9 and S18). Overall, the quantitative secretome profiling identified 1.4- to 2.8-fold fewer number of total proteins compared to the global secretome analysis, which could be due to the difference in the two methodologies and/or complex nature of the quantitative secretome samples, as these were not resolved on SDS-PAGE gel, prior to MS analysis. However, the biological processes enriched remained largely the same in secretomes identified by both global and quantitative proteomic approaches (Tables S8, S9, S17, and S18).
Figure 7.
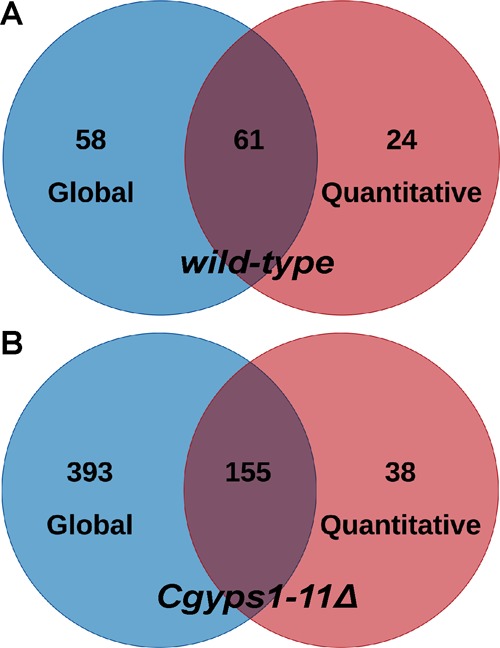
Comparative analysis of global and quantitative secretomes of C. glabrata. Venn diagrams illustrating overlap in proteins identified in global and quantitative secretomes of the wild-type (A) and Cgyps1-11Δ (B) strains.
Further, a comprehensive comparative analysis revealed that a set of 53 proteins was identified in the secretome of both wt and Cgyps1-11Δ mutants by global as well as quantitative secretome profiling (Table S19). Moreover, of these 53 proteins, 25 and 16 proteins showed increased and decreased abundance, respectively, in the Cgyps1-11Δ mutant compared to the wt strain (Tables S15 and S19). Low-abundant proteins in the mutant were represented by cell wall proteins, including CgCwp1, CgCwp2, and CgPir3, and the mucin CgMsb2 (Table S19). The GO analysis of 53 common proteins revealed 13 and 9 proteins to be involved in cell wall organization and carbohydrate metabolism, respectively (Table S19). Altogether, these results indicate a role for CgYapsins in regulation of basic cellular processes, which may in part account for pleiotropic phenotypes associated with loss of CgYapsins.
Next, to determine if relative abundance of proteins can be measured based on the number of total peptides identifed in the global secretome MS data, we calculated the ratio of the total number of peptides obtained for each protein in the Cgyps1-11Δ mutant sample to that in the wt sample (Table S20). We found that of 102 proteins, common between the wt and Cgyps1-11Δ mutant secretomes, 79 proteins showed differential abundance (≥1.5-fold change in the average peptide number ratio) with 65 being high- and 14 being low-abundant proteins (Table S20). Importantly, ∼50% of these proteins (24 up and 9 down) showed different abundance in the quantitative secretome analysis (Table S20), suggesting that the peptide number ratio can be used to determine the protein abundance qualitatively.
Finally, we also determined the relative abundance, using the spectral counting-based approach, of all proteins, identified in global (Tables S21.1 and S21.2) and quantitative (Tables S22.1 and S22.2) secretome analyses. Spectral counting-based relative abundance analysis in global secretomes revealed CgCrh1, CgCwp2, CgScw4, CgBgl2, and CgPir3 to be the five most abundant proteins in the wt secretome (Table S21.1), while CgCwp2, CgYgp1, CgTrx1, CgRpl40b, and CgCis3 were found to be the five most abundant proteins in Cgyps1-11Δ secretome (Table S21.2). Similarly, spectral counting-based relative abundance analysis in quantitative secretomes revealed CgCwp1, CgCwp2, CgCrh1, CgPir3, and CgTos1, and CgCwp1, CgCwp2, CgPir3, CgTos1, and CgHsp150, to be the five most abundant proteins in wt and Cgyps1-11Δ secretomes, respectively (Tables S22.1 and S22.2). In general, a good correlation was found between spectral counting-based and minora node-based label-free quantification of secretome proteins (Tables S14, S15, and S22).
Overall, secretome of the Cgyps1-11Δ mutant, in both global and quantitative proteomic analyses, contained 2- to 5-fold higher number of secretory proteins. In this regard, it is noteworthy that the Cgyps1-11Δ mutant contains lower amount of β-glucan in the cell wall compared to that in the wt strain,15 which may lead to the weak attachment of cell wall proteins. Therefore, protein missorting, stable secreted proteins because of the lack of cell surface-associated proteolytic activity, and/or increased release of proteins due to weak anchoring in the mutant cell wall could all contribute to the large secretome of the Cgyps1-11Δ mutant. To investigate the role of cell wall in modulation of the C. glabrata secretome, we sought to profile global secretomes of Cgyps7Δ and Cgyps2ΔypsCΔ mutants that lack CgYps7 protease and nine proteases (CgYps2-6 and CgYps8–11), respectively. Of note, Cgyps7Δ mutant, like Cgyps1-11Δ mutant, has previously been shown to be sensitive to cell wall stressors.13 Contrarily, the Cgyps2ΔypsCΔ mutant showed wt-like susceptibility to the cell wall stress.13 We first verified the growth attenuation of Cgyps7Δ and Cgyps1-11Δ mutants in the presence of congo red (Figure S3), which binds to β-glucan and chitin, the key fungal cell wall structural components.
Next, we checked the growth profiles of Cgyps7Δ and Cgyps2ΔypsCΔ mutants in YNB medium. The Cgyps2ΔypsCΔ mutant grew similar to wt, while Cgyps7Δ mutant grew slightly more slowly (Figure S1A). However, viability of Cgyps7Δ and Cgyps2ΔypsCΔ cells remained similar to wt cells at all time points, as determined by CFU (Figure S1B), methylene blue staining (Figure S1C), and XTT (Figure S1D) assays. Global secretome analysis identified a total of 59 and 52 proteins in the secretomes of Cgyps7Δ (Table S23) and Cgyps2ΔypsCΔ mutant (Table S24), respectively. Both mutants shared a set of 45 secretory proteins (Figure S4). Further, 55 and 50 proteins were common between wt and Cgyps7Δ, and wt and Cgyps2ΔypsCΔ secretomes, respectively (Figure S4). Similarly, a set of 46 and 44 proteins was common between Cgyps1-11Δ and Cgyps7Δ, and Cgyps1-11Δ and Cgyps2ΔypsCΔ secretomes, respectively (Figure S4). The predicted cellular localization of secretory proteins in Cgyps7Δ and Cgyps2ΔypsCΔ mutants is depicted in Table S25 and 26, respectively. Importantly, CgYps1 and CgYps7 were present in the Cgyps2ΔypsCΔ secretome (Table S24), while the Cgyps7Δ secretome contained seven CgYapsins, CgYps1,2,5,6,9,10, and 11 (Table S23). Functional analysis of Cgyps7Δ and Cgyps2ΔypsCΔ secretomes revealed a large fraction of proteins to belong to fungal-type cell wall organization and carbohydrate metabolic process (Tables S27 and S28). Altogether, the smaller and wt-like secretome of the cell wall stress-sensitive Cgyps7Δ mutant indicates that the larger and differential secretome of the Cgyps1-11Δ mutant is unlikely to be solely due to altered cell wall. These data also suggest that CgYps2–6 and CgYps8–11 proteases are not pivotal to secretome modulation.
CgYps1 and CgYps7 Are Present in the Secretome of C. glabrata
Next, to verify that the Cgyps1-11Δ mutant indeed secretes out a large number of proteins, we prepared the secretome of log-phase wt and Cgyps1-11Δ cultures which had undergone 11 doublings in the minimal YNB medium. SDS-PAGE analysis of equal volumes of secretome fractions of both strains revealed significantly higher amount of proteins in the mutant secretome (Figure 8A), thereby validating the secretome MS data. We also determined the protein profiles of total cell lysates and membrane fractions of wt and Cgyps1-11Δ mutants and found these to be very different from the secretory protein profiles (Figure 8B), suggesting that the secretome of C. glabrata cells contains a distinct set of proteins.
Figure 8.

CgYps1 and CgYps7 are secreted into the medium. (A) Representative SDS-PAGE gel image indicating increased protein secretion into the medium of the Cgyps1-11Δ mutant. The secretomes of wild-type and Cgyps1-11Δ mutants were collected after 11 doublings in the YNB medium, and 50 μL were resolved on a 12% SDS-PAGE gel. Proteins were stained with Coomassie Brilliant Blue (CBB) for visualization. (B) Representative SDS-PAGE gel image depicting the total membrane, cell lysate, and secretory protein profiles of wild-type and Cgyps1-11Δ mutants. Equal volume of secretomes (50 μL) and 100 μg of total membrane and cell lysate were resolved on a 10% SDS-PAGE gel and stained with CBB for visualization. (C) Representative western blot images of CgYps1 and CgYps7 indicating their secretion into the medium of the wild-type strain. Equal volume (50 μL) of secretomes of wild-type and Cgyps1-11Δ strains were loaded on a 10% SDS-PAGE and resolved for 4 h. Proteins were transferred to the polyvinylidene fluoride membrane and probed with anti-CgYps1 and anti-CgYps7 antibodies. CBB-stained SDS-PAGE gels were used as loading control. Of note, the red asterisk marks a nonspecific band seen in the Cgyps1-11Δ secretome.
Further, to check if CgYps1 and CgYps7 are present in the secretome of C. glabrata, we performed immunoblot analysis on the secretome fractions. As shown in Figure 8C, a band of ∼130 kDa, corresponding to CgYps1, and of ∼120 kDa, corresponding to CgYps7, were present in the wt secretome, while the Cgyps1-11Δ mutant secretome, expectedly, showed signal for neither of the protein. A faint nonspecific protein band was observed in the Cgyps1-11Δ secretome in anti-CgYps1 blot (Figure 8C). Of note, both proteins migrated at a higher molecular mass compared to the predicted size of 63.8 and 63.4 kDa for CgYps1 and CgYps7, respectively. The higher molecular weight of CgYps1 and CgYps7 proteins could arise from posttranslational modifications, including β-glucosylation, as reported previously for other cell wall-anchored proteins.57 Altogether, these data suggest that CgYps1 and CgYps7 proteins are released into the external environment. It remains to be determined whether the secretory CgYps1 and CgYps7 forms are proteolytically active.
Effect of the C. glabrata Secretome on the Macrophage Immune Response
Finally, to determine the physiological relevance of the differential secretome of the Cgyps1-11Δ mutant, we incubated the secretomes of both wt and Cgyps1-11Δ strains with PMA-activated human THP-1 macrophages and assessed the macrophage inflammatory cytokine response (Figure 9). Earlier, we have shown that THP-1 cells induced the production of IL-1β upon infection with C. glabratawt cells.15 The IL-1β production was found to be higher upon infection with Cgyps1-11Δ cells.15 Of note, compared to five- to seven-fold replication of wt cells in THP-1 macrophages, the Cgyps1-11Δ mutant is killed in THP-1 cells, which has been attributed to the enhanced IL-1β secretion.15 As shown in Figure 9, incubation of neither wt nor Cgyps1-11Δ secretome with THP-1 macrophages led to elevated production of IL-1β by THP-1 cells. These results suggest that the differential activation of THP-1 macrophages by the Cgyps1-11Δ mutant is not solely dependent upon the proteins secreted by the Cgyps1-11Δ mutant. Together, these data also raise four distinct possibilities. First, C. glabrata infection-induced IL-1β secretion in human THP-1 macrophages is probably not mediated by secretory proteins. Second, the secretory proteins may have lost their host immune response modulation activity during collection and processing of secretome samples. Third, human THP-1 macrophage activation may be triggered by C. glabrata cell wall polysaccharide components, viz., β-glucan, mannan, and chitin. Notably, content of these components were found to be different in the cell walls of wt and Cgyps1-11Δ mutant.15 Fourth, dynamic interactions between live C. glabrata cells and THP-1 macrophages are required for the latter to produce IL-1β. In this context, it is noteworthy that mixed infection of C. glabratawt and Cgyps1-11Δ cells to THP-1 macrophages had an adverse effect on the intracellular replication rate of wt cells, probably owing to relatively higher IL-1β production,15 pointing out a role for either Cgyps1-11Δ cells or secretory components of the Cgyps1-11Δ mutant in controlling the proliferation of wt cells in macrophages. Future investigations will be designed to address the aforementioned possibilities.
Figure 9.
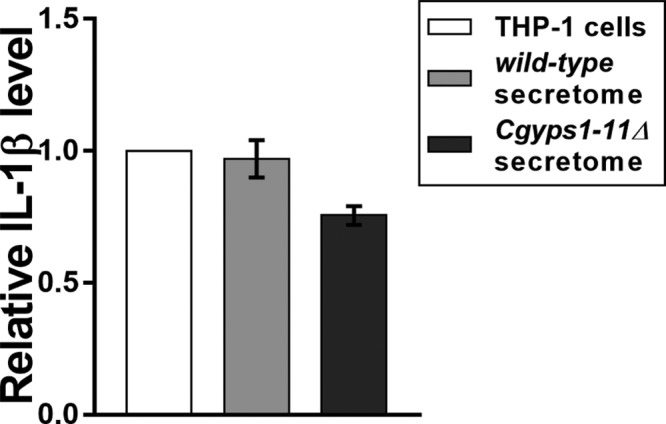
C. glabrata secretome has no effect on the production of the proinflammatory cytokine IL-1β by THP-1 macrophages. PMA-differentiated THP-1 cells were either left untreated or incubated with wild-type and Cgyps1-11Δ secretome for 24 h. Levels of secreted IL-1β were measured in 100 μL of culture media using the BD OptEIA ELISA kit. Data (mean ± standard deviation (SD); n = 2) represent relative amounts of IL-1β produced by secretome-co-incubated THP-1 cells compared to the untreated THP-1 macrophages (considered as 1.0).
Conclusions
Despite C. glabrata being a frequent causal agent of Candida BSIs,1,2,5 its biology and pathogenesis traits remain to be fully elucidated. Using the LC-MS/MS approach, we, here, have identified 119 proteins as constituents of the secretome of C. glabrata. Further, we show, through the FungiFun tool-based analysis, that proteins with aspartyl protease activity and 1,3-β-glucanosyltransferase activity, and proteins with functions in the biogenesis of cell wall and ribosome, represent key components of the secretome. One unexpected finding of our analysis was the presence of eight CgYapsins (CgYps1, 3, 5–7, 9–11) in the C. glabrata secretome. As majority of these enzymes, except for CgYps4 and CgYps11,13 are predicted to be GPI-anchored, their presence in the secretome is consistent with the identification of several other GPI-anchored proteins in the growth medium of C. glabrata and other fungi.28,38,39,44 The mechanisms underlying the presence of GPI-anchored proteins in fungal secretomes include proteolytic cleavage (processing of Flo11 adhesion molecule by Subtilisin-like protease Kex2 in C. albicans and autocatalysis of Yps1 yapsin in S. cerevisiae), release of precursors of cell wall-anchored proteins, cell wall degradation during mother–daughter separation, and routine shedding.28,47,58 The released soluble protein forms may modulate fungal adhesion, protease activity, and cellular signaling pathways.47,48,58 Keeping this in view, it will be intriguing to determine whether CgYapsin release into the external environment stems from a regulated proteolytic cleavage or routine turnover of cell wall proteins.
The six-fold underrepresentation of CgMsb2 in the Cgyps1-11Δ mutant secretome indicates that CgMsb2 could be a substrate of CgYapsins. Our preliminary in silico analysis revealed that the region between 565–590 amino acids in the CgMsb2 protein (936 aa-long) binds to the predicted active site of the CgYps1 enzyme. Future studies will investigate the CgYps1-dependent cleavage of CgMsb2 and its significance in the physiology and virulence of C. glabrata. Further, a decreased abundance of fungal cell wall organization proteins in the Cgyps1-11Δ mutant secretome indicates a pivotal role for CgYapsins in cell wall biogenesis, which is in accordance with our earlier RNA-Seq and cell wall composition data,15 and reported roles for fungal yapsins and C. albicans aspartyl proteases.18,20,23,59 Collectively, these data underscore the conservation of some target proteins among fungal aspartyl proteases.
In conclusion, besides cataloguing the secretome of an avirulent aspartyl protease-deficient C. glabrata strain for the first time, the current study suggests that CgYapsins may modulate the secretome of C. glabrata.
Acknowledgments
The authors are grateful to Dr. Reshma Chowdary Alokam for CgMsb2 structural modeling analysis. They thank Fizza Askari, and S Surya Vamshi and Partha Dey for their help in cytokine measurement and Western blot analysis, respectively. They also thank Anamika Battu and Sandip Patra for their help with serial dilution spotting analysis. They are grateful to Ross Tomaino, Taplin MS facility, and Gagan Deep Jhingan, Vproteomics, for their assistance with MS analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.9b00299.
Figure S1: Similar growth and viability profiles of C. glabratawild-type (wt) and Cgyps1-11Δ strains at the time of secretome collection; Figure S2:CgMSB2 gene is modestly upregulated in the Cgyps1-11Δ mutant; Figure S3:Cgyps1-11Δ and Cgyps7Δ mutants show enhanced sensitivity to the cell wall stressor congo red; Figure S4: comparative analysis of global secretomes of wild-type, Cgyps1-11Δ, Cgyps7Δ, and Cgyps2ΔCΔ strains (Figures S1–S4) (PDF)
Table S1: A list of mass spectrometry parameters used for global secretome analysis. Table S2: A list of mass spectrometry parameters used for quantitative secretome analysis. Table S3: A list of 119 proteins identified in the wild-type secretome in global secretome analysis. Table S4: A list of 548 proteins identified in the Cgyps1-11Δ secretome in global secretome analysis. Table S5: Functional classification of putative GPI-anchored proteins identified in the secretomes of wild-type and Cgyps1-11Δ strains. Table S6: DeepLoc 1.0 web server-based subcellular localization analysis of proteins identified in the wild-type secretome. Table S7: DeepLoc 1.0 web server-based subcellular localization analysis of proteins identified in the Cgyps1-11Δ secretome. Table S8: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories for the wild-type secretome, as determined by the FungiFun tool. Table S9: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories for the Cgyps1-11Δ secretome, as determined by the FungiFun tool. Table S10: Comparative analysis of identified and predicted secretome of C. glabrata. Table S11: A list of 29 proteins identified in the wild-type secretome, that were not predicted to be secretory. Table S12: Comparative analysis of identified secretory proteins in C. glabrata, C. albicans, and S. cerevisiae. Table S13: Analysis of secretory proteins that are unique to C. glabrata. Table S14: A list of 85 proteins identified in the wild-type secretome by label-free quantitative secretome profiling. Table S15: A list of 193 proteins identified in the Cgyps1-11Δ secretome by label-free quantitative secretome profiling. Table S16: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories, as determined by the FungiFun tool, in the set of 114 proteins, that were unique to the Cgyps1-11Δ secretome in quantitative secretome analysis. Table S17: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories, as determined by the FungiFun tool, in the set of 61 proteins, identified in wt secretome by both global and quantitative analysis. Table S18: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories, as determined by the FungiFun tool, in the set of 155 proteins, identified in Cgyps1-11Δ secretome by both global and quantitative analysis. Table S19: A list of 53 common proteins in wild-type and Cgyps1-11Δ secretomes, that were identified by both global and quantitative secretome profiling. Table S20: A list of 79 proteins displaying differential abundance in the Cgyps1-11Δ secretome based on the peptide number ratio. Table S21: Relative abundance analysis, using spectral counting-based approach, of proteins identified in the global secretome. Table S22: Relative abundance analysis, using spectral counting-based approach, of proteins identified in the quantitative secretome. Table S23: A list of 59 proteins identified in the Cgyps7Δ secretome in global secretome analysis. Table S24: A list of 52 proteins identified in the Cgyps2ΔCΔ secretome in global secretome analysis. Table S25: DeepLoc 1.0 web server-based subcellular localization analysis of proteins identified in the Cgyps7Δ secretome. Table S26: DeepLoc 1.0 web server-based subcellular localization analysis of proteins identified in the Cgyps2ΔCΔ secretome. Table S27: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories for the Cgyps7Δ secretome, as determined by the FungiFun tool. Table S28: Enriched GO terms for biological process (BP), cellular component (CC), and molecular function (MF) categories for the Cgyps2ΔCΔ secretome, as determined by the FungiFun tool. (XLSX)
Author Contributions
M.R. and R.K. conceived and designed the study. M.R. and N.K. performed experiments and acquired data. M.R. and R.K. analyzed data, prepared figures, and wrote manuscript.
This work was fully supported by the Wellcome Trust/DBT India Alliance Senior Fellowship [IA/S/15/1/501831] awarded to RK.
The authors declare no competing financial interest.
Supplementary Material
References
- Brown G. D.; Denning D. W.; Gow N. A. R.; Levitz S. M.; Netea M. G.; White T. C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 1–9. 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- Pfaller M. A.; Pappas P. G.; Wingard J. R. Invasive Fungal Pathogens: Current Epidemiological Trends. Clin. Infect. Dis. 2006, 43, S3–S14. 10.1086/504490. [DOI] [Google Scholar]
- Santolaya M. E.; Thompson L.; Benadof D.; Tapia C.; Legarraga P.; Cortés C.; Rabello M.; Valenzuela R.; Rojas P.; Rabagliati R.; et al. A Prospective, Multi-Center Study of Candida Bloodstream Infections in Chile. PLoS One 2019, 14, e0212924 10.1371/journal.pone.0212924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo A. L.; Guimarães T.; Sukienik T.; Pasqualotto A. C.; Andreotti R.; Queiroz-Telles F.; Nouér S. A.; Nucci M. Prognostic Factors and Historical Trends in the Epidemiology of Candidemia in Critically Ill Patients: An Analysis of Five Multicenter Studies Sequentially Conducted over a 9-Year Period. Intensive Care Med. 2014, 40, 1489–1498. 10.1007/s00134-014-3400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A.; Diekema D. J.; Turnidge J. D.; Castanheira M.; Jones R. N. Twenty Years of the SENTRY Antifungal Surveillance Program: Results for Candida Species From 1997-2016. Open Forum Infect. Dis. 2019, 6, S79–S94. 10.1093/ofid/ofy358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna M. T.; Lovero G.; Borghi E.; Amato G.; Andreoni S.; Campion L.; Lo Cascio G.; Lombardi G.; Luzzaro F.; Manso E.; et al. Candidemia in Intensive Care Unit: A Nationwide Prospective Observational Survey (GISIA-3 Study) and Review of the European Literature from 2000 through 2013. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 661–674. [PubMed] [Google Scholar]
- Chakrabarti A.; Sood P.; Rudramurthy S. M.; Chen S.; Kaur H.; Capoor M.; Chhina D.; Rao R.; Eshwara V. K.; Xess I.; et al. Incidence, Characteristics and Outcome of ICU-Acquired Candidemia in India. Intensive Care Med. 2015, 41, 285–295. 10.1007/s00134-014-3603-2. [DOI] [PubMed] [Google Scholar]
- Gabaldón T.; Carreté L. The Birth of a Deadly Yeast: Tracing the Evolutionary Emergence of Virulence Traits in Candida glabrata. FEMS Yeast Res. 2016, 16, fov110. 10.1093/femsyr/fov110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B.; Sherman D.; Fischer G.; Durrens P.; Casaregola S.; Lafontaine I.; De Montigny J.; Marck C.; Neuvéglise C.; Talla E.; et al. Genome Evolution in Yeasts. Nature 2004, 430, 35–44. 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Rodrigues C. F.; Silva S.; Henriques M. Candida glabrata: A Review of Its Features and Resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 673–688. 10.1007/s10096-013-2009-3. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Askari F.; Sahu M.; Kaur R. Candida glabrata: A Lot More Than Meets the Eye. Microorganisms 2019, 7, 39. 10.3390/microorganisms7020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidel P. L.; Vazquez J. A.; Sobel J. D. Candida glabrata: Review of Epidemiology, Pathogenesis, and Clinical Disease with Comparison to C. albicans. Clin. Microbiol. Rev. 1999, 12, 80–96. 10.1128/CMR.12.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R.; Ma B.; Cormack B. P. A Family of Glycosylphosphatidylinositol-Linked Aspartyl Proteases Is Required for Virulence of Candida glabrata. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7628–7633. 10.1073/pnas.0611195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seider K.; Brunke S.; Schild L.; Jablonowski N.; Wilson D.; Majer O.; Barz D.; Haas A.; Kuchler K.; Schaller M.; et al. The Facultative Intracellular Pathogen Candida glabrata Subverts Macrophage Cytokine Production and Phagolysosome Maturation. J. Immunol. 2011, 187, 3072–3086. 10.4049/jimmunol.1003730. [DOI] [PubMed] [Google Scholar]
- Rasheed M.; Battu A.; Kaur R. Aspartyl Proteases in Candida glabrata Are Required for Suppression of the Host Innate Immune Response. J. Biol. Chem. 2018, 293, 6410–6433. 10.1074/jbc.M117.813741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baarlen P.; van Belkum A.; Summerbell R. C.; Crous P. W.; Thomma B. P. H. J. Molecular Mechanisms of Pathogenicity: How Do Pathogenic Microorganisms Develop Cross-Kingdom Host Jumps?. FEMS Microbiol. Rev. 2007, 31, 239–277. 10.1111/j.1574-6976.2007.00065.x. [DOI] [PubMed] [Google Scholar]
- Rapala-Kozik M.; Bochenska O.; Zajac D.; Karkowska-Kuleta J.; Gogol M.; Zawrotniak M.; Kozik A. Extracellular Proteinases of Candida Species Pathogenic Yeasts. Mol. Oral Microbiol. 2018, 113–124. 10.1111/omi.12206. [DOI] [PubMed] [Google Scholar]
- Karkowska-Kuleta J.; Rapala-Kozik M.; Kozik A. Fungi Pathogenic to Humans: Molecular Bases of Virulence of Candida albicans, Cryptococcus neoformans and Aspergillus fumigatus. Acta Biochim. Pol. 2009, 56, 211–224. 10.18388/abp.2009_2452. [DOI] [PubMed] [Google Scholar]
- Bairwa G.; Balusu S.; Kaur R. Aspartyl Proteases in Human Pathogenic Fungi: Roles in Physiology and Virulence. Fungal Cell Wall 2013, 159–197. [Google Scholar]
- Gagnon-Arsenault I.; Tremblay J.; Bourbonnais Y. Fungal Yapsins and Cell Wall: A Unique Family of Aspartic Peptidases for a Distinctive Cellular Function. FEMS Yeast Res. 2006, 6, 966–978. 10.1111/j.1567-1364.2006.00129.x. [DOI] [PubMed] [Google Scholar]
- Bairwa G.; Kaur R. A Novel Role for a Glycosylphosphatidylinositol-Anchored Aspartyl Protease, CgYps1, in the Regulation of PH Homeostasis in Candida glabrata. Mol. Microbiol. 2011, 79, 900–913. 10.1111/j.1365-2958.2010.07496.x. [DOI] [PubMed] [Google Scholar]
- Bairwa G.; Rasheed M.; Taigwal R.; Sahoo R.; Kaur R. GPI (Glycosylphosphatidylinositol)-Linked Aspartyl Proteases Regulate Vacuole Homoeostasis in Candida glabrata. Biochem. J. 2014, 458, 323–334. 10.1042/BJ20130757. [DOI] [PubMed] [Google Scholar]
- Krysan D. J.; Ting E. L.; Abeijon C.; Kroos L.; Fuller R. S. Yapsins Are a Family of Aspartyl Proteases Required for Cell Wall Integrity in Saccharomyces cerevisiae. Eukaryotic. Cell 2005, 4, 1364–1374. 10.1128/EC.4.8.1364-1374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List E. O.; Berryman D. E.; Bower B.; Sackmann-Sala L.; Gosney E.; Ding J.; Okada S.; Kopchick J. J. The Use of Proteomics to Study Infectious Diseases. Drug Targets: Infect. Disord. 2008, 8, 31–45. 10.2174/187152608784139640. [DOI] [PubMed] [Google Scholar]
- Prasad T. S. K.; Harsha H. C.; Keerthikumar S.; Sekhar N. R.; Selvan L. D. N.; Kumar P.; Pinto S. M.; Muthusamy B.; Subbannayya Y.; Renuse S.; et al. Proteogenomic Analysis of Candida glabrata Using High Resolution Mass Spectrometry. J. Proteome Res. 2012, 11, 247–260. 10.1021/pr200827k. [DOI] [PubMed] [Google Scholar]
- Gil-Bona A.; Monteoliva L.; Gil C. Global Proteomic Profiling of the Secretome of Candida albicans Ecm33 Cell Wall Mutant Reveals the Involvement of Ecm33 in Sap2 Secretion. J. Proteome Res. 2015, 14, 4270–4281. 10.1021/acs.jproteome.5b00411. [DOI] [PubMed] [Google Scholar]
- Ranganathan S.; Garg G. Secretome: Clues into Pathogen Infection and Clinical Applications. Genome Med. 2009, 1, 113. 10.1186/gm113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgo A. G.; Heilmann C. J.; Brul S.; de Koster C. G.; Klis F. M. Beyond the Wall: Candida albicans Secret(e)s to Survive. FEMS Microbiol. Lett. 2013, 338, 10–17. 10.1111/1574-6968.12049. [DOI] [PubMed] [Google Scholar]
- Klis F. M.; Brul S. Adaptations of the Secretome of Candida albicans in Response to Host-Related Environmental Conditions. Eukaryotic Cell 2015, 14, 1165–1172. 10.1128/EC.00142-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes D. L.; Wilson D.; Richardson J. P.; Mogavero S.; Tang S. X.; Wernecke J.; Höfs S.; Gratacap R. L.; Robbins J.; Runglall M.; et al. Candidalysin Is a Fungal Peptide Toxin Critical for Mucosal Infection. Nature 2016, 532, 64–68. 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Molero E.; Dekker H. L.; de Boer A. D.; de Groot P. W. J. Identification of Secreted Candida Proteins Using Mass Spectrometry. Methods Mol. Biol. 2016, 79–94. 10.1007/978-1-4939-3052-4_7.. [DOI] [PubMed] [Google Scholar]
- Kuhn D. M.; Balkis M.; Chandra J.; Mukherjee P. K.; Ghannoum M. A. Uses and Limitations of the XTT Assay in Studies of Candida Growth and Metabolism. J. Clin. Microbiol. 2003, 41, 506–508. 10.1128/JCM.41.1.506-508.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A. R.; Peixoto F. P.; Sá-Correia I. Activation of the H+-ATPase in the Plasma Membrane of Cells of Saccharomyces cerevisiae Grown under Mild Copper Stress. Arch. Microbiol. 1998, 171, 6–12. 10.1007/s002030050671. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros J. J.; Tsirigos K. D.; Sønderby C. K.; Petersen T. N.; Winther O.; Brunak S.; von Heijne G.; Nielsen H. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O.; Brunak S.; von Heijne G.; Nielsen H. Locating Proteins in the Cell Using TargetP, SignalP and Related Tools. Nat. Protoc. 2007, 2, 953–971. 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Weig M.; et al. Systematic Identification in Silico of Covalently Bound Cell Wall Proteins and Analysis of Protein-Polysaccharide Linkages of the Human Pathogen Candida glabrata. Microbiology 2004, 150, 3129–3144. 10.1099/mic.0.27256-0. [DOI] [PubMed] [Google Scholar]
- Madinger C. L.; Sharma S. S.; Anton B. P.; Fields L. G.; Cushing M. L.; Canovas J.; Taron C. H.; Benner J. S. The Effect of Carbon Source on the Secretome of Kluyveromyces Lactis. Proteomics 2009, 9, 4744–4754. 10.1002/pmic.200800915. [DOI] [PubMed] [Google Scholar]
- Buerth C.; Heilmann C. J.; Klis F. M.; de Koster C. G.; Ernst J. F.; Tielker D. Growth-Dependent Secretome of Candida utilis. Microbiology 2011, 157, 2493–2503. 10.1099/mic.0.049320-0. [DOI] [PubMed] [Google Scholar]
- Almagro Armenteros J. J.; Sønderby C. K.; Sønderby S. K.; Nielsen H.; Winther O. DeepLoc: Prediction of Protein Subcellular Localization Using Deep Learning. Bioinformatics 2017, 33, 3387–3395. 10.1093/bioinformatics/btx431. [DOI] [PubMed] [Google Scholar]
- Skrzypek M. S.; Binkley J.; Binkley G.; Miyasato S. R.; Simison M.; Sherlock G. The Candida Genome Database (CGD): Incorporation of Assembly 22, Systematic Identifiers and Visualization of High Throughput Sequencing Data. Nucleic Acids Res. 2017, 45, D592–D596. 10.1093/nar/gkw924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis D. O.; Arnaud M. B.; Binkley J.; Shah P.; Skrzypek M. S.; Wymore F.; Binkley G.; Miyasato S. R.; Simison M.; Sherlock G. The Candida Genome Database Incorporates Multiple Candida Species: Multispecies Search and Analysis Tools with Curated Gene and Protein Information for Candida albicans and Candida glabrata. Nucleic Acids Res. 2012, 40, D667–D674. 10.1093/nar/gkr945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priebe S.; Kreisel C.; Horn F.; Guthke R.; Linde J. FungiFun2: A Comprehensive Online Resource for Systematic Analysis of Gene Lists from Fungal Species. Bioinformatics 2015, 31, 445–446. 10.1093/bioinformatics/btu627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead D. A.; Walker J.; Holcombe L.; Gibbs S. R. S.; Yin Z.; Selway L.; Butler G.; Brown A. J. P.; Haynes K. Impact of the Transcriptional Regulator, Ace2, on the Candida glabrata Secretome. Proteomics 2010, 10, 212–223. 10.1002/pmic.200800706. [DOI] [PubMed] [Google Scholar]
- Champer J.; Ito J.; Clemons K.; Stevens D.; Kalkum M. Proteomic Analysis of Pathogenic Fungi Reveals Highly Expressed Conserved Cell Wall Proteins. J. Fungi 2016, 2, 6. 10.3390/jof2010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran M.; Calcagno A.; Findon H.; Bignell E.; Jones M. D.; Warn P.; Denning D. W.; Butler G.; Mühlschlegel Fa.; Haynes K.; et al. Inactivation of Transcription Factor Gene ACE2 in the Fungal Pathogen Candida Glabrata Results in Hypervirulence. Eukaryotic Cell 2004, 3, 546–552. 10.1128/EC.3.2.546-552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S.; Vadaie N.; Chavel C. A.; Birkaya B.; Joshi J.; Grell L.; Cullen P. J. Shedding of the Mucin-like Flocculin Flo11p Reveals a New Aspect of Fungal Adhesion Regulation. Curr. Biol. 2010, 20, 1389–1395. 10.1016/j.cub.2010.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger B. L. Insight into the Antiadhesive Effect of Yeast Wall Protein 1 of Candida albicans. Eukaryotic Cell 2012, 11, 795–805. 10.1128/EC.00026-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos C. M.; de Oliveira H. C.; da Silva J.; de F.; Assato P. A.; Fusco-Almeida A. M.; Mendes-Giannini M. J. S. The Multifaceted Roles of Metabolic Enzymes in the Paracoccidioides Species Complex. Front. Microbiol. 2014, 5, 719. 10.3389/fmicb.2014.00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Park J.; Kim D.; Jung K.; Kang S.; Lee Y. H. Fungal Secretome Database: Integrated Platform for Annotation of Fungal Secretomes. BMC Genomics 2010, 11, 105. 10.1186/1471-2164-11-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum G.; Min X. J. FunSecKB: The Fungal Secretome KnowledgeBase. Database 2011, 2011, bar001. 10.1093/database/bar001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Bona A.; Llama-Palacios A.; Parra C. M.; Vivanco F.; Nombela C.; Monteoliva L.; Gil C. Proteomics Unravels Extracellular Vesicles as Carriers of Classical Cytoplasmic Proteins in Candida albicans. J. Proteome Res. 2015, 14, 142–153. 10.1021/pr5007944. [DOI] [PubMed] [Google Scholar]
- Smeekens J. M.; Xiao H.; Wu R. Global Analysis of Secreted Proteins and Glycoproteins in Saccharomyces cerevisiae. J. Proteome Res. 2017, 16, 1039–1049. 10.1021/acs.jproteome.6b00953. [DOI] [PubMed] [Google Scholar]
- Nimrichter L.; De Souza M. M.; Del Poeta M.; Nosanchuk J. D.; Joffe L.; Tavares P. D. M.; Rodrigues M. L. Extracellular Vesicle-Associated Transitory Cell Wall Components and Their Impact on the Interaction of Fungi with Host Cells. Front. Microbiol. 2016, 7, 1034. 10.3389/fmicb.2016.01034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadaie N.; Dionne H.; Akajagbor D. S.; Nickerson S. R.; Krysan D. J.; Cullen P. J. Cleavage of the Signaling Mucin Msb2 by the Aspartyl Protease Yps1 Is Required for MAPK Activation in Yeast. J. Cell Biol. 2008, 181, 1073–1081. 10.1083/jcb.200704079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri S.; Kumar R.; Chadha S.; Tati S.; Conti H. R.; Hube B.; Cullen P. J.; Edgerton M. Secreted Aspartic Protease Cleavage of Candida albicans Msb2 Activates Cek1 MAPK Signaling Affecting Biofilm Formation and Oropharyngeal Candidiasis. PLoS One 2012, 7, e46020 10.1371/journal.pone.0046020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram A. F.; Kapteyn J. C.; Montijn R. C.; Caro L. H.; Douwes J. E.; Baginsky W.; Mazur P.; van den Ende H.; Klis F. M. Loss of the Plasma Membrane-Bound Protein Gas1p in Saccharomyces cerevisiae Results in the Release of Beta1,3-Glucan into the Medium and Induces a Compensation Mechanism to Ensure Cell Wall Integrity. J. Bacteriol. 1998, 180, 1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon-Arsenault I.; Parisé L.; Tremblay J.; Bourbonnais Y. Activation Mechanism, Functional Role and Shedding of Glycosylphosphatidylinositol-Anchored Yps1p at the Saccharomyces cerevisiae Cell Surface. Mol. Microbiol. 2008, 69, 982–993. 10.1111/j.1365-2958.2008.06339.x. [DOI] [PubMed] [Google Scholar]
- Albrecht A.; Felk A.; Pichova I.; Naglik J. R.; Schaller M.; De Groot P.; MacCallum D.; Odds F. C.; Schäfer W.; Klis F.; et al. Glycosylphosphatidylinositol-Anchored Proteases of Candida albicans Target Proteins Necessary for Both Cellular Processes and Host-Pathogen Interactions. J. Biol. Chem. 2006, 281, 688–694. 10.1074/jbc.M509297200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The global and quantitative secretome mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE34 partner repository with data set identifiers PXD015131, PXD015132, and PXD015147.