

Abstract
Alcohol-related liver disease (ALD) and drug-induced liver injury (DILI) are common causes of severe liver disease, and successful treatments are lacking. Autophagy plays a protective role in both ALD and DILI by selectively removing damaged mitochondria (mitophagy), lipid droplets (lipophagy), protein aggregates and adducts in hepatocytes. Autophagy also protects against ALD by degrading interferon regulatory factor 1 (IRF1) and damaged mitochondria in hepatic macrophages. Specifically, we will discuss selective autophagy for removal of damaged mitochondria and lipid droplets in hepatocytes and autophagy-mediated degradation of IRF1 in hepatic macrophages as protective mechanisms against alcohol-induced liver injury and steatosis. In addition, selective autophagy for removal of damaged mitochondria and protein adducts for protection against DILI is discussed in this review. Development of new therapeutics for ALD and DILI is greatly needed, and selective autophagy pathways may provide promising targets. Drug and alcohol effects on autophagy regulation as well as protective mechanisms of autophagy against DILI and ALD are highlighted in this review.
Keywords: Alcohol, Autophagy, Lipophagy, Liver, Mitophagy, DILI
Graphical Abstract
1. Introduction
Autophagy has been shown to play a protective role against drug-induced liver injury (DILI) and alcohol-related liver disease (ALD). Drug and alcohol effects on autophagy regulation as well as protective mechanisms of autophagy against DILI and ALD are highlighted in this review.
1.1. Drug-induced liver injury (DILI)
DILI is the most common cause of acute liver failure in the United States with cases continuing to rise as more drugs are developed (Lee, 2013; Reuben et al., 2010; Ye et al., 2018). Many drugs cause DILI including chemotherapeutics, antipyretics, non-steroidal anti-inflammatory drugs (NSAIDs), immunosuppressive drugs, antibiotics, and antivirals among others, and over 300 drugs have been linked to liver injury (Bjornsson and Hoofnagle, 2016; Licata, 2016; Ye et al., 2018). Even though occurrence of DILI is rare, it is the most common reason a drug is withdrawn from the market or from clinical trials (Licata, 2016). Parent drugs and their metabolites can cause hepatocyte death and subsequent liver injury by inducing mitochondrial dysfunction, oxidative stress, or inflammation. DILI can be dose-dependent, such as in acetaminophen toxicity, or idiosyncratic / dose-independent (Lee, 2013; Licata, 2016). Unfortunately, the mechanisms for initiation of liver injury by drugs are largely unknown, and management of DILI in patients is challenging due to a lack of biomarkers and tests available for diagnosis. In addition, better treatment options are needed for DILI, which will likely arise from a better understanding of mechanisms involved in initiation of liver injury by various drugs (Licata, 2016; Ye et al., 2018).
1.2. Alcohol-related liver disease (ALD)
ALD is a major cause of liver injury, steatosis, and end-stage liver disease. Alcohol abuse causes liver injury by various mechanisms including oxidative and non-oxidative ethanol metabolism, production of oxidative stress, mitochondrial and lysosomal damage and dysfunction, endoplasmic reticulum stress, inflammation and cytokine release, and initiation of cell death (Gao and Bataller, 2011; Williams et al., 2014). ALD pathogenesis includes fatty liver, alcoholic hepatitis, fibrosis, cirrhosis, and eventual hepatocellular carcinoma. Alcohol metabolism and ALD pathogenesis have been thoroughly reviewed previously (Crabb et al., 2019; Gao and Bataller, 2011; Rehm et al., 2013; Seitz et al., 2018; Shen et al., 2019; Teschke, 2019; Williams et al., 2014) and will not be discussed in detail in this review. Similar to DILI, ALD also lacks good treatment options. Main treatment options for ALD patients include abstinence from alcohol use and liver transplantation. Many transplant programs have removed a previous requirement for ALD patients to be sober for 6 months before receiving liver transplantation due to positive outcomes in patients receiving early liver transplantation. However, only patients with severe disease are eligible for liver transplantation, and 17 to 22% of these patients experience alcohol relapse after transplantation (Diehl, 2002; Godfrey et al., 2019). Therefore, novel therapeutic strategies are necessary to decrease ALD and mortality.
1.3. Autophagy
Autophagy has recently been shown to have roles in both DILI and ALD, and targeting this pathway may provide future therapeutic options for both DILI and ALD patients. Autophagy is an intracellular pathway responsible for maintaining cellular homeostasis by directing cytosolic macromolecules and damaged or excess organelles to lysosomes for degradation. The autophagy pathway is generally considered to be a protective pathway, and it has been shown to play a critical role in maintaining cellular homeostasis in physiological conditions as well as in disease. Regulation of the autophagy pathway has been extensively reviewed and will only be briefly discussed here (Mizushima et al., 2008; Parzych and Klionsky, 2014; Ueno and Komatsu, 2017; Yu et al., 2018).
Initiation of the autophagy pathway involves phagophore formation at the phagophore assembly site (PAS), which recruits other autophagy-related gene (Atg) proteins necessary for autophagosome formation. Phagophore formation is regulated by the Unc-51 like kinase 1 (ULK1) complex, which is composed of ULK1, FAK family-interacting protein of 200 kDa (FIP200), and Atg13 (Ganley et al., 2009; Hosokawa et al., 2009; Jung et al., 2009; Mizushima, 2010). ULK1 and autophagosome formation is positively regulated by AMP-activated protein kinase (AMPK) and negatively regulated by the mechanistic target of rapamycin complex 1 (mTORC1). During low energy conditions, AMPK phosphorylates ULK1 at several sites to activate it, which induces autophagy. When nutrients are plentiful, mTOR phosphorylates ULK1 at S757, inhibiting autophagy (Egan et al., 2011; Kim and Guan, 2011; Kim et al., 2011). Activated ULK1 initiates autophagy via Beclin1 phosphorylation and subsequent activation of the class III phosphoinositide 3-kinase (PtdIns3K) complex to induce autophagosome formation (Russell et al., 2013). After PtdIns3K activation, ubiquitin-like conjugation system complexes Atg12-Atg5-Atg16L1 and microtubule-associated protein light chain 3 (LC3)-Atg7-Atg3 induce phagophore elongation and expansion to form the autophagosome by regulating phosphatidylethanolamine conjugation with LC3, which forms LC3-II. LC3-II is stable on the autophagosome membrane and is commonly used as a marker for autophagy activation (Ichimura et al., 2000; Kabeya et al., 2000; Ohsumi, 2001; Yoshii and Mizushima, 2017). The elongated phagophore then closes around its cargo to form a complete autophagosome. Mechanisms for autophagosome closure are not completely understood but may require the endosomal sorting complex required for transport III (ESCRT-III) subunit CHMP2A, the AAA ATPase VPS4, and VPS37A (Takahashi et al., 2018; Takahashi et al., 2019). Completed autophagosomes fuse with lysosomes to form autolysosomes for cargo degradation. Autophagosome-lysosome fusion requires the GTPase Ras-related protein 7 (Rab7), the SNARE protein syntaxin 17 (STX17), and the lysosomal membrane protein LAMP-2 (Huynh et al., 2007; Itakura et al., 2012; Jager et al., 2004). After fusion, autophagosome inner membrane LC3-II is degraded in the lysosome along with autophagosome cargo (Tanida et al., 2008).
Autophagy has been shown to be both a non-selective and selective process. Non-selective autophagy occurs during nutrient deprivation to degrade cellular contents for energy production. Selective autophagy mediates degradation of specific organelles that are damaged or in excess, such as in mitophagy and lipophagy for selective degradation of mitochondria and lipid droplets (LDs), respectively (Gatica et al., 2018). Several selective autophagy pathways have roles in ALD and / or DILI. Mitophagy and lipophagy in hepatocytes and selective autophagy for removal of IRF1 and mitochondria in hepatic macrophages are protective mechanisms in ALD, and mitophagy and selective autophagy for removal of protein adducts are protective mechanisms in DILI.
1.4. Mitophagy
Mitophagy is a form of selective autophagy specific for degradation of damaged mitochondria. Mitophagy has been shown to be protective against both DILI and alcohol-induced liver injury and steatosis. Selective removal of damaged mitochondria helps maintain a healthy population of mitochondria, which is important for maintaining cellular homeostasis. Removal of damaged mitochondria via mitophagy prevents cell death by decreasing oxidative stress. In addition, maintenance of a healthy mitochondrial population preserves mitochondrial bioenergetics and respiratory chain function. Finally, healthy mitochondria allow for β-oxidation and removal of LDs to prevent steatosis (Ding and Yin, 2012; Williams and Ding, 2015, 2018).
Regulation of the mitophagy pathway has been recently reviewed by our group and others and will not be discussed at length here (Ding and Yin, 2012; Ni et al., 2015; Palikaras et al., 2018; Rodger et al., 2018; Williams and Ding, 2015, 2018). The best characterized mitophagy pathway is the Parkin and phosphatase and tensin homolog (PTEN)-induced kinase (PINK1) pathway. Parkin is an E3 ligase that remains in the cytosol under normal conditions, but it is recruited to damaged or depolarized mitochondria for mitophagy induction (Narendra et al., 2008). PINK1 is required for Parkin recruitment to damaged mitochondria and for activation of its E3 ligase activity (Matsuda et al., 2010). In healthy mitochondria, PINK1 is cleaved by the inner mitochondrial membrane protease PARL, and the truncated form is released into the cytosol for degradation by the proteasome (Jin et al., 2010; Meissner et al., 2011; Yamano and Youle, 2013). When mitochondria are damaged or depolarized, PINK1 becomes stabilized on the mitochondrial membrane and initiates self-phosphorylation to recruit Parkin to the mitochondrial membrane (Okatsu et al., 2012). PINK1 then phosphorylates both Parkin and ubiquitin at the mitochondrial membrane to activate Parkin’s E3 ligase activity and tether it to the mitochondrial membrane. Activated Parkin adds ubiquitin chains onto mitochondrial outer membrane proteins, which are then phosphorylated by PINK1 to additionally recruit and activate Parkin using a feed-forward mechanism (Ordureau et al., 2014; Shiba-Fukushima et al., 2014). Various mitophagy receptor proteins, such as p62/SQSTM1, NDP52, Huntingtin, and optineurin (OPTN) may then be recruited to ubiquitinated mitochondria to help further recruit autophagosomes (Rui et al., 2015; Williams and Ding, 2018). These receptor proteins all have a ubiquitin-associated (UBA) domain for binding to ubiquitinated proteins and an LC3-interacting region (LIR) domain for binding to LC3 on autophagosomes to help initiate autophagosome recruitment and cargo recognition (Birgisdottir et al., 2013).
The Parkin-PINK1 mitophagy pathway is generally considered to be a linear pathway with PINK1 acting upstream of Parkin (Park et al., 2006; Yang et al., 2006). However, PINK1 was recently shown to activate mitophagy alone by ubiquitinating mitochondria and recruiting selective autophagy receptors NDP52 and OPTN (Lazarou et al., 2015). There are also several other Parkin-independent pathways for induction of mitophagy including induction by E3 ligases March5 or Mul1 or mediation of mitophagy by Fun 14 domain containing 1 (Fundc1), Nix, or cardiolipin (Rogov et al., 2014; Williams and Ding, 2018).
1.5. Lipophagy
Lipophagy is a form of selective autophagy specific for degradation of intracellular LDs (Singh et al., 2009) that occurs during nutrient deprivation to provide energy or during cellular stress to remove excess LDs (Kounakis et al., 2019; Schulze et al., 2017b). Lipophagy may require cytosolic triglyceride lipase (ATGL) to initiate lipolysis of large LDs into small LDs to subsequently facilitate their removal by autophagosomes (Sathyanarayan et al., 2017; Schott et al., 2019). In addition, lysosomes can directly enwrap LDs for degradation by micro-lipophagy. After autophagosome or lysosome sequestration, LDs are broken down by lysosomal lipases into fatty acids (Li et al., 2017; Singh et al., 2009)
Exact mechanisms for induction of the lipophagy pathway are still unknown, but several processes important for lipophagy have been discovered. Similar to mitophagy, lipophagy likely involves selective autophagy receptors such as p62, OPTN, Huntingtin, or NDP52 for autophagosome recognition of LDs. The exact receptor necessary has not been identified, but Huntingtin and p62 may act together to initiate autophagosome recruitment to LDs (Kounakis et al., 2019; Rui et al., 2015; Schulze et al., 2017b).
The GTPases Rab7 and Rab10 may have important roles in lipophagy regulation. Rab7 is a regulator of late endocytic and autophagosome trafficking and is associated with the LD membrane (Gutierrez et al., 2004; Jager et al., 2004; Vitelli et al., 1997). Rab7 was shown to possibly recruit autophagosomes and lysosomes to LDs (Schroeder et al., 2015). Rab7 on the LD surface has been shown to co-localize with autophagosomes and lysosomes (Lizaso et al., 2013), and starvation promoted Rab7 activation on the LD surface, which recruited lysosomes to LDs. Rab7 deletion also increased numbers of LDs in hepatoma cells (Schroeder et al., 2015). Schroeder et al. described a “kiss and run” mechanism for Rab7-mediated lipophagy where lysosomes are recruited to LDs, docked for a short period of time, and then depart, suggesting that lysosomes may clear away portions of a large LD at a time (Carmona-Gutierrez et al., 2015; Schroeder et al., 2015).
In addition to Rab7, Rab10 is also on the LD surface and may be involved in lipophagy in hepatocytes. Similar to Rab7, deletion of Rab10 increased numbers of LDs in hepatocytes, and Rab10 on LDs was shown to co-localize with autophagosomes and lysosomes. In addition, starved Huh-7 cells treated with the mTOR inhibitor Torin1 to activate autophagy had increased numbers of Rab10-labeled LDs, further suggesting a role for Rab10 in lipophagy regulation (Li et al., 2016).
Transcription Factor EB (TFEB) also likely plays a role in mediating lipophagy because mice with acute TFEB knock-down had greater steatosis after alcohol treatment compared to control mice (Chao et al., 2018c). TFEB is the master regulator for transcription of genes necessary for lysosome biogenesis and autophagy (Settembre et al., 2011). In addition, Dynamin2 (Dyn2), which is responsible for recycling lysosomes from autolysosomes to maintain autophagy (Schulze et al., 2013) may play a role in lipophagy because inhibition of Dyn2 by alcohol decreased lipophagy in rats, resulting in increased liver steatosis (Li and Ding, 2017). The roles of TFEB and Dyn2 in lipophagy are further discussed in the alcohol section of this review. Overall, the mechanisms regulating lipophagy are becoming more understood, but there is still a lot unknown about regulation of this pathway.
2. Role of Autophagy in Alcohol-induced liver injury and steatosis
Alcohol has been shown to both activate and inhibit autophagy, depending on the alcohol model used. Autophagy was activated in primary mouse hepatocytes and in mice after acute alcohol binge. Alcohol increased LC3-II levels and autophagosome co-localization with lysosomes in mice and primary hepatocytes. LC3-II levels were further increased by alcohol after pre-treatment with chloroquine (CQ) to block lysosome degradation, indicating autophagy flux and activation (Ding et al., 2010; Lin et al., 2013; Ni et al., 2013a; Thomes et al., 2015). Alcohol-induced activation of autophagy in the alcohol binge model was found to be dependent on ethanol metabolism, production of reactive oxygen species (ROS), and inhibition of mTOR (Ding et al., 2010; Lu and Cederbaum, 2015; Scherz-Shouval et al., 2007).
Chronic alcohol feeding was shown to both activate and inhibit autophagy. Two independent studies reported that chronic alcohol feeding activates autophagy. Lin et al. showed increased autophagic flux in mouse livers after chronic alcohol feeding (Lin et al., 2013), and Eid et al. showed increased autophagosome numbers in rat livers after chronic alcohol feeding (Eid et al., 2013). Two independent groups also showed alcohol-induced inhibition of autophagy. Menk et al. showed that chronic alcohol feeding in rats had no effect on LC3-II levels and that p62 levels increased after alcohol feeding, suggesting inhibition of autophagy (Menk et al., 2018). Thomes et al. demonstrated that alcohol feeding in mice increased autophagosome numbers, similar to mice treated with alcohol binge. However, alcohol-fed mice had increased p62 protein expression along with decreased lysosome numbers and decreased levels of autophagosome-lysosome fusion, suggesting autophagy inhibition (Thomes et al., 2015). Autophagy inhibition in this case was due to a lack of available lysosomes for autophagosomes to fuse with to degrade their cargo; this form of autophagy inhibition was recently defined as “insufficient autophagy” (Chao et al., 2018a). Mice treated with the chronic alcohol feeding plus acute binge (Gao-binge) alcohol model also had insufficient autophagy, similar to chronic alcohol-fed mice (Chao et al., 2018c). Even though chronic alcohol feeding has been shown to both activate and inhibit autophagy, chronic alcohol feeding likely inhibits autophagy because it has been reported to cause hepatic protein accumulation and hepatomegaly (Baraona et al., 1975). Alcohol-induced hepatomegaly is likely associated with autophagy inhibition or decreased lysosome function. In support of this, chronic alcohol feeding decreased cathepsin activity in lysosomes (Kharbanda et al., 1995) and reduced lysosomal biogenesis mediated by TFEB (Chao et al., 2018c; Thomes et al., 2015).
Interestingly, TFEB is differentially regulated by alcohol binge and chronic alcohol feeding, which may provide one answer to why these alcohol models have contrasting effects on autophagy activation. Alcohol binge was associated with increased liver autophagosome numbers, lysosome numbers, and TFEB nuclear content and subsequent autophagy activation. Chronic alcohol feeding was also associated with increased autophagosome numbers, but lysosome numbers and TFEB nuclear content were decreased in chronic alcohol fed mice compared to control fed mice, leading to insufficient autophagy and subsequent autophagy inhibition (Thomes et al., 2019; Thomes et al., 2015). Gao-binge alcohol treated mice also had decreased TFEB nuclear content in addition to decreased TFEB expression levels and transcriptional activity (Chao et al., 2018c).
Regulation of alcohol-induced changes in TFEB localization are likely through post-translational modifications of ERK 1/2 and / or mTORC1, which both regulate TFEB (Chao et al., 2018c; Li and Ding, 2016; Thomes et al., 2015). Phosphorylated (inactive) TFEB remains in the cytosol while unphosphorylated (active) TFEB is able to travel to the nucleus to induce gene transcription (Settembre et al., 2012). A recent idea for alcohol-mediated TFEB regulation suggests involvement of the proteasome. Alcohol is known to suppress proteasome activity (Donohue et al., 2007), and the phosphorylated form of TFEB is ubiquitinated by STIP1 homology U-Box containing protein (STUB1) for subsequent degradation by the proteasome (Sha et al., 2017). It is possible that chronic alcohol feeding indirectly inhibits autophagy via suppression of proteasome activity, which may cause an accumulation of phosphorylated TFEB in the cytosol that would prevent entry of unphosphorylated TFEB into the nucleus (Yang et al., 2019).
Even though TFEB regulation varies with alcohol models used in mice, TFEB plays an important protective role in ALD. Mice with acute TFEB knock-down had significantly greater liver injury and steatosis after Gao-binge alcohol treatment compared to control mice, and mice with overexpressed TFEB were protected against Gao-binge alcohol induced liver injury and steatosis (Chao et al., 2018c). Intriguingly, TFEB nuclear content was restored in alcohol-fed rats after withdrawal from alcohol for 7 days, and liver injury and steatosis were reduced (Thomes et al., 2019; Yang et al., 2019). More importantly, nuclear TFEB accumulation was decreased in hepatocytes from human alcoholic hepatitis patients, confirming that TFEB plays an important role in human ALD pathogenesis (Chao et al., 2018c).
It is not clear why various alcohol models regulate autophagy differently. It should be noted that autophagy flux analysis was not always implemented when evaluating alcohol-mediated regulation of autophagy. Due to the dynamic nature of autophagy, measurement of autophagic flux is important for determining autophagy activation or suppression (Klionsky et al., 2012). Future alcohol studies should include autophagy flux assays to determine autophagy activation. In addition, autophagosome-lysosome fusion analysis should be performed to confirm that alcohol-induced autophagy flux produces sufficient autophagy.
Even though autophagy may be regulated differently by chronic and acute alcohol exposure, activation of autophagy is protective against alcohol-induced liver injury and steatosis. Pharmacological activation of autophagy by rapamycin alleviated alcohol-induced steatosis and liver injury in both alcohol binge and chronic alcohol feeding models (Ding et al., 2010; Lin et al., 2013; Lu and Cederbaum, 2015) while pharmacological inhibition of autophagy by 3-methyladeline (3-MA) and CQ exacerbated alcohol-induced hepatocellular apoptosis, steatosis, and liver injury in mice using both the alcohol binge and chronic alcohol feeding models (Ding et al., 2010; Lin et al., 2013; Lu and Cederbaum, 2015). In addition, augmenter of liver regeneration (ALR) was shown to protect against alcohol-induced liver injury by activating autophagy (Liu et al., 2019). ALR is a hepatic growth factor that plays an important role in liver regeneration (Gupta and Venugopal, 2018). Mice overexpressing ALR had decreased liver injury, and ALR knockdown mice had increased liver injury compared to WT mice after alcohol treatment. ALR-mediated protection against alcohol-induced liver injury correlated with autophagy activation (Liu et al., 2019). Therefore, development of therapeutics for autophagy activation may prove useful for treatment of ALD. In addition, development of therapeutics targeted at TFEB regulation may be beneficial for ALD patients.
Interestingly, alcohol-induced autophagy activation was shown to be selective for removing damaged mitochondria (mitophagy) and LDs (lipophagy) in the liver. Alcohol binge in mice did not cause long-lived protein degradation, which is characteristic of non-selective autophagy, but instead enhanced selective degradation of fragmented mitochondria and LDs (Ding et al., 2010; Lin et al., 2013). Alcohol binge also increased degradation of the selective autophagy receptor p62 (Ding et al., 2010; Ding et al., 2011). In addition, chronic alcohol feeding increased autophagic sequestration of mitochondria and LDs in rats (Eid et al., 2013). Therefore, the selective mitophagy and lipophagy pathways are important for protection against ALD pathogenesis and will be further discussed below.
2.1. Mitophagy in ALD
Mitophagy has been shown to be protective against alcohol-induced liver injury and steatosis by removing damaged mitochondria (Ding et al., 2010; Ding et al., 2011). Mechanisms involved in activation of mitophagy after alcohol treatment are not fully understood, but they may involve the PINK1- Parkin signaling pathway. Acute alcohol binge in rats caused Parkin and PINK1 co-localization to mitochondria. Alcohol-treated rats also had increased LC3 puncta formation compared to control rats, and Parkin co-localized with LC3 and mitochondrial markers after alcohol treatment (Eid et al., 2016). In addition, Parkin knockout (KO) mice had increased liver injury and mitochondrial damage and dysfunction compared to Wild-type (WT) mice after treatment with alcohol using both the acute-binge and Gao-binge models. Parkin also translocated to mitochondria after Gao-binge alcohol treatment in mice, suggesting that Parkin-induced mitophagy is activated by Gao-binge alcohol treatment (Williams et al., 2015a). Furthermore, numbers of autophagosomes containing mitochondria were decreased in Parkin KO mice compared to WT mice after alcohol treatment in both acute-binge and Gao-binge alcohol models, suggesting a defect in mitophagy induction in Parkin KO mice. Defective mitophagy in Parkin KO mice likely contributed to their increased liver injury after alcohol treatment (Williams et al., 2015a). Further investigation into alcohol-induced regulation of the mitophagy pathway is needed.
2.2. Lipophagy in ALD
Lipophagy occurs as a protective mechanism to reduce alcohol-mediated steatosis in the liver (Ding et al., 2010; Ding et al., 2011; Eid et al., 2013; Lin et al., 2013). Mechanisms for lipophagy regulation by alcohol are still not completely understood, but they likely involve Rab7 and Dyn2 GTPases. Alcohol was recently shown to inhibit Rab7 activity, which resulted in impaired recruitment of autophagosomes to LDs (Li and Ding, 2017; Schulze et al., 2017a). Hepatocytes from rats fed an alcohol diet for 6 weeks were resistant to starvation-induced lipophagy, likely through alcohol-mediated inhibition of Rab7 activity. Cultured primary hepatocytes from rats treated with alcohol had 80% decreased Rab7 activity, and pharmacological inhibition of Rab7 produced similar resistance to starvation-induced lipophagy as alcohol treatment (Schulze et al., 2017a). The mechanism by which alcohol inhibits Rab7 activity requires further investigation.
Alcohol was also recently found to inhibit activation of Dyn2 (Li and Ding, 2017; Rasineni et al., 2017), which is responsible for recycling lysosomes from autolysosomes to maintain autophagy (Schulze et al., 2013). Inhibition of Dyn2 activity led to decreased lipophagy and subsequent liver steatosis in alcohol-fed rats. Rats were fed alcohol for 6 to 8 weeks, and their isolated hepatocytes were subjected to starvation to activate lipophagy. Triglyceride loss in hepatocytes from alcohol-fed animals was slower during fasting compared to control-diet hepatocytes. Lipophagy was activated in control and alcohol-fed rat livers during fasting because LC3 and p62 levels were increased in liver and LD fractions, and lysosomes colocalized with autophagosomes and LDs. However, lysosome numbers were decreased by 40% in alcohol-fed rats resulting in an accumulation of autophagosomes and insufficient autophagy. The reduction in lysosome availability was at least partly due to an alcohol-mediated inhibition of Dyn2 activity (Rasineni et al., 2017). The mechanism for how alcohol regulates Dyn2 activity is unknown.
TFEB also likely has a role in lipophagy regulation by alcohol because TFEB-induced lysosome biogenesis was decreased by alcohol, and mice with TFEB overexpression were protected against alcohol-induced fatty liver (Chao et al., 2018c; Thomes et al., 2015). TFEB-mediated lysosomal biogenesis can either promote macro-lipophagy (involving the formation of autophagosomes and fusion of autophagosomes with lysosomes) or micro-lipophagy (direct engulfment of LDs by lysosomes) for the removal of LDs to protect against alcohol-induced steatosis. It would be interesting to determine the role of TFEB in these two types of lipophagy using alcohol models in the future. In addition, the effect of alcohol on Rab7 and Dyn2 in vivo should be investigated.
2.3. Hepatic Macrophage Autophagy in ALD
Hepatic macrophages are the resident macrophages in the liver, and hepatic macrophage autophagy has recently been shown to have an important role in alleviation of alcohol-induced liver inflammation, injury, and steatosis. Chronic alcohol feeding was shown to inhibit liver macrophage autophagy because mice fed alcohol for 6 weeks had decreased autophagic flux in liver macrophages compared to macrophages from mice fed control diet (Liang et al., 2019). However, macrophage autophagy is important for preventing alcohol-induced liver injury and steatosis because myeloid-specific Atg7 KO mice co-treated with chronic alcohol feeding and LPS had increased liver injury, steatosis, and cell death compared to WT mice (Liang et al., 2019). Mechanistically, macrophage autophagy prevented accumulation of damaged mitochondria and ROS production, resulting in decreased activation of the NLRP3 inflammasome and inhibition of liver injury and steatosis (Liang et al., 2019). Additionally, macrophage autophagy promoted p62-mediated degradation of IRF1, which inhibited transcription of CCL5 and CXCL10, two chemokines known for promoting liver inflammation (Liang et al., 2019). Interestingly, myeloid-specific Atg5 KO mice had similar levels of liver injury, inflammation, and steatosis as WT mice after Gao-binge alcohol treatment (Denaes et al., 2016). It is unknown why myeloid-specific Atg5 KO and Atg7 KO mice behave differently after alcohol treatment, but it may also be due to variations in alcohol models used for these mice.
The Cannabinoid receptor 2 (CB2) has recently been shown to have a protective role in ALD by activating autophagy of hepatic macrophages. Cannabinoid receptors are G-protein coupled receptors that have been shown to play a protective role against liver injury. They are mostly expressed in immune cells (Mallat et al., 2013). CB2 in hepatic macrophages was shown to alleviate alcohol-induced inflammation and steatosis by preventing macrophage activation via induction of macrophage autophagy in a heme-oxygenase-1 (HO-1) dependent pathway (Denaes et al., 2016). Myeloid-specific CB2 KO mice treated with Gao-binge alcohol had increased steatosis, pro-inflammatory gene expression, and neutrophil recruitment compared to WT mice. In addition, Gao-binge treated mice given a CB2 agonist had decreased pro-inflammatory gene expression and steatosis compared to Gao-binge treated mice not given a CB2 agonist. Protection against alcohol-induced steatosis and inflammation provided by the CB2 agonist was lost in Atg5 myeloid-specific KO mice, emphasizing the need for macrophage autophagy in CB2-mediated protection from alcohol (Denaes et al., 2016). Further investigation into the effects of CB2 receptor agonists on alcohol-induced liver injury and steatosis may be beneficial for future therapeutic development for ALD. In addition, it would be interesting to further investigate whether the combination of CB2 agonists with other known autophagy inducers such as mTOR inhibitors in hepatic macrophage autophagy prevents alcohol-induced liver injury, inflammation, and steatosis.
3. Autophagy in DILI
3.1. Acetaminophen (APAP)
APAP overdose is responsible for most cases of acute liver failure in the United States (Craig et al., 2010; Larson et al., 2005). APAP metabolism has been extensively reviewed and will not be discussed in detail here (McGill and Jaeschke, 2013; Ramachandran and Jaeschke, 2018). Briefly, APAP is metabolized by cytochrome p450s, mainly by Cyp2e1, to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI). After therapeutic doses of APAP, NAPQI is bound and detoxified by glutathione (GSH). However, APAP overdose leads to depletion of GSH, which allows for NAPQI binding to proteins and subsequent mitochondrial dysfunction, hepatocyte necrosis, liver injury and possible liver failure (McGill and Jaeschke, 2013; Ramachandran and Jaeschke, 2018). The only current therapeutic option for APAP overdose is N-acetylcysteine (NAC), which is a prodrug for GSH synthesis. The downfall to NAC treatmentis that it must be given early after overdose to replenish GSH supply before NAPQI has the opportunity to bind to proteins. If NAC is not administered early enough, the only other therapeutic option for APAP-induced acute liver failure is liver transplantation (Corcoran et al., 1985; Craig et al., 2010). Since APAP is one of the most widely-used antipyretic drugs in the United States (Herndon and Dankenbring, 2014), a better understanding of mechanisms leading to APAP-induced liver injury and development of therapeutic options for preventing APAP-induced acute liver failure are needed.
The role of autophagy in APAP-induced liver injury was recently reviewed in detail by our group (Chao et al., 2018b) and will be briefly summarized here. Autophagy has been shown to be both activated and inactivated by APAP. Autophagy is activated by APAP in mice, as demonstrated by increased autophagic flux in GFP-LC3 mice treated with APAP (Ni et al., 2012). The mechanism for APAP-induced activation of autophagy in hepatocytes is still uncertain but may be via inhibition of mTORC1 at early time points (Ni et al., 2012). Adiponectin was also shown to protect against APAP-induced liver injury by activating autophagy via AMPK and ULK1 (Lin et al., 2014). Interestingly, APAP was also recently shown to inhibit autophagy. Kang et al. showed impaired autophagic flux after APAP treatment in mice (Kang et al., 2019). APAP also inhibited autophagy in a model of non-alcoholic fatty liver disease (NAFLD), which led to aggravated disease in mice (Shi et al., 2019). Reasons for discrepancies in APAP-induced activation or inhibition of autophagy are unknown but may involve different mouse models used and / or differences in time points of autophagic flux assessment.
Regardless if autophagy is activated or inhibited by APAP, activation of autophagy protects against APAP-induced liver injury. Inhibition of autophagy by CQ or 3-MA both increased APAP-mediated necrosis in mouse hepatocytes while co-treatment and post-treatment with the autophagy activator rapamycin decreased APAP-mediated hepatocyte necrosis. Furthermore, mice co-treated with APAP and CQ had increased liver injury compared to APAP treated mice, and co-treatment with APAP and rapamycin inhibited APAP-induced liver injury (Ni et al., 2012). In addition, treatment with various drugs in addition to rapamycin have been shown to protect against APAP-induced hepatotoxicity via autophagy activation. Similar to alcohol-fed mice, ALR treatment protected mice from APAP-induced liver injury by activating autophagy (Hu et al., 2019). Treatment with the resveratrol derivative pterostilbene (Kang et al., 2019) and with IL-22 (Mo et al., 2018) both protected against APAP-induced liver injury by activating autophagy. Therefore, autophagy is overall protective against APAP-mediated liver injury, likely via selective removal of APAP adducts and damaged mitochondria, as discussed below.
Formation of APAP-adducts, particularly mitochondrial protein adducts, during APAP overdose is thought to contribute to liver injury during APAP overdose (McGill et al., 2012; Qiu et al., 1998; Ramachandran and Jaeschke, 2018; Xie et al., 2015), and autophagy was suggested to remove and degrade APAP adducts as a protective mechanism (Ni et al., 2016). APAP-adducts co-localized with autophagosomes and lysosomes in APAP-treated primary mouse hepatocytes, and autophagosomes and autolysosomes isolated from APAP-treated mouse livers contained APAP-adducts. Torin-1 induced activation of autophagy decreased blood levels of APAP-adducts in mice treated with APAP, which correlated with decreased liver injury and hepatocellular necrosis. In addition, autophagy inhibition by leupeptin and CQ both increased APAP-adduct levels, liver injury, and hepatocellular necrosis in APAP-treated mice (Ni et al., 2016). The specific mechanisms for selective removal of adducts by autophagy require further investigation, but p62 likely plays a role in targeting APAP adducts for recognition by the autophagosome. p62 co-localized with APAP-adducts in primary mouse hepatocytes treated with APAP. In addition, p62 depleted hepatocytes had reduced clearance of APAP-adducts and increased necrosis compared to p62-expressing hepatocytes after APAP treatment (Ni et al., 2016).
Autophagy has also been shown to be protective against APAP-induced liver injury via selective removal of damaged mitochondria by mitophagy. APAP-induced mitochondrial dysfunction and oxidative stress play a key role in mediating APAP-induced liver injury (Jaeschke et al., 2012). Therefore, timely removal of damaged mitochondria by mitophagy is an important protective mechanism against APAP-induced liver injury. APAP-induced autophagy in primary mouse hepatocytes caused sequestration of mitochondria in autophagosomes and degradation of mitochondrial proteins, suggesting activation of mitophagy by APAP overdose (Ni et al., 2012; Ni et al., 2013b; Wang et al., 2019; Williams et al., 2015b).
The mechanism for APAP-induced activation of mitophagy is not completely understood but may involve Parkin and PINK1. Parkin translocated to mitochondria after APAP treatment, which correlated with increased ubiquitination of mitochondrial proteins (Wang et al., 2019; Williams et al., 2015b). Parkin translocation to mitochondria after APAP treatment was dependent on PINK1 (Wang et al., 2019). Mice with acute knockdown of Parkin had increased liver injury after APAP treatment compared to control mice (Williams et al., 2015b). Parkin and PINK1 double KO mice also had increased liver injury and mortality after APAP treatment compared to WT mice (Wang et al., 2019), suggesting an importance for the Parkin / PINK1 mitophagy pathway in alleviating APAP-induced liver injury. However, Parkin KO mice were surprisingly protected against APAP-mediated liver injury (Wang et al., 2019; Williams et al., 2015b), and PINK1 KO mice had decreased liver injury after APAP treatment compared to WT mice (Wang et al., 2019). Mitophagy was significantly decreased in Parkin and PINK1 double KO mice after APAP treatment, but Parkin KO and PINK1 KO mice still had active mitophagy (Wang et al., 2019; Williams et al., 2015b). These data suggest that PINK1 and Parkin may compensate for each other to activate mitophagy. In addition, other mediators of mitophagy may be activated in the absence of Parkin and / or PINK1 in response to APAP overdose.
Parkin KO mice also had other mechanisms unrelated to mitophagy that protected them from APAP-induced liver injury, such as increased hepatocyte proliferation and decreased JNK activation after APAP treatment compared to WT mice (Williams et al., 2015b). JNK activation is well known to exacerbate APAP-induced liver injury (Gunawan et al., 2006; Hanawa et al., 2008), and increased hepatocyte proliferation likely helped Parkin KO mice recover faster from APAP-induced hepatocyte necrosis than WT mice. Interestingly, JNK activation was increased in PINK1 KO mice compared to WT mice after APAP treatment (Wang et al., 2019). Proliferation wasn’t investigated in PINK1 KO mice after APAP treatment, but PINK1 also has a role in mediating proliferation (O’Flanagan and O’Neill, 2014).
Mitochondrial spheroids may also have a protective role against APAP-induced liver injury (Ding et al., 2012). Mitochondrial spheroids are mitochondria with cup-like morphology with their lumen connected to the cytosol by a small opening. Mitochondrial spheroids can enwrap cellular contents, such as lipid droplets or other mitochondria. Mitochondrial spheroids co-localize with lysosomes, but their ability to degrade their contents requires further investigation (Ni et al., 2013b). Together, it is clear that timely removal of damaged mitochondria either via PINK1-Parkin dependent or independent mitophagy plays a critical role in protection against APAP-induced liver injury. Future studies are needed to determine whether removal of damaged mitochondria by mitophagy would also be beneficial for hepatocyte proliferation and liver regeneration.
3.2. Efavirenz
Efavirenz (EFV) is a commonly used non-nucleoside reverse transcriptase inhibitor for AIDS treatment. EFV is generally considered to be a safe drug, but it has been linked to mitochondrial depolarization and damage, oxidative stress production, hepatocyte cell death, and liver injury (Apostolova et al., 2011b; Blas-Garcia et al., 2014; Bumpus, 2011; Chen et al., 2014). EFV induces mitophagy in human hepatocytes, and EFV-induced mitophagy is assumed to be a protective mechanism because pharmacological inhibition of autophagy increased EFV-induced hepatocellular apoptosis, which was likely due to a build-up of damaged mitochondria (Apostolova et al., 2011a, b).
The exact mechanism for EFV-induced liver injury is not completely understood, but it may involve cell death caused by autophagic stress. Autophagic stress occurs when the autophagic degradation capacity for damaged mitochondria by mitophagy is exceeded, resulting in a build-up of damaged and dysfunctional mitochondria and subsequent cell death. EFV was shown to induce mitophagy when human hepatocytes were given a clinically-relevant dose of 25 μM. However, when human hepatocytes were given a 50 μM dose of EFV, autophagic flux was blocked and cell death increased, likely due to levels of damaged mitochondria exceeding the threshold for successful degradation by mitophagy (Apostolova et al., 2011a, b).
Even though clinically relevant doses of EFV are considered safe and induce mitophagy, AIDS patients are frequently treated with multiple drugs for co-infections, and other drugs used for treatment may also lead to mitochondrial damage and hepatocyte death. Therefore, use of EFV in combination with other hepatocyte-harming drugs may produce results similar to the 50 μM dose of EFV, leading to autophagic stress, cell death and subsequent liver injury (Apostolova et al., 2011a).
In addition to mitophagy induction by EFV, the autophagy adaptor protein p62 was shown to play a protective role against EFV-induced oxidative stress and inflammation independent of the autophagy pathway. Alegre et al. recently reported that EFV treatment in Hep3B cells upregulated transcription of the autophagy adaptor protein p62, and increased p62 levels were independent of autophagy regulation. Authors concluded that increased p62 transcription was induced by EFV-mediated mitochondrial damage. In addition, they showed that EFV treatment in Hep3B cells activated the NLRP3 inflammasome, which was exacerbated by p62 silencing. Interestingly, p62 silencing increased EFV-induced ROS production but had no effect on cell death. However, pharmacological inhibition of autophagy alone and in combination with p62 silencing in Hep3B cells increased EFV-induced cell death, confirming a protective role for autophagy against cell death during EFV treatment (Alegre et al., 2018). It would be valuable to study the effects of EFV treatment on mitophagy and p62 regulation in vivo in the future.
3.3. Diclofenac
Diclofenac is a commonly used NSAID for treatment of pain and swelling in rheumatic diseases (Derry et al., 2012). The cause of diclofenac-induced liver injury is not completely understood, but it likely involves drug-induced mitochondrial damage and dysfunction (Gomez-Lechon et al., 2003; Helfgott et al., 1990; Masubuchi et al., 2002; Ramachandran et al., 2018).
Autophagy, particularly mitophagy, was recently shown to have a potential role in protection against diclofenac-induced hepatotoxicity. Kang et al. showed that diclofenac treatment of hepatocytes induced mitophagy, and that co-incubation of hepatocytes with AMPK activator 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) for autophagy induction inhibited mitochondrial depolarization and hepatocyte death. However, determination of autophagy and mitophagy activation was dependent on LC3-II and Parkin levels alone without autophagy flux analysis. Therefore, it is unclear if protection by AICAR was via autophagy induction or by another mechanism. Treatment of hepatocytes with rapamycin did not inhibit diclofenac-induced mitochondrial depolarization or cell death, suggesting that mTOR inhibition-induced autophagy may not be a protective mechanism against diclofenac-induced cell death (Kang et al., 2019). However, autophagy stress may also play a role in diclofenac-mediated liver injury as in EFV discussed above. AICAR was likely protective via another autophagy-independent mechanism, such as by promoting mitochondrial fusion (Kang et al., 2016). Due to diclofenac causing mitochondrial damage and dysfunction, the role of mitophagy in protection against diclofenac-induced hepatotoxicity should still be further investigated.
3.4. Cisplatin
Cisplatin is a widely used chemotherapeutic. Although rare, cisplatin can cause hepatocellular death and subsequent hepatotoxicity. Hepatotoxicity of cisplatin is dose-dependent and may lead to decreasing dosage used or halting treatment in cancer patients. The mechanism for cisplatin-mediated liver injury is unknown (Qi et al., 2019). Cisplatin may mediate liver injury by inhibiting autophagy. Qu et al. showed that cisplatin inhibited autophagy because rats treated with cisplatin had decreased LC3-II protein expression and increased p62 protein expression in their livers. Cisplatin treatment in rats also led to upregulation of the NLRP3 inflammasome, release of pro-inflammatory cytokines and subsequent liver injury. NLRP3 activation was suggested to be due to suppression of autophagy (Qu et al., 2018).
In contrast to Qu et al., Chen’s group showed that cisplatin induces autophagy in HepG2 cells (Li et al., 2019; Liu et al., 2018). Cisplatin increased protein and puncta levels of LC3-II and decreased protein levels of p62. Cisplatin also increased protein levels of phosphorylated AMPK and decreased phosphorylated levels of mTOR in HepG2 cells, suggesting autophagy activation via mTOR inhibition (Li et al., 2019; Liu et al., 2018). Interestingly, cisplatin slightly decreased viability in human hepatocytes, but it did not affect autophagy (Li et al., 2019).
It is not clear why these studies show differences in autophagy regulation by cisplatin. It may be due to differences in models used. For example, Qu et al. used an in vivo rat model while Chen’s group used HepG2 cells and hepatocytes. It could also be due to different cells used. Chen’s group used both a cancer cell line and normal hepatocytes, which may behave differently in regards to autophagy regulation by cisplatin. It is important to note that autophagy flux analysis with a lysosome inhibitor was not performed for any of the above studies, leaving interpretation of cisplatin-induced autophagy regulation open for debate (Li et al., 2019; Liu et al., 2018; Qu et al., 2018).
4. Concluding Remarks
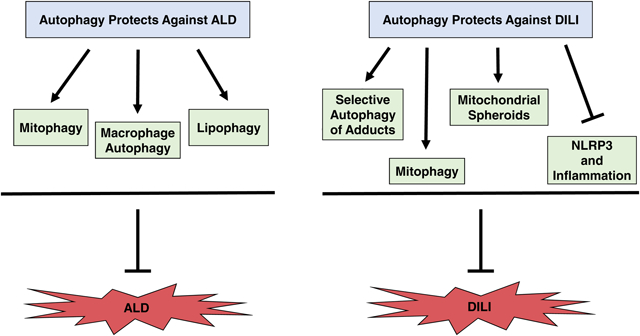
Current research progress has significantly increased our understanding of molecular mechanisms involved in autophagy-mediated protection against DILI and ALD, but many questions still remain. For example, different models of ALD and DILI produce contrasting effects on autophagy activation status, which may be clarified by assessing autophagy flux and performing autophagosome-lysosome fusion analysis to ensure autophagy sufficiency in future studies. In addition, assessment of autophagy flux at multiple time points rather than one snap-shot time point may provide a better understanding of autophagy regulation by alcohol and DILI-causing drugs. Moreover, future DILI in vivo studies would help elucidate mechanisms involved in induction of liver injury by various drugs. Nevertheless, current evidence generally supports autophagy as a protective mechanism against DILI and ALD, particularly via upregulation of selective autophagy pathways. The role of autophagy in ALD and DILI has been summarized in Figure 1. Further investigation into mechanisms involved in autophagy regulation by DILI-causing drugs and alcohol may provide better therapeutic options for ALD and DILI patients.
Figure 1. Autophagy protects against ALD and DILI.


A) Autophagy protects against ALD by selectively removing damaged mitochondria, excess LDs, and hepatic macrophages. Alcohol was shown to both induce and inhibit autophagy depending on the alcohol model used. Mitophagy induction during excessive alcohol use degrades damaged mitochondria, which prevents ROS production and subsequent cell death and also inhibits steatosis by maintaining a healthy population of mitochondria capable of β-oxidation. Alcohol-induced mitophagy induction is not completely understood but may rely on the PINK1-Parkin mitophagy pathway. Parkin-independent mechanisms for alcohol-induced mitophagy activation may also exist. Alcohol-induced lipophagy prevents liver steatosis by selectively degrading lipid droplets. Mechanisms involved in alcohol-induced lipophagy are becoming clearer and may involve TFEB, Dyn2, and / or Rab7. In addition, lysosomes may directly enwrap LDs via micro-lipophagy. Macrophage autophagy prevents accumulation of damaged mitochondria and ROS, which decreases NLRP3 inflammasome activation and subsequent liver injury. Macrophage autophagy also promotes p62-mediated degradation of IRF1, which inhibits transcription of chemokines CCL5 and CXL10, two important initiators of inflammation. Mechanisms for hepatic macrophage autophagy are not completely understood, but CB2 may have a role in its induction. Overall, selective autophagy pathways are important for protection against ALD. B) Autophagy protects against DILI initiated by APAP, EFV, Diclofenac, and Cisplatin. APAP was shown to both induce and inhibit autophagy. Discrepancies regarding the effect of APAP on autophagy activation may be due to differences in models used or in time points of autophagy flux assessment. Autophagy protects against APAP-induced liver injury by degrading damaged mitochondria by mitophagy and selectively removing and degrading APAP-adducts. APAP-induced mitophagy likely involves the PINK1-Parkin pathway, but Parkin-independent mechanisms for mitophagy induction may also exist. The mechanism for selective autophagy of APAP adducts is not completely understood, but the adaptor protein p62 may play a role in recognition of APAP adducts by autophagosomes. Mitochondrial spheroids may also protect against APAP-induced liver injury, which requires further investigation. EFV and diclofenac induce mitophagy, which protects against DILI by removing damaged mitochondria. The mechanisms for mitophagy induction by EFV and diclofenac have not been investigated. Cisplatin was shown to both induce and inhibit autophagy. Discrepancies in the effect of cisplatin on autophagy activation status may be due to differences in models or cells used. Inhibition of autophagy by cisplatin causes NLRP3 activation and subsequent liver injury. Autophagy activation during cisplatin use may protect against liver injury by inhibiting activation of NLRP3. Overall, autophagy protects against DILI by selective removal of damaged mitochondria and protein adducts and by preventing inflammation by reducing NLRP3 activation.
Highlights.
Selective autophagy may provide therapeutic targets for ALD and DILI.
Mitophagy, lipophagy, and macrophage autophagy protect against ALD.
Mitophagy and selective autophagy for adducts protect against DILI.
Autophagy activity requires assessment of autophagic flux and sufficiency.
Acknowledgments
The studies were supported in part by R37 AA020518, R37 AA020518-08S1, R01 DK102142, U01 AA024733, R21 AA027250 (W.X.D).
Abbreviations
- 3-MA
3-methyladeline
- AICAR
5-Aminoimidazole-4-carboxamide ribonucleotide
- ALD
Alcohol-related liver disease
- ALR
augmenter of liver regeneration
- AMPK
AMP-activated protein kinase
- APAP
acetaminophen
- Atg
autophagy-related gene
- ATGL
cytosolic triglyceride lipase
- CB2
cannabinoid receptor 2
- CQ
chloroquine
- DILI
drug-induced liver injury
- Dyn2
Dynamin 2
- EFV
efavirenz
- ESCRT-III
endosomal sorting complex required for transport III
- Fip200
FAK family-interacting protein of 200 kDA
- Fundc1
Fun14 domain containing 1
- GSH
glutathione
- HO-1
heme-oxygenase 1
- IRF1
interferon regulatory factor 1
- KO
knock out
- LC3
microtubule-associated protein light chain 3
- LD
Lipid droplet
- LIR
LC3-interacting region
- MTORC1
mechanistic target of rapamycin complex 1
- NAC
N-acetylcysteine
- NAFLD
non-alcoholic fatty liver disease
- NAPQI
N-acetyl-p-benzoquinone imine
- NSAID
non-steroidal anti-inflammatory drug
- OPTN
optineurin
- PAS
phagophore assembly site
- PINK1
phosphatase and tensin homolog (PTEN)-induced kinase
- Ptdlns3k
class III phosphoinositide 3-kinase
- Rab
ras-related protein
- ROS
reactive oxygen species
- STUB1
STIP1 homology U-Box containing protein
- STX17
syntaxin 17
- TFEB
transcription factor EB
- UBA
ubiquitin-associated
- ULK1
Unc-51 like kinase 1
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alegre F, Moragrega AB, Polo M, Marti-Rodrigo A, Esplugues JV, Blas-Garcia A, Apostolova N, 2018. Role of p62/SQSTM1 beyond autophagy: a lesson learned from drug-induced toxicity in vitro. British journal of pharmacology 175, 440–455. 10.1111/bph.14093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV, 2011a. Autophagy as a rescue mechanism in efavirenz-induced mitochondrial dysfunction: a lesson from hepatic cells. Autophagy 7, 1402–1404. 10.4161/auto.7.11.17653 [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV, 2011b. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology 54, 1009–1019. 10.1002/hep.24459 [DOI] [PubMed] [Google Scholar]
- Baraona E, Leo MA, Borowsky SA, Lieber CS, 1975. Alcoholic hepatomegaly: accumulation of protein in the liver. Science 190, 794–795. 10.1126/science.1198096 [DOI] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T, 2013. The LIR motif - crucial for selective autophagy. Journal of cell science 126, 3237–3247. 10.1242/jcs.126128 [DOI] [PubMed] [Google Scholar]
- Bjornsson ES, Hoofnagle JH, 2016. Categorization of drugs implicated in causing liver injury: Critical assessment based on published case reports. Hepatology 63, 590–603. 10.1002/hep.28323 [DOI] [PubMed] [Google Scholar]
- Blas-Garcia A, Polo M, Alegre F, Funes HA, Martinez E, Apostolova N, Esplugues JV, 2014. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: a comparison with efavirenz. The Journal of antimicrobial chemotherapy 69, 2995–3000. 10.1093/jac/dku262 [DOI] [PubMed] [Google Scholar]
- Bumpus NN, 2011. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicology and applied pharmacology 257, 227–234. 10.1016/j.taap.2011.09.008 [DOI] [PubMed] [Google Scholar]
- Carmona-Gutierrez D, Zimmermann A, Madeo F, 2015. A molecular mechanism for lipophagy regulation in the liver. Hepatology 61, 1781–1783. 10.1002/hep.27738 [DOI] [PubMed] [Google Scholar]
- Chao X, Ni HM, Ding WX, 2018a. Insufficient autophagy: a novel autophagic flux scenario uncovered by impaired liver TFEB-mediated lysosomal biogenesis from chronic alcohol-drinking mice. Autophagy 14, 1646–1648. 10.1080/15548627.2018.1489170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao X, Wang H, Jaeschke H, Ding WX, 2018b. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver international: official journal of the International Association for the Study of the Liver 38, 1363–1374. 10.1111/liv.13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, … Ding WX, 2018c. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 155, 865–879 e812. 10.1053/j.gastro.2018.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Melchior WB Jr., Wu Y, Guo L, 2014. Autophagy in drug-induced liver toxicity. Journal of food and drug analysis 22, 161–168. 10.1016/j.jfda.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran GB, Racz WJ, Smith CV, Mitchell JR, 1985. Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. The Journal of pharmacology and experimental therapeutics 232, 864–872 [PubMed] [Google Scholar]
- Crabb DW, Im GY, Szabo G, Mellinger JL, Lucey MR, 2019. Diagnosis and Treatment of Alcohol-Related Liver Diseases: 2019 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 10.1002/hep.30866 [DOI] [PubMed] [Google Scholar]
- Craig DG, Lee A, Hayes PC, Simpson KJ, 2010. Review article: the current management of acute liver failure. Alimentary pharmacology & therapeutics 31, 345–358. 10.1111/j.1365-2036.2009.04175.x [DOI] [PubMed] [Google Scholar]
- Denaes T, Lodder J, Chobert MN, Ruiz I, Pawlotsky JM, Lotersztajn S, Teixeira-Clerc F, 2016. The Cannabinoid Receptor 2 Protects Against Alcoholic Liver Disease Via a Macrophage Autophagy-Dependent Pathway. Scientific reports 6, 28806. 10.1038/srep28806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry S, Rabbie R, Moore RA, 2012. Diclofenac with or without an antiemetic for acute migraine headaches in adults. The Cochrane database of systematic reviews, CD008783. 10.1002/14651858.CD008783.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl AM, 2002. Liver disease in alcohol abusers: clinical perspective. Alcohol 27, 7–11. 10.1016/s0741-8329(02)00204-5 [DOI] [PubMed] [Google Scholar]
- Ding WX, Guo F, Ni HM, Bockus A, Manley S, Stolz DB, … Yin XM, 2012. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. The Journal of biological chemistry 287, 42379–42388. 10.1074/jbc.M112.413682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, … Yin XM, 2010. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 139, 1740–1752. 10.1053/j.gastro.2010.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Yin XM, 2011. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 7, 248–249. 10.4161/auto.7.2.14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Yin XM, 2012. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biological chemistry 393, 547–564. 10.1515/hsz-2012-0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue TM Jr., Cederbaum AI, French SW, Barve S, Gao B, Osna NA, 2007. Role of the proteasome in ethanol-induced liver pathology. Alcoholism, clinical and experimental research 31, 1446–1459. 10.1111/j.1530-0277.2007.00454.x [DOI] [PubMed] [Google Scholar]
- Egan D, Kim J, Shaw RJ, Guan KL, 2011. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644. 10.4161/auto.7.6.15123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid N, Ito Y, Horibe A, Otsuki Y, 2016. Ethanol-induced mitophagy in liver is associated with activation of the PINK1-Parkin pathway triggered by oxidative DNA damage. Histology and histopathology 31, 1143–1159. 10.14670/HH-11-747 [DOI] [PubMed] [Google Scholar]
- Eid N, Ito Y, Maemura K, Otsuki Y, 2013. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. Journal of molecular histology 44, 311–326. 10.1007/s10735-013-9483-x [DOI] [PubMed] [Google Scholar]
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X, 2009. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The Journal of biological chemistry 284, 12297–12305. 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Bataller R, 2011. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585. 10.1053/j.gastro.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ, 2018. Cargo recognition and degradation by selective autophagy. Nature cell biology 20, 233–242. 10.1038/s41556-018-0037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey EL, Stribling R, Rana A, 2019. Liver Transplantation for Alcoholic Liver Disease: An Update. Clinics in liver disease 23, 127–139. 10.1016/j.cld.2018.09.007 [DOI] [PubMed] [Google Scholar]
- Gomez-Lechon MJ, Ponsoda X, O’Connor E, Donato T, Castell JV, Jover R, 2003. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochemical pharmacology 66, 2155–2167. 10.1016/j.bcp.2003.08.003 [DOI] [PubMed] [Google Scholar]
- Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N, 2006. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131, 165–178. 10.1053/j.gastro.2006.03.045 [DOI] [PubMed] [Google Scholar]
- Gupta P, Venugopal SK, 2018. Augmenter of liver regeneration: A key protein in liver regeneration and pathophysiology. Hepatology research: the official journal of the Japan Society of Hepatology 48, 587–596. 10.1111/hepr.13077 [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, Colombo MI, 2004. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. Journal of cell science 117, 2687–2697. 10.1242/jcs.01114 [DOI] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N, 2008. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. The Journal of biological chemistry 283, 13565–13577. 10.1074/jbc.M708916200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfgott SM, Sandberg-Cook J, Zakim D, Nestler J, 1990. Diclofenac-associated hepatotoxicity. Jama 264, 2660–2662 [PubMed] [Google Scholar]
- Herndon CM, Dankenbring DM, 2014. Patient perception and knowledge of acetaminophen in a large family medicine service. Journal of pain & palliative care pharmacotherapy 28, 109–116. 10.3109/15360288.2014.908993 [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, … Mizushima N, 2009. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell 20, 1981–1991. 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Sun H, Deng WY, Huang WQ, Liu Q, 2019. Augmenter of Liver Regeneration Protects Against Acetaminophen-Induced Acute Liver Injury in Mice by Promoting Autophagy. Shock 52, 274–283. 10.1097/SHK.0000000000001250 [DOI] [PubMed] [Google Scholar]
- Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S, 2007. LAMP proteins are required for fusion of lysosomes with phagosomes. The EMBO journal 26, 313–324. 10.1038/sj.emboj.7601511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, … Ohsumi Y, 2000. A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N, 2012. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A, 2012. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug metabolism reviews 44, 88–106. 10.3109/03602532.2011.602688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL, 2004. Role for Rab7 in maturation of late autophagic vacuoles. Journal of cell science 117, 4837–4848. 10.1242/jcs.01370 [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ, 2010. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. The Journal of cell biology 191, 933–942. 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, … Kim DH, 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular biology of the cell 20, 1992–2003. 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, … Yoshimori T, 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal 19, 5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang KY, Shin JK, Lee SM, 2019. Pterostilbene protects against acetaminophen-induced liver injury by restoring impaired autophagic flux. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 123, 536–545. 10.1016/j.fct.2018.12.012 [DOI] [PubMed] [Google Scholar]
- Kang SW, Haydar G, Taniane C, Farrell G, Arias IM, Lippincott-Schwartz J, Fu D, 2016. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PloS one 11, e0165638. 10.1371/journal.pone.0165638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr., 1995. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochimica et biophysica acta 1245, 421–429. 10.1016/0304-4165(95)00121-2 [DOI] [PubMed] [Google Scholar]
- Kim J, Guan KL, 2011. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell cycle 10, 1337–1338. 10.4161/cc.10.9.15291 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL, 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology 13, 132–141. 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, … Zuckerbraun B, 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounakis K, Chaniotakis M, Markaki M, Tavernarakis N, 2019. Emerging Roles of Lipophagy in Health and Disease. Frontiers in cell and developmental biology 7, 185. 10.3389/fcell.2019.00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, … Acute Liver Failure Study, G., 2005. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372. 10.1002/hep.20948 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, … Youle RJ, 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WM, 2013. Drug-induced acute liver failure. Clinics in liver disease 17, 575–586, viii. 10.1016/j.cld.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ding WX, 2016. A Gene Transcription Program Decides the Differential Regulation of Autophagy by Acute Versus Chronic Ethanol? Alcoholism, clinical and experimental research 40, 47–49. 10.1111/acer.12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ding WX, 2017. Impaired Rab7 and Dynamin2 Block Fat Turnover by Autophagy in Alcoholic Fatty Livers. Hepatology communications 1, 473–476. 10.1002/hep4.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang X, Yang X, Liu J, Li L, Ma W, Chen M, 2019. Differential effects of ginkgol C17:1 on cisplatin-induced cytotoxicity: Protecting human normal L02 hepatocytes versus sensitizing human hepatoma HepG2 cells. Oncology letters 17, 3181–3190. 10.3892/ol.2019.9974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zong WX, Ding WX, 2017. Recycling the danger via lipid droplet biogenesis after autophagy. Autophagy 13, 1995–1997. 10.1080/15548627.2017.1371394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, … McNiven MA, 2016. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Science advances 2, e1601470. 10.1126/sciadv.1601470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Zhong Z, Kim SY, Uchiyama R, Roh YS, Matsushita H, … Seki E, 2019. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. The Journal of biological chemistry 294, 12359–12369. 10.1074/jbc.RA119.007409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata A, 2016. Adverse drug reactions and organ damage: The liver. European journal of internal medicine 28, 9–16. 10.1016/j.ejim.2015.12.017 [DOI] [PubMed] [Google Scholar]
- Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, … Yin XM, 2013. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. Journal of hepatology 58, 993–999. 10.1016/j.jhep.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Wu F, Lin S, Pan X, Jin L, Lu T, … Li X, 2014. Adiponectin protects against acetaminophen-induced mitochondrial dysfunction and acute liver injury by promoting autophagy in mice. Journal of hepatology 61, 825–831. 10.1016/j.jhep.2014.05.033 [DOI] [PubMed] [Google Scholar]
- Liu J, Li Y, Yang X, Dong Y, Wu J, Chen M, 2018. Effects of ginkgol C17:1 on cisplatin-induced autophagy and apoptosis in HepG2 cells. Oncology letters 15, 1021–1029. 10.3892/ol.2017.7398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Xie P, Li W, Wu Y, An W, 2019. Augmenter of Liver Regeneration Protects against Ethanol-Induced Acute Liver Injury by Promoting Autophagy. The American journal of pathology 189, 552–567. 10.1016/j.ajpath.2018.11.006 [DOI] [PubMed] [Google Scholar]
- Lizaso A, Tan KT, Lee YH, 2013. beta-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy 9, 1228–1243. 10.4161/auto.24893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cederbaum AI, 2015. Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity. Biomolecules 5, 2659–2674. 10.3390/biom5042659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat A, Teixeira-Clerc F, Lotersztajn S, 2013. Cannabinoid signaling and liver therapeutics. Journal of hepatology 59, 891–896. 10.1016/j.jhep.2013.03.032 [DOI] [PubMed] [Google Scholar]
- Masubuchi Y, Nakayama S, Horie T, 2002. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 35, 544–551. 10.1053/jhep.2002.31871 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, … Tanaka K, 2010. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. The Journal of cell biology 189, 211–221. 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H, 2013. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharmaceutical research 30, 2174–2187. 10.1007/s11095-013-1007-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H, 2012. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicology and applied pharmacology 264, 387–394. 10.1016/j.taap.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK, 2011. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. Journal of neurochemistry 117, 856–867. 10.1111/j.1471-4159.2011.07253.x [DOI] [PubMed] [Google Scholar]
- Menk M, Graw JA, Poyraz D, Mobius N, Spies CD, von Haefen C, 2018. Chronic Alcohol Consumption Inhibits Autophagy and Promotes Apoptosis in the Liver. International journal of medical sciences 15, 682–688. 10.7150/ijms.25393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, 2010. The role of the Atg1/ULK1 complex in autophagy regulation. Current opinion in cell biology 22, 132–139. 10.1016/j.ceb.2009.12.004 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ, 2008. Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo R, Lai R, Lu J, Zhuang Y, Zhou T, Jiang S, … Xie Q, 2018. Enhanced autophagy contributes to protective effects of IL-22 against acetaminophen-induced liver injury. Theranostics 8, 4170–4180. 10.7150/thno.25798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ, 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology 183, 795–803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX, 2012. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55, 222–232. 10.1002/hep.24690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Du K, You M, Ding WX, 2013a. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. The American journal of pathology 183, 1815–1825. 10.1016/j.ajpath.2013.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y, … Ding WX, 2016. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. Journal of hepatology 65, 354–362. 10.1016/j.jhep.2016.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Ding WX, 2015. Mitochondrial dynamics and mitochondrial quality control. Redox biology 4, 6–13. 10.1016/j.redox.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Jaeschke H, Ding WX, 2013b. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox biology 1, 427–432. 10.1016/j.redox.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flanagan CH, O’Neill C, 2014. PINK1 signalling in cancer biology. Biochimica et biophysica acta 1846, 590–598. 10.1016/j.bbcan.2014.10.006 [DOI] [PubMed] [Google Scholar]
- Ohsumi Y, 2001. Molecular dissection of autophagy: two ubiquitin-like systems. Nature reviews. Molecular cell biology 2, 211–216. 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, … Matsuda N, 2012. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nature communications 3, 1016. 10.1038/ncomms2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, … Harper JW, 2014. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Molecular cell 56, 360–375. 10.1016/j.molcel.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N, 2018. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nature cell biology 20, 1013–1022. 10.1038/s41556-018-0176-2 [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, … Chung J, 2006. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. 10.1038/nature04788 [DOI] [PubMed] [Google Scholar]
- Parzych KR, Klionsky DJ, 2014. An overview of autophagy: morphology, mechanism, and regulation. Antioxidants & redox signaling 20, 460–473. 10.1089/ars.2013.5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F, 2019. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chemical research in toxicology 32, 1469–1486. 10.1021/acs.chemrestox.9b00204 [DOI] [PubMed] [Google Scholar]
- Qiu Y, Benet LZ, Burlingame AL, 1998. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. The Journal of biological chemistry 273, 17940–17953. 10.1074/jbc.273.28.17940 [DOI] [PubMed] [Google Scholar]
- Qu X, Gao H, Tao L, Zhang Y, Zhai J, Song Y, Zhang S, 2018. Autophagy inhibition-enhanced assembly of the NLRP3 inflammasome is associated with cisplatin-induced acute injury to the liver and kidneys in rats. Journal of biochemical and molecular toxicology, e22208. 10.1002/jbt.22228 [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2018. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene expression 18, 19–30. 10.3727/105221617X15084371374138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Visschers RGJ, Duan L, Akakpo JY, Jaeschke H, 2018. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. Journal of clinical and translational research 4, 75–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasineni K, Donohue TM Jr., Thomes PG, Yang L, Tuma DJ, McNiven MA, Casey CA, 2017. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatology communications 1, 501–512. 10.1002/hep4.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm J, Samokhvalov AV, Shield KD, 2013. Global burden of alcoholic liver diseases. Journal of hepatology 59, 160–168. 10.1016/j.jhep.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Reuben A, Koch DG, Lee WM, Acute Liver Failure Study, G., 2010. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology 52, 2065–2076. 10.1002/hep.23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodger CE, McWilliams TG, Ganley IG, 2018. Mammalian mitophagy - from in vitro molecules to in vivo models. The FEBS journal 285, 1185–1202. 10.1111/febs.14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V, 2014. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Molecular cell 53, 167–178. 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, … Zhang S, 2015. Huntingtin functions as a scaffold for selective macroautophagy. Nature cell biology 17, 262–275. 10.1038/ncb3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, … Guan KL, 2013. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature cell biology 15, 741–750. 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyanarayan A, Mashek MT, Mashek DG, 2017. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell reports 19, 1–9. 10.1016/j.celrep.2017.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z, 2007. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO journal 26, 1749–1760. 10.1038/sj.emboj.7601623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, McNiven MA, 2019. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. The Journal of cell biology 218, 3320–3335. 10.1083/jcb.201803153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA, 2015. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61, 1896–1907. 10.1002/hep.27667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze RJ, Rasineni K, Weller SG, Schott MB, Schroeder B, Casey CA, McNiven MA, 2017a. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatology communications 1, 140–152. 10.1002/hep4.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze RJ, Sathyanarayan A, Mashek DG, 2017b. Breaking fat: The regulation and mechanisms of lipophagy. Biochimica et biophysica acta. Molecular and cell biology of lipids 1862, 1178–1187. 10.1016/j.bbalip.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, McNiven MA, 2013. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. The Journal of cell biology 203, 315–326. 10.1083/jcb.201306140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, … Tsukamoto H, 2018. Alcoholic liver disease. Nature reviews. Disease primers 4, 16. 10.1038/s41572-018-0014-7 [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, … Ballabio A, 2011. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, … Ballabio A, 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal 31, 1095–1108. 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT, 2017. STUB1 regulates TFEB-induced autophagy-lysosome pathway. The EMBO journal 36, 2544–2552. 10.15252/embj.201796699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen NT, Salajegheh A, Brown RS Jr., 2019. A Call to Standardize Definitions, Data Collection, and Outcome Assessment to Improve Care in Alcohol-Related Liver Disease. Hepatology 70, 1038–1044. 10.1002/hep.30587 [DOI] [PubMed] [Google Scholar]
- Shi C, Xue W, Han B, Yang F, Yin Y, Hu C, 2019. Acetaminophen aggravates fat accumulation in NAFLD by inhibiting autophagy via the AMPK/mTOR pathway. European journal of pharmacology 850, 15–22. 10.1016/j.ejphar.2019.02.005 [DOI] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, … Imai Y, 2014. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS genetics 10, e1004861. 10.1371/journal.pgen.1004861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, … Czaja MJ, 2009. Autophagy regulates lipid metabolism. Nature 458, 1131–1135. 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, … Wang HG, 2018. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nature communications 9, 2855. 10.1038/s41467-018-05254-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Liang X, Hattori T, Tang Z, He H, Chen H, … Wang HG, 2019. VPS37A directs ESCRT recruitment for phagophore closure. The Journal of cell biology 218, 3336–3354. 10.1083/jcb.201902170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E, 2008. LC3 and Autophagy. Methods in molecular biology 445, 77–88. 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- Teschke R, 2019. Alcoholic Liver Disease: Current Mechanistic Aspects with Focus on Their Clinical Relevance. Biomedicines 7 10.3390/biomedicines7030068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomes PG, Rasineni K, Yang L, Donohue TM Jr., Kubik JL, McNiven MA, Casey CA, 2019. Ethanol withdrawal mitigates fatty liver by normalizing lipid catabolism. American journal of physiology. Gastrointestinal and liver physiology 316, G509–G518. 10.1152/ajpgi.00376.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr., 2015. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcoholism, clinical and experimental research 39, 2354–2363. 10.1111/acer.12904 [DOI] [PubMed] [Google Scholar]
- Ueno T, Komatsu M, 2017. Autophagy in the liver: functions in health and disease. Nature reviews. Gastroenterology & hepatology 14, 170–184. 10.1038/nrgastro.2016.185 [DOI] [PubMed] [Google Scholar]
- Vitelli R, Santillo M, Lattero D, Chiariello M, Bifulco M, Bruni CB, Bucci C, 1997. Role of the small GTPase Rab7 in the late endocytic pathway. The Journal of biological chemistry 272, 4391–4397. 10.1074/jbc.272.7.4391 [DOI] [PubMed] [Google Scholar]
- Wang H, Ni HM, Chao X, Ma X, Rodriguez YA, Chavan H, … Ding WX, 2019. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox biology 22, 101148. 10.1016/j.redox.2019.101148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ding WX, 2015. A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease. Biomolecules 5, 2619–2642. 10.3390/biom5042619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ding WX, 2018. Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biological chemistry 399, 147–178. 10.1515/hsz-2017-0228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Manley S, Ding WX, 2014. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World journal of gastroenterology 20, 12908–12933. 10.3748/wjg.v20.i36.12908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ni HM, Ding Y, Ding WX, 2015a. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. American journal of physiology. Gastrointestinal and liver physiology 309, G324–340. 10.1152/ajpgi.00108.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ni HM, Haynes A, Manley S, Li Y, Jaeschke H, Ding WX, 2015b. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice. The Journal of biological chemistry 290, 10934–10946. 10.1074/jbc.M114.602284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, … Jaeschke H, 2015. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicology and applied pharmacology 289, 213–222. 10.1016/j.taap.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Youle RJ, 2013. PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. 10.4161/auto.24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yang C, Thomes PG, Kharbanda KK, Casey CA, McNiven MA, Donohue TM Jr., 2019. Lipophagy and Alcohol-Induced Fatty Liver. Frontiers in pharmacology 10, 495. 10.3389/fphar.2019.00495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, … Lu B, 2006. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proceedings of the National Academy of Sciences of the United States of America 103, 10793–10798. 10.1073/pnas.0602493103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Nelson LJ, Gomez Del Moral M, Martinez-Naves E, Cubero FJ, 2018. Dissecting the molecular pathophysiology of drug-induced liver injury. World journal of gastroenterology 24, 1373–1385. 10.3748/wjg.v24.i13.1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Mizushima N, 2017. Monitoring and Measuring Autophagy. International journal of molecular sciences 18 10.3390/ijms18091865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Chen Y, Tooze SA, 2018. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215. 10.1080/15548627.2017.1378838 [DOI] [PMC free article] [PubMed] [Google Scholar]