

Abstract
Objective
Sympathetic nervous system (SNS) overactivity is a risk factor for insulin resistance and cardiovascular disease (CVD). We evaluated the impact of bromocriptine‐QR, a dopamine‐agonist antidiabetes medication, on elevated resting heart rate (RHR) (a marker of SNS overactivity in metabolic syndrome), blood pressure (BP) and the relationship between bromocriptine‐QR's effects on RHR and HbA1c in type 2 diabetes subjects.
Design and Subjects
RHR and BP changes were evaluated in this post hoc analysis of data from a randomized controlled trial in 1014 type 2 diabetes subjects randomized to bromocriptine‐QR vs placebo added to standard therapy (diet ± ≤2 oral antidiabetes medications) for 24 weeks without concomitant antihypertensive or antidiabetes medication changes, stratified by baseline RHR (bRHR).
Results
In subjects with bRHR ≥70 beats/min, bromocriptine‐QR vs placebo reduced RHR by −3.4 beats/min and reduced BP (baseline 130/79; systolic, diastolic, mean arterial BP reductions [mm Hg]: −3.6 [P = .02], −1.9 [P = .05], −2.5 [P = .02]). RHR reductions increased with higher baseline HbA1c (bHbA1c) (−2.7 [P = .03], −5 [P = .002], −6.1 [P = .002] with bHbA1c ≤7, >7, ≥7.5%, respectively] in the bRHR ≥70 group and more so with bRHR ≥80 (−4.5 [P = .07], −7.8 [P = .015], −9.9 [P = .005]). Subjects with bRHR <70 had no significant change in RHR or BP. With bHbA1c ≥7.5%, %HbA1c reductions with bromocriptine‐QR vs placebo were −0.50 (P = .04), −0.73 (P = .005) and −1.22 (P = .008) with bRHR <70, ≥70 and ≥80, respectively. With bRHR ≥70, the magnitude of bromocriptine‐QR‐induced RHR reduction was an independent predictor of bromocriptine‐QR's HbA1c lowering effect.
Conclusion
Bromocriptine‐QR lowers elevated RHR with concurrent decrease in BP and hyperglycaemia. These findings suggest a potential sympatholytic mechanism contributing to bromocriptine‐QR's antidiabetes effect and potentially its previously demonstrated effect to reduce CVD events.
Keywords: dopamine, sympathetic nervous system, type 2 diabetes
Sympathetic nervous system (SNS) overactivity is well‐known to potentiate insulin resistance and cardiovascular disease and central dopamine agonism is a known mechanism for reducing sympathetic tone. This study evaluated (a) the effect of circadian‐timed bromocriptine‐QR (B‐QR), a unique quick‐release formulation of bromocriptine, a sympatholytic dopamine D2 receptor agonist that is approved for the treatment of type 2 diabetes (T2D), on elevated resting heart rate (RHR), a widely acknowledged peripheral marker of elevated SNS activity in metabolic syndrome patients that has been shown to be associated with increased insulin resistance and cardiovascular risk, and (b) the relationship between B‐QR's impact on elevated RHR and its glycemic control effect in T2D subjects. The results demonstrate that B‐QR significantly reduces elevated RHR while simultaneously lowering blood pressure and the magnitude of RHR reduction (which is also determined by how elevated the heart rate is at baseline and baseline HbA1c, ie, greater RHR reduction with higher baseline RHR and higher baseline HbA1c) is a predictor of bromocriptine‐QR's effect to lower HbA1c in subjects with elevated RHR and poor glycemic control (ie, the greater the RHR reduction, the greater the HbA1c reduction).

1. INTRODUCTION
The sympathetic nervous system (SNS) plays an important role in maintaining normal cardiovascular homeostasis and health by regulating systemic vascular resistance, blood pressure (BP), heart rate, cardiac output and normal vascular endothelial function in response to a multitude of acute environmental, physical and mental status alterations.1, 2 It also regulates normal glucose homeostasis by enhancing hepatic glucose output and adipose‐free fatty acid mobilization during fasting periods of the day and in circumstances such as acute hypoglycaemia or prolonged starvation.3, 4, 5, 6, 7, 8 However, chronic overactivity of the SNS leads to cardiovascular as well as metabolic adverse effects.9, 10, 11, 12, 13, 14, 15, 16, 17, 18 Cardiovascular adverse effects of chronically elevated sympathetic tone include vasoconstriction facilitating increased BP, an overactivated renin‐angiotensin system potentiating increased BP, increased heart rate and most importantly inflammation and reactive oxygen species (ROS) generation in the micro‐ and macro‐vasculature as well as within the myocardium itself potentiating arterial stiffness, myocardial apoptosis and myocardial ischaemia/reperfusion injury.2, 10, 15, 16, 17, 18, 19, 20, 21, 22, 23 Less well recognized are the adverse metabolic effects of chronically increased SNS activity, which include increased hepatic gluconeogenesis, decreased hepatic glucose disposal, increased free fatty acid (FFA) mobilization from the adipose tissue, ROS generation and inflammation in adipose and liver, and decreased blood flow to muscle, all potentiating insulin resistance in those tissues and beta cell dysfunction resulting from inflammatory factors, ROS, lipotoxicity and glucotoxicity.10, 12, 13, 16, 18, 24, 25, 26
Elevated resting heart rate (RHR) can reflect elevated central sympathetic‐to‐parasympathetic activity balance27, 28 and has been shown to be a common occurrence in insulin resistance syndrome, independent of high BP or obesity.29, 30, 31, 32 Most importantly, in the insulin resistance syndrome, elevated RHR values are significantly correlated with other measures of SNS activity such as muscle sympathetic nerve activity and serum noradrenaline levels.12, 16, 18, 29 While clinically RHR between 60 and 100 beats per minute (BPM) is considered the “normal” range for RHR and RHR ≥100 is used as the criteria for defining tachycardia, a large body of evidence from epidemiological and clinical studies suggests that increasing RHR within the “normal” range is associated with increased cardiometabolic risk, particularly above 70‐80 beats per minute (BPM). Such elevated RHR has been associated with insulin resistance,26, 33 altered beta cell function,34 impaired glucose regulation and increased risk of developing type 2 diabetes mellitus35, 36, 37 as well as increased cardiovascular disease (CVD) risk38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56 and mortality.38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 51, 52, 54, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65
Bromocriptine‐QR (B‐QR), a quick‐release formulation of micronized bromocriptine, is the only sympatholytic dopamine‐agonist US FDA‐approved for the treatment of type 2 diabetes. In several preclinical66, 67, 68, 69, 70, 71, 72 and clinical studies,73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84 bromocriptine administration has repeatedly been demonstrated to reduce measures of elevated SNS activity such as reduction of elevated sympathetic outflow, plasma norepinephrine, BP and/or conversion of nondipper profile of circadian mean arterial pressure to a dipper profile. A critical aspect of dopaminergic control of autonomic function is via circadian modulation of the central biological clock pacemaker circuit (circadian neuronal afferent signals to and including the suprachiasmatic nuclei [SCN]) (see below).
The biological clock pacemaker circuit (circadian efferent signals to and including the SCN) for the body is a primary regulator of autonomic balance in the body.85, 86, 87 A diminution of the circadian peak in dopaminergic input signalling to this SCN clock system (at daily waking from the sleep cycle) is coupled to and potentiates an increase in hypothalamic pre‐autonomic neuronal activities that lead to overactivation of the SNS and metabolic syndrome in animals.88 The circadian‐timed administration of either dopamine agonist systemically or dopamine to the SCN clock area in insulin‐resistant animals to induce (mimic) the normal circadian peak of dopaminergic activity at the SCN pacemaker that is diminished in insulin resistance states has been observed to reduce chronic overactivity of SNS pre‐autonomic neurons in the hypothalamus and measures of subsequent chronic activation of peripheral sympathetic tone in insulin‐resistant states.66, 88, 89, 90, 91, 92, 93 Moreover, reduction of brain dopamine synthesis in healthy humans for just a couple of days induces peripheral insulin resistance.94, 95
Circadian‐timed (onset of daily waking) administration of the antidiabetes agent B‐QR, a quick‐release formulation of micronized bromocriptine, to re‐establish the normal circadian peak of central nervous system (CNS) dopaminergic activity that is diminished in insulin‐resistant states among mammals has been observed to improve insulin sensitivity and reduce CVD events in type 2 diabetes.96, 97 Therefore, the possibility exists that such B‐QR therapy may reduce elevated RHR, a measure of SNS tone in insulin resistance syndrome,29 that may in part explain the agent's impact to improve glycaemic control96 and reduce adverse cardiovascular outcomes in type 2 diabetes.98, 99, 100, 101 However, the impact of circadian B‐QR therapy on the pathophysiological parameter of elevated RHR and, importantly, its relation to glycaemic control in type 2 diabetes has never been investigated. The primary aim of this present study was twofold: (a) to investigate the effect of B‐QR on elevated RHR in type 2 diabetes subjects and (b) to assess the nature of any inter‐relationship between B‐QR's impact to reduce elevated RHR and to reduce HbA1c in these subjects.
2. METHODS
2.1. Study subjects and design
The study population (N = 1014) of type 2 diabetes subjects was derived from the Cycloset Safety Trial (CST). The study protocol and design for the CST have been previously described in detail.98, 102 Briefly, the CST was a multicenter, placebo‐controlled, double‐blind, parallel‐group safety and efficacy study in outpatient type 2 diabetes subjects recruited from general practice and diabetes clinics across 74 clinical centres in the United States and Puerto Rico. Subjects were between the ages of 30 and 80 years and had a body mass index <43 kg/m2, with established type 2 diabetes by ADA 2003 criteria and HbA1c ≤10.0%. Subjects with New York Heart Classifications I and II congestive heart failure (CHF) were allowed to participate, as were subjects with a history of myocardial infarction (MI) or coronary revascularization occurring >6 months before enrolment. Subjects were required to have maintained a stable diabetes treatment regimen for ≥30 days prior to randomization, consisting of lifestyle interventions of medical nutrition therapy and appropriately prescribed physical activity with or without oral antihyperglycaemic agents (≤2) or insulin either alone or in combination with 1 oral antihyperglycaemic agent.
Following randomization (2:1 active agent vs placebo), the study drug (B‐QR or placebo) was titrated from an initial starting dose of 1 tablet daily (0.8 mg B‐QR per tablet or matching placebo) by increasing the daily dose by 1 tablet per week until a maximum tolerated daily dose between 2 and 6 tablets once daily (1.6‐4.8 mg B‐QR/day) was achieved. The study drug was taken with the morning meal, within 2 hours of waking. Subjects were required to continue their established antihyperglycaemic treatments during the first 3 months of the study. However, the dosages of the oral agents or insulin could be modified as deemed appropriate by the study site investigator. After 3 months, alterations in the diabetes treatment regimen were allowed, if deemed necessary by the study site investigator, as long as these changes did not result in a final regimen that exceeded two oral agents or insulin plus one oral agent, exclusive of the study drug. Due to the above study design, the prespecified statistical analysis plan for the CST102 had specified that data at 24 weeks of treatment be considered for efficacy analyses and therefore were used to assess B‐QR's effects on dysglycemia and RHR in the present study.
The study protocol was approved by site‐specific or central institutional review boards, and all subjects provided written informed consent to participate in the study before enrolment. The study was conducted in accordance with the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) 2004 guidelines. This current study and analyses are original and different from any previously reported results from the CST.
The study population for the present study consisted of all subjects from the CST who completed 24 weeks of study drug treatment with no concomitant hypertension medication changes (to avoid confounding arising from BP and/or heart rate effects from changes in concomitant antihypertensive medications during this period) and no antidiabetes medication changes (since the original CST protocol had allowed for changes in dosages and regimens of antidiabetes medications, to avoid any potential confounding arising from the possibility of such diabetes medication changes affecting RHR or BP changes). Subjects on insulin therapy were excluded from this population given the limitations of the database in clearly determining insulin dose changes (and hence ensuring that there were no changes in concomitant diabetes therapies) and also more importantly to control for the potential effects of insulin itself and changes in insulin dose on sympathetic activity given that insulin acts centrally to increase SNS activity.103, 104 A total of 1014 subjects (642 B‐QR, 372 placebo) meeting the above criteria constituted the study population for this study.
Resting heart rate was derived from 12‐lead electrocardiograms (ECGs) obtained at baseline and at 24 weeks. BP and HbA1c measurements were obtained at the baseline and 24‐week study visits.
2.2. Statistical analyses
To assess the effects of treatment with B‐QR on RHR and if baseline RHR influences this effect and determine if there is a RHR threshold at which the treatment effect might first occur, a 2‐way analysis of variance was performed to test the interaction of treatment arm (B‐QR vs placebo) and baseline RHR subgroup (stratified as RHR <60, 60‐69, 70‐79 and ≥80) as two independent variables/factors and change in RHR from baseline to week 24 as the outcome variable. The treatment effects within each RHR subgroup were further analysed with t tests. Paired sample t tests were used to assess within‐treatment group changes and Student's t test for between‐group differences. Based on the findings from these initial analyses, that indicated baseline RHR ≥70 as the threshold above which an effect of B‐QR in lowering RHR was evident (Table 2), all further analyses were stratified by a baseline RHR cut‐off of <70 vs ≥70 or ≥80 BPM.
Table 2.
Effects of bromocriptine‐QR vs placebo treatment for 24 wk on resting heart rate stratified by baseline resting heart rate
| Baseline RHR subgroups | Bromocriptine‐QR (B‐QR) | B‐QR within group change | Placebo (PL) | PL within group change | B‐QR vs PL between‐group difference | ||
|---|---|---|---|---|---|---|---|
| Baseline | After 24 wk of treatment | Baseline | After 24 wk of treatment | ||||
|
RHR <60 N = 294 (174 B‐QR; 120 P) |
54.1 ± 0.3 | 57.5 ± 0.6 |
3.4 P < .001 |
55.0 ± 0.3 | 58.3 ± 0.6 |
3.3 P < .001 |
0.1 P = .90 |
|
RHR 60‐69 N = 348 (225 B‐QR; 123 P) |
64.4 ± 0.2 | 64.7 ± 0.5 |
0.3 P = .61 |
64.6 ± 0.3 | 65.3 ± 0.7 |
0.8 P = .26 |
−0.5 P = .55 |
|
RHR 70‐79 N = 248 (158 B‐QR; 90 P) |
74.1 ± 0.2 | 70.8 ± 0.7 |
−3.3 P < .001 |
73.4 ± 0.3 | 72.5 ± 0.9 |
−0.9 P = .30 |
−2.4 P = .027 |
|
RHR ≥80 N = 124 (85 B‐QR; 39 P) |
86.5 ± 0.7 | 78.9 ± 1.1 |
−7.6 P < .001 |
85.4 ± 0.8 | 82.7 ± 1.6 |
−2.7 P = .07 |
−4.9 P = .01 |
Data shown as mean ± standard error of mean.
Abbreviation: RHR, resting heart rate.
In addition to changes in RHR, changes in systolic BP (SBP), diastolic BP (DBP) and mean arterial pressure (MAP) from baseline to 24 weeks were analysed in the study population stratified by baseline RHR < or ≥70 BPM. Multivariable linear regression analyses stratified by RHR <70 and ≥70 BPM were performed with change in RHR from baseline to 24 weeks as the outcome variable and treatment with B‐QR vs placebo as a covariate along with age, gender, race, BMI, duration of diabetes, baseline HbA1c, baseline RHR, baseline and changes in SBP and DBP as other covariates included to further evaluate the effect of B‐QR treatment on RHR after controlling for potential effects of these other factors on RHR.
Further analyses were then performed to evaluate (a) the relationships between baseline RHR as well as baseline HbA1c and the change in RHR associated with B‐QR therapy (vs placebo) and (b) the relationship between B‐QR's impact on elevated RHR and its glycaemic control effect.
To evaluate the relationships between baseline RHR, baseline HbA1c and B‐QR's impact on RHR, the changes in RHR with B‐QR vs placebo were analysed in the baseline RHR <70, ≥70, and ≥80 subgroups stratified by baseline HbA1c (baseline HbA1c ≤7, >7 and ≥7.5).
The nature of the relationship between (a) baseline RHR and B‐QR's antidiabetes effect and (b) B‐QR's impact on elevated RHR and its antidiabetes effect each was analysed in those subjects with suboptimal glycaemic control (HbA1c ≥7.5%) at baseline (N = 198:125 B‐QR, 73 placebo) as described below.
To evaluate if baseline RHR impacts the glycaemic control effects of B‐QR, the change in HbA1c from baseline to week 24 with B‐QR vs placebo was analysed in subjects with baseline HbA1c ≥7.5 stratified by baseline RHR <70, ≥70, and ≥80.
The relationship between study drug‐induced change in elevated RHR and change in HbA1c was analysed in subjects with baseline HbA1c ≥7.5 and baseline RHR ≥70 using Pearson correlation as well as multivariable linear regression with change in HbA1c from baseline to week 24 as the outcome variable and change in RHR as a covariate along with age, gender, race, baseline HbA1c and other concomitant diabetes medications (metformin, SU and/or TZD, each coded as 0 = no and 1 = yes) as the other variables included in the analysis.
All statistical analyses were performed using SPSS software (Build 1.0.0.1012; IBM Corp). The significance level was set at P < .05. Data are presented as mean ± standard error of the mean (SEM) except categorical variables shown as numbers and per cent.
3. RESULTS
3.1. Baseline characteristics
The baseline characteristics of the study population are shown in Table 1. Study subjects in the B‐QR and placebo treatment arms were well matched at baseline within each RHR subgroup and overall; besides the expected difference in mean RHR, there were no major differences in the baseline characteristics of subjects across the different RHR subgroups. The average blood pressure control was good in all groups but close to 70% of the subjects had a history of hypertension in each group and were on antihypertensive medications. In this regard, it should be re‐emphasized that concomitant antihypertensive medication changes did not occur during the course of this study per the study inclusion criteria described in Section 2.
Table 1.
Baseline characteristics of the study population
| Baseline RHR <70 | Baseline RHR ≥70 | Baseline RHR ≥80 | ||||
|---|---|---|---|---|---|---|
| B‐QR (n = 399) | Placebo (n = 243) | B‐QR (n = 243) | Placebo (n = 129) | B‐QR (n = 85) | Placebo (n = 39) | |
| Age (y) | 61 ± 0.5 | 61 ± 0.6 | 58 ± 0.6 | 59 ± 0.9 | 59 ± 1.1 | 56 ± 1.4 |
| Gender (% male) | 61 | 60 | 56 | 47 | 48 | 41 |
| Race (% Caucasian) | 63 | 70 | 65 | 64 | 62 | 69 |
| BMI (kg/m2) | 31.7 ± 0.3 | 32.2 ± 0.3 | 32.9 ± 0.3 | 32.1 ± 0.5 | 32.2 ± 0.6 | 32.5 ± 0.9 |
| Duration of diabetes (y) | 5.5 ± 03 | 6.6 ± 0.4* | 6.2 ± 0.3 | 6.1 ± 0.5 | 7.3 ± 0.7 | 5.9 ± 0.8 |
| HbA1c (%) | 6.64 ± 0.04 | 6.69 ± 0.06 | 6.93 ± 0.07 | 6.92 ± 0.10 | 7.05 ± 0.11 | 6.84 ± 0.19 |
| Fasting glucose (mg/dL) | 133 ± 1.6 | 133 ± 2.0 | 143 ± 2.7 | 137 ± 3.3 | 148 ± 5.1 | 136 ± 6.2 |
| Baseline RHR (bpm) | 60 ± 0.3 | 60 ± 0.4 | 78 ± 0.5 | 77 ± 0.6 | 86.5 ± 0.7 | 85.4 ± 0.8 |
| Systolic BP (mm Hg) | 130 ± 0.7 | 130 ± 0.8 | 130 ± 0.8 | 128 ± 1.1 | 131 ± 1.5 | 125 ± 1.9* |
| Diastolic BP (mm Hg) | 77 ± 0.4 | 77 ± 0.6 | 79 ± 0.6 | 78 ± 0.7 | 78 ± 1.0 | 77 ± 1.4 |
| eGFR (mL/min/1.73 m2) | 66 ± 0.6 | 67 ± 0.7 | 67 ± 0.8 | 66 ± 1.1 | 66 ± 1.4 | 67 ± 1.8 |
| Serum creatinine (mg/dL) | 1.1 ± 0.01 | 1.1 ± 0.01 | 1.1 ± 0.01 | 1.1 ± 0.02 | 1.1 ± 0.02 | 1.1 ± 0.02 |
| Hypertension history (% yes) | 69 | 70 | 72 | 72 | 72 | 67 |
Data shown as mean ± standard error of mean.
Abbreviations: BMI, body mass index; BP, blood pressure; eGFR, estimated glomerular filtration rate; RHR, resting heart rate.
P < .05 for between‐treatment group (B‐QR vs placebo) within specified baseline RHR category.
3.2. RHR threshold for effect of B‐QR
Two‐way analysis of variance to explore the effect of treatment with B‐QR (vs placebo) and baseline RHR on the change in RHR from baseline to week 24 revealed statistically significant interaction between the effects of treatment arm and baseline RHR subgroup on the change in RHR (F [3, 1006] 3.3, P = .02), with significant changes in RHR with B‐QR therapy seen only in the RHR subgroups with baseline RHR ≥70 BPM (see Table 2 for details). In the baseline RHR between 70 and 79 subgroup, the RHR change from week 0 to week 24 was −3.3 BPM (P < .001) within the B‐QR treated group and −0.9 (P = .3) within the placebo group yielding a between‐group difference of −2.4 BPM (P = .027). In the baseline RHR ≥80 BPM subgroup, RHR decreased significantly by −7.6 BPM (P < .001) in the B‐QR‐treated group, while the mean RHR change of −2.7 BPM in the placebo group was not statistically significant (P = .07) yielding a between‐treatment group difference of −4.9 BPM RHR reduction with B‐QR relative to placebo (P = .01). There were no significant changes in RHR with either B‐QR or placebo in the subgroups with baseline RHR <60 or 60‐69 (ie RHR <70). Baseline RHR < or ≥70 was therefore used as the cut‐off for the subsequent analyses as described below.
3.3. Effects of B‐QR on RHR and BP stratified by baseline RHR ≥70/<70
Among subjects with baseline RHR ≥70 (N = 372:243 B‐QR, 129 placebo), RHR decreased from baseline to week 24 on average by −4.8 BPM in the B‐QR treated group and by −1.5 in the placebo‐treated group yielding a between‐group difference of −3.4 BPM (P = .001) (see Table 3 for more details). B‐QR therapy relative to placebo also reduced SBP by −3.6 mm Hg (P = .02), DBP by −1.9 mm Hg (P = .05) and MAP by −2.5 mm Hg (P = .02) (see Table 3 for more details).
Table 3.
Effects of bromocriptine‐QR vs placebo treatment for 24 wk on resting heart rate and blood pressure in subjects with baseline RHR ≥70
| Bromocriptine‐QR (B‐QR) (N = 243) | B‐QR within group change | Placebo (PL) N = 129 | PL within group change | B‐QR vs PL between‐group difference | |||
|---|---|---|---|---|---|---|---|
| Baseline | After 24 wk of treatment | Baseline | After 24 wk of treatment | ||||
| RHR (BPM) | 78.4 ± 0.5 | 73.6 ± 0.6 |
−4.8 P < .001 |
77.0 ± 0.6 | 75.6 ± 0.9 |
−1.5 P = .06 |
−3.4 P = .001 |
| Systolic BP (mm Hg) | 129.9 ± 0.9 | 127.1 ± 0.9 |
−2.8 P = .002 |
127.7 ± 1.1 | 128.5 ± 1.3 |
0.8 P = .6 |
−3.6 P = .02 |
| Diastolic BP (mm Hg) | 79.2 ± 0.6 | 76.8 ± 0.6 |
−2.4 P < .001 |
78.0 ± 0.7 | 77.5 ± 0.8 |
−0.5 P = .5 |
−1.9 P = .05 |
| MAP (mm Hg) | 96.1 ± 0.6 | 93.6 ± 0.6 |
−2.6 P < .001 |
94.6 ± 0.7 | 94.5 ± 0.8 |
−0.05 P = .9 |
−2.5 P =.02 |
Data shown as mean ± standard error of mean.
Abbreviations: BP, blood pressure; MAP, mean arterial pressure; RHR, resting heart rate.
Among subjects with RHR <70 at baseline, there was no reduction in RHR within either treatment group and no significant difference in the RHR change from baseline with B‐QR therapy relative to placebo (between‐treatment group difference in change in RHR from baseline −0.4, P = .5).
Multivariable regression analysis demonstrated that treatment with B‐QR (vs placebo) is a significant independent predictor (P = .001) of change in RHR, after adjusting for other factors including age, gender, race, BMI, duration of diabetes, baseline HbA1c, and baseline as well as change in SBP and DBP, in subjects with baseline RHR ≥70 but not in those with baseline RHR <70.
3.4. Effect of baseline HbA1c on B‐QR's impact on RHR
The mean change in RHR from baseline to week 24 with B‐QR therapy vs placebo among subjects with baseline RHR ≥70 when stratified by baseline HbA1c was −2.7 BPM (P = .03) in subjects with baseline HbA1c ≤7, −5.0 BPM (P = .002) in subjects with baseline HbA1c >7 and −6.1 BPM (P = .002) in those with baseline HbA1c ≥7.5 (Figure 1; Table 4).
Figure 1.
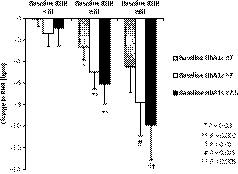
Bromocriptine‐QR vs placebo reduces elevated resting heart rate (between‐group difference in change from baseline) as a function of baseline resting heart and baseline haemoglobin A1c. Data shown as mean ± standard error of mean. RHR, resting heart rate
Table 4.
Resting heart rate changes with bromocriptine‐QR vs placebo stratified by baseline resting heart rate and baseline haemoglobin A1c
| Baseline RHR groups stratified by baseline HbA1c | Bromocriptine‐QR (B‐QR) | Placebo (P) |
Between‐treatment group difference in RHR change Week 0‐24 (P‐value) |
||||
|---|---|---|---|---|---|---|---|
| RHR at baseline |
RHR change Week 0‐24 |
RHR at baseline |
RHR change Week 0‐24 |
||||
| Baseline RHR <70 BPM | |||||||
| HbA1c ≤7 (N = 295 B‐QR, 177 P) | 60 ± 0.4 | 1.7 ± 0.4*** | 60 ± 0.4 | 1.7 ± 0.5*** | 0.04 ± 0.7 (P = .95) | ||
| HbA1c >7 (N = 103 B‐QR, 66 P) | 61 ± 0.6 | 1.4 ± 0.8 | 60 ± 0.6 | 2.8 ± 0.9** | −1.4 ± 1.2 (P = .25) | ||
| HbA1c ≥7.5 (N = 60 B‐QR, 41 P) | 60 ± 0.8 | 1.1 ± 1.1 | 61 ± 0.8 | 2.0 ± 1.2 | −0.9 ± 1.7 (P = .6) | ||
| Baseline RHR ≥70 BPM | |||||||
| HbA1c ≤7 (N = 148 B‐QR, 87 P) | 78 ± 0.6 | −5.2 ± 0.8*** | 77 ± 0.7 | −2.5 ± 0.9** | −2.7 ± 1.2 (P = .03) | ||
| HbA1c >7 (N = 95 B‐QR, 42 P) | 79 ± 0.8 | −4.3 ± 0.9*** | 77 ± 0.9 | 0.7 ± 1.1 | −5.0 ± 1.6 (P = .002) | ||
| HbA1c ≥7.5 (N = 61 B‐QR, 31 P) | 79 ± 1.0 | −4.5 ± 1.2*** | 77 ± 1.2 | 1.6 ± 1.2 | −6.1 ± 1.9 (P = .002) | ||
| Baseline RHR ≥80 BPM | |||||||
| HbA1c ≤7 (N = 47 B‐QR, 29 P) | 86 ± 0.0.9 | −8.6 ± 1.6*** | 86 ± 0.7 | −4.1 ± 1.8* | −4.5 ± 2.4 (P = .07) | ||
| HbA1c >7 (N = 38 B‐QR, 10 P) | 87 ± 1.1 | −6.5 ± 1.5*** | 85 ± 2.2 | 1.3 ± 1.7 | −7.8 ± 3.1 (P = .015) | ||
| HbA1c ≥7.5 (N = 24 B‐QR, 9 P) | 86 ± 1.4 | −8.1 ± 1.9*** | 85 ± 2.4 | 1.8 ± 1.9 | −9.9 ± 3.3 (P = .005) | ||
Data shown as mean ± standard error of mean.
Abbreviations: HbA1c, haemoglobin A1c; RHR, resting heart rate.
P < .05 for within‐treatment group change in RHR.
P ≤ .01 for within‐treatment group change in RHR.
P ≤ .001 for within‐treatment group change in RHR.
The magnitude of the RHR reductions from baseline with B‐QR vs placebo was even greater when the analyses were limited to subjects with elevated baseline RHR ≥80 (N = 124:85 B‐QR, 39 placebo), with a mean change in RHR from baseline to week 24 with B‐QR therapy vs placebo of −4.5 BPM (P = .07) in those with baseline HbA1c ≤7, −7.8 BPM (P = .015) in those with baseline HbA1c >7 and −9.9 BPM (P = .005) in those with baseline HbA1c ≥7.5 (Figure 1; Table 4).
There was no significant change in RHR among subjects with baseline RHR <70 regardless of baseline HbA1c (RHR change week 0 to week 24 with B‐QR therapy vs placebo −0.04 [P = .95], −1.4 [P = .26] and −0.9 [P = .59] BPM in subjects with baseline HbA1c ≤7, >7, and ≥7.5, respectively).
3.5. Effect of baseline RHR on B‐QR's glycaemic effect
To assess the relationship between baseline RHR and the antidiabetes effect of B‐QR, the change in HbA1c from baseline to week 24 with B‐QR therapy vs placebo was analysed in subjects with poor glycaemic control (defined as baseline HbA1c ≥7.5%) at baseline (N = 198:125 B‐QR, 73 placebo), stratified by baseline RHR. HbA1c reduction as a function of baseline RHR demonstrated HbA1c reductions with B‐QR vs placebo as follows: RHR <70 BPM: −0.50 (P = .04); RHR ≥70 BPM: −0.73 (P = .005), ≥80 BPM: −1.22 (P = .008) (Figure 2).
Figure 2.
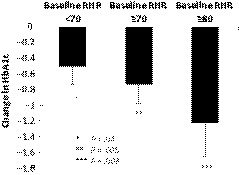
Glycaemic control effect of bromocriptine‐QR vs placebo (between‐group difference in change from baseline HbA1c) in T2DM subjects with suboptimal glycaemic control (baseline HbA1c ≥7.5) stratified by baseline resting heart rate. Data shown as mean ± SEM. RHR, resting heart rate
3.6. Relationship between B‐QR‐induced change in RHR and B‐QR's antidiabetes effect
The relationship between the B‐QR induced change in RHR and change in HbA1c was analysed in the subjects with baseline RHR ≥70 BPM and baseline HbA1c ≥7.5. The change (decrease) in RHR from baseline to week 24 significantly positively correlated with the change (decrease) in HbA1c (Pearson r = .40, P = .001) in subjects treated with B‐QR but not placebo. Multivariable regression analysis with change in HbA1c from baseline to week 24 as the outcome variable and change in RHR as a covariate along with age, gender, race, baseline HbA1c, and other concomitant diabetes medications (metformin, SU and/or TZD) as the other variables included in the analysis demonstrated that among the B‐QR treated subjects with baseline RHR ≥70 BPM and baseline HbA1c ≥7.5, the magnitude of RHR reduction is a significant independent predictor of B‐QR's effect on reducing HbA1c (β .42; P = .001) (see Table 5 for more details).
Table 5.
Relationship of treatment‐induced change in resting heart rate with change in haemoglobin A1c
|
Multivariable regression analysis Outcome: HbA1c change from baseline to week 24 with study drug treatment | ||||
|---|---|---|---|---|
| Covariates/Predictors | Bromocriptine‐QR | Placebo | ||
| Standardized β | P‐value | Standardized β | P‐value | |
| Age | −.06 | .66 | .36 | .15 |
| Gender (0 = female, 1 = male) | −.16 | .23 | −.28 | .18 |
| Race (0 = non‐Caucasian, 1 = Caucasian) | .23 | .06 | .03 | .89 |
| Baseline HbA1c | .01 | .92 | −.14 | .51 |
| Concomitant treatment with metformin (0 = no, 1 = yes) | −.12 | .34 | −.03 | .90 |
| Concomitant treatment with a SU (0 = no, 1 = yes) | .02 | .90 | −.13 | .58 |
| Concomitant treatment with a TZD (0 = no, 1 = yes) | −.10 | .42 | −.35 | .13 |
| Change in RHR from baseline to week 24 with study drug treatment | .42 | .001 | .13 | .57 |
Abbreviations: HbA1c, haemoglobin A1c; RHR, resting heart rate.
4. DISCUSSION
Circadian‐timed treatment of type 2 diabetes subjects with B‐QR reduced an elevated RHR (≥70 BPM or ≥80 BPM) by between approximately 3‐10 BPM relative to placebo dependent upon the baseline RHR and HbA1c level in the study population. The magnitude of this RHR reduction was greater the more elevated the baseline RHR above 70 BPM, with greater reductions seen with baseline RHR ≥80. There was no such B‐QR impact to reduce RHR in subjects with baseline RHR below 70 BPM. That is, the B‐QR influence to reduce RHR was only observable if the RHR was elevated to a range above 70 BPM, a threshold that has in previous studies been associated with increased risk of insulin resistance, metabolic syndrome, type 2 diabetes and adverse CVD outcomes.46, 47, 49, 50, 51, 61, 65 The B‐QR‐induced reduction in RHR was accompanied also by a concurrent reduction in BP, suggesting an influence to reduce central elevated sympathetic drive to the cardiovascular system.66, 73, 74, 76, 84 Importantly, the magnitude of the B‐QR effect to reduce elevated RHR increased also with increasing HbA1c level at baseline which may reflect higher levels of underlying elevated sympathetic activity contributing to both the elevated RHR and higher A1c in these subsets. In addition, the degree of B‐QR's impact to reduce elevated RHR was an independent predictor of its effect to reduce HbA1c among subjects with poor glycaemic control (baseline HbA1c ≥7.5). To our knowledge, this is the first demonstration of such an effect on RHR with any FDA‐approved antidiabetes medication. This interaction suggests that B‐QR therapy may be targeting an aetiologic factor of both elevated RHR and dysglycemia. This aetiologic factor likely is elevated SNS activity, an autonomic imbalance pathology known to both increase RHR and potentiate insulin resistance and dysglycemia.10, 12, 13, 26 Such a SNS target of B‐QR would be consistent with the reported sympatholytic mechanism of action of circadian‐timed B‐QR therapy to improve glycaemic control and reduce CVD risk in type 2 diabetes subjects.96, 105 However, although SNS innervation of the myocardium is extensive and exerts a prominent control of myocardial function,106 reduced parasympathetic drive to the heart may also contribute to elevated RHR 107 and central dopamine action can function to reverse this vagal imbalance as well,108 an effect which may also participate in the observed B‐QR effects on elevated RHR in this study.
It is important to appreciate in general and relative to the present investigation in particular that a large body of evidence indicates that while tachycardia is usually defined as heart rate ≥100 BPM, RHR thresholds substantially lower than this traditional tachycardia criterion are also associated with significant increased cardiovascular risks.39, 40, 41, 46, 47, 49, 50, 51, 53, 54, 61, 62, 65 Accumulating evidence from a multitude of large longitudinal epidemiological studies and clinical trials indicate that chronically elevated RHR over a threshold of approximately the >70‐80 BPM range is significantly associated with and is a predictor of increased cardiometabolic risk (insulin resistance, metabolic syndrome, type 2 diabetes and CVD) as well as both cardiovascular and all‐cause mortality and such associations have been reported in general healthy populations as well as in those with hypertension, coronary artery disease or heart failure.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 48, 51, 52, 53, 54, 55, 57, 58, 59, 60, 62, 63, 64, 109, 110 RHR ≥70 BPM has therefore been used as the cut‐off for defining elevated RHR in previous clinical studies having identified increased CVD risk above this threshold.45, 46, 47, 49, 50, 51, 53, 61, 62, 65, 111 Consequently, in the context of these reported findings, the reduction of elevated RHR above 70‐80 BPM by 6‐10 BPM with circadian‐timed B‐QR therapy in type 2 diabetes subjects with poor glycaemic control (HbA1c ≥7.5) in the present study can be clinically meaningful. These findings also may relate to (and provide a theoretical mechanistic basis [via reducing elevated SNS tone] for) the 40%‐50% reduction in CVD outcomes observed with this therapy in the type 2 diabetes population.98, 99, 100, 101
The observation that (a) the higher the RHR the greater the B‐QR induced reduction in RHR and (b) the magnitude of the elevated RHR reduction with B‐QR is an independent predictor of the magnitude of HbA1c reduction with the therapy in type 2 diabetes subjects whose glycaemia is poorly controlled is an interesting and potentially clinically important finding as it may help identify “best responders” to the therapy. An understanding of why the magnitude of the B‐QR effect to reduce RHR predicts the magnitude of its effect to reduce HbA1c in type 2 diabetes subjects whose glycemia is poorly controlled may best be obtained by (a) appreciating the relationship between RHR and SNS tone on the one hand and the influence of chronic elevated SNS activity upon cardiometabolic health and glycaemic control on the other and (b) realizing the sympatholytic nature of circadian B‐QR therapy upon chronically elevated SNS tone in insulin‐resistant states as follows. Importantly, in insulin resistance syndrome, elevated RHR (over 70‐80 BPM) is a marker of an increase in cardiac SNS dominance either in absolute terms or in relative terms of SNS/parasympathetic nervous system (PSNS) activity balance 27, 28, 106 and SNS overactivity is considered to be the most likely central mechanism to explain the association between elevated RHR and adverse cardiometabolic outcomes.12, 18, 26, 37 While the autonomic imbalance of elevated SNS and depressed PSNS activities to the heart each can contribute to elevated RHR, available evidence suggests that the elevated RHR association with insulin resistance syndrome is most closely coupled to overactive SNS tone that involves several metabolic tissues in addition to the heart.10, 12, 16, 17, 18, 19, 24, 25, 37, 112, 113 Both elevated RHR33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 51, 52, 53, 54, 55, 56, 57 and elevated SNS tone9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 114 are associated with and predict the future onset of CVD, insulin resistance, metabolic syndrome and type 2 diabetes. The association of elevated RHR with development of type 2 diabetes has been mainly attributed to increased insulin resistance secondary to elevated SNS activity,12, 16, 26, 35, 36, 37 although elevated RHR has also been reported to be a predictor of beta cell dysfunction and consequent impaired glucose regulation independent of the level of insulin sensitivity.34
Chronic SNS overactivity is an important pathophysiological phenomenon that potentiates hypertension, vasoconstriction and vascular insulin resistance,20, 115, 116, 117, 118 vascular oxidative stress,19, 119 endothelial dysfunction,19, 120, 121, 122 myocardial oxidative and nitrative stress and apoptosis,15 and renal renin‐angiotensin system overactivation,20, 21, 22, 23 as well as metabolic changes such as increased adipose inflammation and lipolysis3, 24, 123, 124 (inducing hyperaemia, a potent stimulus for central activation of SNS tone125, 126), increased hepatic oxidative stress, inflammation, lipotoxicity, glucose output, insulin resistance,66, 127 ectopic fat deposition24, 66 and muscle insulin resistance.128, 129, 130 Collectively, these pathophysiologies underlie development of vascular stiffness and arteriosclerosis, atherosclerosis and atherosclerosis progression, cardiac remodelling, occurrence of myocardial ischaemia and arrhythmias, reduced left ventricular function, kidney dysfunction, hypertension and metabolic derangements of obesity, dyslipidemia, metabolic syndrome and type 2 diabetes,10, 11, 12, 13, 41, 114, 131 occurrences often associated with elevated RHR.10, 11, 12, 13, 18, 26, 36, 37, 41, 114, 131
Long‐term bromocriptine therapy is well known to reduce elevated sympathetic tone in hypertensive animals and humans.66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 132 The present study extends the specifics of these findings on vascular hemodynamics and uncovers an important relationship between B‐QR impact on RHR and glycaemic control in type 2 diabetes subjects. Although acute peripheral effects of bromocriptine can function to inhibit noradrenaline release from sympathetic neurons, studies of chronic bromocriptine impact on elevated sympathetic tone implicate a dominant central mechanism of action in this regard.66, 74, 82 Insights into how circadian‐timed B‐QR therapy operates to simultaneously alleviate elevated RHR and dysglycemia in type 2 diabetes subjects may be derived from studies of hypothalamic biological clock circuitry control of autonomic balance and the neuroendocrine axis and dopamine's influence on this system as follows. The CNS biological clock circuitry centred on the SCN co‐ordinates autonomic and endocrine system modulation of biochemical metabolic events in the liver, adipose, muscle and other peripheral tissues including the vasculature and heart85, 86, 133, 134, 135 to generate a whole‐body metabolism and to synchronize/co‐ordinate metabolism within the individual to the cyclic environment (eg, circadian variations in vascular tone and heart rate associated with the sleep/wake cycle, co‐ordination of fuel mobilization with the daily sleep‐fasting period, and anabolic fuel storage processes with wake‐feeding periods of the day). The SCN is the seat of the autonomic nervous system, sending direct and indirect signals to multiple CNS (eg, hypothalamic) centres that are pre‐autonomic fibres regulating sympathetic/parasympathetic activity balance from moment to moment and rhythmically over the course of the day.85, 86, 87, 88, 135, 136, 137
A series of studies have demonstrated that a diminution of circadian peak dopaminergic activity at the SCN area signals the SCN to send neural messages to several brain centres to activate sympathetic tone and alter glucose and FFA sensing in a manner that potentiates glucose intolerance, insulin resistance, and leptin resistance.96 Elements of the western lifestyle including high fat/sugar diets, psychological stress and altered sleep/wake architecture all diminish brain dopamine activity and are strongly associated with insulin resistance syndrome.138, 139, 140, 141, 142 Reinstatement of the circadian peak in brain dopaminergic activity in insulin resistance syndrome attenuates these brain (SCN) neural signalling pathways that potentiate the syndrome.96 Such a reinstatement of CNS dopaminergic effect would be expected to manifest decreases in elevated RHR and dysglycemia in type 2 diabetes subjects as observed herein with circadian‐timed B‐QR therapy. Moreover, such B‐QR treatment would also be expected and has in fact been observed to reduce elevated plasma triglyceride and FFA levels and insulin resistance in type 2 diabetes.96
Circadian‐timed morning administration of B‐QR produces a brief pulse of dopaminergic activity to the body, including the CNS, that would reverse the diminished morning dopaminergic signalling to the CNS143 (including the biological clock SCN) of the type 2 diabetes patient and resultantly reduce overactivity of pre‐autonomic hypothalamic neurons in the brain areas that stimulate increased SNS outflow to the periphery in the manner described above. It should be realized that circadian‐timed B‐QR therapy may act to reduce sympathetic tone by multiple distinct mechanisms including (a) the above‐described action at dopamine receptors at the biological clock (SCN) to reduce its activation of hypothalamic pre‐autonomic sympathetic fibres,72, 144 (b) dopamine action directly on paraventricular nuclei pre‐autonomic sympathetic fibres to inhibit their SNS activation 93 and (c) peripheral action directly on postganglionic‐presynaptic sympathetic fibres to inhibit their noradrenaline release.75, 76, 145 It should be appreciated, however, that peripheral effects of dopamine agonism may themselves be regulated by CNS dopamine function.146 The absence of any significant effect of B‐QR on RHR or BP in those type 2 diabetes subjects with RHR <70 suggests that the “resetting” effect of timed B‐QR therapy may correct SNS hyperactivity or SNS‐to‐PSNS dominance to the heart responsible for increased RHR, but does not affect normal RHR/BP in the absence of vascular SNS hyperactivity, while SNS activity may still be elevated and impacted elsewhere (eg, liver, adipose) in the insulin‐resistant body.
The limitations of this study include the absence of any additional direct measures of SNS tone, the lack of physical activity/fitness data on the study subjects which may influence RHR and glycaemic control, and the lack of measures of insulin sensitivity to assess correlations between RHR and insulin action. The present findings however suggest that such future studies are warranted.
5. CONCLUSION
The present study has demonstrated that circadian‐timed bromocriptine‐QR therapy significantly reduces elevated (but not normal) RHR and blood pressure in type 2 diabetes subjects, the magnitude of which RHR reduction is positively correlated to each of the baseline elevated RHR and HbA1c level. This impact of B‐QR to reduce elevated RHR is an independent predictor of its impact to reduce elevated HbA1c (ie the greater the RHR reduction, the greater the HbA1c reduction). These findings lend further support to the reported bromocriptine‐QR mechanism of improving glycaemic control in type 2 diabetes in part via reduction of elevated SNS tone.105 These findings also suggest that type 2 diabetes maximum responder populations to bromocriptine‐QR may be those subjects with elevated (>~80 BPM) RHR or other markers of elevated SNS tone. The impact of B‐QR to reduce elevated RHR (and the antecedent elevated SNS tone) provides a potential contributing mechanism for the observed marked reduction in CVD outcomes with B‐QR therapy.98, 99, 100, 101
CONFLICT OF INTEREST
BC and ME are employed by VeroScience LLC. AV has received grant support from VeroScience. AHC is employed by and has equity interests in VeroScience LLC.
AUTHOR CONTRIBUTIONS
BC made substantial contributions to the conception/design of the work, analysis and interpretation of data, and drafting the manuscript. AV contributed to the interpretation of the data and critically revising the manuscript for important intellectual content. ME contributed to analysis of the data and drafting the manuscript. AHC made substantial contributions to the conception/design of the work, interpretation of data, and drafting the manuscript. All authors have read and approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
The Cycloset Safety Trial (CST) study protocol was approved by site‐specific or central institutional review boards, and all subjects provided written informed consent to participate in the study before enrolment. The study was conducted in accordance with the International Council for harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) 2004 guidelines. This current post hoc study and analyses are original and different from any previously reported results from the CST.
Chamarthi B, Vinik A, Ezrokhi M, Cincotta AH. Circadian‐timed quick‐release bromocriptine lowers elevated resting heart rate in patients with type 2 diabetes mellitus. Endocrinol Diab Metab. 2020;3:e00101 10.1002/edm2.101
Trial Registration: ClinicalTrials.gov Identifier: NCT00377676. Registered 18 September 2006.
Funding information
This study was funded by VeroScience, LLC and S2 Therapeutics, Inc.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Westfall TC, Westfall DP. Neurotransmission: the autonomic and somatic motor nervous systems In: Brunton LL, ed. Goodman and Gillman's the Pharmacological Basis of Therapeutics, 12 ed. McGraw Hill Medical; 2011:171‐218. [Google Scholar]
- 2. Bruno RM, Ghiadoni L, Seravalle G, Dell'oro R, Taddei S, Grassi G. Sympathetic regulation of vascular function in health and disease. Front Physiol. 2012;3:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steffens AB, Scheurink AJ, Luiten PG, Bohus B. Hypothalamic food intake regulating areas are involved in the homeostasis of blood glucose and plasma FFA levels. Physiol Behav. 1988;44:581‐589. [DOI] [PubMed] [Google Scholar]
- 4. Shimazu T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia. 1981;20:343‐356. [PubMed] [Google Scholar]
- 5. Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3:286‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev. 1987;3:185‐206. [DOI] [PubMed] [Google Scholar]
- 7. van den Hoek AM, van Heijningen C, Schroder‐van der Elst JP, et al. Intracerebroventricular administration of neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes. 2008;57:2304‐2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimazu T. Reciprocal innervation of the liver: its significance in metabolic control. Adv Metab Disord. 1983;10:355‐384. [DOI] [PubMed] [Google Scholar]
- 9. Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac beta‐adrenergic receptors, and heart failure. Circulation. 2000;101:1634‐1637. [DOI] [PubMed] [Google Scholar]
- 10. Lambert GW, Straznicky NE, Lambert EA, Dixon JB, Schlaich MP. Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther. 2010;126:159‐172. [DOI] [PubMed] [Google Scholar]
- 11. Grassi G, Quarti‐Trevano F, Seravalle G, Dell'Oro R. Cardiovascular risk and adrenergic overdrive in the metabolic syndrome. Nutr Metab Cardiovasc Dis. 2007;17:473‐481. [DOI] [PubMed] [Google Scholar]
- 12. Wulsin LR, Horn PS, Perry JL, Massaro JM, D'Agostino RB. Autonomic imbalance as a predictor of metabolic risks, cardiovascular disease, diabetes, and mortality. J Clin Endocrinol Metab. 2015;100:2443‐2448. [DOI] [PubMed] [Google Scholar]
- 13. Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. 2015;2015:341583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carnethon MR, Golden SH, Folsom AR, Haskell W, Liao D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: the Atherosclerosis Risk In Communities study, 1987–1998. Circulation. 2003;107:2190‐2195. [DOI] [PubMed] [Google Scholar]
- 15. Hu A, Jiao X, Gao E, et al. Chronic beta‐adrenergic receptor stimulation induces cardiac apoptosis and aggravates myocardial ischemia/reperfusion injury by provoking inducible nitric‐oxide synthase‐mediated nitrative stress. J Pharmacol Exp Ther. 2006;318:469‐475. [DOI] [PubMed] [Google Scholar]
- 16. Egan BM. Insulin resistance and the sympathetic nervous system. Curr Hypertens Rep. 2003;5:247‐254. [DOI] [PubMed] [Google Scholar]
- 17. Esler M. The sympathetic nervous system in hypertension: back to the future? Curr Hypertens Rep. 2015;17:11. [DOI] [PubMed] [Google Scholar]
- 18. Tentolouris N, Argyrakopoulou G, Katsilambros N. Perturbed autonomic nervous system function in metabolic syndrome. Neuromolecular Med. 2008;10:169‐178. [DOI] [PubMed] [Google Scholar]
- 19. Bleeke T, Zhang H, Madamanchi N, Patterson C, Faber JE. Catecholamine‐induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res. 2004;94:37‐45. [DOI] [PubMed] [Google Scholar]
- 20. Seravalle G, Grassi G. Sympathetic nervous system, hypertension, obesity and metabolic syndrome. High Blood Press Cardiovasc Prev. 2016;23:175‐179. [DOI] [PubMed] [Google Scholar]
- 21. DiBona GF. Sympathetic nervous system and the kidney in hypertension. Curr Opin Nephrol Hypertens. 2002;11:197‐200. [DOI] [PubMed] [Google Scholar]
- 22. Fisher JP, Young CN, Fadel PJ. Central sympathetic overactivity: maladies and mechanisms. Auton Neurosci. 2009;148:5‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology (Bethesda). 2007;22:310‐319. [DOI] [PubMed] [Google Scholar]
- 24. Geerling JJ, Boon MR, Kooijman S, et al. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. J Lipid Res. 2014;55:180‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lambert E, Straznicky N, Sari CI, et al. Dyslipidemia is associated with sympathetic nervous activation and impaired endothelial function in young females. Am J Hypertens. 2013;26:250‐256. [DOI] [PubMed] [Google Scholar]
- 26. Facchini FS, Stoohs RA, Reaven GM. Enhanced sympathetic nervous system activity. the linchpin between insulin resistance, hyperinsulinemia, and heart rate. Am J Hypertens. 1996;9:1013‐1017. [DOI] [PubMed] [Google Scholar]
- 27. Cooley RL, Montano N, Cogliati C, et al. Evidence for a central origin of the low‐frequency oscillation in RR‐interval variability. Circulation. 1998;98:556‐561. [DOI] [PubMed] [Google Scholar]
- 28. Monfredi O, Lyashkov AE, Johnsen A‐B, et al. Biophysical characterization of the underappreciated and important relationship between heart rate variability and heart rate. Hypertension. 2014;64:1334‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quarti Trevano F, Seravalle G, Macchiarulo M, et al. Reliability of heart rate as neuroadrenergic marker in the metabolic syndrome. J Hypertens. 2017;35:1685‐1690. [DOI] [PubMed] [Google Scholar]
- 30. Brown MR, Fisher LA, Spiess J, Rivier C, Rivier J, Vale W. Corticotropin‐releasing factor: actions on the sympathetic nervous system and metabolism. Endocrinology. 1982;111:928‐931. [DOI] [PubMed] [Google Scholar]
- 31. Fisher LA, Jessen G, Brown MR. Corticotropin‐releasing factor (CRF): mechanism to elevate mean arterial pressure and heart rate. Regul Pept. 1983;5:153‐161. [DOI] [PubMed] [Google Scholar]
- 32. Troisi RJ, Weiss ST, Parker DR, Sparrow D, Young JB, Landsberg L. Relation of obesity and diet to sympathetic nervous system activity. Hypertension. 1991;17:669‐677. [DOI] [PubMed] [Google Scholar]
- 33. Jiang X, Liu X, Wu S, et al. Metabolic syndrome is associated with and predicted by resting heart rate: a cross‐sectional and longitudinal study. Heart. 2015;101:44‐49. [DOI] [PubMed] [Google Scholar]
- 34. Bonnet F, Empana JP, Natali A, et al. Elevated heart rate predicts beta cell function in non‐diabetic individuals: the RISC cohort. Eur J Endocrinol. 2015;173:409‐415. [DOI] [PubMed] [Google Scholar]
- 35. Shigetoh Y, Adachi H, Yamagishi S‐I, et al. Higher heart rate may predispose to obesity and diabetes mellitus: 20‐year prospective study in a general population. Am J Hypertens. 2009;22:151‐155. [DOI] [PubMed] [Google Scholar]
- 36. Bemelmans RHH, Wassink AMJ, van der Graaf Y, et al. Risk of elevated resting heart rate on the development of type 2 diabetes in patients with clinically manifest vascular diseases. Eur J Endocrinol. 2012;166:717‐725. [DOI] [PubMed] [Google Scholar]
- 37. Aune D, Ó Hartaigh B, Vatten LJ. Resting heart rate and the risk of type 2 diabetes: a systematic review and dose–response meta‐analysis of cohort studies. Nutr Metab Cardiovasc Dis. 2015;25:526‐534. [DOI] [PubMed] [Google Scholar]
- 38. Bohm M, Reil JC, Deedwania P, Kim JB, Borer JS. Resting heart rate: risk indicator and emerging risk factor in cardiovascular disease. Am J Med. 2015;128:219‐228. [DOI] [PubMed] [Google Scholar]
- 39. Ho JE, Larson MG, Ghorbani A, et al. Long‐term cardiovascular risks associated with an elevated heart rate: the Framingham Heart Study. J Am Heart Assoc. 2014;3:e000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fox K, Borer JS, Camm AJ, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823‐830. [DOI] [PubMed] [Google Scholar]
- 41. Tadic M, Cuspidi C, Grassi G. Heart rate as a predictor of cardiovascular risk. Eur J Clin Invest. 2018;48(3). https://orcid.org/10.1111/eci.12892 [DOI] [PubMed] [Google Scholar]
- 42. Caetano J, Delgado AJ. Heart rate and cardiovascular protection. Eur J Intern Med. 2015;26:217‐222. [DOI] [PubMed] [Google Scholar]
- 43. Cooney MT, Vartiainen E, Laatikainen T, Juolevi A, Dudina A, Graham IM. Elevated resting heart rate is an independent risk factor for cardiovascular disease in healthy men and women. Am Heart J. 2010;159:612‐619.e3. [DOI] [PubMed] [Google Scholar]
- 44. Arnold JM, Fitchett DH, Howlett JG, Lonn EM, Tardif JC. Resting heart rate: a modifiable prognostic indicator of cardiovascular risk and outcomes? Can J Cardiol. 2008;24(Suppl A):3A‐8A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Böhm M, Swedberg K, Komajda M, et al. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo‐controlled trial. Lancet. 2010;376:886‐894. [DOI] [PubMed] [Google Scholar]
- 46. Fox K, Bousser M‐G, Amarenco P, et al. Heart rate is a prognostic risk factor for myocardial infarction: a post hoc analysis in the PERFORM (Prevention of cerebrovascular and cardiovascular Events of ischemic origin with teRutroban in patients with a history oF ischemic strOke or tRansient ischeMic attack) study population. Int J Cardiol. 2013;168:3500‐3505. [DOI] [PubMed] [Google Scholar]
- 47. Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left‐ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817‐821. [DOI] [PubMed] [Google Scholar]
- 48. Tverdal A, Hjellvik V, Selmer R. Heart rate and mortality from cardiovascular causes: a 12 year follow‐up study of 379,843 men and women aged 40–45 years. Eur Heart J. 2008;29:2772‐2781. [DOI] [PubMed] [Google Scholar]
- 49. Nikolovska Vukadinović A, Vukadinović D, Borer J, et al. Heart rate and its reduction in chronic heart failure and beyond. Eur J Heart Fail. 2017;19:1230‐1241. [DOI] [PubMed] [Google Scholar]
- 50. Swedberg K, Komajda M, Böhm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet. 2010;376:875‐885. [DOI] [PubMed] [Google Scholar]
- 51. Woodward M, Webster R, Murakami Y, et al. The association between resting heart rate, cardiovascular disease and mortality: evidence from 112,680 men and women in 12 cohorts. Eur J Prev Cardiol. 2014;21:719‐726. [DOI] [PubMed] [Google Scholar]
- 52. Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long‐term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967‐974. [DOI] [PubMed] [Google Scholar]
- 53. Khan H, Kunutsor S, Kalogeropoulos AP, et al. Resting heart rate and risk of incident heart failure: three prospective cohort studies and a systematic meta‐analysis. J Am Heart Assoc. 2015;4:e001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang S‐L, Wang C‐L, Wang P‐L, et al. Resting heart rate associates with one‐year risk of major adverse cardiovascular events in patients with acute coronary syndrome after percutaneous coronary intervention. Exp Biol Med (Maywood). 2016;241:478‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Castagno D, Skali H, Takeuchi M, et al. Association of heart rate and outcomes in a broad spectrum of patients with chronic heart failure: results from the CHARM (Candesartan in Heart Failure: Assessment of Reduction in Mortality and morbidity) program. J Am Coll Cardiol. 2012;59:1785‐1795. [DOI] [PubMed] [Google Scholar]
- 56. Vazir A, Claggett B, Cheng S, et al. Association of resting heart rate and temporal changes in heart rate with outcomes in participants of the atherosclerosis risk in communities study. JAMA Cardiol. 2018;3:200‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bemelmans RHH, van der Graaf Y, Nathoe HM, et al. The risk of resting heart rate on vascular events and mortality in vascular patients. Int J Cardiol. 2013;168:1410‐1415. [DOI] [PubMed] [Google Scholar]
- 58. Custodis F, Roggenbuck U, Lehmann N, et al. Resting heart rate is an independent predictor of all‐cause mortality in the middle aged general population. Clin Res Cardiol. 2016;105:601‐612. [DOI] [PubMed] [Google Scholar]
- 59. Greenland P, Daviglus ML, Dyer AR, et al. Resting heart rate is a risk factor for cardiovascular and noncardiovascular mortality: the Chicago Heart Association Detection Project in Industry. Am J Epidemiol. 1999;149:853‐862. [DOI] [PubMed] [Google Scholar]
- 60. Alhalabi L, Singleton MJ, Oseni AO, Shah AJ, Zhang ZM, Soliman EZ. Relation of higher resting heart rate to risk of cardiovascular versus noncardiovascular death. Am J Cardiol. 2017;119:1003‐1007. [DOI] [PubMed] [Google Scholar]
- 61. Disegni E, Goldbourt U, Reicher‐Reiss H, et al. The predictive value of admission heart rate on mortality in patients with acute myocardial infarction. SPRINT Study Group. Secondary Prevention Reinfarction Israeli Nifedipine Trial. J Clin Epidemiol. 1995;48:1197‐1205. [DOI] [PubMed] [Google Scholar]
- 62. Nauman J, Janszky I, Vatten LJ, Wisloff U. Temporal changes in resting heart rate and deaths from ischemic heart disease. JAMA. 2011;306:2579‐2587. [DOI] [PubMed] [Google Scholar]
- 63. Kristal‐Boneh E, Silber H, Harari G, Froom P. The association of resting heart rate with cardiovascular, cancer and all‐cause mortality. Eight year follow‐up of 3527 male Israeli employees (the CORDIS Study). Eur Heart J. 2000;21:116‐124. [DOI] [PubMed] [Google Scholar]
- 64. Gillman MW, Kannel WB, Belanger A, D'Agostino RB. Influence of heart rate on mortality among persons with hypertension: the Framingham Study. Am Heart J. 1993;125:1148‐1154. [DOI] [PubMed] [Google Scholar]
- 65. Pittaras AM, Faselis C, Doumas M, et al. Heart rate at rest, exercise capacity, and mortality risk in veterans. Am J Cardiol. 2013;112:1605‐1609. [DOI] [PubMed] [Google Scholar]
- 66. Ezrokhi M, Luo S, Trubitsyna Y, Cincotta AH. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol Metab Syndr. 2014;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oguro M, Takeda K, Itoh H, et al. Role of sympathetic nerve inhibition in the vasodepressor effect of bromocriptine in normotensive and hypertensive rats. Jpn Circ J. 1992;56:943‐949. [DOI] [PubMed] [Google Scholar]
- 68. Kanayama Y, Kohno M, Takaori K, Itoh S, Yasunari K, Takeda T. Involvement of sympathetic nervous system inhibition in the hypotensive effect of bromocriptine in spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1987;14:141‐144. [DOI] [PubMed] [Google Scholar]
- 69. Lahlou S, Duarte GP. Hypotensive action of bromocriptine in the DOCA‐salt hypertensive rat: contribution of spinal dopamine receptors. Fundam Clin Pharmacol. 1998;12:599‐606. [DOI] [PubMed] [Google Scholar]
- 70. Kaya D, Ellidokuz E, Onrat E, Ellidokuz H, Celik A, Kilit C. The effect of dopamine type‐2 receptor blockade on autonomic modulation. Clin Auton Res. 2003;13:275‐280. [DOI] [PubMed] [Google Scholar]
- 71. Lahlou S, Demenge P. Contribution of spinal dopamine receptors to the hypotensive action of bromocriptine in rats. J Cardiovasc Pharmacol. 1991;18:317‐325. [DOI] [PubMed] [Google Scholar]
- 72. Falk RH, Desilva RD, Lown B. Reduction in vulnerability to ventricular fibrillation by bromocriptine, a dopamine agonist. Cardiovasc Res. 1981;15:175‐180. [DOI] [PubMed] [Google Scholar]
- 73. Ziegler MG, Lake CR, Williams AC, Teychenne PF, Shoulson I, Steinsland O. Bromocriptine inhibits norepinephrine release. Clin Pharmacol Ther. 1979;25:137‐142. [DOI] [PubMed] [Google Scholar]
- 74. Franchi F, Lazzeri C, Barletta G, Ianni L, Mannelli M. Centrally mediated effects of bromocriptine on cardiac sympathovagal balance. Hypertension. 2001;38:123‐129. [DOI] [PubMed] [Google Scholar]
- 75. Mannelli M, Ianni L, Lazzeri C, et al. In vivo evidence that endogenous dopamine modulates sympathetic activity in man. Hypertension. 1999;34:398‐402. [DOI] [PubMed] [Google Scholar]
- 76. Mannelli M, Delitala G, De feo LM, et al. Effects of different dopaminergic antagonists on bromocriptine‐induced inhibition of norepinephrine release. J Clin Endocrinol Metab. 1984;59:74‐78. [DOI] [PubMed] [Google Scholar]
- 77. Kolloch R, Kobayashi K, DeQuattro V. Dopaminergic control of sympathetic tone and blood pressure: evidence in primary hypertension. Hypertension. 1980;2:390‐394. [DOI] [PubMed] [Google Scholar]
- 78. Sowers JR. Dopaminergic control of circadian norepinephrine levels in patients with essential hypertension. J Clin Endocrinol Metab. 1981;53:1133‐1137. [DOI] [PubMed] [Google Scholar]
- 79. Sowers JR, Stern N, Nyby MD, Jasberg KA. Dopaminergic regulation of circadian rhythms of blood pressure, renin and aldosterone in essential hypertension. Cardiovasc Res. 1982;16:317‐323. [DOI] [PubMed] [Google Scholar]
- 80. Mejia‐Rodriguez O, Alvarez‐Aguilar C, Ledesma‐Ramirez M, Paniagua‐Sierra R. Therapeutic effect of bromocriptine together with the established treatment for hypertension in patients undergoing peritoneal dialysis. Proc West Pharmacol Soc. 2004;47:122‐124. [PubMed] [Google Scholar]
- 81. Durrieu G, Senard JM, Tran MA, Rascol A, Montastruc JL. Effects of levodopa and bromocriptine on blood pressure and plasma catecholamines in parkinsonians. Clin Neuropharmacol. 1991;14:84‐90. [DOI] [PubMed] [Google Scholar]
- 82. Montastruc JL, Chamontin B, Rascol A. Parkinson's disease and hypertension: chronic bromocriptine treatment. Neurology. 1985;35:1644‐1647. [DOI] [PubMed] [Google Scholar]
- 83. Degli Esposti E, Sturani A, Santoro A, Zuccala A, Chiarini C, Zucchelli P. Effect of bromocriptine treatment on prolactin, noradrenaline and blood pressure in hypertensive haemodialysis patients. Clin Sci (Lond). 1985;69:51‐56. [DOI] [PubMed] [Google Scholar]
- 84. Luchsinger A, Velasco M, Urbina A, et al. Comparative effects of dopaminergic agonists on cardiovascular, renal, and renin‐angiotensin systems in hypertensive patients. J Clin Pharmacol. 1992;32:55‐60. [DOI] [PubMed] [Google Scholar]
- 85. Kalsbeek A, Palm IF, La Fleur SE, et al. SCN outputs and the hypothalamic balance of life. J Biol Rhythms. 2006;21:458‐469. [DOI] [PubMed] [Google Scholar]
- 86. Buijs RM, la Fleur SE, Wortel J, et al. The suprachiasmatic nucleus balances sympathetic and parasympathetic output to peripheral organs through separate preautonomic neurons. J Comp Neurol. 2003;464:36‐48. [DOI] [PubMed] [Google Scholar]
- 87. Kalsbeek A, Bruinstroop E, Yi CX, Klieverik LP, La Fleur SE, Fliers E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann N Y Acad Sci. 2010;1212:114‐129. [DOI] [PubMed] [Google Scholar]
- 88. Luo S, Zhang Y, Ezrokhi M, Li Y, Tsai TH, Cincotta AH. Circadian peak dopaminergic activity response at the biological clock pacemaker (suprachiasmatic nucleus) area mediates the metabolic responsiveness to a high‐fat diet. J Neuroendocrinol. 2018;30(1). 10.1111/jne.12563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin‐releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology. 2000;71:68‐78. [DOI] [PubMed] [Google Scholar]
- 90. Luo S, Ezrokhi M, Trubitsyna Y, Li Y, Cincotta AH. Elevation of norepinephrine (NE) activity at the ventromedial hypothalamus (VMH) of normal rats induces the obese hypertensive insulin restsant state without altered feeding. Diabetes. 2015;64(Suppl 1):A540. [Google Scholar]
- 91. Luo S, Meier AH, Cincotta AH. Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology. 1998;68:1‐10. [DOI] [PubMed] [Google Scholar]
- 92. Ezrokhi M, Trubitsyna Y, Luo S, Cincotta AH. Timed dopamine agonist treatment ameliorates both vascular nitrosative /oxidative stress pathology and aortic stiffness in arteriosclerotic, hypertensive SHR rats. Diabetes. 2010;59(Suppl 1):A67. [Google Scholar]
- 93. Zheng H, Liu X, Li Y, Mishra PK, Patel KP. Attenuated dopaminergic tone in the paraventricular nucleus contributing to sympathoexcitation in rats with type 2 diabetes. Am J Physiol Regul Integr Comp Physiol. 2014;306:R138‐R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. ter Horst KW, Lammers NM, Trinko R, et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci Transl Med. 2018;10(442). pii: eaar3752. 10.1126/scitranslmed.aar3752 [DOI] [PubMed] [Google Scholar]
- 95. Caravaggio F, Borlido C, Hahn M, et al. Reduced insulin sensitivity is related to less endogenous dopamine at D2/3 receptors in the ventral striatum of healthy nonobese humans. Int J Neuropsychopharmacol. 2015;18:pyv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Raskin P, Cincotta AH. Bromocriptine‐QR therapy for the management oif type 2 diabetes mellitus: developmental basis and therapeutic profile summary. Expert Rev Endocrinol Metab. 2016;11:113‐148. [DOI] [PubMed] [Google Scholar]
- 97. Shivaprasad C, Kalra S. Bromocriptine in type 2 diabetes mellitus. Indian J Endocrinol Metab. 2011;15:S17‐S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gaziano JM, Cincotta AH, O'Connor CM, et al. Randomized clinical trial of quick‐release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33:1503‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gaziano JM, Cincotta AH, Vinik A, Blonde L, Bohannon N, Scranton R. Effect of bromocriptine‐QR (a quick‐release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J Am Heart Assoc. 2012;1:e002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chamarthi B, Gaziano JM, Blonde L, et al. Timed bromocriptine‐QR therapy reduces progression of cardiovascular disease and dysglycemia in subjects with well‐controlled type 2 diabetes mellitus. J Diabetes Res. 2015;2015:157698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chamarthi B, Ezrokhi M, Rutty D, Cincotta AH. Impact of bromocriptine‐QR therapy on cardiovascular outcomes in type 2 diabetes mellitus subjects on metformin. Postgrad Med. 2016;128:761‐769. [DOI] [PubMed] [Google Scholar]
- 102. Scranton RE, Gaziano JM, Rutty D, Ezrokhi M, Cincotta A. A randomized, double‐blind, placebo‐controlled trial to assess safety and tolerability during treatment of type 2 diabetes with usual diabetes therapy and either cycloset or placebo. BMC Endocr Disord. 2007;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stocker SD, Gordon KW. Glutamate receptors in the hypothalamic paraventricular nucleus contribute to insulin‐induced sympathoexcitation. J Neurophysiol. 2015;113:1302‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti‐insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol. 2013;304:H1538‐H1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Defronzo RA. Bromocriptine: a sympatholytic, d2‐dopamine agonist for the treatment of type 2 diabetes. Diabetes Care. 2011;34:789‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zaglia T, Mongillo M. Cardiac sympathetic innervation, from a different point of (re)view. J Physiol. 2017;595:3919‐3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Levy MN, Martin PJ. Autonomic control of cardiac conduction and automaticity In: Shepherd JT, Vatner SF, eds. Nervous Control of the Heart. Amsterdam: Harwood Academic Publishers; 1996:201‐226. [Google Scholar]
- 108. Dyavanapalli J, Byrne P, Mendelowitz D. Activation of D2‐like dopamine receptors inhibits GABA and glycinergic neurotransmission to pre‐motor cardiac vagal neurons in the nucleus ambiguus. Neuroscience. 2013;247:213‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Böhm M, Borer J, Ford I, et al. Heart rate at baseline influences the effect of ivabradine on cardiovascular outcomes in chronic heart failure: analysis from the SHIFT study. Clin Res Cardiol. 2013;102:11‐22. [DOI] [PubMed] [Google Scholar]
- 110. Grassi G, Arenare F, Quarti‐Trevano F, Seravalle G, Mancia G. Heart rate, sympathetic cardiovascular influences, and the metabolic syndrome. Prog Cardiovasc Dis. 2009;52:31‐37. [DOI] [PubMed] [Google Scholar]
- 111. Fox K, Komajda M, Ford I, et al. Effect of ivabradine in patients with left‐ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trials. Eur Heart J. 2013;34:2263‐2270. [DOI] [PubMed] [Google Scholar]
- 112. Liao D, Sloan RP, Cascio WE, et al. Multiple metabolic syndrome is associated with lower heart rate variability. The Atherosclerosis Risk in Communities Study. Diabetes Care. 1998;21:2116‐2122. [DOI] [PubMed] [Google Scholar]
- 113. Pikkujamsa SM, Huikuri HV, Airaksinen KE, et al. Heart rate variability and baroreflex sensitivity in hypertensive subjects with and without metabolic features of insulin resistance syndrome. Am J Hypertens. 1998;11:523‐531. [DOI] [PubMed] [Google Scholar]
- 114. Grassi G, Seravalle G, Mancia G. Sympathetic activation in cardiovascular disease: evidence, clinical impact and therapeutic implications. Eur J Clin Invest. 2015;45:1367‐1375. [DOI] [PubMed] [Google Scholar]
- 115. Mancia G, Grassi G. The autonomic nervous system and hypertension. Circ Res. 2014;114:1804‐1814. [DOI] [PubMed] [Google Scholar]
- 116. Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res. 2015;116:976‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vollenweider P, Randin D, Tappy L, Jequier E, Nicod P, Scherrer U. Impaired insulin‐induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest. 1994;93:2365‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jamerson KA, Julius S, Gudbrandsson T, Andersson O, Brant DO. Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension. 1993;21:618‐623. [DOI] [PubMed] [Google Scholar]
- 119. Yasunari K, Matsui T, Maeda K, Nakamura M, Watanabe T, Kiriike N. Anxiety‐induced plasma norepinephrine augmentation increases reactive oxygen species formation by monocytes in essential hypertension. Am J Hypertens. 2006;19:573‐578. [DOI] [PubMed] [Google Scholar]
- 120. Spieker LE, Hürlimann D, Ruschitzka F, et al. Mental stress induces prolonged endothelial dysfunction via endothelin‐A receptors. Circulation. 2002;105:2817‐2820. [DOI] [PubMed] [Google Scholar]
- 121. Hijmering ML, Stroes ES, Olijhoek J, Hutten BA, Blankestijn PJ, Rabelink TJ. Sympathetic activation markedly reduces endothelium‐dependent, flow‐mediated vasodilation. J Am Coll Cardiol. 2002;39:683‐688. [DOI] [PubMed] [Google Scholar]
- 122. Ghiadoni L, Donald AE, Cropley M, et al. Mental stress induces transient endothelial dysfunction in humans. Circulation. 2000;102:2473‐2478. [DOI] [PubMed] [Google Scholar]
- 123. Wang YY, Lin SY, Chuang YH, Chen CJ, Tung KC, Sheu WH. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am J Physiol Endocrinol Metab. 2011;300:E155‐E163. [DOI] [PubMed] [Google Scholar]
- 124. Kuo LE, Czarnecka M, Kitlinska JB, Tilan JU, Kvetnansky R, Zukowska Z. Chronic stress, combined with a high‐fat/high‐sugar diet, shifts sympathetic signaling toward neuropeptide Y and leads to obesity and the metabolic syndrome. Ann N Y Acad Sci. 2008;1148:232‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Florian JP, Pawelczyk JA. Non‐esterified fatty acids increase arterial pressure via central sympathetic activation in humans. Clin Sci (Lond). 2009;118:61‐69. [DOI] [PubMed] [Google Scholar]
- 126. Gadegbeku CA, Dhandayuthapani A, Sadler ZE, Egan BM. Raising lipids acutely reduces baroreflex sensitivity. Am J Hypertens. 2002;15:479‐485. [DOI] [PubMed] [Google Scholar]
- 127. Wang YY, Lin SY, Chuang YH, Sheu WH, Tung KC, Chen CJ. Activation of hepatic inflammatory pathways by catecholamines is associated with hepatic insulin resistance in male ischemic stroke rats. Endocrinology. 2014;155:1235‐1246. [DOI] [PubMed] [Google Scholar]
- 128. Julius S, Gudbrandsson T, Jamerson K, Tariq Shahab S, Andersson O. The hemodynamic link between insulin resistance and hypertension. J Hypertens. 1991;9:983‐986. [DOI] [PubMed] [Google Scholar]
- 129. Lembo G, Capaldo B, Rendina V, et al. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am J Physiol. 1994;266:E242‐E247. [DOI] [PubMed] [Google Scholar]
- 130. Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest. 1990;85:1844‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mancia G, Bousquet P, Elghozi JL, et al. The sympathetic nervous system and the metabolic syndrome. J Hypertens. 2007;25:909‐920. [DOI] [PubMed] [Google Scholar]
- 132. Hoekfelt B. Dopamine receptor stimulants in hypertension (discussion). Acta Med Scand. 1977(Suppl);606:100. [DOI] [PubMed] [Google Scholar]
- 133. Scheer FA, Ter Horst GJ, van Der Vliet J, Buijs RM. Physiological and anatomic evidence for regulation of the heart by suprachiasmatic nucleus in rats. Am J Physiol Heart Circ Physiol. 2001;280:H1391‐H1399. [DOI] [PubMed] [Google Scholar]
- 134. Buijs FN, Cazarez F, Basualdo MC, et al. The suprachiasmatic nucleus is part of a neural feedback circuit adapting blood pressure response. Neuroscience. 2014;266:197‐207. [DOI] [PubMed] [Google Scholar]
- 135. Warren WS, Champney TH, Cassone VM. The suprachiasmatic nucleus controls the circadian rhythm of heart rate via the sympathetic nervous system. Physiol Behav. 1994;55:1091‐1099. [DOI] [PubMed] [Google Scholar]
- 136. Bartness TJ, Song CK, Demas GE. SCN efferents to peripheral tissues: implications for biological rhythms. J Biol Rhythms. 2001;16:196‐204. [DOI] [PubMed] [Google Scholar]
- 137. Buijs RM, Chun SJ, Niijima A, Romijn HJ, Nagai K. Parasympathetic and sympathetic control of the pancreas: a role for the suprachiasmatic nucleus and other hypothalamic centers that are involved in the regulation of food intake. J Comp Neurol. 2001;431:405‐423. [DOI] [PubMed] [Google Scholar]
- 138. Alghasham A, Rasheed N. Stress‐mediated modulations in dopaminergic system and their subsequent impact on behavioral and oxidative alterations: an update. Pharm Biol. 2014;52:368‐377. [DOI] [PubMed] [Google Scholar]
- 139. Davis JF, Tracy AL, Schurdak JD, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci. 2008;122:1257‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159:1193‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Volkow ND, Tomasi D, Wang G‐J, et al. Evidence that sleep deprivation downregulates dopamine D2R in ventral striatum in the human brain. J Neurosci. 2012;32:6711‐6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Volkow ND, Wang G‐J, Telang F, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. NeuroImage. 2008;42:1537‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dunn JP, Kessler RM, Feurer ID, et al. Relationship of dopamine type 2 receptor binding potential with fasting neuroendocrine hormones and insulin sensitivity in human obesity. Diabetes Care. 2012;35:1105‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Vila E, Badia A, Jane F. Effects of bromocriptine on catecholamine receptors mediating cardiovascular responses in the pithed rat. J Auton Pharmacol. 1985;5:125‐130. [DOI] [PubMed] [Google Scholar]
- 145. Mannelli M, Lazzeri C, Ianni L, et al. Dopamine and sympathoadrenal activity in man. Clin Exp Hypertens. 1997;19:163‐179. [DOI] [PubMed] [Google Scholar]
- 146. Cincotta A. Hypothalamic role in the insulin resistance syndrome In: Hansen B, Shaffrir E, eds. Insulin Resistance and Insulin Resistance Syndrome. London: Taylor and Francis; 2002:271‐312. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.