

Abstract
The development of new anti-cancer therapeutic agents is necessary to improve antitumor efficacy and reduce toxicities. Here we report using a systematic anticancer drug screening approach we developed previously, to concurrently screen colon and glioma cancer cell lines for 2000 compounds with known bioactivity and 1920 compounds with unknown activity. The hits specific to each tumor cell line were then selected, and further tested with the same cells transfected with EGFP (Enhanced Green Fluorescent Protein) alone. By comparing the percentage of signal reduction from the same cells transfected with the sensor-conjugated reporter system; hits preferably causing apoptosis were identified. Among the known lead compounds, many cardiac glycosides used as cardiotonic drugs were found to effectively and specifically kill colon cancer cells, while statins (hypolipidemic agents) used as cholesterol lowering drugs were relatively more effective in killing glioma cells.
Keywords: Drug discovery, colorectal cancer, glioma, carditonic, HTS, statins
1. INTRODUCTION
Cancer is already the second leading cause of death in the US, and is anticipated to become the leading cause of mortality within the next several years. After decades of systematic research and development, a pool of chemotherapeutic agents has been established for the treatment of human cancers [1]. While most of those agents work mainly by impairing mitosis and targeting fast-dividing cells [2], they also have a host of serious side effects. Therefore, the need for anticancer agents that are both more effective and less toxic remains as high as ever [3]. Current approaches to drug discovery have increasingly embraced target-based methods, which include target identification, target validation, hit discovery and lead compound optimization. A target is usually defined as a single gene, gene product or molecular mechanism that has been identified based on genetic analysis or biological observations. The advantages of the target-based identification approach include the ability to apply molecular and chemical knowledge to investigate specific molecular hypotheses, higher throughput and the ability to use small-molecule high throughput screening strategies as well as biologic-based approaches, such as identifying monoclonal antibodies [4]. However, the effectiveness of the targeted-based approach has been questioned [5], despite its emergence as the dominant paradigm, it has posted the lowest rate of new drug approvals in generations [6]. A recent study showed the contribution of target-based screening to first-in-class small molecule drug discovery led to only 17 FDA approvals, a number far less than the 28 approvals contributed by phenotypic approaches. In the subcategory of cancer drugs, in spite of overwhelming efforts and popularity, the target-based approach barely produced more approved drugs [4]. This apparent lack of productivity can be explained mainly by a fundamental pitfall in target-based approaches. Because of the difficulty involved in fully recognizing all disease-relevant targets, hypotheses often stem from prior biases. As a result, targets are selected based on what seems available, but with incomplete knowledge. Further, target validation is typically performed in limited or semi-artificial scenarios. Notably, it is also likely that the selected target plays an essential role in normal cell functions. Furthermore, tumorigenesis does not always result from a single static target. Rather, the triggering or key target may also be obscured by other secondary, but more dominant targets as the tumor advances. Therefore, a compelling rationale exists for the use of alternative approaches to improve screening procedures and increase success rates [7]. Recently, we developed a systematic anticancer drug screening reporter system based on controlled and self-amplified protein degradation.8 In this new approach, the sensor-conjugated reporter system reduces optical fluorescent signals and can effectively detect apoptosis, growth arrest and cell death. When used in high throughput screening (HTS), the approach demonstrated conclusively that it was simple, cost effective and exhibited high efficacy with a very low false positive rate. Here, we report data collected using this approach to concurrently screen about 4000 compounds against colon and glioma cancer cell lines. The preliminary hits were tested with the same cell lines transfected with EGFP alone or with the sensor-conjugated reporter system, by comparing the percentage of signal reduction in the optical readout between the same cell lines with different transfectants or between two different cell lines. Through these comparisons, we were able to identify hits which typically caused apoptosis as well as those specific to each tumor cell line. Although many of the hits have already been reported or possess limited clinical value, several cardiac glycosides used as cardiotonic drugs were found to be not only very powerful, but also to be specific for killing colon cancer cells. In contrast, statins (hypolipidemic agents) used as cholesterol lowering drugs are relatively more effective to kill glioma cells.
2. MATERIALS AND METHOD
2.1. Cell Culture
Rat glioma C6, 9L cells and breast tumor MDA-231 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA), and human colon cancer cells DLD1, SW620, Difi, Lim405, HCT116 and Km12 were kindly provided by Dr. Robert J Coffey (Vanderbilt University, Nashville, TN). All cells were cultured in DMEM medium supplemented with 10% FBS and 50 units/ml penicillin and 50μg/ml streptomycin (Invitrogen, CA) under standard culture conditions in a humidified incubator maintained at 5% CO2 and 37°C.
2.2. Compound Library
The compounds tested in this study are managed and distributed by the Vanderbilt HTS core facility (http://www.vanderbilt.edu/hts/). Six 384-well plates (1,920 compounds) are part of the VICB collection (sources from ChemDiv and ChemBridge), are chosen to maximally represent the chemical diversity of the larger ChemDiv and ChemBridge compound collections which includes natural products, natural product derivatives and synthetic compounds. In addition, 2,000 compounds with known bioactivity known as the Spectrum Collection were obtained from Mircrosource Discovery Systems, Inc. (Gaylordsville, CT) in seven 384 well plates. This library consists of 60% drug components; 25% natural products and 15% other bioactive components.
2.3. Assay Procedure
HTS assays were performed based on a reporter system we developed [8] to detect cellular apoptosis, growth arrest and cell death based on controlled and self-amplified protein degradation. The key component of the reporter system is an apoptotic sensor as a part of a chimerical fused fluorescent protein, consisting of a peptide sequence DEVD (Asp-Glu-Val-Asp)-linked procaspase 3, ubiquitin (Ub), and a strong consensus sequence of N-degron. This non-conventional signal loss approach has been demonstrated to provide excellent sensitivity and thus it is suited for 386-well HTS platforms. The simplicity eliminates operational errors and variations introduced by multiple steps; and because there is no need for any substrate, it’s also cost effective.
2.4. High Throughput Screening Assay
Prior to HTS assays, C6 and DLD1 cells were stably transfected with the reporter system as previously described [8]. The transfected C6 (1000) or DLD1 (500) cells were seeded in multiple 386-well plates for concurrent and comparative drug selection in 30μl of complete growth medium and allowed to settle overnight. About twenty-four hours after seeding, compounds from the Vanderbilt HTS compound library were prepared with each compound at a stock concentration of 10mM in DMSO, and then diluted to 40μM in growth media. 10μl of each compound was then transferred into cell plates to yield a final screening concentration of 10μM by using a high-precision multi-well pipetting instrument (Velocity 11 Bravo) within the HTS facility. No-treatment and vehicle-treated (DMSO alone) controls were setup against each of the cell lines in the first and last two columns of each 384-well plate per standard HTS protocols. After 2 days, the cell media was removed and fluorescence was measured per well from the top by a plate reader (Synergy 2, BioTek, Winooski, VT).
2.5. Confirmation and Counter-Screen of Preliminary Hits
Positive hits based on signal reductions greater than 6x standard deviations (based on 4 columns of 16 control wells) were further confirmed by repeating the screening at a single concentration, but against four cell plates. Two plates were identical to the initial screening, seeded with C6 and DLD1 stably transfected with the sensor-conjugated reporter vector; two additional plates were also setup with C6 and DLD1 stably transfected with EGFP alone. Tumor specific hits against glioma and colorectal cancer cells were selected based on comparing signal differences. The percentage changes from cells expressing sensor-conjugated reporter to those from cells expressing EGFP alone were compared and analyzed to select pro-apoptotic hits.
2.6. Characterization of the IC50 of the Hit Compounds
IC50 is defined here as cell growth inhibition by each hit compound measured at the end of 2 days of treatment with cells stably transfected with the reporter system. The data was calculated by fitting dose-response curves. When IC50 was determined, the selected hit compounds were serially diluted with complete medium to obtain a concentration range from 30μM to 1.5nM. 10μl of this dilution was added to each well containing 30μl of medium and cells. After 2 days, the cell media was dumped, and fluorescence was measured per well from the top by a plate reader.
2.7. Fluorescence Microscopy of Live Cells
EGFP or sensor-reporter transfected cells were seeded in the wells of 384-well black plates for HTS, after the screening experiments (preliminary, confirmation or IC50 determination). The cells were directly examined under a standard fluorescence microscope or imaged using an automated fluorescence microscope in the HTS facility (Molecular Devices ImageXpress Micro XL) with cells covered by a thin layer of PBS.
2.8. Effects of Selective Hits on Other Cell Types
Selective hits were purchased from Aldrich Market Select (with Vitas-M Laboratory, Ltd. Sigma, and ChemBridge Corporation as the suppliers), and were prepared in DMSO at 20mM stock. Eight cell lines, two glioma cells (C6, 9L); five colorectal cancer lines (Difi, Lim405, HCT116, SW620 and KM12), and one breast carcinoma cells MDA231; were plated in multiple wells (1×103 cells per well) of 96-well plates. 24 hours after the seeding, the selective hits (Lovastatin, Simvastatin, and VU0008642 as glioma specific, Neriifolin, Strophanthidin, and VU0008957 as colorectal cancer cells specific) were added into the wells of the plates. After 3 days additional incubation, cell numbers were determined by characterizing the DNA contents with CyQuant reagent (Invitrogen). For the same setups with compound treatments, an apoptosis assay based on Caspase 3/7 activity was also performed (caspase-Glo kit, Promega, Madison, WI) after 24 hours treatment.
2.9. Data Processing
Raw fluorescence values were compared to the average of multiple controls (64 wells each plate with no drug treatment); hits in screening runs were identified as compounds showing repressing activity greater than six standard deviations from the control population. IC50 was estimated from dose-response curves by using GraphPad Prism software (GraphPad Software, La Jolla, CA).
3. RESULTS
3.1. High Throughput Screening of the Chemical Library Against Glioma and Colorectal Cancer Cells
Compound libraries (3920 compounds total) were screened against glioma (C6) and colorectal cancer cells (DLD1) concurrently by using a reporter system we developed8, which differ from conventional approach in 1) no need for any external assay reagent; 2) with few steps; 3) more sensitive by using signal reduction for positive hits. The initial screen with 10μM final concentrations against both cells lines yielded 242 hits, and the degree of inhibition for the first 1,000 compounds from both libraries for C6 cells appears in supplemental data Fig. (1). Hits were selected based on fluorescence strength, which is equivalent to 6x standard deviation below control average. As expected, the percentage of hits is much higher in the Spectrum library than those from unbiased libraries. Specifically, 216 hits were confirmed after the second confirmatory screening, 152 hits (approximately 7.6%) coming from the Spectrum library, while 64 (approximately 3.3%) were from the VICB library. The confirmed hits are summarized in the supplemental data (Supplemental Table 1). Considering possible instrumental errors, the data from this larger-scale screening demonstrate further that our methodology produced a very low false positive rate. Notably, the confirmatory screening might produce more errors than preliminary screening because the operation to pick out testing compounds, as suggested by that some of antineoplastic hits were mistakenly eliminated. Beyond hits related to alkylating agents known to target DNA synthesis and mitosis, there are antibacterial, antifungal, antiprotozoal, antiamebic or insecticidal compounds serve primarily to block protein synthesis. Other toxic chemicals, such as irritants, herbicides, antivirals, chelating agents or anti-infective compounds were noted as well. Clearly, the hits also include target-specific compounds such as calcium channels or NF-kappaB blockers, heat shock protein, Na+/K+ pump or TrkA (neurotrophic tyrosine kinase receptor type 1) receptor inhibitors.
3.2. The Most Effective Compounds in Killing Both Cell Lines
Most of the hit compounds are toxic chemicals in the category of antiviral, antibacterial, antifungal or anti-infection drugs, such as hinokitiol or oxyquinoline hemisulfate (see supplemental Table 2). One of few exceptions is Teniposide, a strong chemotherapeutic agent, which causes dose-dependent single-and double-stranded breaks in DNA and DNA-protein cross-links, and was mainly used previously in the treatment of childhood acute lymphocytic leukemia (ALL). Another is Nisoldipine, a calcium channel blocker of the dihydropyridine class, used to treat high blood pressure by relaxing blood vessels. This compound was also selected out when we screened the NIH clinical library [8]; the potential application of this class of molecules as anti-tumor drugs may merit further investigation in the future.
3.3. Colorectal Cancer and Glioma-Specific Lead Compounds
During the initial and confirmatory screening runs, two tumor cell lines with different tissue origins were evaluated concurrently against the same compound library. We define a compound as being relatively tumor specific when it attains less than a 15% inhibition rate in one cell line but achieves a rate greater than 50% in the second cell line (with compound concentration of 10μM and treatment time of 48 hours). Overall, more than 90% of these tumor specific hits can be confirmed on the second run. The selective inhibition of these hits was further confirmed by their similar actions on cells that expressed EGFP alone (equivalent to native tumor cells). The top nine hits specific to C6 glioma cells are listed in Table 1; three of which are antihyperlipidemic (simvastatin, lovastatin, rosuvastatin), inhibit HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase, the rate-controlling enzyme in the mevalonate pathway that produces cholesterol and other isoprenoids. Therefore, they are clinically used for the reduction of lipid or cholesterol levels in the blood. Another glioma-specific inhibitor is lefunomide (a pyrimidine synthesis inhibitor), an antirheumatic drug used to treat active, moderate to severe rheumatoid arthritis and psoriatic arthritis [9]. Chlorambucil, a nitrogen mustard alkylating agent that can be administered orally, is a chemotherapy drug used primarily in the treatment of chronic lymphocytic leukemia. Menadione, a synthetic chemical compound sometimes used as a nutritional supplement because of its vitamin K activity, is an analog of 1,4-naphthoquinone with a methyl group in the 2-position. Of late, menadione, in combination with vitamin C, has undergone study for its potential as a treatment for prostate cancer [10]. The last three compounds in the table, VU0042151, VU0042244, VU0008642 have not been characterized and have unknown function.
Table 1.
Top 9 Hits Relatively Specific to C6 Glioma Cells.
| VU ID | Formula | Chemical name | Therapeutic Use |
|---|---|---|---|
| VU0239489 | C12H9F3N2O2 | Lefunomide | PDGF receptor blocker |
| VU0243381 | C25H38O5 | Simvastatin | HMGCoA reductase inhibitor |
| VU0239686 | C24H36O5 | Lovastatin | HMGCoA reductase inhibitor |
| VU0239761 | C14H19Cl2NO2 | Chlorambucil | Alkylating Agent |
| VU0243301 | C22H28FN3O6S | Rosuvastatin | HMGCoA reductase inhibitor |
| VU0243203 | C11H8O2 | Menadione | Prothrombogenic agent |
| VU0042151 | C17H13ClN2O2S2 | VU0042151 | |
| VU0042244 | C18H15ClN2O2S2 | VU0042244 | |
| VU0008642 | C15H8BrF3N2O | VU0008642 |
The top 15 hits specific to colorectal cancer DLD1 cells are listed in Table 2. Most of these are cardiotonic steroids, the cardiac glycoside has been used to treat or prevent cardiac arrhythmias and increase cardiac output. The identified hits are Convallatoxin, Cymarin, Oleandrin, Gitoxigenin, Gitoxin, Digitoxin, Digoxin, Emicymarin, Sarmentogenin, Neriifolin and Strophanthidin. While most of these are natural products, some are isometric to each other or are derivatives of others. For instance, digitoxigenin can be derived from the hydrolysis of digitoxin. These compounds may have the ability to inhibit the proliferation of tumor cells and stimulate apoptosis due to high concentration of intracellular calcium that occurs via inhibition of the Na/K-ATPase pump. Oleandrin has also been reported to suppress the activation of NF-kappa B and AP-1 and c-Jun NH2-terminal kinase [11]. The Oleandrin-mediated suppression of NF-kappa B was not restricted to human epithelial cells as it has also been observed in human lymphoid, insect and murine macrophage cells [12]. The known compound on the list unrelated to a cardiotonic is Astemizole, a histamine H1-receptor antagonist that also acts as a functional inhibitor of sphingomyelinase [13]. Since histamine favors the proliferation of normal and malignant cells, antihistaminic drugs can inhibit tumor cell proliferation. Astemizole has gained enormous interest since it also targets important proteins involved in cancer progression, namely, ether à-go-go 1 (Eag1, a voltage-gated, K(+)-selective channel) and the Eag-related gene (Erg) potassium channels. Eag1 is thought to be an important marker and a therapeutic target for several different cancers. Astemizole can inhibit Eag1 and Erg channel activity and decrease tumor cell proliferation both in vitro and in vivo [14]. The last three compounds that appear in Table 2 are new, uncharacterized compounds. Structural analysis of these compounds, suggest that some may be a ligand to the EGF receptor and can act as a CDK2 inhibitor.
Table 2.
Top 15 Hits Relatively Specific to DLD1 Colon Cancer Cells.
| VU ID | Formula | Chemical Name | Therapeutic Use |
|---|---|---|---|
| VU0243959 | C25H32O7 | Strophanthidinic lactate | Cardiotonic |
| VU0243643 | C29H42O10 | Convallatoxin | Cardiotonic |
| VU0243985 | C30H44O9 | Cymarin | Cardiotonic |
| VU0243649 | C32H48O9 | Oleandrin | Cardiotonic |
| VU0243891 | C27H38O7 | Gitoxigenin diacetate | Cardiotonic |
| VU0243780 | C41H64O14 | Gitoxin | Cardiotonic |
| VU0243072 | C41H64O13 | Digitoxin | Cardiotonic |
| VU0243073 | C41H64O14 | Digoxin | Cardiotonic |
| VU0244322 | C23H34O5 | Sarmentogenin | Cardiotonic |
| VU0243685 | C30H46O8 | Neriifolin | Cardiotonic |
| VU0239915 | C28H31FN4O | Astemizole | H1 antihistamine |
| VU0243719 | C23H32O6 | Strophanthidin | Cardiotonic |
| VU0042387 | C22H27ClN2O3 | VU0042387 | |
| VU0007775 | C18H17N5O | VU0007775 | |
| VU0008957 | C23H21FN2O | VU0008957 |
3.4. Selection of Pro-Apoptotic Hits
Preliminary hits were further screened against the same two cell lines but were transfected with only EGFP. The resultant effects were compared to those obtained from cell lines transfected with the sensor-conjugated reporter systems. Those hits show dramatic effects on cells with sensor-conjugated reporter systems compared to those with EGFP alone. As a result, they will be considered as pro-apoptotic hits. The data show that more than 90% of the hits produced greater signal reduction in the reporter system than those in EGFP alone. This suggests that apoptosis plays some role in the action of majority hits. Based on the magnitude of the differences, the top 12 hits that are the most apoptotic for both C6 and DLD1 cell lines are listed in Table 3. Unfortunately, most of these are toxic chemicals within the category of antiviral, antibacterial, antifungal or anti-infection drugs. Erysolin is very effective in inducing apoptosis and cell death and has been reported for many colon cancel lines [15]. Dactinomycin is used to treat Wilms and Ewings tumors, testicular cancer, sarcomas, and other types of cancers. Pristimerol is listed in PubChem and other databases as having no bioactivity; however, its analogs (celastrols) were found to be powerful inducers for heat shock proteins, which are directly related to tumor cell proliferation [16]. Celastrols has been reported to show antibacterial properties at 30μg/ml [17]. By applying the same approach, we analyzed the hits unique to each cell lines and the resultant data show that apoptosis plays a role in all of these hits.
Table 3.
Top Apoptotic Hits to Both C6 Glioma and DLD1 Colon Cancer Cells.
| VU ID | Formula | Chemical Name | Therapeutic Use |
|---|---|---|---|
| VU0243589 | C19H14O3 | Rosolic acid | Diagnostic acid |
| VU0244412 | C9H11N3O4 | Cyclocytidine hydrochloride | Antineoplastic |
| VU0243606 | C8Cl4N2 | Tetrachloroisophthalonitrile | Antifungal |
| VU0243609 | C10H12O2 | Hinokitiol | Antifungal, insecticide |
| VU0243952 | C30H42O4 | Pristimerol | |
| VU0243623 | C6H11NO2S2 | Erysolin | Antiproliferative |
| VU0244478 | C22H30Cl2N10 | Chlorhexidine | Antibacterial, disinfectant |
| VU0244428 | C26H56N10 | Alexidine hydrochloride | Antibacterial |
| VU0243096 | C13H6Cl6O2 | Hexachlorophene | Antiinfective (topical) |
| VU0244355 | C25H30N3 | Gentian violet | Antibacterial, anthelmintic |
| VU0239513 | C9H5I2NO | Iodoquinol | Antiamebic |
| VU0243287 | C64H88N10O16 | Dactinomycin | Antineoplastic |
3.5. IC50 for Selective Hits
IC50 was determined by using a drug concentration range from 30μM to about 15nM. For statins which are relative specific to Glioma cells, they have higher IC50 (at μM range) as shown in Fig. (2). The estimated IC50 for Lovastatin and Simvastatin on C6 cells are 230nM and 900nM, respectively. However, cardiac glycosides have a lower IC50 for DLD1 cells; the IC50 for Neriifolin and Strophanthidin on DLD1 cells are 25nM and 250nM respectively. Since cardiac glycosides are structurally related, we estimated all their IC50 by using our sensor-conjugated reporter system with 48 hours treatment. The results are summarized in Table 4. The lowest IC50 is convallatoxin, about 10nM, neriifolin and oleandrin are also effective. The least effective one is digoxin. Interestingly, strophanthidinic lactone is about eight times more potent than strophanthidin, so this information may provide useful insights for future drug design and modification.
Fig. (2).

Fluorescent microscopic images of C6 and DlD1 cells after been treated with selective cell-specific hits. EGFP transfected C6 or DLD1 cells were treated with or without 10μM of selected compounds for 48 hours, and then examined and imaged under a fluorescent microscope. The images at 20x amplification were taken from cells of control (no added compound, DMSO only), and three selected C6 specific hits -Lovastatin, Simvastatin, and VU0008642-as labeled (Top panel, first row are C6 cells, and second row is DLD1 cells) (A); The same setup as Panel A, but drug compounds used are Neriifolin, Strophanthidin, and VU0008957, which are more specific to DLD1 cells (B).
Table 4.
IC50 of Cardiac Glycosides on Colorectal DLD-1 cells.
| Name | IC50(μM) | Structure |
|---|---|---|
| OLEANDRIN | 0.03 | 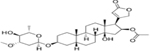 |
| GITOXIGENIN | 0.34 | 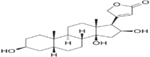 |
| GITOXIN | 2 | |
| DIGITOXIN | 0.3 | |
| DIGOXIN | 7.5 | |
| NERIIFOLIN | 0.025 | 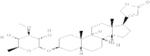 |
| STROPHANTHIDIN* | 0.25 | 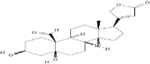 |
| CONVALLATOXIN | 0.01 |  |
| CYMARIN | 0.1 |  |
Strophanthidinic lactone has an IC50 of 0.04μM.
3.6. Validation of Selected Hits in Other Cancer Cell Lines for Specificity and Potency
Based on their commercial availability, three lead compounds that relate specifically to glioma (Lovastatin, Simvastatin and VU0008642) or to colorectal cancer cells (Neriifolin, Strophanthidin and VU0008957) were selected for further confirmation. To rule out the possibility that the effects of these compounds on the target cells were solely caused by the reporter senor aggregated action, the cell images were observed following 2 days of treatment on EGFP alone-transfected cells. As shown in the top panel of Fig. (2), based on the factors of cell number and fluorescence brightness, Lovastatin, Simvastatin, and VU0008642 indeed imparted a greater inhibitory effect on growth among C6 glioma cells (top row) than on DLD1 cells. Alternatively, Neriifolin, Strophanthidin and VU0008957 exhibited significant growth inhibition on DLD1 cells but produced minimal effects on C6 glioma cells (Panel B of Fig. (2). To test their specificity and the effect on the proliferation of target cells after a longer course of treatment, these six compounds were used to treat eight cell lines: two glioma cells (C6, 9L), five colorectal cancer lines (Difi, Lim405, HCT116, SW620 and KM12) and one breast carcinoma cells MDA231. Of those compounds, 10μM of each was added to multiple wells of the target cells and treated for 3 days. Following the treatment, cell numbers were quantified using the CyQUANT cell proliferation assay kit, which measures DNA content. As shown in the top three graphs of Fig. (3), although Lovastatin, Simvastatin and VU0008642 showed greater inhibition on gliomas (C6 and 9L, the 2nd cluster), those compounds also showed inhibitory effects on colon and breast cancer cells after 3 days of treatment. Interestingly, while Neriifolin, Strophanthidin and VU0008957 showed surprisingly small effects on glioma cells after 3 days of treatment, they produced an inhibition rate of greater than 75% on all colon cancer cells tested (bottom three graphs of Fig. 3). These 3 compounds also exhibited significant antitumor activity among breast cancer MDA231 cells, most likely because they share epithelial origins. Notably, as determined by the caspase-Glo kit (data not shown), all compounds slightly increased cellular apoptotic activities in the target cells after 24 hours. Specifically, the degree of caspase activation by the compounds is roughly proportional to their potential to inhibit cell proliferation.
Fig. (3).
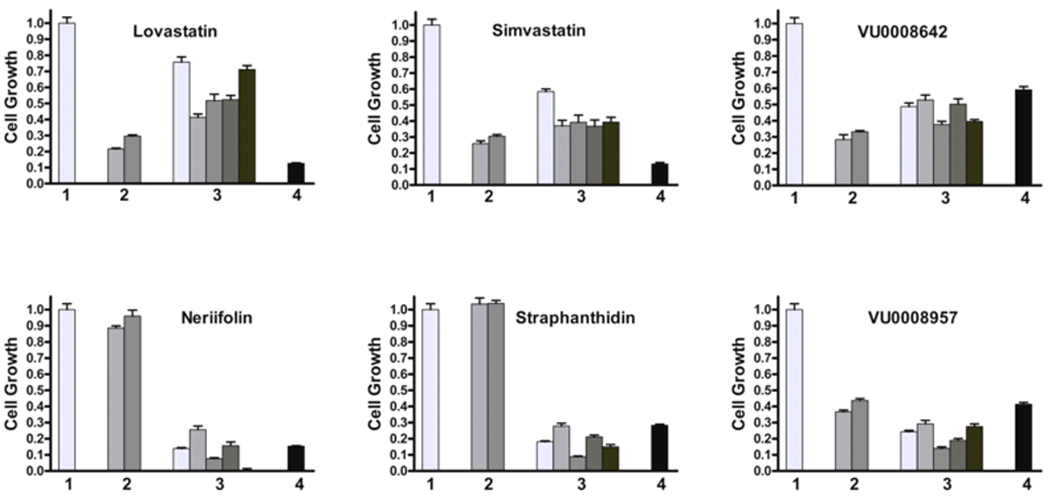
Test tumour specific hits on the growth inhibition of cancer cells from same or different tissue origins. Along with controls, six compounds in two groups were used to treat eight cell lines, two glioma cells (C6, 9L); five colorectal cancer lines (Difi, Lim405, HCT116, SW620 and KM12), and one breast carcinoma cells MDA231; all cells (1×103) were plated in multiple wells of multiple 96-well plates. 24 hours after seeding, DMSO or 10μM of these compound were added to multiple wells of these cells and treated for 3 days, the cell proliferation were quantified by using CyQUANT cell proliferation assay. No drug treatment (DMSO alone) group was normalized as 1 (standard derivation was calculated from all 9 cell lines) and given as single bar in cluster 1, data from two glioma cells (C6, 9L) were plotted in cluster 2, data from five colorectal cells (from left to right, are Difi, Lim405, HCT116, SW620 and KM12 respectively) were plotted in cluster 3; the data for MDA-231 were plotted in cluster 4. All the data from treatment group were normalized against their individual cell line’s no treatment controls. Drug compounds used are Lovastatin, Simvastatin, and VU0008642 as glioma specific, Neriifolin, Strophanthidin, and VU0008957 as colorectal cancer cells specific. Each data point was the average from 6 wells; standard deviations were shown as error bars.
4. DISCUSSION
Gliomas are characterized by acquired genetic mutations. For instance, tumor suppressor protein 53 (p53) is an early mutation. It is believed that subsequent events are caused by mutations found in phosphatase, tensin homolog (PTEN), epidermal growth factor receptor (EGFR) and other genes. Recently, mutations in isocitrate dehydrogenases (IDH1 and IDH2) were found in 40% of gliomas and in 100% of the 1p/19q codeleted gliomas, mutations that are inversely correlated to grade. The IDH1 mutation is a strong and independent predictor of survival regardless of grade [18]. The IDH1 and IDH2 genes are involved in the citrate cycle in mitochondria and play a key role in the metabolism of fat and cholesterol. As a result, they are likely indirectly involved in the alteration of glycolysis and apoptosis. Here, we found that Lovastatin and Simvastatin, members of the statins family, can inhibit glioma proliferation and induce their apoptosis. Statins have been recognized as inhibitors to HMG-CoA reductase and can reduce cholesterol biosynthesis. Moreover, they may relate directly or indirectly to the regulation of IDHs. Clinical studies have shown that statin use can reduce cancer-related mortality and it is believed that reduced cholesterol availability constrained the cellular proliferation necessary for cancer growth and metastasis [19]. It has been reported that several natural (lovastatin, simvastatin and pravastatin) and synthetic (cerivastatin and atorvastatin) statins exert a cytotoxic effect on human lymphoblasts and myeloma tumor cells by activating the mitochondrial pathway of apoptosis [20]. These data suggest the potential application of statins in the treatment of certain types of tumors when used alone or in combination with other chemotherapeutic or molecular-targeted agents or with radiotherapy or chemopreventive therapy [21]. C6 is morphologically similar to GBM when injected into the brain of neonatal rats and it has been widely used as an experimental model system for glioblastoma growth and invasion [22]. These features will provide an ideal small animal model with which we can study the effectiveness of statins and other selected lead compounds as anticancer drugs in vivo.
The colorectal adenocarcinoma cell line DLD-1 used in our preliminary HTS screening carries a p53 mutation (C -> T mutation which results in Ser -> Phe at position 241) [23]. This also proved positive for keratin and multiple oncogenes, such as c-myc, K-ras, H-ras, N-ras, myb, sis and fos; however, it proved negative for N-myc [24]. Thus, DLD-1 represents both a malignant and metastatic colorectal cell. The other commonly mutated gene in colorectal cancer is APC, TGF-β (or the downstream protein, SMAD) [25]. By comparative screening against glioma cells, most known lead compounds specific to colon cancer cells that we discovered are cardiac glycosides, which target to the sodium pump (Na+/K+-ATPase) on cell membranes. These cardiac steroids have been widely used in the treatment of congestive heart failure. The altered expression of sodium pump subunits has been reported in different cancer types. In non-small cell lung cancers (NSCLCs), glioblastomas (GBMs), melanomas and kidney tumors, the cancer cells overexpress the alpha-1 subunit of the sodium pump compared to corresponding normal tissues. In human colorectal cancers, however, it was reported that the alpha 3-isoform is upregulated, yet the alpha 1-isoform is actually downregulated [26]. We observed that these cardiac glycosides were highly potent to all of the colon cell lines we tested but had far less effect on glioma. This could suggest that these compounds have a high selective affinity to alpha 3-isoform or that they act through other mechanisms of which we are unaware.
CONCLUSION
Despite the hundreds of ongoing clinical trials for anticancer drugs, most trials for novel drug treatments have failed due to unexpected side effects and lack of efficacy. Therefore, drugs developed previously that possess novel antitumor properties may offer viable and cost-effective alternatives for treating cancer. The implication that statins and cardiotonic steroids can be employed as anticancer drugs may merit further investigation. Currently, the development of these compounds as anticancer agents has been impaired by their potential to induce cardiovascular side effects. As a result, efforts should be made to modify these molecules to reduce or mitigate their side effects and improve their anticancer ability [27]. Our IC50 data for various carditonic steroids on colon cancer cells will provide insight on the relationship between the potency and their molecular structures. Other lead compounds discovered from libraries with unknown compounds will be subjected to future characterization. Such additional investigations may provide clues about a new class of molecules capable of attacking colorectal cancers.
Supplementary Material
Fig. (1).
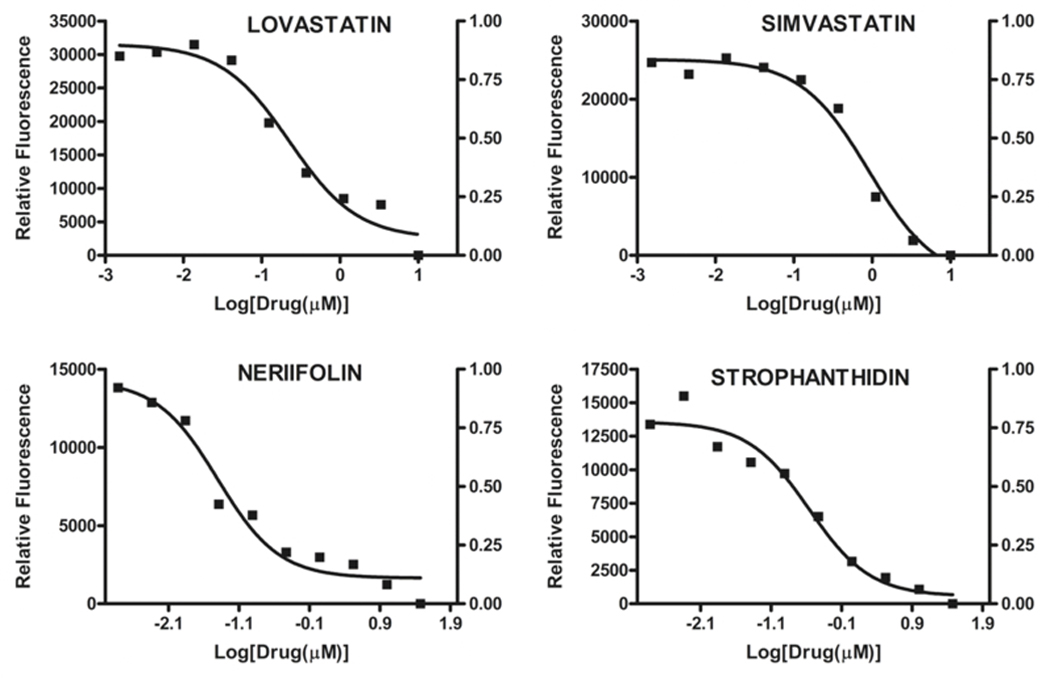
IC-50 estimation of selective hits from dose response curve. Reporter-expressing C6(1×103 cells) and DLD1 cells (500 cells) were plated in multiple wells of several 384-well plates. 24 hours after seeding, the tested compounds were serially diluted 1:3 with complete medium by 9 times (for a concentration range from 30μM to 1.5nM). After 2 days’ incubation, the cell media was dumped, fluorescence was measured per well from the top by a plate reader. Each data point represents the average of a triplicate reading, data fit was carried out by GraphPad. The estimated IC-50 for Lovastatin and Simvastatin on C6 cells are 230nM and 900nM, respectively. Neriifolin Strophanthidin on DLD1 cells are 25nM and 250nM, respectively.
ACKNOWLEDGEMENTS
We thank Drs. D. Mi and J. Bauer for helps in HTS execution, and Drs. R. Coffey and C. Manning for colon cancer cell lines. This research was partially supported by grants from the National Cancer Institute 1P50CA128323 and U24CA126588 to Dr. John Gore.
Biography
Jingping Xie

John C. Gore

Footnotes
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- [1].Calles ECA Chemotherapeutic Agents and Their Uses, Dosages, and Toxicities In: Cancer Management: A Multidisciiplinary Approach, Medical, Surgical. and Radiaton Oncology, 13 edition ed.; Pazdur, R., Ed. Cmp; : 2011; [Google Scholar]; (b) Jessup JM; McGinnis LS; Winchester DP; Eyre H; Fremgen A; Murphy GP; Menck HR Clinical highlights from the National Cancer Data Base: 1996. CA Cancer J. Clin, 1996, 46(3), 185–192. [DOI] [PubMed] [Google Scholar]
- [2].Calvo CTE Principles of Oncologic Pharmacotherapy. 11 ed.; 2008. [Google Scholar]
- [3].Mans DRA; da Rocha AB; Schwartsmann G. Anti-cancer drug discovery and development in brazil: targeted plant collection as a rational strategy to acquire candidate anti-cancer compounds. Oncologist, 2000, 5(3), 185–198. [DOI] [PubMed] [Google Scholar]
- [4].Swinney DC; Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov, 2011, 10(7), 507–519. [DOI] [PubMed] [Google Scholar]
- [5].Sams-Dodd F. Is poor research the cause of the declining productivity of the pharmaceutical industry? An industry in need of a paradigm shift. Drug Discov. Today, 2013, 18 (5–6), 211–217. [DOI] [PubMed] [Google Scholar]
- [6].Williams M. Productivity shortfalls in drug discovery: contributions from the preclinical sciences? J. Pharmacol. Exp. Ther, 2011, 336(1), 3–8. [DOI] [PubMed] [Google Scholar]
- [7].Reaume AG Drug repurposing through nonhypothesis driven phenotypic screening. Drug Discov. Today, 2011, 8(3), 4. [Google Scholar]
- [8].Xie J; Wang C; Virostko J; Manning HC; Pham W; Bauer J; Gore JC A novel reporter system for molecular imaging and high-throughput screening of anticancer drugs. Chembiochem, 2013, 14(12), 1494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pinto P; Dougados M. Leflunomide in clinical practice. Acta Reumatol. Port, 2006, 31(3), 215–224. [PubMed] [Google Scholar]
- [10].Jamison JM; Gilloteaux J; Taper HS; Summers JL Evaluation of the in vitro and in vivo antitumor activities of vitamin C and K-3 combinations against human prostate cancer. J. Nutr, 2001, 131(1), 158S–160S. [DOI] [PubMed] [Google Scholar]
- [11].Manna SK; Sah NK; Newman RA; Cisneros A; Aggarwal BB Oleandrin suppresses activation of nuclear transcription factor-kappaB, activator protein-1, and c-Jun NH2-terminal kinase. Cancer Res., 2000, 60(14), 3838–3847. [PubMed] [Google Scholar]
- [12].Sreenivasan Y; Sarkar A; Manna SK Oleandrin suppresses activation of nuclear transcription factor-kappa B and activator protein-1 and potentiates apoptosis induced by ceramide. Biochem. Pharmacol, 2003, 66(11), 2223–2239. [DOI] [PubMed] [Google Scholar]
- [13].Kornhuber J; Muehlbacher M; Trapp S; Pechmann S; Friedl A; Reichel M; Muhle C; Terfloth L; Groemer TW; Spitzer GM; Liedl KR; Gulbins E; Tripal P. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE, 2011, 6(8), e23852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Garcia-Quiroz J; Camacho J. Astemizole: an old anti-histamine as a new promising anti-cancer drug. Anticancer Agents Med. Chem, 2011, 11(3), 307–314. [DOI] [PubMed] [Google Scholar]
- [15].Kim MJ; Kim SH; Lim S-J, Comparison of the Apoptosis-inducing Capability of Sulforaphane Analogues in Human Colon Cancer Cells. Anticancer Res., 2010, 30(9), 3611–3619. [PubMed] [Google Scholar]
- [16].Westerheide SD; Bosman JD; Mbadugha BNA; Kawahara TLA; Matsumoto G; Kim S; Gu W; Devlin JP; Silverman RB; Morimoto RI Celastrols as Inducers of the Heat Shock Response and Cytoprotection. J. Biological. Chem, 2004, 279 (53), 56053–56060. [DOI] [PubMed] [Google Scholar]
- [17].Lopez MR; de Leon L; Moujir L. Antibacterial properties of phenolic triterpenoids against Staphylococcus epidermidis. Planta Med., 2011, 77(7), 726–729. [DOI] [PubMed] [Google Scholar]
- [18].Ducray F; Idbaih A; Wang XW; Cheneau C; Labussiere M; Sanson M. Predictive and prognostic factors for gliomas. Expert Rev. Anticancer Ther, 2011, 11(5), 781–789. [DOI] [PubMed] [Google Scholar]
- [19].Nielsen SF; Nordestgaard BG; Bojesen SE Statin use and reduced cancer-related mortality. New Eng. J. Med, 2012, 367(19), 1792–1802. [DOI] [PubMed] [Google Scholar]
- [20].Cafforio P; Dammacco F; Gernone A; Silvestris F, Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis, 2005, 26(5), 883–891. [DOI] [PubMed] [Google Scholar]
- [21].Chan KKW; Oza AM; Siu LL, The Statins as Anticancer Agents. Clin. Cancer Res, 2003, 9(1), 10–19. [PubMed] [Google Scholar]
- [22].Grobben B; De Deyn PP; Slegers H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res., 2002, 310 (3), 257–270. [DOI] [PubMed] [Google Scholar]
- [23].Rodrigues NR; Rowan A; Smith ME; Kerr IB; Bodmer WF; Gannon JV; Lane DP p53 mutations in colorectal cancer. Proc. Natl. Acad Sci. USA, 1990, 87(19), 7555–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Trainer DL; Kline T; McCabe FL; Faucette LF; Feild J; Chaikin M; Anzano M; Rieman D; Hoffstein S; Li DJ; et al. , Biological characterization and oncogene expression in human colorectal carcinoma cell lines. Int. J. Cancer, 1988, 41(2), 287–296. [DOI] [PubMed] [Google Scholar]
- [25].Markowitz SD; Bertagnolli MM Molecular basis of colorectal cancer. New Eng. J. Med, 2009, 361(25), 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sakai H; Suzuki T; Maeda M; Takahashi Y; Horikawa N; Minamimura T; Tsukada K; Takeguchi N. Up-regulation of Na(+),K(+)-ATPase alpha 3-isoform and down-regulation of the alpha1-isoform in human colorectal cancer. FEBS Lett., 2004, 563(1–3), 151–154. [DOI] [PubMed] [Google Scholar]
- [27].Mijatovic T; Dufrasne F; Kiss R, Cardiotonic steroids-mediated targeting of the Na(+)/K(+)-ATPase to combat chemoresistant cancers. Curr. Med. Chem, 2012, 19(5), 627–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.