

Abstract
As many as five members of the APOBEC3 family of DNA cytosine deaminases are capable of inhibiting HIV-1 replication by deaminating viral cDNA cytosines and interfering with reverse transcription. HIV-1 counteracts restriction with the virally encoded Vif protein, which forms a hybrid ubiquitin ligase complex that directly binds APOBEC3 enzymes and targets them for proteasomal degradation. APOBEC3H (A3H) is unique among family members by dimerization through cellular and viral duplex RNA species. RNA binding is required for localization of A3H to the cytoplasmic compartment, for efficient packaging into nascent HIV-1 particles, and ultimately for effective virus restriction activity. Here we compared wild-type human A3H and RNA binding-defective mutants to ask whether RNA may be a factor in the functional interaction with HIV-1 Vif. We used structural modeling, immunoblotting, live cell imaging, and split green fluorescence protein (GFP) reconstitution approaches to assess the capability of HIV-1 Vif to promote the degradation of wild-type A3H in comparison to RNA binding-defective mutants. The results combined to show that RNA is not strictly required for Vif-mediated degradation of A3H, and that RNA and Vif are likely to bind this single domain DNA cytosine deaminase on physically distinct surfaces. However, a subset of the results also indicated that the A3H degradation process may be affected by A3H protein structure, subcellular localization, and differences in the constellation of A3H interaction partners suggesting additional factors may also influence the fate and functionality of this host-pathogen interaction.
Keywords: APOBEC3H, DNA cytosine deaminase, HIV-1 Vif, Subcellular localization, Vif-APOBEC interaction
Graphical Abstract
Introduction
The human APOBEC3 family consists of seven members, namely APOBEC3A (A3A), APOBEC3B (A3B), APOBEC3C (A3C), APOBEC3D (A3D), APOBEC3F (A3F), APOBEC3G (A3G), and APOBEC3H (A3H) (reviewed by [1-3]). These enzymes have all exhibited single-stranded DNA (ssDNA) cytosine deamination activity in a variety of assays and are expressed at varying levels in the cytoplasmic compartment of different cell types (an exception being the predominantly nuclear A3B). At least five of these deaminases, A3C-H, are expressed in CD4-positive T lymphocytes and capable of restricting the replication of HIV-1 through deamination-dependent and deamination-independent mechanisms. Virus restriction minimally requires nucleic acid binding activity in order to enter viral particles (RNA), deaminate viral cDNA replication intermediates (DNA), and impede reverse transcription (RNA and DNA). The virion infectivity factor, Vif, provides the primary APOBEC3 counterdefense mechanism for HIV-1 and related lentiviruses (reviewed by [1-3]). Vif heterodimerizes with the cellular transcription co-factor CBF-β (core binding factor-beta) and nucleates the assembly of an E3 ubiquitin ligase complex that directly binds and targets APOBEC3 proteins for polyubiquitination and proteasomal degradation [4-11]. Mutagenesis studies have shed light on some of the molecular determinants of the APOBEC3-Vif interaction. For instance, residue F39 of Vif is specifically required for interacting with A3H because changing it to valine results in a separation-of-function variant that still degrades A3G and A3F but no longer degrades A3H [12, 13]. For A3H, α3 and α4 helices have been implicated in binding to Vif with D121K on α4 conferring resistance to HIV-1 Vif-mediated degradation [14-19].
RNA has multiple roles in the antiviral activities of APOBEC3 enzymes. First, RNA binding is a key determinant in cytoplasmic localization of several APOBEC3 family members [20-26]. The clearest example is human A3H where disruption of RNA binding residues causes a simultaneous disruption of cytoplasmic localization [24, 27]. Similar observations have been made for various A3G and A3F mutants but the precise RNA binding mechanism has yet to be determined for these related enzymes [25, 28, 29]. Second, RNA binding is required for the packaging of A3F, A3G, and A3H into viral particles and subsequent antiviral activity [27, 30-40]. In particular, CLIP-seq (cross-linking immunoprecipitation coupled to next generation sequencing) experiments with A3F, A3G, and A3H have indicated preferential binding to viral RNA over cellular RNA, which is likely to facilitate packaging into virions [39]. Last, but not least, a proportion of the DNA deaminase-independent activities of A3F, A3G, and A3H can be attributed to the RNA binding properties of these enzymes exerting a steric block to DNA synthesis by the viral reverse transcriptase [27, 41-48].
Multiple A3H crystal structures have combined to demonstrate an evolutionarily conserved dimerization mechanism mediated by duplex RNA [27, 49, 50]. Conserved structural elements in A3H create an extensive, positively charged patch that binds strongly to duplex RNA. For human A3H, the RNA binding region is comprised of amino acids in loop 1 (L1 R18), loop 7 (L7 H114 and W115), and α-helix 6 (α6 R171, A172, R175, R176, and R179) [27]. Amino acid substitutions of several of these residues (R18E, H114A, W115A, A172E, RR175/6EE, and R179E) cause perturbations in cytoplasmic localization, encapsidation, and Vif-deficient HIV-1 restriction [24, 27]. Moreover, a human A3H triple mutant, R115A, R175E, and R176E, lacks all of these activities and is also monomeric and fully defective for binding to duplex RNA [27].
Here, we investigated the role of RNA in HIV-1 Vif-mediated degradation of human A3H. First, immunoblots of cellular extracts were used to compare the Vif-susceptibility of WT A3H and a panel of mutants with substitutions of one or more amino acids implicated in binding to duplex RNA. Second, live cell imaging approaches were used to quantify the kinetics of Vif-mediated degradation of WT A3H versus select RNA binding-defective mutants. Finally, a split GFP reconstitution system was used to show that HIV-1 Vif is not capable of disrupting RNA-mediated A3H dimerization in living cells. Overall, the results of these experiments support a model in which Vif targets a surface on A3H that is distinct from the duplex RNA binding region.
Results
Structures of A3H in complex with RNA
The x-ray structures of human, chimpanzee, and pig-tailed macaque A3H revealed a novel dimerization mechanism in which two monomers are united by binding to opposing sides of the same duplex RNA [27, 49, 50] (human A3H ribbon schematic in Fig. 1a and a superposition of the three structures in Fig. 1b). Several residues in A3H loop 1, loop 7, and helix 6 combine to bind duplex RNA, including a loop 7 tryptophan residue from each protomer that stacks with RNA nucleobases (W115 in each species’ enzyme; amino acid positions shaded red in Fig. 1a and shown by alignment in Fig. 1c). Mutating these residues in human A3H compromises RNA binding and dimerization and, surprisingly, results in elevated ssDNA cytosine deamination activity [27]. For instance, a human A3H triple mutant with W115A in loop 7 and R175E and R176E in helix 6 has a monomeric size exclusion profile and a large increase in ssDNA deaminase activity [27]. This triple mutant combines amino acid substitutions that individually cause defects in cytoplasmic localization, packaging into nascent HIV-1 particles, and restricting the infectivity of Vif-deficient HIV-1 virions, which are all metrics of duplex RNA binding activity [27]. Interestingly, the duplex RNA binding domain is located near the predicted HIV-1 Vif binding surface of human A3H, which prior studies have shown requires amino acids in helices 3 and 4 including D121 [14, 16, 19] (positions shaded yellow in Fig. 1d; considered again below and in Discussion).
Fig. 1. Structures of A3H in complex with RNA.

(a) X-ray structure of human A3H in complex with duplex RNA showing loop 1, loop 7, and α-helix 6 interacting with duplex RNA (ribbon schematic of PDB ID 6B0B). Positions of conserved RNA-interacting residues are shaded red.
(b) Superposition of human (blue; PDB ID 6B0B), chimpanzee (green; PDB ID 5Z98), and pig-tailed macaque (beige; PDB ID 5W3V) A3H structures.
(c) Partial amino acid sequence alignment of the loop 7 and α-helix 6 regions of human, chimpanzee, and pig-tailed macaque A3H proteins. Conserved RNA-interacting residues are highlighted in red.
(d) A3H structure with yellow highlights for previously identified Vif-interacting residues including D121. The orientation is similar to those in the bottom of panel (a) and panel (b) to facilitate comparisons.
A subset of A3H RNA-binding residues are required for functional interaction with Vif
Most naturally occurring A3H variants including haplotype II are sensitive to Vif-mediated degradation [12, 13, 15, 16, 18, 51, 52]. The WT construct used here is A3H haplotype II splice variant 1-183. To ask whether RNA binding-defective proteins are susceptible or resistant to Vif-mediated degradation, a panel of mutants was co-transfected into 293T cells with 0.5 μg or 1.0 μg of HIV-1 LAI Vif (WT) or a degradation-defective mutant (F39V) and, after a 48-hour incubation, protein levels were analyzed by immunoblotting whole cell extracts (representative blots in Fig. 2a with quantification in 2b). As controls, expression of WT Vif decreases levels of WT A3H as well as those of an A3H E56A catalytic mutant by approximately 50%, and Vif F39V is defective in WT A3H degradation (similar to previous observations [15, 27]). As an additional control, A3H D121K resists degradation as evidenced by similar band intensities with WT Vif and the F39V mutant (again similar to previous observations [16, 19]).
Fig. 2. Vif susceptibility of human A3H RNA binding-defective mutants.

(a) Immunoblots of 293T cells expressing WT A3H or the indicated mutants, together with empty vector (−), HIV-1 LAI Vif, or a F39V derivative. A3H was detected using an anti-A3H antibody and Vif was detected using an anti-HA antibody. Anti-α-tubulin was used as a loading control.
(b) A bar graph quantifying immunoblot data from panel (a) and from an independent experiment (not shown). The A3H level for each construct is normalized to that of the corresponding empty vector control. The mean and difference between two independent experiments are shown. The dashed line represents level of sensitivity of WT A3H to Vif.
(c) Immunoblots of 293T cells expressing WT A3H or the indicated mutants, together with empty vector (−), HIV-1 IIIB-derived Hyper Vif (F39, H48, EKGE60-63) or Hypo Vif (V39, N48, GDAK60-63) with the same antibodies as in panel (a).
(d) A structural depiction of human A3H residues required (red) or dispensable (orange) for functional interaction with HIV-1 Vif.
In comparison, A3H mutants with single amino acid substitutions of residues implicated in RNA binding showed either Vif-sensitive or Vif-resistant phenotypes. These different phenotypes were even observed for mutants within the same surface region of the protein. For instance, A3H helix 6 mutants A172E, R175E, and R179E showed a WT-like Vif sensitivity, whereas R176E appeared fully resistant to Vif-mediated degradation (representative blots in Fig. 2a with quantification in 2b). A3H loop 7 mutants, H114A and W115A, showed completely resistant phenotypes, whereas A3H loop 1 R18E retained a WT-like Vif sensitivity. Based on the single amino acid substitution mutant phenotypes, not surprisingly, the monomeric A3H triple mutant was also resistant to Vif-mediated degradation and this property was unchanged by adding an E56A catalytic glutamate substitution. Similar results were obtained for the Vif protein of a different HIV-1 strain (IIIB) with previously demonstrated hyperfunctional amino acid substitutions [13] showing strong degradation of WT A3H and little degradation activity with the monomeric A3H triple mutant (Fig. 2c). These additional results are notable because the hyperfunctional Vif protein caused degradation of almost all cellular A3H and even showed partial activity against A3H D121K (all results in this panel are in comparison to a Vif variant, hypo, that has F39V as above and no A3H degradation activity as reported previously [13, 51]). These results are summarized by color coding phenotypes of these mutants on the surface of the 3-dimensional structure of A3H with RNA (Fig. 2d).
RNA-binding mutants of A3H are degraded less efficiently by HIV-1 Vif
We next used live cell fluorescent video microscopy to measure the degradation kinetics of fluorescently-tagged A3H at a single-cell level during HIV-1 infection. HeLa cell lines stably expressing WT or mutant A3H fused to the C-terminus of a yellow fluorescent protein (YFP-A3H) were generated and then infected with a replication-defective HIV-1 construct that, following integration into host cell genomic DNA, expresses mCherry as a reporter protein (MOI = 0.5). WT A3H and the Vif-resistant derivative D121K variant each localized to cytoplasmic and nucleolar compartments, as reported [17, 24, 27, 48, 53]. The RNA binding-defective A3H triple mutant showed compromised cytoplasmic localization and appeared predominantly nuclear, also as reported [24, 27]. As expected, WT LAI Vif expressed from the virus at the same time as the mCherry marker caused the degradation of WT A3H (representative image in Fig. 3a and supplemental movie S1). The WT YFP-A3H fluorescence intensity declined at a relative rate of approximately 10% per hour (Fig. 3c). In comparison, both the YFP-A3H-D121K control and the RNA binding-defective triple mutant were resilient to degradation by LAI Vif (representative image in Fig. 3a, quantification in 3c, and supplemental movie S2). As an additional control, infection of the same cells with an HIV-1 construct encoding Vif-F39V did not cause a reduction in fluorescence of any forms of A3H (images not shown; quantification summarized in Fig. 3c). These single-cell results are consistent with the immunoblot data described above using LAI Vif and untagged WT A3H.
Fig. 3. The degradation kinetics of human A3H and key mutants.

(a) Representative images of HeLa cells stably expressing WT or mutant A3H fused to the C-terminus of a YFP before infection or 25-hour post-infection with an HIV-1 LAI mCherry reporter virus (WT Vif; MOI = 0.5). Parallel cultures were also infected with a reporter virus expressing LAI Vif F39V as a negative control (images not shown). Cells circled with dashed lines became infected as evidenced by mCherry expression. The scale bar at the upper right corner represents 10 μm. Also see supplemental movies S1-2 and S5-6.
(b) Representative images of HeLa cells stably expressing WT or mutant A3H fused to the C-terminus of a YFP before infection or 16-hour post-infection with an HIV-1 NL4-3 mCherry reporter virus (Hyper-Vif; MOI = 0.5). Parallel cultures were also infected with a reporter virus expressing Hypo-Vif as a negative control (images not shown). Cells circled with dashed became infected as evidenced by mCherry expression. The scale bar at the upper right corner represents 10 μm. Also see supplemental movies S3-4 and S7-8.
(c) Quantification of the rate of A3H degradation (change in YFP fluorescence) of a minimum of 10 infected cells representing each of the indicated conditions (mean +/− SD).
(d) Flow cytometry quantification of the relative MFI of YFP-A3H in HeLa cells infected under the same conditions as panels (a-b). Data were normalized by setting the post-infection MFI of each Vif F39V or Hypo-Vif reaction to 100%.
Additional live cell fluorescent imaging experiments were done with a set of HIV-1 NL4-3 mCherry reporter viruses encoding either Hyper- or Hypo-Vif (variants described above and, notably, NL4-3 Vif and IIIB Vif have identical amino acid sequences). We observed that HIV-1 NL4-3 mCherry Hyper-Vif not only degraded WT YFP-A3H (supplemental movie S3, as expected, but it also triggered the degradation of YFP-A3H-D121K and the YFP-A3H-triple mutant (supplemental movie S4), albeit with slower kinetics (representative image in Fig. 3b and quantification in 3c). Infection by HIV-1 NL4-3 mCherry Hypo-Vif did not reduce levels of any form of A3H (Fig. 3c). These single cell imaging results were confirmed by flow cytometric quantification of the median fluorescent intensity (MFI) of the YFP (A3H) signal in infected cells 48 hours post-infection (Fig. 3d). Taken together, these real-time imaging data showed that the A3H RNA binding-defective mutant is less susceptible to HIV-1 Vif-mediated degradation compared to WT A3H. However, it can still be degraded by HIV-1 NL4-3 derived Hyper-Vif (but not Hypo-Vif), suggesting that the RNA-binding defective mutant of A3H is still capable of interacting with Hyper-Vif and, importantly, that RNA may not be essential for a functional interaction to occur between Vif and A3H.
A split GFP system to study A3H dimerization in living cells
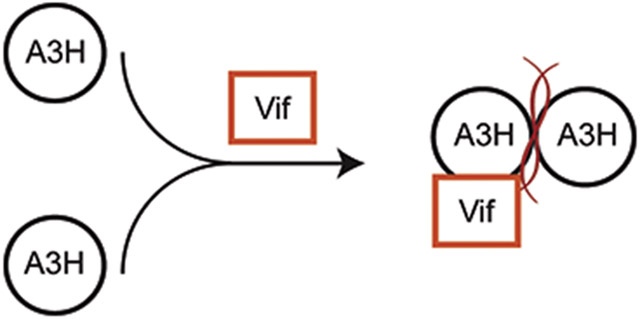
To be able to study A3H dimerization in living cells, a split fluorescence system was designed in which one A3H protomer was N-terminally tagged with GFP β-strands 1-10 (GFP1-10) and the second with GFP β-strand 11 (GFP11) plus an extended flexible linker. Based on prior reports with other proteins including various homo- and hetero-dimerization partners (reviewed in [54, 55]) and structural data from our group and others [27, 49, 50], efficient GFP reconstitution would only be expected following A3H dimerization through duplex RNA (schematic in Fig. 4a). Indeed, a robust GFP signal was observed in both the cytoplasmic compartment and in nucleoli following co-transfection of 293T cells with these constructs (Fig. 4b-c). By flow cytometry, more than 70% of transfected mCherry-positive cells (co-transfection marker) were GFP-positive with MFI more than 20-fold higher than negative controls (Fig. 4c). Moreover, transfection of either construct alone failed to emit fluorescence (not shown), and similarly tagged A3A constructs also failed to reconstitute strong GFP signal (Fig. 4b-c). The latter negative result was expected because A3A is monomeric in living cells and it also behaves as a monomer under most conditions in vitro [56-58] (although A3A dimerization has been reported under some conditions in vitro [59]). As further validation of the robustness of this system, A3H-D121K yielded strong GFP signal similar to WT A3H, and the RNA binding-defective triple mutant (W115A, R175E, R176E) was indistinguishable from A3A (Fig. 4b-c). These results indicated that the split GFP reconstitution system can be used as a quantitative measure of A3H dimerization in living cells.
Fig. 4. A split GFP system to study A3H dimerization in living cells.

(a) A schematic of the split GFP system for A3H dimerization. One A3H construct is fused to GFP β-strand 1-10 (GFP1-10) and the other to β-strand 11 (GFP11). GFP Reconstitution occurs upon duplex RNA-mediated dimerization of A3H.
(b) Representative fluorescent microscopy images of 293T cells expressing split GFP constructs of the indicated A3s. A mCherry plasmid was included as a co-transfection control. The scale bar represents 20 μm.
(c) Flow cytometry profiles of the same cells as in panel (b). The MFI is shown for the whole mCherry-positive population, and the percentages of GFP-negative and GFP-positive cells are included at the bottom left and right, respectively (gates based on untransfected 293T cells analyzed in parallel).
Vif overexpression does not interfere with A3H-mediated dimerization
We next asked whether HIV-1 IIIB Vif N48H, previously reported to degrade WT A3H but not cause cell cycle arrest [60, 61], is capable of interfering with A3H dimerization using the split GFP system. In one scenario, Vif may bind to A3H regardless of its dimerization status, and if so, the GFP fluorescence signal should be unaffected by the presence of Vif (top schematic in Fig. 5a). Alternatively, Vif may intercept A3H prior to binding RNA, prevent dimerization, and lead to diminished GFP fluorescence reconstitution (bottom schematic in Fig. 5a).
Fig. 5. Vif does not interfere with A3H dimerization in the split GFP system.

(a) Models for Vif interaction with A3H. The top schematic depicts Vif binding to a distinct surface on A3H (predicted to have no interference in GFP reconstitution), whereas the bottom schematic depicts Vif competing with RNA for A3H (interference in GFP reconstitution).
(b) Immunoblots of 293T cells expressing A3A, A3H, or the indicated A3H mutants, together with empty vector (−) or HIV-1 IIIB Vif N48H +/− MG132. A3H was detected using an anti-A3H antibody and Vif was detected using an anti-HA antibody. Anti-α-tubulin was used as a loading control. A bar graph is shown above the immunoblots for quantification of flow cytometry data from panel (c) and from an independent experiment not shown. The MFI level of cells of each indicated condition is normalized to A3A in the absence of Vif or MG132. The mean and difference between two independent experiments are shown.
(c) Flow cytometry profiles of 293T cells expressing the indicated A3 and Vif construct (+/− MG132). Each profile is labeled as in Fig. 4c.
To distinguish between these possibilities, 293T cells were co-transfected with the same panel of split GFP constructs as above together with Vif N48H or an empty vector control. Each reaction also included an mCherry expression vector as a transfection control. Following a 32-hour incubation period, one set of reactions was treated for 16 hours with MG132 and the parallel set was untreated, and then all reactions were analyzed in parallel by immunoblotting and flow cytometry (Fig. 5b-c). Similar to results presented above in Fig. 2, in the absence of MG132, Vif triggered a reduction in WT A3H levels but did not affect the A3H RNA binding-defective triple mutant (W115A R175E R176E) or Vif-resistant A3H-D121K. This result is supported by flow cytometry where an MFI reduction of 4-fold was observed (representative flow cytometry histograms in Fig. 5c and duplicate experiments averaged in the bar graph in the top part of Fig. 5b).
MG132 was included in the experiment to prevent A3H proteasomal degradation and facilitate quantification of the dimerization phenotype. Interestingly, the dimerization potential of WT A3H was unaffected by Vif under these conditions, as evidenced by statistically indistinguishable MFIs (representative flow cytometry histograms in Fig. 5c and duplicate experiments averaged in the bar graph in the top part of Fig. 5b). It is worth noting that the lack of GFP reconstitution by the A3H triple mutant is likely due to a failure to dimerize through RNA and not to lower expression levels, because MG132 treatment caused an increase in steady-state levels of this mutant to near those of WT A3H [49] (compare lanes 3, 7, 11, and 15 in Fig. 5b) and its GFP MFI remained approximately 10-fold lower (representative flow cytometry histograms in Fig. 5c and duplicate experiments averaged in the bar graph in the top part of Fig. 5b). These results combined to indicate that the dimerization status of A3H is unlikely to be affected by the functional interaction with Vif.
Discussion
RNA plays essential roles in the dimerization, subcellular localization, and antiviral functions of A3H. Here we address whether the RNA bound by A3H has a role in HIV-1 Vif-mediated degradation. Immunoblot results with a panel of RNA binding mutants were somewhat inconclusive with some A3H mutants resisting Vif and others maintaining WT levels of sensitivity (Fig. 2). The Vif-resistance of an RNA binding-defective triple mutant of A3H was confirmed by imaging A3H degradation in living cells, but observations with a hyper-functional Vif variant indicated that even this mutant could be targeted for degradation (albeit at an approximately 4-fold slower rate than WT A3H; Fig. 3). A split GFP system enabled asking whether Vif might compete with the same surface as duplex RNA and interfere with A3H dimerization (Fig. 4). These results combined to favor a model in which the Vif and RNA binding surfaces of A3H are physically distinct. For instance, even under conditions of proteasome inhibition to prevent A3H degradation, Vif did not have the capability to alter the dimerization potential of WT A3H.
As confirmed here and demonstrated in prior studies, α helix 4 including residue D121 is essential for A3H degradation by HIV-1 Vif [14-19]. However, the hyper-Vif variant could still trigger the degradation of the D121K mutant suggesting that this Vif protein may have a greater conformational flexibility and/or recognize a slightly different overall surface than the Vif proteins from HIV-1 IIIB/NL4-3 and HIV-1 LAI. Subcellular localization and/or as-yet-unidentified binding partners might also play a role in the observed resistance of the subset of RNA binding-defective mutants to Vif-mediated degradation. For instance, cytoplasmic A3H was degraded at a faster rate than nucleolar A3H (e.g., residual nucleolar A3H is evident in Fig. 3a-b in the presence of WT Vif and hyper-Vif, respectively; also see supplemental movies S1 and S3). Moreover, several of the RNA binding-defective mutants, including the triple mutant, tended to accumulate at higher levels in the nuclear compartment including the nucleoli, and this mislocalization likely also contributed to the overall degree of Vif resistance (particularly in immunoblots of whole cell lysates). Taken together with our prior localization studies, the RNA binding-defective mutants that are more nuclear also tend to be more resistant to Vif (and those that are cytoplasmic are more sensitive) [24, 27]. It is therefore reasonable to postulate that the binding of Vif to CBF-β immediately post-translation and the assembly of a large >200 kDa E3 ligase complex hinders Vif from accessing the nuclear compartment.
These results are also sensible from practical and evolutionary perspectives. Recent structural, biophysical, and functional studies have indicated that duplex RNA is required for A3H to package from cytoplasmic sites into assembling viral particles and also for deamination-independent restriction of virus replication [27, 49, 50, 62]. The massive molar excess of structured RNA in cells would make it difficult for Vif to compete for the same binding surface of A3H. Therefore, it is likely that the HIV-1 Vif protein evolved to avoid competition with RNA and bind directly to a distinct A3H surface. Thus, Vif may have evolved the potential to degrade both monomeric as well as RNA-dimerized forms of cellular A3H and thereby maintain the upper hand in counteracting restriction.
Materials and Methods
APOBEC3 expression constructs
The constructs expressing intron-containing WT, E56A, H114A, W115A, R175E, R176E, W115A/R175E/R176E, R18E, A172E, and R179E huA3H haplotype II in pcDNA3.1(+) (Invitrogen) have been reported [27]. Other mutants of A3H (D121K and E56A/W115A/R175E/R176E) within the same backbone were made by site-directed mutagenesis and verified by Sanger sequencing (primer sequences available on request). Retroviral transduction vectors encoding YFP fused to A3H (haplotype II) and mutants were generated by subcloning SYFP2 from SYFP2-C1 (Addgene #22878) into pcDNA3.1 plasmids containing A3H using NheI and KpnI restriction cut sites followed by transfer into MigR1-derived simple retroviral vector using MluI and MfeI cut sites. The split GFP system used in this study was based on a previous system [63] with minor modifications. A gblock (IDT) encoding GFP β-strand 11 (GFP11) followed by a 27 amino-acid flexible linker was ordered and cloned into the previously reported pcDNA3.1(+) construct of WT A3H protein to the N-termini [27]. A ClaI restriction site was added in between GFP11 and the linker. GFP β-strand 1-10 (GFP1-10) was PCR amplified and cloned in place of GFP11 to make the GFP1-10 construct.
HIV-1 and Vif expression constructs
The lentiviral Vif protein from HIV-1 IIIB (EU541617) was codon optimized (GenScript Corp) and cloned into pVR1012 with a C-terminal HA tag using SalI and BamHI. The Vif protein from HIV-1 LAI (1102247D) was cloned into pVR1012 with a C-terminal HA tag using SalI and NotI. F39V mutant of HIV-1 LAI Vif, Hypo-variant (F39V), N48H, and Hyper-variant (N48H, GDAK60-63EKGE) of HIV-1 IIIB Vif were made by site-directed mutagenesis and verified by Sanger sequencing (primer sequences available on request). HIV-1 NL4-3 mCherry reporter viruses were generated by subcloning Hypo- and Hyper-Vif encoding segments from IIIB infectious molecular clones [64] into a NL4-3 E-R-mCherry full-length reporter virus plasmid [65] using AgeI and SalI cut sites. HIV-1 LAI mCherry reporter viruses were generated by transferring the mCherry reporter cassette from pRGH (NIH ARP #12427) into a previously described LAI infectious molecular clone using BlpI and XhoI cut sites. LAI F39V was generated by site-directed mutagenesis. All plasmids were confirmed by restriction digestion and Sanger sequencing.
Cell lines
293T cells were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S). HeLa cells were maintained at 37°C and 5% CO2 in DMEM supplemented with 10% FBS, 1% P/S, and 1% GlutaMAX.
Vif degradation experiments
293T cells were transfected with WT or mutant pcDNA3.1(+)-A3H, along with 500 or 1000 ng pVR1012- HIV-1 LAI Vif WT/F39V-HA (Fig. 2a), or 50 or 100 ng pVR1012- HIV-1 IIIB Hyper-/Hypo-Vif (Fig. 2c) or empty vector, using Transit-LT1 (Mirus). The following amounts of A3H were transfected: 25 ng of WT, D121K, E56A, H114A, R175E, R18E, and A172E, 100 ng of R179E, and 200 ng of W115A, R176E, W115A/R175E/R176E, and E56A/W115A/R175E/R176E. Empty vector is added to equalize the amounts of total DNA. After 48 hours, cells were harvested for immunoblot analysis.
Immunoblotting experiments
Cell lysates were prepared by resuspension of washed cell pellets directly in 2.5× Laemmli sample buffer. Proteins were separated using discontinuous sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). HA-tagged A3 proteins were detected using monoclonal mouse anti-HA (BioLegend #MMS-101P). A3H proteins were detected using polyclonal rabbit anti-A3H (Novus #NBP1-91682). Tubulin was detected using a monoclonal mouse anti-α-Tubulin antibody (Covance #MMS-489P). Immunoblots were quantified using ImageStudio.
Live cell fluorescent imaging
Reporter viruses were produced in 293T cells by co-transfection with VSV-G to generate pseudo-typed single cycle infectious virus. Viral inoculum was harvested at 48 hours post-transfection and passed through a 0.45-μm syringe filter. HeLa YFP-A3H stable cells were plated at 20,000 cells per well of an ibidi μ-well slide (ibidi GmbH) in 200 μL of Fluorobrite DMEM (Thermo Fisher Scientific) supplemented with 10% FBS, 1% GlutaMAX, 1% P/S, and 100 mM HEPES. Cells were infected at a MOI of 0.5 infectious units per cell with 2 μg/mL polybrene. Live cell fluorescent video microscopy was performed as previously described [65] beginning at 12 hours after addition of virus and a multi-color image was captured every 60 minutes (mCherry, YFP, and DAPI) for up 40 hours post-infection. All images were processed and analyzed using FIJI/ImageJ2 [69]. Briefly, regions of interest (ROIs) were drawn around whole single cells based on YFP-A3H image and around nuclei of those cells based on DAPI in every frame of live cell movies. Cytoplasmic fluorescence was quantified by subtracting nuclear from whole cell integrated density (IntDen). The cytoplasmic YFP and whole cell mCherry fluorescence were normalized to each single cell’s value at the beginning of each movie. Rate of change in YFP (showing YFP-A3H degradation) was quantified for the period 5 hours following detection of mCherry.
Split GFP experiments
293T cells were transfected with 100 ng GFP1-10 plasmids, 25 ng GFP11 plasmids, 500 ng of HIV-1 IIIB Vif N48H-HA or empty vector, and 100 ng mCherry plasmid. Cells were treated with MG132 at a final concentration of 2 μM at 32 hours and subjected to analyses by fluorescence imaging, flow cytometry, and immunoblotting at 48 hours post-transfections. Cells were treated with NucBlue live cell stain (Invitrogen) at 37°C for 30 minutes for DAPI (4’,6’-diamidino-2-phenylindole) staining prior to imaging. Images were collected using a Nikon inverted Ti-E deconvolution microscope. A fraction of the cells was resuspended in phosphate-buffered saline (PBS) with 1 mM ethylenediaminetetraacetic acid (EDTA) in preparation for flow cytometry. GFP and mCherry fluorescence was measured on a BD FACS Canto II flow cytometer. Data were analyzed using FlowJo flow cytometry analysis software (version 8.8.6). The fraction of GFP+ cells and median fluorescence intensity (MFI) of all transfected (mCherry-positive) cells was quantified for each sample. The remaining cells were prepared for immunoblotting.
Sequence alignments.
All amino acid sequences were aligned using Clustal Omega. The amino acid sequences of human A3H, chimpanzee A3H, and pig-tailed macaque A3H correspond to GenBank accession number ACK77775.1, PDB ID 5Z98_A, and PDB ID 5W3V_A, respectively.
Supplementary Material
(S1) YFP-A3H (green) infected with HIV-1 LAI mCherry reporter viruses encoding WT Vif (red).
(S2) YFP-A3H-triple mutant (green) infected with HIV-1 LAI mCherry reporter viruses encoding WT Vif (red).
(S3) YFP-A3H (green) infected with HIV-1 NL4-3 mCherry encoding Hyper-Vif (red).
(S4) YFP-A3H-triple mutant (green) infected with HIV-1 NL4-3 mCherry encoding Hyper-Vif (red).
(S5) YFP-A3H (green) infected with HIV-1 LAI mCherry reporter viruses encoding F39V Vif (red).
(S6) YFP-A3H-triple mutant (green) infected with HIV-1 LAI mCherry reporter viruses encoding F39V Vif (red).
(S7) YFP-A3H (green) infected with HIV-1 NL4-3 mCherry encoding Hypo-Vif (red).
(S8) YFP-A3H-triple mutant (green) infected with HIV-1 NL4-3 mCherry encoding Hypo-Vif (red).
Highlights.
Vif degrades a subset of RNA binding-defective A3H mutants
RNA-mediated A3H dimerization is not hindered by Vif
Vif and RNA are likely to bind physically distinct surfaces of A3H
Acknowledgements
This work was supported by NIAID R37 AI064046 (to R.S.H.), NIGMS R01 GM118000 (to R.S.H. and H.A.), and NIGMS R01 GM118047 (to H.A.). J.W. received partial salary support from the University of Minnesota Graduate School, Interdisciplinary Doctoral Fellowship. D.J.S. received partial salary support from the University of Minnesota Craniofacial Research Training (MinnCResT) program (NIH T90DE022732). R.S.H. is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished University McKnight Professor, and an Investigator of the Howard Hughes Medical Institute. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: pRGH-WT (#12427) from Ivan Sadowski and Viviana Simon. Microscopy imaging and analysis was performed at the University Imaging Centers, University of Minnesota.
Abbreviations:
- A3
APOBEC3
- A3H
APOBEC3H
- CBF-β
core binding factor-beta
- GFP
green fluorescence protein
- HA
hemagglutinin
- ssDNA
single-stranded DNA
- Vif
virion infectivity factor
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
R.S.H. is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc. H.A. is a consultant of ApoGen Biotechnologies Inc. The other authors have no conflicts to declare.
Ethics Approval and Consent to Participate
There are no ethical considerations related to this work.
Consent for Publication
Not applicable.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- [1].Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi DF, Izumi T, Pathak VK. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol. 2014;426:1220–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nakano Y, Aso H, Soper A, Yamada E, Moriwaki M, Juarez-Fernandez G, Koyanagi Y, Sato K. A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology. 2017;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. [DOI] [PubMed] [Google Scholar]
- [5].Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. [DOI] [PubMed] [Google Scholar]
- [6].Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. [DOI] [PubMed] [Google Scholar]
- [7].Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. [DOI] [PubMed] [Google Scholar]
- [8].Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. [DOI] [PubMed] [Google Scholar]
- [9].Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. [DOI] [PubMed] [Google Scholar]
- [10].Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2011;481:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2011;481:376–379. [DOI] [PubMed] [Google Scholar]
- [12].Binka M, Ooms M, Steward M, Simon V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol. 2012;86:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, Brown WL, Reilly C, Emerman M, Harris RS. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet. 2014;10:e1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakashima M, Tsuzuki S, Awazu H, Hamano A, Okada A, Ode H, Maejima M, Hachiya A, Yokomaku Y, Watanabe N, Akari H, Iwatani Y. Mapping region of human restriction factor APOBEC3H critical for interaction with HIV-1 Vif. J Mol Biol. 2017;429:1262–1276. [DOI] [PubMed] [Google Scholar]
- [15].Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol. 2009;83:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol. 2010;84:1902–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li MM, Emerman M. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J Virol. 2011;85:8197–8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol. 2011;85:3142–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ooms M, Letko M, Simon V. The structural interface between HIV-1 Vif and human APOBEC3H. J Virol. 2017;91:e02289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem. 2006;281:29105–29119. [DOI] [PubMed] [Google Scholar]
- [21].Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol. 2007;81:2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Izumi T, Burdick R, Shigemi M, Plisov S, Hu WS, Pathak VK. Mov10 and APOBEC3G localization to processing bodies is not required for virion incorporation and antiviral activity. J Virol. 2013;87:11047–11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Phalora PK, Sherer NM, Wolinsky SM, Swanson CM, Malim MH. HIV-1 replication and APOBEC3 antiviral activity are not regulated by P bodies. J Virol. 2012;86:11712–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Salamango DJ, Becker JT, McCann JL, Cheng AZ, Demir Ö, Amaro RE, Brown WL, Shaban NM, Harris RS. APOBEC3H subcellular localization determinants define zipcode for targeting HIV-1 for restriction. Mol Cell Biol. 2018;38:e00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Stenglein MD, Matsuo H, Harris RS. Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J Virol. 2008;82:9591–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wichroski MJ, Robb GB, Rana TM. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006;2:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shaban NM, Shi K, Lauer KV, Carpenter MA, Richards CM, Salamango D, Wang J, Lopresti MW, Banerjee S, Levin-Klein R, Brown WL, Aihara H, Harris RS. The antiviral and cancer genomic DNA deaminase APOBEC3H is regulated by an RNA-mediated dimerization mechanism. Mol Cell. 2018;69:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fukuda H, Li S, Sardo L, Smith JL, Yamashita K, Sarca AD, Shirakawa K, Standley DM, Takaori-Kondo A, Izumi T. Structural determinants of the APOBEC3G N-terminal domain for HIV-1 RNA association. Front Cell Infect Microbiol. 2019;9:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kouno T, Luengas EM, Shigematsu M, Shandilya SM, Zhang J, Chen L, Hara M, Schiffer CA, Harris RS, Matsuo H. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat Struct Mol Biol. 2015;22:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Apolonia L, Schulz R, Curk T, Rocha P, Swanson CM, Schaller T, Ule J, Malim MH. Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PLoS Pathog. 2015;11:e1004609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bogerd HP, Cullen BR. Single-stranded RNA facilitates nucleocapsid: APOBEC3G complex formation. RNA. 2008;14:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004;279:33177–33184. [DOI] [PubMed] [Google Scholar]
- [33].Huthoff H, Malim MH. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and virion encapsidation. J Virol. 2007;81:3807–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schafer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–168. [DOI] [PubMed] [Google Scholar]
- [36].Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, Freed EO, Hu WS, Pathak VK. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem. 2004;279:35822–35828. [DOI] [PubMed] [Google Scholar]
- [37].Wang T, Tian C, Zhang W, Luo K, Sarkis PT, Yu L, Liu B, Yu Y, Yu XF. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol. 2007;81:13112–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang T, Tian C, Zhang W, Sarkis PT, Yu XF. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J Mol Biol. 2008;375:1098–1112. [DOI] [PubMed] [Google Scholar]
- [39].York A, Kutluay SB, Errando M, Bieniasz PD. The RNA binding specificity of human APOBEC3 proteins resembles that of HIV-1 nucleocapsid. PLoS Pathog. 2016;12:e1005833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol. 2004;78:12058–12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bélanger K, Langlois MA. RNA-binding residues in the N-terminus of APOBEC3G influence its DNA sequence specificity and retrovirus restriction efficiency. Virology. 2015;483:141–148. [DOI] [PubMed] [Google Scholar]
- [42].Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4:e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol. 2013;87:1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kobayashi T, Koizumi Y, Takeuchi JS, Misawa N, Kimura Y, Morita S, Aihara K, Koyanagi Y, Iwami S, Sato K. Quantification of deaminase activity-dependent and -independent restriction of HIV-1 replication mediated by APOBEC3F and APOBEC3G through experimental mathematical investigation. J Virol. 2014;88:5881–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Morse M, Huo R, Feng Y, Rouzina I, Chelico L, Williams MC. Dimerization regulates both deaminase-dependent and deaminase-independent HIV-1 restriction by APOBEC3G. Nat Commun. 2017;8:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. [DOI] [PubMed] [Google Scholar]
- [48].Ooms M, Majdak S, Seibert CW, Harari A, Simon V. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J Virol. 2010;84:7961–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Matsuoka T, Nagae T, Ode H, Awazu H, Kurosawa T, Hamano A, Matsuoka K, Hachiya A, Imahashi M, Yokomaku Y, Watanabe N, Iwatani Y. Structural basis of chimpanzee APOBEC3H dimerization stabilized by double-stranded RNA. Nucleic Acids Res. 2018;46:10368–10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bohn JA, Thummar K, York A, Raymond A, Brown WC, Bieniasz PD, Hatziioannou T, Smith JL. APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat Commun. 2017;8:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe. 2013; 14:411–421. [DOI] [PubMed] [Google Scholar]
- [52].Li MM, Wu LI, Emerman M. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J Virol. 2010;84:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA, Harris RS. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun. 2016;7:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Romei MG, Boxer SG. Split green fluorescent proteins: scope, limitations, and outlook. Annu Rev Biophys. 2019;48:19–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Foglieni C, Papin S, Salvade A, Afroz T, Pinton S, Pedrioli G, Ulrich G, Polymenidou M, Paganetti P. Split GFP technologies to structurally characterize and quantify functional biomolecular interactions of FTD-related proteins. Sci Rep. 2017;7:14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi K, Demir O, Carpenter MA, Wagner J, Kurahashi K, Harris RS, Amaro RE, Aihara H. Conformational switch regulates the DNA cytosine deaminase activity of human APOBEC3B. Sci Rep. 2017;7:17415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li J, Chen Y, Li M, Carpenter MA, McDougle RM, Luengas EM, Macdonald PJ, Harris RS, Mueller JD. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J Mol Biol. 2014;426:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Byeon IJ, Ahn J, Mitra M, Byeon CH, Hercik K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun. 2013;4:1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kouno T, Silvas TV, Hilbert BJ, Shandilya SMD, Bohn MF, Kelch BA, Royer WE, Somasundaran M, Kurt Yilmaz N, Matsuo H, Schiffer CA. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat Commun. 2017;8:15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ooms M, Letko M, Binka M, Simon V. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS One. 2013;8:e57744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhao K, Du J, Rui Y, Zheng W, Kang J, Hou J, Wang K, Zhang W, Simon VA, Yu XF. Evolutionarily conserved pressure for the existence of distinct G2/M cell cycle arrest and A3H inactivation functions in HIV-1 Vif. Cell Cycle. 2015;14:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mitra M, Singer D, Mano Y, Hritz J, Nam G, Gorelick RJ, Byeon IJ, Gronenborn AM, Iwatani Y, Levin JG. Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology. 2015;12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, Huang B. Versatile protein tagging in cells with split fluorescent protein. Nat Commun. 2016;7:11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Haché G, Shindo K, Albin JS, Harris RS. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr Biol. 2008;18:819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Becker JT, Sherer NM. Subcellular localization of HIV-1 gag-pol mRNAs regulates sites of virion assembly. J Virol. 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(S1) YFP-A3H (green) infected with HIV-1 LAI mCherry reporter viruses encoding WT Vif (red).
(S2) YFP-A3H-triple mutant (green) infected with HIV-1 LAI mCherry reporter viruses encoding WT Vif (red).
(S3) YFP-A3H (green) infected with HIV-1 NL4-3 mCherry encoding Hyper-Vif (red).
(S4) YFP-A3H-triple mutant (green) infected with HIV-1 NL4-3 mCherry encoding Hyper-Vif (red).
(S5) YFP-A3H (green) infected with HIV-1 LAI mCherry reporter viruses encoding F39V Vif (red).
(S6) YFP-A3H-triple mutant (green) infected with HIV-1 LAI mCherry reporter viruses encoding F39V Vif (red).
(S7) YFP-A3H (green) infected with HIV-1 NL4-3 mCherry encoding Hypo-Vif (red).
(S8) YFP-A3H-triple mutant (green) infected with HIV-1 NL4-3 mCherry encoding Hypo-Vif (red).