

Abstract
In the study of multivalent interactions at interfaces, as occur for example at cell membranes, the density of the ligands or receptors displayed at the interface plays a pivotal role, affecting both the overall binding affinities and the valencies involved in the interactions. In order to control the ligand density at the interface, several approaches have been developed, and they concern the functionalization of a wide range of materials. Here, different methods employed in the modification of surfaces with controlled densities of ligands are being reviewed. Examples of such methods encompass the formation of self‐assembled monolayers (SAMs), supported lipid bilayers (SLBs) and polymeric layers on surfaces. Particular emphasis is given to the methods employed in the study of different types of multivalent biological interactions occurring at the functionalized surfaces and their working principles.
Keywords: multivalency, surfaces, ligand density, monolayers, interfaces
Play it again, SAM: An overview of various surface functionalization methods that allow control over the ligand density is provided, with a focus on the study of multivalent interactions at two‐dimensional interfaces.

1. Introduction
Multivalency is a fundamental principle involved in a wide variety of biological systems, such as protein−protein interactions, cellular recognition and modulation of cell signaling.1 Several weak, non‐covalent and reversible interactions, for example between proteins and carbohydrates, can provide an overall strong and highly specific binding when occurring in a multivalent fashion. In this way, enhancement of binding affinities of several orders of magnitude is generally achieved.2
The enhancement of the affinity due to multivalent interactions plays an important role in the improvement of selectivity in the target recognition in biological processes.3 Pathogens, for example, take advantage of multiple protein−carbohydrate interactions for cell adhesion which is the onset for the invasion or infection of the host cell.4 One of the most studied multivalent systems is the influenza virus, which binds multivalently with its hemagglutinin (HA) protein to sialic acid residues expressed on the cell surface in the first stage of the infection (Figure 1A). The cholera toxin (CT) secreted by Vibrio cholerae, composed of one toxic A subunit and five identical B subunits responsible of the uptake of the toxin, binds multivalently to GM1 residues of epithelial cells in the small intestine facilitating endocytosis (Figure 1B).5 More examples have been extensively reported in literature.1a, 6
Figure 1.
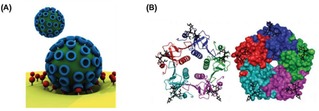
Examples of biological multivalent systems. (A) Schematic representation of the multivalent interaction of a virus with a cell membrane. Adapted from ref. [1b] with permission from John Wiley and Sons. (B) Structure of the pentavalent B subunits of cholera toxin binding to five mannose residues. Reproduced from ref. [5b] by permission of The Royal Society of Chemistry.
The study and the quantification of multivalent interactions is important in order to obtain molecular insight into the biological systems in which they are involved. For this reason numerous synthetic systems have been developed to investigate multivalency and its principles. Some contributions in this field have promoted the understanding of multivalent interactions both in solution and at surfaces in a quantitative manner.7
Applications of multivalency principles have been investigated in several fields. Multivalent molecules have been synthesized, for example, to induce cellular responses.8 Application of strong multivalent receptor−protein interactions have been employed for the preparation of inhibitors of pathogens such as Escherichia coli 9 or influenza.10 Other multivalent molecules, such as dendrimers, have been extensively used in nanomedicine.11 A large number of multivalent structures, employing scaffolds to which multiple ligands are anchored, have been synthesized, with varying size, shape, valency and physical characteristics for a wide range of applications.12
Here, a general overview of the surface functionalization methods employed for the study of multivalent interactions at (two‐dimensional) interfaces is provided, with a focus on the methods that allow control over the ligand density. Modification of nanoparticle surfaces with controlled ligand density, although largely reported in literature,13 is beyond the scope of this review. In the first part, surface modification methods employed for the control of the ligand density is discussed. The second part aims at providing an overview of multivalent systems investigated at interfaces.
2. Multivalency at Surfaces
Although less investigated than multivalent systems in solution, multivalent interactions at interfaces are particularly important.14 Surfaces modified with several monovalent ligands or receptors can be considered as multivalent platforms at which multivalent interactions can be studied.14 Therefore, interactions at surfaces (e. g. of cell membranes, self‐assembled monolayers and lipid membranes) can be investigated in order to better understand multivalency principles and their effect on biological systems.
Several studies of multivalent principles at interfaces have been reported. A large contribution was given by Reinhoudt and Huskens in this field by their studies of multivalent systems binding at β‐cyclodextrin self‐assembled monolayers (CD SAMs).15 In their studies, these so‐called molecular printboards have been developed for the selective binding of molecules with defined valencies and binding affinities. Moreover, models have been described for the interpretation of the binding of multivalent molecules to interface‐immobilized receptors.
Concomitant to the study of the fundamental principles of multivalency, the application of multivalent interactions at interfaces has been largely investigated. Multivalent binding of molecules and proteins at functionalized surfaces has been used in nanotechnology for the fabrication of dynamic nanochips or biosensors. Reversible DNA binding on surfaces was achieved by using photolabile multivalent dendrons, which release DNA upon UV irradiation due to degradation and charge‐switching multivalency.16 In another example, Fragoso et al. immobilized Cytochrome c at an electrode surface via multivalent supramolecular interactions.17 Tkac and coworkers developed lectin biosensors for the detection of glycoproteins with a detection limit in the femtomolar range (Figure 2A).18 More recently, Chen and coworkers reported a dynamic supramolecular multivalent platform capable of specifically capturing and releasing bacteria (Figure 2B).19 Numerous glycan microarrays have been developed for the selective modification of surfaces with monovalent or multivalent ligands in order to bind carbohydrate binding proteins (CBP) in a multivalent fashion.20 In this way, the binding affinities and selectivities of CBPs for specific receptors have been investigated.
Figure 2.
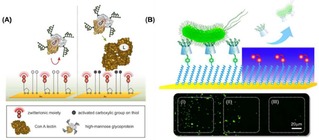
(A) Schematic representation of a self‐assembled monolayer of 11‐mercaptoundecanoic formed on a gold surface, and subsequent activation of the −COOH group for the immobilization of lectins for the detection of glycoproteins. Adapted with permission from ref. [18]. Copyright 2013 American Chemical Society. (B) Localized release of bacteria on the Au surface: area (I) refers to the region without UV irradiation, area (II) to the boundary region, and area (III) to the region irradiated with UV. Adapted with permission from ref. [19]. Copyright 2017 American Chemical Society.
2.1. Role of the Ligand Density on the Surface
In the study of multivalent biological systems, bioactive surfaces need to meet specific requirements to provide an optimal performance of interaction. First of all, surfaces must provide a high and well‐controlled binding capacity of the biomolecules, which must prevent, at the same time, their denaturation upon binding. Secondly, non‐specific interactions of biomolecules need to be prevented. Therefore, adequate surface modifications that provide antifouling properties are required.21 Additionally, ligand−receptor interactions are known to be strongly dependent on a threshold ligand density.22 In multivalent interactions, the average density of ligands on the surface needs to match the density of binding sites of the receptors in order to provide an efficient recognition.23 In the case of low ligand densities, average inter‐ligand distances are too large and only weak monovalent interactions occur. When ligand densities reach a minimum value (threshold), multivalent interactions can occur, providing a stronger and more stable binding. For this reason, the surface ligand density appears to be a fundamental parameter that needs to be taken into account for the achievement of optimal interactions at the interface.
The importance of the control of the ligand density at surfaces was discussed by Kiessling and coworkers with an illustrative example in their study of the interaction of concanavalin (ConA) with mannose‐functionalized surfaces.24 ConA, a tetrameric lectin with a binding affinity for mannose residues and presenting binding sites with a distance of approximately 6.5 nm, can bind divalently to ligand‐functionalized surfaces. In this study, the authors observed that the adsorption of ConA on gold surfaces, detected by SPR, was strongly dependent on the saccharide density at the interface (Figure 3A). Specifically, a small adsorption of proteins onto the surface was observed at low ligand surface densities, for which the average carbohydrate spacing was larger than 6.5 nm. The reduced adsorption was attributed to a weak monovalent ligand−receptor interaction. By increasing the carbohydrate density on the surface (i. e. with an average distance of less than 6.5 nm), a much higher protein adsorption was obtained owing to the occurring multivalent interaction. A further increase of the ligand density, however, did not result in an increased amount of protein bound to the surface. Due to the rigidity, the low valency and large spacing of ConA, several carbohydrates remained unbound at the surface.
Figure 3.
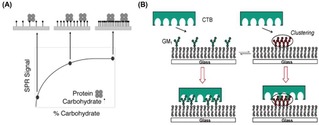
(A) Schematic model of the binding of ConA on mannose‐modified surfaces. In case of low mannose densities, the average distance between residues is too large to enable ConA to bind with two residues. At an intermediate carbohydrate coverage, the distance between mannose residues allows the protein to interact with multiple carbohydrates on the surface. A high ligand density does not lead to a further increase of the amount of protein bound to the surface. Reprinted with permission from ref. [24]. Copyright 2003 American Chemical Society. (B) Schematic representation of the inhibition of CTB binding caused by clustering of GM1. Reprinted with permission from ref. [25]. Copyright 2007 American Chemical Society.
Additional effects such as steric hindrance or electrostatic repulsion between ligands on the surface may affect the overall ligand−receptor interaction, leading to alteration or even a decrease of binding affinities at increasing ligand densities. Cremer and coworkers, for example, have reported an effect of ligand clustering in the binding of the cholera toxin B subunit (CTB) with ganglioside GM1 at a supported lipid bilayer (SLB). By varying the concentration of GM1 from 0.02 to 10.0 mol% in the phospholipid membrane, the binding of cholera toxin proteins was weakened upon increase of the ligand density (Figure 3B).25
3. Methods for Controlling the Surface Density
Several approaches for the functionalization of surfaces with a wide range of ligands have been reported. Self‐assembled monolayers (SAMs), supported lipid bilayers (SLBs) and different types of polymers have been extensively employed for the functionalization of substrates with ligands such as carbohydrates, peptides or DNA strands. Among all the surface modification strategies, three main conceptual approaches can be used for the modification of surfaces with a precise control over the ligand density displayed at the interface: i) mixing of active and inactive ligands; ii) using pre‐functionalized polymers or proteins; iii) controlling surface modification in three‐dimensional structures. In this section, some examples for each of the mentioned method are reported. A brief overview of the different types of chemistry employed for the modification of surfaces is also provided.
3.1. Mixing of Molecules in Solution
Among the different approaches generally employed for the control over the ligand density on the surface, the mixing of functionalized and non‐functionalized molecules in solution prior to the functionalization of surfaces is by far the most used. In particular, two main approaches can be used for a controlled modification of substrates: Surfaces can be modified in a first step with active functional groups such as N‐hydroxysuccinimide (NHS) or click chemistry groups, which subsequently react with ligands bearing proper chemical modification. Alternatively, ligand‐modified molecules can be directly reacted onto the surface. In both cases, the molar ratio of functionalized molecules in the mixture is correlated (although not necessarily in a linear fashion) to the density of ligands displayed at the surface. This method is mainly used with SAMs, SLBs and protein‐modified surfaces.
3.2. Self‐Assembled Monolayers (SAMs)
One of the most commonly employed surface modification methods for the study of interactions at interfaces is the formation of SAMs.26 SAMs are well‐known two‐dimensional nanostructures formed by the ordered assembly of molecules onto a large variety of solid surfaces.27 The two most commonly used classes of SAMs are the sulfur‐containing molecules (sulfides, disulfides and thiols) on gold (and other noble metal) substrates and the alkylsilanes on oxide surfaces. Due to the ease of preparation, the high stability, and the possibility of modifying substrates with desired properties, SAMs have been largely employed for the modification of surfaces for a wide range of applications. Detailed descriptions of the properties and applications of SAMs have been exhaustively illustrated in several reviews.27, 28
The formation of SAMs of sulfur‐containing molecules on gold is an exquisite method for the modification of surfaces with controlled ligand density.29 The most commonly used method to form SAMs consists of the mixing of alkanethiols in different ratios with thiols containing a functional group, such as NHS or maleimide. Subsequently, ligands bearing complementary functional groups (i. e. amino groups or thiols) are reacted onto the thiol‐modified substrates. Alternatively, in order to improve the antifouling properties of the SAMs, thiols containing poly(ethylene glycol) (PEG) chains are used in the mixtures. Gold surfaces are therefore incubated with these solutions in order to form monolayers with a stochastic display of functional groups. By varying the molar fraction of thiols bearing functional groups in the thiol mixture, it is possible to accurately tune the ligand density on the surface in the second step. This method has been used for a large variety of applications such as the formation of micro‐arrays and (bio)sensors for multivalent interactions.30
Impedimetric glycan biosensors have been developed using mixtures of thiols on gold substrates by Hushegyi et al.31 Mixed SAMs of sialic acid residues were made with controlled ligand densities. Specifically, mixtures of 11‐mercaptoundecanoic acid (MUA) with 6‐mercaptoexanol were used for the functionalization of gold substrates, with MUA molar fractions ranging from 5 % to 50 %. After activation of the carboxylic acid groups of MUA with EDC/NHS, the formed esters were reacted with amine‐terminated glycans. By using the same approach, Magnusson and coworkers functionalized surfaces with controllable densities of modified chemoattractant peptides.32 Solutions of maleimide‐terminated and OEG‐containing alkyldisulfides were used for the formation of SAMs on gold (Figure 4A). Cys‐terminated ligands were subsequently bound on the surface by coupling with maleimide. In another example, Mrksich and coworkers formed SAMs on gold using thiols presenting maleimide groups for the anchoring of carbohydrates and peptides.33
Figure 4.
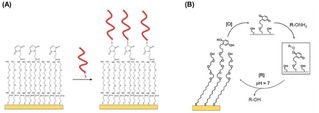
(A) Immobilization of Cys‐modified peptides on gold surfaces modified with SAMs formed from maleimide and OEG‐containing thiols. The maleimide allows the specific functionalization with peptides whereas the OEG improves the antifouling properties of the surface. Adapted with permission from ref. [32]. Copyright 2008 American Chemical Society. (B) Schematic representation of electrochemical oxidation [O] of hydroquinone to quinone and subsequent reaction with a soluble oxyamine‐tagged ligand (RONH2) to give the redox‐active oxime conjugate on the surface. Adapted from ref. [34] with permission from John Wiley and Sons.
Yousaf and Mrksich pioneered the formation of electroactive SAMs for the controlled functionalization of surfaces with ligands.34 Hydroquinone‐functionalized thiols, which can be chemically or electrochemically oxidized to benzoquinone, were employed for the modification of surfaces. After oxidation, the obtained quinone monolayer can subsequently react, for example, with cyclo‐pentadiene‐modified peptides by a Diels‐Alder reaction,35 or with oxyamine ligands to give a redox‐active oxime conjugate36 on the surface (Figure 4B). In this way, a large variety of biologically active ligands can be reversibly anchored to the surface. Surfaces were, for example, functionalized using high‐throughput microarray technology with controlled densities of FLAG or RGD peptide ligands. After the immobilization of the thiolated hydroquinone, the hydroquinone headgroups were converted to quinone by electrochemical oxidation at 750 mV. In this way the quinone monolayer was reacted with an aminooxy‐functionalized peptide. The molar fraction of hydroquinone‐functionalized thiols in the formation of the SAM regulates the peptide ligand density at the interface.
By exploiting click chemistry reactions, Dubacheva et al. reported the formation of SAMs composed of mixed pegylated thiols with and without azide functional groups for the modification of surfaces with controlled ferrocene (Fc) or adamantane (Ad) densities.37 Alkyne derivatives of the guests were added to the surface by exploiting the azide−alkyne click reaction. Control of the guest density was achieved by tuning the fraction of azide‐modified thiols mixed with the regular PEG thiol in the first step. The variation of the functional group densities from 0.5 to 330 pmol/cm2 led to average distances between neighboring ferrocenes ranging from 18 to 0.7 nm, respectively.
With a different approach, SAMs can be directly formed with ligand‐functionalized thiols. In this approach, a chemical modification of the ligand is performed prior to adsorption onto the surface. In this way, Houseman et al. functionalized surfaces with controlled densities of Gly−Arg−Gly−Asp−Ser (GRGDS) peptide ligands by exploiting the formation of alkanethiol‐based SAMs on gold.38
The formation of silane monolayers on glass substrates is a valid alternative for the functionalization of surfaces with ligands. Compared to thiol‐based SAMs, silane‐terminated monolayers have a higher chemical stability, thus allowing a broader range of chemical reactions on surfaces.39 However, silanes present the disadvantage of being highly reactive. Moreover, the control over ligand density appears to be less predictable for silane‐based monolayers than for thiols on gold. A binary mixed SAM consisting of 3‐aminopropyltriethoxysilane (APTES) and octadecyltrimethoxysilane (ODS) showed that APTES is significantly enriched in the mixed SAM compared to the starting solution.40 Similarly, when amino‐terminated silanes and methyl‐terminated silanes were mixed in different ratios to form SAMs on aluminum oxide surfaces by co‐adsorption of the silanes, the composition of the monolayer, as studied with AFM, contact angle, XPS and mechanical tests, indicated that the amine‐terminated silanes adsorbed two times faster than the methyl‐terminated silanes.41 An efficient modification of substrates with controlled ligand densities was achieved by Wayment et al. by mixing a low concentration of (3‐aminopropyl)triethoxysilane (APTES) and a much larger concentration of (2‐cyanoethyl)triethoxysilane (CETES). The low amino group density on the surface due to the presence of APTES, allowed the modification of surfaces with a very low fraction of biotin moieties (<10−7).42
3.3. Supported Lipid Bilayers (SLBs)
Although extensively used for the study of biological interactions at interfaces, SAMs resemble only remotely real biological membranes and lack some typical membrane properties, such as membrane fluidity, which is essential to mimic mobility and ligand reorganization occurring at cell membranes upon interactions.43 In this regard, the formation of SLBs has emerged as a valid alternative method for the modification of surfaces for biological studies.44 SLBs are two‐dimensional fluid platforms consisting of phospholipids retaining some of the relevant properties of cell membranes.45 Phospholipid vesicles, under specific conditions, rupture on activated hydrophilic surfaces such as mica, glass and silicon, forming a notably stable lipid bilayer.44a The presence of a thin water layer (approx. 10 Å thick)46 between the formed lipid bilayer and the underlying surface allows the mobility of the lipids on the surface providing fluidity to SLBs. In particular, the fluidity of the system can be regulated by varying the chemical composition of the SLB.47 The mobility of the SLB is an additional key feature in the modification of surfaces with ligands as, compared to SAMs, it allows dynamic clustering of receptors, which can effectively compensate for a low ligand density at the interface.
SLBs have been reported to present excellent antifouling properties, preventing the nonspecific adsorption of proteins and cells onto their surface.48 Methods for the introduction of ligands/receptors of choice in the SLB have been reported in literature.45 Briefly, lipids modified with a particular functional moiety can be added to the lipid mixture during the vesicle preparation and thus get displayed on the surface after the formation of the SLB. Subsequently, as in the case of SAMs, modified ligands that react or interact with these functional groups can be anchored onto the surface. In a different approach, lipids modified synthetically with ligands before SLB formation can be directly added to the lipid mixture before the formation of the SLB. In both approaches, controlling the molar fraction of functionalized lipids in the lipid mixture provides an exquisite method to control the surface density.
Several ligands and receptors have been anchored to SLBs with tunable densities by exploiting both non‐covalent and covalent interactions. Biotinylated lipids can be introduced in SLBs by mixing 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) or 1‐palmitoyl‐2‐oleoyl‐glycero‐3‐phosphocholine (POPC) lipids with biotinylated phosphatidylethanolamine (biotin−DOPE) during the preparation of vesicles.49 Streptavidin (SAv) can, therefore, be used as a linker for the further functionalization of surfaces with biotinylated linkers by exploiting the strong non‐covalent biotin−SAv interaction.50 Koçer and Jonkheijm, for example, varied the amounts of DOPE−biotin (between 0.01 and 1 mol%) in fluid DOPC and non‐fluid DPPC‐based SLBs in order to functionalize surfaces with varying RGD ligand densities.51 Alternatively, lipid molecules containing a nitrilotriacetic acid (NTA) head group can be doped into the SLB, and proteins modified with histidine tags can be chelated with the NTA lipids in the presence of Ni2+ ions.52 Multiple histidine are typically added to ensure, at the same time, the stability of the attachment of the proteins and their proper orientation at the interface. Lipids modified with two NTA moieties (bis‐NTA) were synthesized by Piehler and coworkers for the modification of SLBs with histidine‐tagged proteins.53
In the case of covalent modification of SLBs, lipids containing reactive functional groups, as for example maleimide, can be incorporated into the bilayer for the binding of complementary, e. g. thiol, functionalized molecules or proteins containing cysteine residues. Thid et al. used vesicles doped with 0–5 % maleimide‐terminated lipids for the formation of SLBs on SiO2 substrates, which were subsequently functionalized with IKVAV‐containing peptides.54 A different approach consists of the direct modification of biomolecules with a lipid that can be inserted into the SLB. Control of the surface ligand density can be quantitatively achieved by adjusting the molar fraction of modified lipids in the lipid mixture during the preparation of the vesicle. Synthetic glycolipids have been used, for example, for the introduction of controlled densities of glycans in SLBs.25, 55, 56
As a valid alternative to SLBs, supported lipid monolayers (SLMs) can be also employed for the modification of surfaces with ligands. Methods as Langmuir‐Blodgett or Langmuir‐Schaefer can be used for the formation of monolayers.57 Alternatively, SLMs can be formed from the rupture of lipid vesicles on hydrophobic self‐assembled monolayers.58 Octadecanethiol on gold or octadecyltrichlorosilane on glass surfaces are two typical examples of hydrophobic monolayers used for hybrid bilayer formation, owing to the possibility of forming highly ordered and well‐packed monolayers.59
Kiessling and coworkers developed an SLM in order to control the mannose ligand density on surfaces (Figure 5).60 In their studies, POPC liposomes containing different ratios of synthetic glycolipids bearing mannose groups where added on gold surfaces pre‐functionalized with alkanethiols. As in the case of SLBs, by tuning the molar ratio of synthetic glycolipids in the mixture with POPC during the formation of liposomes, it was possible to control the density of mannose exposed on the surface.
Figure 5.
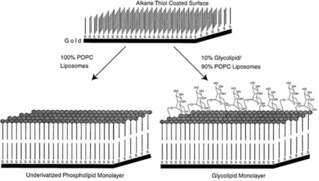
Scheme illustrating control over ligand density using supported lipid monolayers. Adapted with permission from ref. [60]. Copyright 1998 American Chemical Society.
3.4. Formation of Protein Layers
The formation of protein layers on surfaces has been also employed for the control of the surface density on the surface. In particular, the functionalization of substrates with SAv has been largely used for the modification of surfaces with biotinylated receptors. Techniques such as biolayer interferometry (BLI), for example, employ SAv‐covered surfaces for the modification of surfaces with controlled densities of saccharides. By controlling the molar ratio of biotinylated ligands with biotinylated dummy molecules, it is possible to tune the ligand density displayed on the surface. Xiong et al. functionalized surfaces with varying densities of sialic acid residues this way.61 The formation of polymer layers onto surfaces represents a valid intermediate degree of dynamic ligand display at the interface as, despite the lack of mobility, many conformations of the ligands are accessible.
3.5. Pre‐Modified Polymers and Proteins
The use of modified polymers or proteins represent an alternative way to functionalize surfaces with control over the exposed ligand density. In particular, polymers and proteins can be modified with specific ligands prior to their adsorption. The modification and the inter‐ligand spacing on the polymer backbone or the protein provides thereby control of the ligand density displayed at the interface.
Several examples of polymers and polyelectrolytes which allow an easy and controlled modification have been reported. Among several types of polymers, poly‐l‐lysine (PLL) has been extensively employed for the modification of surfaces with ligands. Owing to its positive charge at physiological pH, PLL can adsorb spontaneously from aqueous solutions on negatively charged substrates (such as glass, titanium, niobium oxide) by electrostatic interactions, thus forming molecular monolayers.62 At the same time, the presence of terminal amino groups present at the lateral chains allows easy modification of the polymer.62, 63 PLL can be, for example, grafted with (PEG) chains by an NHS coupling reaction to generate PLL‐graft‐PEG (PLL‐g‐PEG), and the grafting ratio can be easily controlled during the synthesis step. PLL‐g‐PEG has been generated carrying additional biologically relevant ligands such as peptides,64 biotin65 or NTA66 at the termini of the PEG side chains allowing the display of these ligands on the functionalized surface. The control of the grafting ratio of the copolymer leads to the possibility of tailoring the ligand density on the surface. Among several reported examples, Barth et al. showed the possibility of tuning the saccharide density on the surface by using mannoside‐functionalized PLL‐g‐PEG.67 In this study, a series of PLL‐g‐PEGs was synthesized containing either mono or oligo‐mannosides, and these polymers were assembled on Nb2O5‐coated glass surfaces. The mannose surface density was hereby varied between 0 and 26 pmol/cm2.
More recently, Duan and coworkers employed oligo(ethylene glycol) (OEG)‐grafted PLL (PLL−OEG) for the fabrication of a bio‐functionalized film on nano‐bioFETs.68 OEG and OEG−biotin moieties were grafted in different ratios to produce PLL‐g‐OEG‐biotin containing different ligand densities. The total degree of functionalization of the PLL was varied to study the optimal functionalization to obtain both stability of the PLL on the surface and maximal adsorption of SAv. In our group, maleimide (Mal) groups were used instead of biotin in order to generate substrates modified with tunable densities of functional groups.69 By exploiting the coupling between maleimide and thiolated peptide nucleic acid (PNA), PNA probes were displayed on the surface to form a biorecognition surface that allows the detection of complementary DNA (cDNA). The responses for cDNA hybridization were found to depend on the PNA probe density displayed at the interface which was set during the preceding PLL synthesis step, by controlling the amount of Mal in the polymer.
Another intriguing example of control over ligand spacing using polypeptides is given in the work of Lin et al.70 Here, a polyproline helix type II (PPII), which is a non‐charged polypeptide, was used as a scaffold for the binding of proteins onto surfaces. PPII was selectively modified at one side with glycans, whereas the other side was modified with fluorous groups which allowed the anchoring of the peptide on fluorous slides (Figure 6A). By tuning the number and the position of lateral fluorous chains, the display of the polymers on the surface was controlled. The surface density control was achieved by diluting the functionalized scaffold with unfunctionalized ones.70
Figure 6.
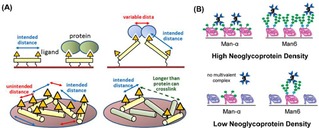
(A) Schematic representation of different arrangements of polyproline scaffolds on fluorous surfaces for the control of carbohydrate densities. Reprinted with permission from ref. [70]. Copyright 2017 American Chemical Society. (B) Different binding modes of ConA proteins to substrates with high and low neoglycoprotein density. Reprinted with permission from ref. [23]. Copyright 2010 American Chemical Society.
Alternatively, neoglycoproteins have been used to control the density of ligands on surfaces. Gildersleeve and coworkers have presented the use of glycoproteins bearing mannose residues for the modification of surfaces (Figure 6B).23 With this method, mannose residues with different lengths were displayed on the surfaces to bind multivalently ConA proteins. Ligand density control was achieved by mixing BSA with glycoproteins in solution prior to adsorption onto the surface.
Another powerful method to functionalize surfaces with biological ligands with controlled density is represented by the immobilization of single‐strand DNA onto the surface and the subsequent hybridization with ligand‐modified complementary oligonucleotides. In this regard, Niemeyer and co‐workers have provided significant contributions. For example, microarrays of several cell‐specific ligands have been immobilized by DNA hybridization to study the selective interactions of cells.71 With a similar approach Chevolot et al. produced DNA‐based carbohydrate biochips for the selective recognition of lectins.72
3.6. Polymer Brushes
An attractive alternative approach to engineer surfaces with controlled ligand density is represented by the use of polymer brushes. These polymers consists of either block copolymers or end‐grafted polymers which are tethered to a surface at one end, allowing the formation of coatings with desirable thickness in a nanometric range.73 The other end of the polymer chain, instead, presents a functionality which can influence the property of the surface and can allow secondary functionalization of substrates. Several reviews have been reported, thoroughly describing properties and applications of this class of polymers.74 By reacting or anchoring ligands such as carbohydrates or peptides to the functional groups displayed at polymer brushes, it is possible to modify surfaces with ligands with controlled density.75
Haag and coworkers reported a polyglycerol (PG)‐based block copolymer, synthesized from PG and a poly(allyl glycidyl ether) (PAGE), which was used for the functionalization of a large variety of surfaces with controlled RGD peptide densities (Figure 7).76 Interestingly, the addition of azide groups in the PG copolymer can allow secondary modification of the polymer with RGD by exploiting the strain‐promoted cyclo‐addition. By mixing azide‐terminated with bromide‐terminated polymers it was possible to tune the grafting density of RGD on the surface.
Figure 7.
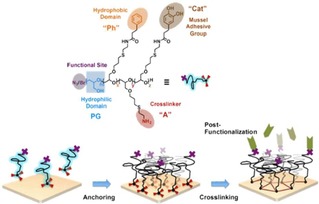
Representation of surface modification with PG‐based amphiphilic block copolymers. The PG catechol groups contribute to binding to polar surfaces, while the phenyl groups are used for anchoring to non‐polar surfaces. The N3 or Br terminal groups allow secondary modification of the surfaces. Adapted with permission from ref. [74]. Copyright 2017 American Chemical Society.
In a different example, polymer brushes were grafted from a surface by using surface‐initiated atom transfer radical polymerization (SI‐ATRP). Non‐fouling poly(2‐hydroxyethyl methacrylate) (PHEMA) and poly(poly(ethylene glycol)methacrylate) (PPEGMA) brushes were prepared on glass and silicon substrates with thicknesses between 20 and 150 nm with controlled RGD densities.74a Post‐modification of the surface, performed with different concentrations of RGD‐based peptide ligands in solution, led to ligand surface densities ranging between 0.5–12 pmol/cm2.
Shi et al. presented the use of polymer brushes in combination with non‐covalent host‐guest interactions for the control of lysine ligand densities.77 Lysine‐functionalized surfaces were prepared by integrating lysine‐modified CD derivatives by host‐guest interactions onto adamantyl‐pre‐modified copolymer brushes. Control of the localized and average lysine density at the surface was achieved by changing the lysine valency on the CD scaffolds and by diluting lysine‐modified CD with pure CD, respectively (Figure 8). The influence of surface presentation of lysine residues and their local density in the binding affinity of plasminogen (Plg) was investigated by using lysine‐modified polymer brushes. As a main conclusion, the displacement and the density of the lysines was found to affect not only the binding affinities but also the valency of the overall interactions. Copolymer surfaces modified with heptavalent lysines [CD(Lys)7] showed higher Plg adsorption and higher Plg binding affinities compared to the monovalent ones [CD(Lys)1].
Figure 8.
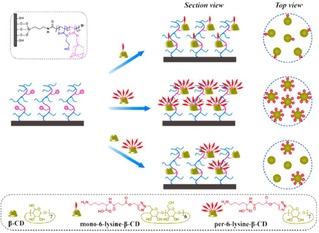
Schematic representation of the modulation of localized and average lysine densities of surfaces by host‐guest interactions using polymer brushes. Adamantyl‐modified polymer brushes are modified with lysine‐functionalized cyclodextrins bearing different numbers of lysines in order to produce different surface ligand densities. Adapted with permission from ref. [75]. Copyright 2015 American Chemical Society.
3.7. Formation of 3D Layers
While the formation of monolayers and bilayers are the most used methods for the functionalization of surfaces with ligands, these platforms allow only a 2D arrangement of the ligands on surfaces. However, the formation of a 3D environment, which resembles the in vivo conditions of biological interactions, can be particularly important in the study of biological systems. Cells, for examples, have shown different responses when placed in a 3D environment compared to 2D.29, 78
In order to achieve a surface modification with 3D structures, glycodendrimers have been anchored onto surfaces. Pieters and coworkers anchored glycodendrimer‐based glycan microarrays on porous aluminum oxide.79 Specifically, alkyne‐functionalized dendrimers were anchored on a maleimide‐functionalized surface via an amine functional group linked to the core of the dendrimers. Subsequently, azide‐functionalized carbohydrates were linked onto the surface by copper‐catalyzed click chemistry. By controlling the valency of the carbohydrate‐modified dendrimers, the ligand density at the surfaces was varied.
Alternatively, microarrays based on end‐point immobilization of oriented glycopolymers have been employed to mimic natural cell surface glycans in 3D. Sun and coworkers presented an O‐cyanate chain‐end functionalized glycopolymer for the modification of surfaces with glycans.80 Glycopolymers were pre‐complexed with boronic acid ligands composed of varying lengths and then immobilized by isourea‐bond formation at high pH onto an amine‐functionalized glass slide. After the immobilization, the boronic acid ligands were released from the immobilized glycopolymers at a reduced pH to generate the oriented and density‐controlled glycopolymer microarray.
In a different approach, Musah et al. immobilized polyacrylamide‐based hydrogels on a glass surface with varying densities of RGD peptides.81 Specifically, hydrogels bearing succinimidyl ester groups were used for the modification of surfaces. After the formation of the hydrogel, the activated esters were reacted with a mixture of an amine bearing a maleimide group and glucamine. While the former was used for peptide attachment by the coupling with a cysteine residue, the latter was employed as inert component. The ratio of glucamine and the maleimide‐containing amine was varied in order to tune the peptide density. In another study by Murphy et al., PEG‐based hydrogels where modified with varying densities of RGD peptides.82 The ligand density on the surfaces was varied by mixing RGD and RDG (inactive peptide) hydrogels, while keeping the total peptide concentration constant.
4. Multivalent Studies at Functionalized Platforms
A large number of multivalent systems such as molecules, proteins, viruses or cells has been investigated at ligand‐functionalized platforms. CD‐modified surfaces have been used, for example, for the binding of multivalent polymers, and SAMs have been used for the formation of glycan arrays for the study of the interactions of proteins and cells. In this section, an overview of some relevant examples reported in the literature is provided. Examples of platforms with control over the ligand density are here reported, in which the density‐dependent, multivalent affinity and binding behavior has been investigated. Examples of multivalent reversible binding are also discussed in this section.
4.1. Binding of Molecules and Polymers at Interfaces
The multivalent binding of several types of molecules at interfaces has been reported. Both small oligovalent ligands and large polyvalent polymers and dendrimers have been investigated, showing strong binding affinities for ligand‐modified surfaces. The improved binding affinities and stability of the molecules on the surface, in comparison to the monovalent parent interaction, have been found to correlate with the multivalent nature of the interactions. Several examples of synthetic multivalent host‐guest interactions at interfaces have been reported by Huskens and coworkers, providing a detailed understanding of the thermodynamic contributions of multivalent binding at interfaces.83 Stronger binding affinities were observed for multivalent interactions at the interface compared to the equivalent systems in solution due to a local high concentration (also called effective molarity) of receptors created by surface immobilization.
In a recent report, the binding of fluorescent dye‐labelled multivalent azopyridine molecules to gold and plastic substrates was investigated by Valderrey et al.84 In this work, heteroternary host‐guest complexes of multivalent azopyridines with methyl viologen/cucurbit[8]uril inclusion complexes were formed at viologen‐functionalized surfaces. Surface binding constants of multivalent ligands, determined by SPR, showed binding affinities that were two orders of magnitude higher than that of the monovalent one. Interestingly, supramolecular exchange experiments performed by patterning of mono and divalent molecules on surfaces, showed a substitution of the monovalent guest by the multivalent ones.
The dynamic binding of multivalent redox‐active ferrocenyl dendrimers at β‐CD monolayers was investigated by Nijhuis et al.85 The dendrimers of higher generations formed kinetically stable supramolecular assemblies at the CD surface presenting up to eight multivalent interacting pairs. Desorption of the dendrimers was achieved by electrochemical oxidation of the Fc end groups of the dendrimers which led to an efficient unbinding of the molecules from the host surface. Desorption and re‐adsorption of the dendrimers were repeated several times without significant decomposition of the system.
Several examples of the binding of polyvalent polymers at modified platforms have been reported. Polymer systems have been used as models for the investigation of biological interactions. The multivalent interaction of p‐tert‐butylphenyl or adamantyl‐functionalized poly(iso‐butene‐alt‐maleic acid)s at CD SAMs, leading to thermodynamically and kinetically stable multivalent assemblies, was reported by Crespo‐Biel et al.86 The polymer concentration and the nature or number of the functionalization did not affect the adsorption notably, nor did addition of monovalent competitors in solution lead to measurable polymer desorption, showing the strength of the overall interaction. The polymers were found to adsorb in a conformationally fully unwound fashion, leading to very thin (<1 nm) layers with practically complete usage of the polymer‐attached guest sites.
Dubacheva et al. reported the interaction of CD‐modified hyaluronan (HA) polymers with ferrocene (Fc) or adamantane (Ad)‐modified surfaces (see above).37 The variation of Ad and Fc densities allowed the study of the superselective binding behavior of the multivalent polymers. In this work, an analytical model was developed that provides quantitative predictions of the tuning of the superselective binding properties of the polymer, based on molecular characteristics such as affinity and valency. The effect of the ligand mobility at surfaces to the superselective binding was recently investigated.87 The binding of multivalent CD‐modified HA polymers was found to be enhanced and shifted to lower receptor densities at fluid interfaces (based on SLBs) compared to immobile ones (based on SAMs).
4.2. Binding of Proteins and Peptides at Interfaces
Multivalent interactions at interfaces of a wide range of proteins have been investigated, which generally showed a strong dependence of the overall binding affinity on the surface ligand density. One of the most studied polyvalent proteins is ConA. The weak binding affinity of ConA to mono‐saccharide ligands (K a∼104 M−1), enforced by multivalent interactions, together with its tetrameric structure, makes ConA an ideal model for the study of protein−carbohydrate interactions at the interface.88 Hereby some of the most relevant examples of studies of the binding of ConA at surfaces are reported.
Sato et al. designed a lectin‐recognizing molecular interface for ConA by using SAMs consisting of 12‐mercaptododecyl β‐maltoside (MalC12SH) and OH‐terminated thiols (HOCnSH, of varying length) as filling molecules (Figure 9A).89 Variations of both ligand density and dummy thiol length led to the optimization of the protein−carbohydrate interaction. Specifically, the enhancement of the valency of the interaction was controlled by creating a significant height difference between the saccharide and the terminus of the filling molecule. An optimal ligand density was found at 10 % of MalC12SH in the mixed monolayer. Kiessling and coworkers investigated the multivalent binding of ConA at SLMs, where the best sensitivity of the interaction was achieved with 10 % of functionalization with mannose in the total lipid mixture.60 Alternatively, a mannose‐containing cross‐linked polyacrylamide (c‐PAAm) was employed as a surface‐immobilized polyvalent ligand for the multivalent recognition of ConA on surfaces.90 With this approach, high‐affinity binding (∼106 M−1) and highly sensitive detection were achieved.
Figure 9.
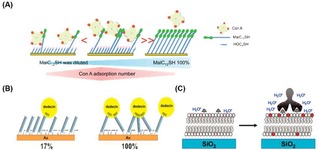
Multivalent binding of proteins on ligand‐presenting platforms. A) Schematic representation of binding of ConA for different densities of β‐maltoside. Different densities of saccharide‐modified thiols in the total thiol mixture causes a difference in the binding valency and affinity of ConA. Reprinted with permission from ref. [87]. Copyright 2012 American Chemical Society. (B) Schematic representation of the formation of a flavin‐modified dsDNA layer. The use of DNA mixtures with and without flavin allows the variation of flavin density on the surface, leading to different valency in the overall interaction with dodecin proteins. Adapted with permission from ref. [91]. Copyright 2015 American Chemical Society. (C) Binding of an ant‐biotin antibody on a biotin‐modified SLB resulting in an increased fluorescence of Texas Red DHPE‐lipids. Adapted with permission from ref. [92]. Copyright 2009 American Chemical Society.
A wide range of multivalent protein−carbohydrate interactions at interfaces has been investigated. SAMs containing mannose, lactose, or α‐Gal trisaccharide were used in the study of specific carbohydrate−protein interactions with ConA, ECL−lactose and anti‐Gal, respectively. Overall binding constants for these interactions were determined by QCM, SPR and electrochemistry studies.91
The multivalent binding of the Lens culinaris (LENS) lectin was investigated on mannose‐functionalized crystalline Si(111) surfaces.92 After the modification of the surfaces with glycan residues by “click” coupling, the multivalent interactions were investigated by quantitative attenuated total reflectance Fourier transform infrared spectroscopy (ATR‐FTIR) and atomic force microscopy (AFM). The variation of the ligand density at the surface (varied from 7.2×1012 to 1.2×1014 molecules per cm2) showed an effect on the protein binding, and optimal binding was achieved at a fraction of 10 mol% of mannose residues on the surface. At a lower mannose density (1 mol%), only monovalent interactions occurred, which resulted in a decrease of protein adsorption. At high ligand density (100 %) instead, limitations in protein adsorption were observed as a consequence of steric hindrance between mannose residues, as the average inter‐ligand density (0.91 nm) resulted to be lower than the size of the carbohydrate recognition domain (1.4 nm).92
Nöll and coworkers reported the multivalent interaction of the flavoprotein dodecin with a flavin‐terminated DNA monolayer.93 Surfaces were modified with controlled densities of flavin by using single‐stranded DNA (ssDNA) which was stably anchored on a gold surface using three dithiane groups. In a following step, a complementary flavin‐modified ssDNA was added for hybridization. By mixing flavin‐free and flavin‐modified complementary DNA in different ratios, it was possible to tune the ligand density displayed at the interface. The density was found to influence the valency of the dodicin interaction at the interface. Low flavin surface coverages (<17 %) led to weak monovalent binding with the protein, while at high densities (>31 %) multiple binding events allowed a more stable multivalent binding (Figure 9B). At high flavin coverage up to three binding pockets were estimated to be accessible for each dodecin due to the octahedral arrangement of the six dodecin binding pockets. By using multivalent interactions, dodecin proteins were also tested for the generation of stable sandwich‐type flavin−apododecin−flavin architectures on surfaces.93
The multivalent interaction of anti‐biotin antibodies and cholera toxin B subunits with biotin and GM1‐modified surfaces, respectively, was presented by the group of Cremer.94 SLBs were doped with pH‐sensitive ortho‐Texas Red‐DHPE lipids, which fluoresce at acidic pH and become non‐fluorescent at higher pH. The binding of a negatively charged protein, causing a local decrease of pH at the interface, affected the fluorescence at the SLB (Figure 9C). By following the increase in fluorescence intensity upon binding of the protein, equilibrium dissociation constants were obtained with affinities in the nM range.
Cremer and coworkers inserted 2,4‐dinitrophenyl (DPN) in the SLBs, and studied the effect of the density of DPN to the binding with their associated IgG antibodies by using a high‐throughput microfluidic device.95 By mixing DPN‐conjugated lipids with egg phosphatidylcholine (egg−PC) in different ratios in the vesicle preparation step, the DPN density in the lipid membranes was varied from 0.1 to 5.0 mol%. Interestingly, the results showed that the density affects the affinity of the interaction. The apparent dissociation constant, K Dapp, between DNP and the antibodies, obtained with epifluorescence microscopy, increased by about a factor of 10 by increasing the DNP density from 0.1 to 3.75 mol%, while higher densities did not lead to a further increased affinity.
Joubert et al. formed microarrays based on poly(bis‐SorbPC) lipids doped with GM1 lipids for the study of the interaction with the cholera toxin protein.96 After UV‐initiated polymerization, air stable poly(lipid) bilayer microarrays were obtained. GM1 molar ratios in the lipid mixture were varied from 0 to 10 % leading to varying adsorption of labeled CTB proteins. The extent of binding of CTB in each spot, detected by fluorescence microscopy, was correlated to the mole percentage of GM1. CTB proteins were successfully removed from surfaces by exposure of GM1‐modified arrays to denaturants. In this way the regeneration of the arrays was achieved, and the CTB binding capability was confirmed after multiple regeneration cycles.
Brock and coworkers investigated the binding selectivity of oligo‐histidines to immobilized multiple NTA moieties.97 By means of microarrays, mono‐, bis‐, tris‐ and tetrakis‐NTA chelators were spotted at different surface densities. The ability of histidine‐based multivalent binders to discriminate fluorescently labelled hexa and decahistidine peptides was tested. This work showed that, when both peptides were incubated together, an increased affinity of decahistidine was observed compared to hexahistidine, while also showing a strong dependence on the chelator density. Binding assays by dual‐color total internal reflection fluorescence spectroscopy revealed active exchange of His6 by His10, thus confirming the high selectivity towards His10.
4.3. Binding of Viruses and Virus‐Like Particles at Interfaces
Interactions of viruses with receptor‐modified surfaces have been widely investigated. Functionalization of surfaces with precise control of the ligand densities was achieved for the development of biosensing surfaces able to detect low concentrations of virus in solution. Quantification of the interaction of several types of viruses was also achieved. Mixed SAMs with control over the density of sialic acid residues were developed by Hushegyi et al. (see above) to form impedimetric glycan biosensors for the detection of the influenza A virus (IAV) hemagglutinins (HAs) in the attomolar range.31 Aptamer‐based sensors (aptasensors) were developed by Bai and coworkers for the detections of IAV.98 The aptamer surface density appeared to affect the binding affinity between surface‐anchored aptamers and viruses, showing a more than 100 times higher sensitivity when the surface density was increased from 4.8×1011 to 14×1011 molecules/cm2 (Figure 10A). Alternatively, a mucin‐mimetic glycopolymer‐based microarray was employed for the study on the binding of IAV with sialic acid receptors (Figure 10B).99 Printing of azide‐modified glycopolymers on cyclooctyne‐coated surfaces produced microarrays with increasing glycopolymer densities. H1N1 and H3N2 viruses were incubated on the platform, showing selectivity for specific glycans. Reversible binding of viral proteins and viruses was obtained by using pH‐switchable monolayers. Recently, Sellergren and coworkers reported the interaction of the H5N1 (A/Anhui/2005) influenza virus on a sialic acid‐modified reversible SAM (rSAM).100
Figure 10.
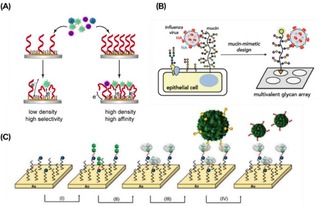
A) Representation of the interaction of IAV with aptamer‐modified sensors in which the density of aptamers on the surface affects the binding affinity of viruses. Adapted from ref. [96] with permission from Elsevier. (B) Schematic representation of the interaction of IAV with a mucin‐mimetic microarray. Reproduced from ref. [97] by permission of The Royal Society of Chemistry. (C) Schematic representation of stepwise assembly of the CB[8] monolayers and subsequent multivalent interaction of modified CCMV viruses. Reproduced from ref. [99] by permission of The Royal Society of Chemistry.
Weineisen et al. reported the controlled and switchable immobilization of modified viruses on surfaces.101 Azobenzene‐modified Cowpea chlorotic mottle virus (CCMV) was multivalently bound to surfaces by the formation of a photoresponsive, hetero‐ternary complex between azobenzene, cucurbit[8]uril (CB[8]) and methylviologen (MV). The association constant (K a=1.4×106 M−1) obtained for this system was found to be several orders of magnitude higher than that of a single interaction (K a=3×103 M−1),102 showing the multivalent nature of the binding. Subsequent irradiation of the surface with UV light for 5 min caused a trans‐cis isomerization of the azobenzene moieties resulting in the release of the viruses.
Höök and coworkers presented a study of a multivalent model system for the investigation of virus binding at interfaces. In order to mimic virion association to a cell membrane, small lipid vesicles (100 nm diameter) were used. The binding of the vesicles to an SLB was achieved through multiple cholesterol‐based DNA linker molecules. Total internal reflection microscopy was used to track single attached vesicles, which showed that the variation of the numbers of linking DNA tethers led to variation of the vesicle diffusion coefficient on the surface.103
We recently reported the multivalent binding of recombinant hemagglutinin (rHA) protein clusters at sialoglycan‐functionalized SLBs.104 Affinities and selectivities for binding at receptor‐modified platforms were observed for HA particles from different virus variants and at varying receptor densities using QCM. The small size of the clusters and their limited number of HA trimers allowed an accurate estimation of the interaction area and the valency in binding to the receptor surface. A low nanomolar overall affinity was obtained, which was achieved with 6–9 HA‐sugar molecular interaction pairs, thus presenting a weak multivalent binding and a rapid association/dissociation behavior.
4.4. Binding of Cells and Bacteria at Interfaces
A large variety of platforms has been modified with receptors for the investigation of the interactions of cells and bacteria at surfaces. SAMs of alkanethiols were formed on gold surfaces presenting RGD peptides for the study of the attachment of cells. The microenvironment in which the ligands are displayed affects the attachment and the morphology of the adhering cells.38 In other studies, SLBs functionalized with 19‐mer peptides containing the IKVAV sequence were used to investigate the attachment of PC12105 and AHP106 cells on the surface. A nonlinear correlation was observed between the density of IKVAV presented on the SLB and the number of attached AHP cells, showing a threshold of ligand density necessary for cell attachment. The effect of the ligand density on the cell behavior has also been reported in other reviews.29, 107
An example of multivalent interactions of bacteria at the interface was presented by Guo and coworkers, in which SLB‐based microarrays were developed for the adhesion of E. coli.56 The density of mannose on the surface was varied over 2 orders of magnitude (between 0.002–0.3 molecules per nm2), showing that the FimH adhesion protein of E. coli changes avidity from monovalent to multivalent as the density of mannose increases (Figure 11). The same interaction was studied by Van Weerd et al., utilizing an SLB‐based platform on which a continuous, locked‐in mannose gradient was formed.108 This study demonstrated the specific binding of FimH proteins and the selective binding above a threshold density of mannose. Binding affinities corresponding to a K d of 0.9×10−21 M were obtained, confirming the multivalent nature of the interaction, as monovalent interactions have been reported to be in the μM range.109
Figure 11.
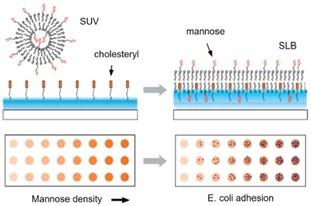
Formation of a mannose‐presenting SLB surface, made from unilamellar vesicles, and a schematic illustration of a glycan density gradient microarray for studying pathogen adhesion. Glycan density on the surface can be tuned by varying the molar ratio of the glycan‐functionalized lipid in the mixture during the preparation of the vesicles. Adapted with permission from ref. [56]. Copyright 2009 American Chemical Society.
5. Conclusions and Outlook
The ligand density displayed at the interface is a fundamental parameter in the study of the multivalent systems. The variation of the density of ligands appears to influence the valency involved in the multivalent interactions and, therefore, the overall binding affinity and selectivity. Here, a review of the surface modification methods employed in the functionalization of surfaces has been provided.
The first part has focused on the different chemical approaches employed for the modification of surfaces for the control of the ligand density. SAMs, SLBs, modified polymers and proteins have been extensively used for a controlled functionalization of surfaces with ligands. Inter‐ligand distances on surfaces that match spacing of the receptor binding sites of proteins generally improve the binding of proteins. Moreover, the importance of a ligand threshold density has been shown, as a minimum density of ligands is required to provide strong multivalent interactions.
In the second part of this review, examples of multivalent systems have been discussed. The binding of multivalent molecules, proteins, viruses and cells have been extensively investigated. The development of methodologies for studying multivalent interactions at interfaces through density variation is important for a detailed understanding of the relevant molecular aspects of the interaction.
The use of platforms for the study of biological interactions at interfaces, despite the increase in the last years, is still very limited. However, the development of such platforms that allow selective biological recognition events and their quantification is essential for a wide range of applications. For example, biosensors for the identification of pathogens such as bacteria and viruses could provide timely treatments of patients after an infection. Sensors for the quantification of virus interactions with cell receptors can be useful for the development of virus warning systems in the prevention of epidemics or pandemics. Alternatively, platforms can be employed for the development and testing of new drugs. A future synergy of chemists, biologists, biochemists and biophysicists in this field can contribute to a fast outgrowth of platforms able to investigate a wide range of biological interactions.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Daniele Di Iorio (1990) studied Chemistry at Sapienza University of Rome where he received both his Bachelor's degree in 2013 and his Master's degree (summa cum laude) in 2015 working under the supervision of Prof. Mario Barteri. In September 2019 he received his PhD at the University of Twente in the Molecular Nanofabrication group under the supervision of Prof. Jurriaan Huskens. The aim of his project was to study multivalent interactions at interfaces in biological systems by exploiting the functionalization of surfaces.

Biographical Information
Jurriaan Huskens (1968) studied chemical engineering at the Eindhoven University of Technology, and obtained his PhD (1994) at the Delft University of Technology with Herman van Bekkum. After postdoctoral stays with Dean Sherry (UT Dallas) and Manfred Reetz (MPI Kohlenforschung), he became assistant professor (1998) with David Reinhoudt at the University of Twente, where he became full professor “Molecular Nanofabrication” in 2005. He received the Unilever Research Award 1990, a Marie Curie fellowship (1997), the Gold Medal 2007 of the Royal Netherlands Chemical Society, and a Fellowship from the Institute of Advanced Study, Durham University, UK (2019). Present research interests encompass: supramolecular chemistry at interfaces, supramolecular materials, multivalency, nanofabrication, and solar fuels. He is (co)author of about 400 refereed research papers and five patents.

Acknowledgements
This work was financially supported by the Marie Curie Innovative Training Network MULTI‐APP (No. 642793) and by the Netherlands Organization for Scientific Research (NWO), TOP grant 715.015.001.
D. Di Iorio, J. Huskens, ChemistryOpen 2020, 9, 53.
Dedicated to Professor Jean‐Marie Lehn for his life‐long contributions to supramolecular science, on the occasion of his 80th birthday.
References
- 1.
- 1a. Mammen M., Choi S.-K., Whitesides G. M., Angew. Chem. Int. Ed. 1998, 37, 2754–2794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2908–2953; [Google Scholar]
- 1b. Fasting C., Schalley C. A., Weber M., Seitz O., Hecht S., Koksch B., Dernedde J., Graf C., Knapp E.-W., Haag R., Angew. Chem. Int. Ed. 2012, 51, 10472–10498; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10622–10650; [Google Scholar]
- 1c. Mulder A., Huskens J., Reinhoudt D. N., Org. Biomol. Chem. 2004, 2, 3409–3424. [DOI] [PubMed] [Google Scholar]
- 2. Mahon E., Barboiu M., Org. Biomol. Chem. 2015, 13, 10590–10599. [DOI] [PubMed] [Google Scholar]
- 3. Kim Y., Cao Z., Tan W., Proc. Natl. Acad. Sci. 2008, 105, 5664–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu W., Pieters R. J., Expert Opin. Drug Discovery 2019, 14, 387–395. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Merritt E. A., Sarfaty S., Akker F. V. D., L′Hoir C., Martial J. A., Hol W. G. J., Protein Sci. 1994, 3, 166–175; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Branson T. R., Turnbull W. B., Chem. Soc. Rev. 2013, 42, 4613–4622. [DOI] [PubMed] [Google Scholar]
- 6. Bernardi A., Jiménez-Barbero J., Casnati A., De Castro C., Darbre T., Fieschi F., Finne J., Funken H., Jaeger K.-E., Lahmann M., Lindhorst T. K., Marradi M., Messner P., Molinaro A., Murphy P. V., Nativi C., Oscarson S., Penadés S., Peri F., Pieters R. J., Renaudet O., Reymond J.-L., Richichi B., Rojo J., Sansone F., Schäffer C., Turnbull W. B., Velasco-Torrijos T., Vidal S., Vincent S., Wennekes T., Zuilhof H., Imberty A., Chem. Soc. Rev. 2013, 42, 4709–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Mulder A., Auletta T., Sartori A., Del Ciotto S., Casnati A., Ungaro R., Huskens J., Reinhoudt D. N., J. Am. Chem. Soc. 2004, 126, 6627–6636; [DOI] [PubMed] [Google Scholar]
- 7b. Huskens J., Prins L. J., Haag R., Ravoo B. J., Multivalency: Concepts, Research & Applications., Wiley, 2018. [Google Scholar]
- 8. Kiessling L. L., Gestwicki J. E., Strong L. E., Angew. Chem. Int. Ed. 2006, 45, 2348–2368; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2408–2429. [Google Scholar]
- 9. Gouin S. G., Wellens A., Bouckaert J., Kovensky J., ChemMedChem 2009, 4, 749–755. [DOI] [PubMed] [Google Scholar]
- 10. Papp I., Sieben C., Sisson A. L., Kostka J., Böttcher C., Ludwig K., Herrmann A., Haag R., ChemBioChem 2011, 12, 887–895. [DOI] [PubMed] [Google Scholar]
- 11. Rolland O., Turrin C.-O., Caminade A.-M., Majoral J.-P., New J. Chem. 2009, 33, 1809–1824. [Google Scholar]
- 12.
- 12a. Lundquist J. J., Toone E. J., Chem. Rev. 2002, 102, 555–578; [DOI] [PubMed] [Google Scholar]
- 12b. Baldini L., Casnati A., Sansone F., Ungaro R., Chem. Soc. Rev. 2007, 36, 254–266. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Wang X., Ramström O., Yan M., Anal. Chem. 2010, 82, 9082–9089; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Takae S., Akiyama Y., Otsuka H., Nakamura T., Nagasaki Y., Kataoka K., Biomacromolecules 2005, 6, 818–824; [DOI] [PubMed] [Google Scholar]
- 13c. Elias D. R., Poloukhtine A., Popik V., Tsourkas A., Nanomed-Nanotechnol. 2013, 9, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huskens J., Curr. Opin. Chem. Biol. 2006, 10, 537–543. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Huskens J., Deij M. A., Reinhoudt D. N., Angew. Chem. Int. Ed. 2002, 41, 4467–4471; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4647–4651; [Google Scholar]
- 15b. Huskens J., Mulder A., Auletta T., Nijhuis C. A., Ludden M. J. W., Reinhoudt D. N., J. Am. Chem. Soc. 2004, 126, 6784–6797; [DOI] [PubMed] [Google Scholar]
- 15c. Onclin S., Mulder A., Huskens J., Ravoo B. J., Reinhoudt D. N., Langmuir 2004, 20, 5460–5466; [DOI] [PubMed] [Google Scholar]
- 15d. Auletta T., Dordi B., Mulder A., Sartori A., Onclin S., Bruinink C. M., Péter M., Nijhuis C. A., Beijleveld H., Schönherr H., Vancso G. J., Casnati A., Ungaro R., Ravoo B. J., Huskens J., Reinhoudt D. N., Angew. Chem. Int. Ed. 2004, 43, 369–373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 373–377. [Google Scholar]
- 16. Kostiainen M. A., Smith D. K., Ikkala O., Angew. Chem. Int. Ed. 2007, 46, 7600–7604; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7744–7748. [Google Scholar]
- 17. Fragoso A., Caballero J., Almirall E., Villalonga R., Cao R., Langmuir 2002, 18, 5051–5054. [Google Scholar]
- 18. Bertok T., Klukova L., Sediva A., Kasák P., Semak V., Micusik M., Omastova M., Chovanová L., Vlček M., Imrich R., Vikartovska A., Tkac J., Anal. Chem. 2013, 85, 7324–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhan W., Wei T., Cao L., Hu C., Qu Y., Yu Q., Chen H., ACS Appl. Mater. Interfaces 2017, 9, 3505–3513. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Narla S. N., Nie H., Li Y., Sun X.-L., Glycoconjugate J. 2015, 32, 483–495; [DOI] [PubMed] [Google Scholar]
- 20b. Park S., Gildersleeve J. C., Blixt O., Shin I., Chem. Soc. Rev. 2013, 42, 4310–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Q., Zhang Y., Wang H., Brash J., Chen H., Acta Biomater. 2011, 7, 1550–1557. [DOI] [PubMed] [Google Scholar]
- 22. Martinez-Veracoechea F. J., Frenkel D., Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10963–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y., Li Q., Rodriguez L. G., Gildersleeve J. C., J. Am. Chem. Soc. 2010, 132, 9653–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith E. A., Thomas W. D., Kiessling L. L., Corn R. M., J. Am. Chem. Soc. 2003, 125, 6140–6148. [DOI] [PubMed] [Google Scholar]
- 25. Shi J., Yang T., Kataoka S., Zhang Y., Diaz A. J., Cremer P. S., J. Am. Chem. Soc. 2007, 129, 5954–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wink T., van Zuilen S. J., Bult A., van Bennekom W. P., Analyst 1997, 122, 43R–50R. [DOI] [PubMed] [Google Scholar]
- 27. Love J. C., Estroff L. A., Kriebel J. K., Nuzzo R. G., Whitesides G. M., Chem. Rev. 2005, 105, 1103–1170. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Onclin S., Ravoo B. J., Reinhoudt D. N., Angew. Chem. Int. Ed. 2005, 44, 6282–6304; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6438–6462; [Google Scholar]
- 28b. Ulman A., Chem. Rev. 1996, 96, 1533–1554. [DOI] [PubMed] [Google Scholar]
- 29. Satav T., Huskens J., Jonkheijm P., Small 2015, 11, 5184–5199. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Love K. R., Seeberger P. H., Angew. Chem. Int. Ed. 2002, 41, 3583–3586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3733–3736; [Google Scholar]
- 30b. Zhi Z.-L., Laurent N., Powell A. K., Karamanska R., Fais M., Voglmeir J., Wright A., Blackburn J. M., Crocker P. R., Russell D. A., Flitsch S., Field R. A., Turnbull J. E., ChemBioChem 2008, 9, 1568–1575; [DOI] [PubMed] [Google Scholar]
- 30c. Frasconi M., Mazzei F., Ferri T., Anal. Bioanal. Chem. 2010, 398, 1545–1564. [DOI] [PubMed] [Google Scholar]
- 31. Hushegyi A., Bertok T., Damborsky P., Katrlik J., Tkac J., Chem. Commun. 2015, 51, 7474–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wetterö J., Hellerstedt T., Nygren P., Broo K., Aili D., Liedberg B., Magnusson K.-E., Langmuir 2008, 24, 6803–6811. [DOI] [PubMed] [Google Scholar]
- 33. Houseman B. T., Gawalt E. S., Mrksich M., Langmuir 2003, 19, 1522–1531. [Google Scholar]
- 34. Chan E. W. L., Park S., Yousaf M. N., Angew. Chem. Int. Ed. 2008, 47, 6267–6271; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6363–6367. [Google Scholar]
- 35.
- 35a. Yousaf M. N., Houseman B. T., Mrksich M., Angew. Chem. Int. Ed. 2001, 40, 1093–1096; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1127–1130; [Google Scholar]
- 35b. Yousaf M. N., Houseman B. T., Mrksich M., Proc. Natl. Acad. Sci. 2001, 98, 5992–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoover D. K., Lee E.-j., Chan E. W. L., Yousaf M. N., ChemBioChem 2007, 8, 1920–1923. [DOI] [PubMed] [Google Scholar]
- 37. Dubacheva G. V., Curk T., Auzély-Velty R., Frenkel D., Richter R. P., Proc. Natl. Acad. Sci. 2015, 112, 5579–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Houseman B. T., Mrksich M., Biomaterials 2001, 22, 943–955. [DOI] [PubMed] [Google Scholar]
- 39. Haensch C., Hoeppener S., Schubert U. S., Chem. Soc. Rev. 2010, 39, 2323–2334. [DOI] [PubMed] [Google Scholar]
- 40. Tong Y., Tyrode E., Osawa M., Yoshida N., Watanabe T., Nakajima A., Ye S., Langmuir 2011, 27, 5420–5426. [DOI] [PubMed] [Google Scholar]
- 41. Lee I., Wool R. P., Thin Solid Films 2000, 379, 94–100. [Google Scholar]
- 42. Wayment J. R., Harris J. M., Anal. Chem. 2006, 78, 7841–7849. [DOI] [PubMed] [Google Scholar]
- 43.H. Jung, A. D. Robison, P. S. Cremer, J. Struct. Biol 2009, 168. [DOI] [PMC free article] [PubMed]
- 44.
- 44a. Richter R. P., Bérat R., Brisson A. R., Langmuir 2006, 22, 3497–3505; [DOI] [PubMed] [Google Scholar]
- 44b. Loose M., Schwille P., J. Struct. Biol. 2009, 168, 143–151. [DOI] [PubMed] [Google Scholar]
- 45. van Weerd J., Karperien M., Jonkheijm P., Adv. Healthcare Mater. 2015, 4, 2743–2779. [DOI] [PubMed] [Google Scholar]
- 46. Cremer P. S., Boxer S. G., J. Phys. Chem. B 1999, 103, 2554–2559. [Google Scholar]
- 47. Seu K. J., Cambrea L. R., Everly R. M., Hovis J. S., Biophys. J. 2006, 91, 3727–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Glasmästar K., Larsson C., Höök F., Kasemo B., J. Colloid Interface Sci. 2002, 246, 40–47. [DOI] [PubMed] [Google Scholar]
- 49. Kaizuka Y., Douglass A. D., Varma R., Dustin M. L., Vale R. D., Proc. Natl. Acad. Sci. 2007, 104, 20296–20301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. González M., Bagatolli L. A., Echabe I., Arrondo J. L., Argaraña C. E., Cantor C. R., Fidelio G. D., J. Biol. Chem. 1997, 272, 11288–11294. [DOI] [PubMed] [Google Scholar]
- 51. Koçer G., Jonkheijm P., Adv. Healthcare Mater. 2017, 6, 1600862. [DOI] [PubMed] [Google Scholar]
- 52. Nye J. A., Groves J. T., Langmuir 2008, 24, 4145–4149. [DOI] [PubMed] [Google Scholar]
- 53. Lata S., Gavutis M., Piehler J., J. Am. Chem. Soc. 2006, 128, 6–7. [DOI] [PubMed] [Google Scholar]
- 54. Thid D., Bally M., Holm K., Chessari S., Tosatti S., Textor M., Gold J., Langmuir 2007, 23, 11693–11704. [DOI] [PubMed] [Google Scholar]
- 55. Shen L., Wang Y., Lin C.-I., Liu H.-w., Guo A., Zhu X. Y., ACS Chem. Biol. 2014, 9, 1877–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhu X. Y., Holtz B., Wang Y., Wang L.-X., Orndorff P. E., Guo A., J. Am. Chem. Soc. 2009, 131, 13646–13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stottrup B. L., Veatch S. L., Keller S. L., Biophys. J. 2004, 86, 2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gray J. C., Webster C. I., Williams D. H., Packman L. C., Cooper M. A., Nucleic Acids Res. 2000, 28, 1618–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.
- 59a. Castellana E. T., Cremer P. S., Surf. Sci. Rep. 2006, 61, 429–444; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59b. Babayco C. B., Turgut S., Smith A. M., Sanii B., Land D., Parikh A. N., Soft Matter 2010, 6, 5877–5881. [Google Scholar]
- 60. Mann D. A., Kanai M., Maly D. J., Kiessling L. L., J. Am. Chem. Soc. 1998, 120, 10575–10582. [Google Scholar]
- 61.
- 61a. Xiong X., Coombs P. J., Martin S. R., Liu J., Xiao H., McCauley J. W., Locher K., Walker P. A., Collins P. J., Kawaoka Y., Skehel J. J., Gamblin S. J., Nature 2013, 497, 392–396; [DOI] [PubMed] [Google Scholar]
- 61b. Xiong X., Martin S. R., Haire L. F., Wharton S. A., Daniels R. S., Bennett M. S., McCauley J. W., Collins P. J., Walker P. A., Skehel J. J., Gamblin S. J., Nature 2013, 499, 496–499. [DOI] [PubMed] [Google Scholar]
- 62. Kenausis G. L., Vörös J., Elbert D. L., Huang N., Hofer R., Ruiz-Taylor L., Textor M., Hubbell J. A., Spencer N. D., J. Phys. Chem. B 2000, 104, 3298–3309. [Google Scholar]
- 63. Huang N.-P., Michel R., Voros J., Textor M., Hofer R., Rossi A., Elbert D. L., Hubbell J. A., Spencer N. D., Langmuir 2001, 17, 489–498. [Google Scholar]
- 64. VandeVondele S., Vörös J., Hubbell J. A., Biotechnol. Bioeng. 2003, 82, 784–790. [DOI] [PubMed] [Google Scholar]
- 65. Huang N.-P., Vörös J., De Paul S. M., Textor M., Spencer N. D., Langmuir 2002, 18, 220–230. [Google Scholar]
- 66. Zhen G., Falconnet D., Kuennemann E., Vörös J., Spencer N. D., Textor M., Zürcher S., Adv. Funct. Mater. 2006, 16, 243–251. [Google Scholar]
- 67. Barth K. A., Coullerez G., Nilsson L. M., Castelli R., Seeberger P. H., Vogel V., Textor M., Adv. Funct. Mater. 2008, 18, 1459–1469. [Google Scholar]
- 68. Duan X., Mu L., Sawtelle S. D., Rajan N. K., Han Z., Wang Y., Qu H., Reed M. A., Adv. Funct. Mater. 2015, 25, 2279–2286. [Google Scholar]
- 69. Movilli J., Rozzi A., Ricciardi R., Corradini R., Huskens J., Bioconjugate Chem. 2018, 29, 4110–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lin T.-H., Lin C.-H., Liu Y.-J., Huang C. Y., Lin Y.-C., Wang S.-K., ACS Appl. Mater. Interfaces 2017, 9, 41691–41699. [DOI] [PubMed] [Google Scholar]
- 71.
- 71a. Schroeder H., Ellinger B., Becker C. F. W., Waldmann H., Niemeyer C. M., Angew. Chem. Int. Ed. 2007, 46, 4180–4183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4258–4261; [Google Scholar]
- 71b. Arrabito G., Reisewitz S., Dehmelt L., Bastiaens P. I., Pignataro B., Schroeder H., Niemeyer C. M., Small 2013, 9, 4243–4249. [DOI] [PubMed] [Google Scholar]
- 72. Chevolot Y., Bouillon C., Vidal S., Morvan F., Meyer A., Cloarec J.-P., Jochum A., Praly J.-P., Vasseur J.-J., Souteyrand E., Angew. Chem. Int. Ed. 2007, 46, 2398–2402; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2450–2454. [Google Scholar]
- 73. Senaratne W., Andruzzi L., Ober C. K., Biomacromolecules 2005, 6, 2427–2448. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Tugulu S., Silacci P., Stergiopulos N., Klok H.-A., Biomaterials 2007, 28, 2536–2546; [DOI] [PubMed] [Google Scholar]
- 74b. Zhao B., Brittain W. J., Prog. Polym. Sci. 2000, 25, 677–710; [Google Scholar]
- 74c. Minko S., J. Macromol. Sci. C 2006, 46, 397–420. [Google Scholar]
- 75.
- 75a. Jiang H., Xu F.-J., Chem. Soc. Rev. 2013, 42, 3394–3426; [DOI] [PubMed] [Google Scholar]
- 75b. Edmondson S., Osborne V. L., Huck W. T. S., Chem. Soc. Rev. 2004, 33, 14–22. [DOI] [PubMed] [Google Scholar]
- 76. Yu L., Cheng C., Ran Q., Schlaich C., Noeske P.-L. M., Li W., Wei Q., Haag R., ACS Appl. Mater. Interfaces 2017, 9, 6624–6633. [DOI] [PubMed] [Google Scholar]
- 77. Shi X., Zhan W., Chen G., Yu Q., Liu Q., Du H., Cao L., Liu X., Yuan L., Chen H., Langmuir 2015, 31, 6172–6178. [DOI] [PubMed] [Google Scholar]
- 78. Tibbitt M. W., Anseth K. S., Biotechnol. Bioeng. 2009, 103, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.
- 79a. Branderhorst H. M., Ruijtenbeek R., Liskamp R. M. J., Pieters R. J., ChemBioChem 2008, 9, 1836–1844; [DOI] [PubMed] [Google Scholar]
- 79b. Parera Pera N., Branderhorst H. M., Kooij R., Maierhofer C., van der Kaaden M., Liskamp R. M. J., Wittmann V., Ruijtenbeek R., Pieters R. J., ChemBioChem 2010, 11, 1896–1904. [DOI] [PubMed] [Google Scholar]
- 80. Narla S. N., Sun X.-L., Lab Chip 2012, 12, 1656–1663. [DOI] [PubMed] [Google Scholar]
- 81. Musah S., Morin S. A., Wrighton P. J., Zwick D. B., Jin S., Kiessling L. L., ACS Nano 2012, 6, 10168–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nguyen E. H., Zanotelli M. R., Schwartz M. P., Murphy W. L., Biomaterials 2014, 35, 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.
- 83a. Perl A., Gomez-Casado A., Thompson D., Dam H. H., Jonkheijm P., Reinhoudt D. N., Huskens J., Nat. Chem. 2011, 3, 317–322; [DOI] [PubMed] [Google Scholar]
- 83b. Gomez-Casado A., Dam H. H., Yilmaz M. D., Florea D., Jonkheijm P., Huskens J., J. Am. Chem. Soc. 2011, 133, 10849–10857. [DOI] [PubMed] [Google Scholar]
- 84.V. Valderrey, M. Wiemann, P. Jonkheijm, S. Hecht, J. Huskens, ChemPlusChem, 0. [DOI] [PubMed]
- 85.
- 85a. Nijhuis C. A., Huskens J., Reinhoudt D. N., J. Am. Chem. Soc. 2004, 126, 12266–12267; [DOI] [PubMed] [Google Scholar]
- 85b. Nijhuis C. A., Yu F., Knoll W., Huskens J., Reinhoudt D. N., Langmuir 2005, 21, 7866–7876. [DOI] [PubMed] [Google Scholar]
- 86. Crespo-Biel O., Péter M., Bruinink C. M., Ravoo B. J., Reinhoudt D. N., Huskens J., Chem. Eur. J. 2005, 11, 2426–2432. [DOI] [PubMed] [Google Scholar]
- 87. Dubacheva G. V., Curk T., Frenkel D., Richter R. P., J. Am. Chem. Soc. 2019, 141, 2577–2588. [DOI] [PubMed] [Google Scholar]
- 88. Weatherman R. V., Kiessling L. L., J. Org. Chem. 1996, 61, 534–538. [DOI] [PubMed] [Google Scholar]
- 89. Sato Y., Yoshioka K., Murakami T., Yoshimoto S., Niwa O., Langmuir 2012, 28, 1846–1851. [DOI] [PubMed] [Google Scholar]
- 90. Yu L., Huang M., Wang P. G., Zeng X., Anal. Chem. 2007, 79, 8979–8986. [DOI] [PubMed] [Google Scholar]
- 91. Zhang Y., Luo S., Tang Y., Yu L., Hou K.-Y., Cheng J.-P., Zeng X., Wang P. G., Anal. Chem. 2006, 78, 2001–2008. [DOI] [PubMed] [Google Scholar]
- 92. Yang J., Chazalviel J.-N., Siriwardena A., Boukherroub R., Ozanam F., Szunerits S., Gouget-Laemmel A. C., Anal. Chem. 2014, 86, 10340–10349. [DOI] [PubMed] [Google Scholar]
- 93. Gutiérrez Sánchez C., Su Q., Schönherr H., Grininger M., Nöll G., ACS Nano 2015, 9, 3491–3500. [DOI] [PubMed] [Google Scholar]
- 94. Jung H., Robison A. D., Cremer P. S., J. Am. Chem. Soc. 2009, 131, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang T., Baryshnikova O. K., Mao H., Holden M. A., Cremer P. S., J. Am. Chem. Soc. 2003, 125, 4779–4784. [DOI] [PubMed] [Google Scholar]
- 96. Joubert J. R., Smith K. A., Johnson E., Keogh J. P., Wysocki V. H., Gale B. K., Conboy J. C., Saavedra S. S., ACS Appl. Mater. Interfaces 2009, 1, 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. André T., Reichel A., Wiesmüller K.-H., Tampé R., Piehler J., Brock R., ChemBioChem 2009, 10, 1878–1887. [DOI] [PubMed] [Google Scholar]
- 98. Bai C., Lu Z., Jiang H., Yang Z., Liu X., Ding H., Li H., Dong J., Huang A., Fang T., Jiang Y., Zhu L., Lou X., Li S., Shao N., Biosens. Bioelectron. 2018, 110, 162–167. [DOI] [PubMed] [Google Scholar]
- 99. Huang M. L., Cohen M., Fisher C. J., Schooley R. T., Gagneux P., Godula K., Chem. Commun. 2015, 51, 5326–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yeung S. Y., Mucha A., Deshmukh R., Boutrus M., Arnebrant T., Sellergren B., ACS Cent. Sci. 2017, 3, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weineisen N. L., Hommersom C. A., Voskuhl J., Sankaran S., Depauw A. M. A., Katsonis N., Jonkheijm P., Cornelissen J. J. L. M., Chem. Commun. 2017, 53, 1896–1899. [DOI] [PubMed] [Google Scholar]
- 102. Sankaran S., van Weerd J., Voskuhl J., Karperien M., Jonkheijm P., Small 2015, 11, 6187–6196. [DOI] [PubMed] [Google Scholar]
- 103. Block S., Zhdanov V. P., Höök F., Nano Lett. 2016, 16, 4382–4390. [DOI] [PubMed] [Google Scholar]
- 104. Di Iorio D., Verheijden M. L., van der Vries E., Jonkheijm P., Huskens J., ACS Nano 2019, 13, 3413–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Svedhem S., Dahlborg D., Ekeroth J., Kelly J., Höök F., Gold J., Langmuir 2003, 19, 6730–6736. [Google Scholar]
- 106. Thid D., Holm K., Eriksson P. S., Ekeroth J., Kasemo B., Gold J., J. Biomed. Mater. Res. Part A 2008, 84A, 940–953. [DOI] [PubMed] [Google Scholar]
- 107. Koçer G., Jonkheijm P., Adv. Healthcare Mater. 2018, 7, 1701192. [DOI] [PubMed] [Google Scholar]
- 108. van Weerd J., Sankaran S., Roling O., Sukas S., Krabbenborg S., Huskens J., le Gac S., Ravoo B. J., Karperien M., Jonkheijm P., Adv. Mater. Interfaces 2016, 3, 1600055. [Google Scholar]
- 109. Bouckaert J., Mackenzie J., De Paz J. L., Chipwaza B., Choudhury D., Zavialov A., Mannerstedt K., Anderson J., Piérard D., Wyns L., Seeberger P. H., Oscarson S., De Greve H., Knight S. D., Mol. Microbiol. 2006, 61, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]