

Abstract
When administered intravenously, various particles and nanomedicines activate complement, potentially leading to infusion reactions and other adverse drug reactions. Particles form within formulations of therapeutic proteins due to stresses incurred during shipping, handling and administration to patients. In this study, IVIg solutions were stored in multiple types of vials and prefilled syringes and exposed to agitation and freeze thawing stresses to generate particles. The stressed samples were added to human serum to determine whether these particles activated complement. Sub-visible IVIg particles ranging in size between 2 and 10 microns activated complement in a fashion that was linearly dependent on the number of particles dosed, whereas little correlation was found between doses of larger particles (>10 microns) and complement activation. Activation of complement by sub-visible particles of IVIg followed the alternative pathway, as shown by the release of complement cascade factor Bb and the production of the anaphylatoxins C3a and C5a without generation of C4a. The number and the morphology of sub-visible particles formed depended on the applied stress, formulation and on the container material. But the capacity of the 2-10 micron-sized particles to activate complement in human serum appeared to depend only on particle concentration.
Introduction
Therapeutic proteins offer effective treatments for a wide range of human diseases. However, in a fraction of patients, the safety and efficacy of these medicines are compromised by adverse drug reactions (ADRs) that include unwanted immune responses, infusion reactions, anaphylaxis, and even death1-13. Some of these ADRs are quite common. For example, hypersensitivity reactions following the first exposure to rituximab are reported in about half of patients with B-cell malignancies14, 14.5% of patients were reported to experience severe infusion reactions in response to the first administration of cetuximab15 and, 12.3% of patients receiving infusions of the anti-TNF-α antibody infliximab experienced infusion reactions16. Anaphylaxis, hypersensitivity and increased (undesired) antibody responses all can result from complement activation. Complement is normally activated as a biological defense mechanism against viral and microbial challenges, but nanoparticles in intravenously-administered drug formulations can also activate the complement cascade.17-23 Once the complement cascade is activated, it can have strong pro-inflammatory and anaphylactoid outcomes22. The strength of these responses is driven largely by the extents to which C3a and C5a, the two anaphylatoxins of the complement system, are produced. These two signaling molecules have the capacity to cause blood vessel dilation, modulate cytokine production, cause histamine release and oxidative burst, and act as signals for chemotaxis of innate immune cells19, 24, 25.
Although complement activation-driven actions can be very helpful for fighting localized microbial infections, they can be detrimental if they occur systemically, as happens in complement activation-related pseudoallergy (CARPA). CARPA is an infusion reaction that has been associated with intravenous administration of various liposomal26, 27 and micellar formulations of drugs, as well as to administration of nanoparticles17 and therapeutic antibodies22, 28-30. CARPA can provoke severe cardiopulmonary responses including anaphylactic cardiogenic shock and death22, 28.
Therapeutic proteins readily aggregate to form soluble oligomers and sub-visible particles during production, shipping, storage and administration to patients31. These aggregates are a risk factor for unwanted immune responses, which can lead to loss of therapeutic efficacy and other adverse reactions13, 32-34. Protein aggregation is often initiated by interactions with interfaces to which proteins are exposed, such as air-water, container-water and ice-water interfaces. These interfacial stresses can be magnified by handling. For example, aggregation of proteins at the container-water interfaces found in silicone oil-lubricated syringes can be greatly accelerated by agitation35, 36 and levels of particles within therapeutic protein formulations may be dramatically increased by freeze-thawing37, 38.
We hypothesized that, like some nanoparticulate drugs, the particles generated by handling of protein formulations may be capable of activating complement. To test this hypothesis, we first conducted accelerated stability experiments to generate particles within a polyclonal antibody formulation (intravenous immunoglobulin, IVIg) by subjecting the antibody formulation to freeze-thawing stresses in multiple types of vials or to stresses caused by agitation in multiple types of prefilled syringes. We then tested whether these handling-induced particles were capable of activating complement when added to human serum samples. Complement activation was measured using enzyme-linked immunosorbent assays (ELISA) to quantify the levels of C3a and C5a, two key signaling molecules for the pro-inflammatory properties of complement. In addition, we measured levels of C4a and Bb to probe which complement pathways might be activated. Production of C4a is a marker of complement activation by the classical or lectin pathways, whereas production of Bb is indicative of complement activation via the alternative pathway.
Materials and Methods
Materials used were of USP grade or higher. Intravenous immunoglobulin (IVIG; GAMMAGARD LIQUID®, Shire US Inc., Lexington, MA) was purchased from Wardenburg Pharmacy at the University of Colorado at Boulder. Chemicals purchased from Sigma Aldrich (St. Louis, Missouri) included sodium phosphate monobasic, sodium phosphate dibasic, and glycine. Chemicals purchased from Fisher Scientific (Waltham, Massachusetts) included polysorbate 20 (Tween 20™, N.F., Multi-Compendial, J.T. Baker), 10X phosphate buffered saline, and HyClone™ water for injection (WFI). Siliconized glass syringes were BD Hypak SCF 1mL long 27G1/2 (BD Medical-Pharmaceutical Systems, Franklin Lakes, NJ). SiOPlas™ syringes (1ml), (SiO2 Medical Products, Inc., Auburn, AL) were composed of cyclic olefin polymer (COP) syringe barrels whose interior surfaces were coated with a silica-based barrier coating system and an organosiloxane lubricant, both applied by plasma enhanced chemical vapor deposition39. SiOPlas™ vials (6ml) were composed of COP that was coated on the vial interior with a barrier coating system. 6 mL Ompi EZ-fill® borosilicate glass vials (Schott, AG) were provided by SiO2 Medical Products, Inc. (Auburn, AL).
Protein Formulation
Prior to use, IVIg was formulated at a protein concentration of 1 mg/mL in either phosphate-buffered saline at pH 7.4 (PBS), or in 250mM glycine pH 4.25 with 0.02% (v/v) polysorbate 20 (PS20). To remove any pre-existing particulates, GAMMAGARD LIQUID® samples containing 100 mg/mL immunoglobulin G in 250 mM glycine were centrifuged at 20,000 × g for 20 minutes at 4°C and the supernatant was used as a stock solution. The IVIg stock solution was diluted 1:100 into either 0.22 micron filtered PBS pH 7.4 or 250 mM glycine pH 4.25 with 0.02% (v/v) polysorbate 20 to yield a final concentration of 1 mg/mL IVIg.
Accelerated stress testing of IVIg formulations using agitation
Siliconized glass and SiOPlas™ syringes were filled with 1mg/mL IVIg in 250mM glycine pH 4.25 with 0.02% (v/v) PS20. Syringes were filled so that a uniform headspace gap of 4mm was present between the liquid and the stopper (Datwyler Pharma Packaging, Pennsauken, NJ). These syringes were rotated end-over end at room temperature for 10 days, causing the air bubble in the headspace gap to move from one end of syringe to other end with each rotation. In a few of the syringes that were tested, the air bubble became lodged at one end of the syringe and failed to move as the syringe was rotated. These syringes were excluded from further analysis. After the 10 days of end-over-end rotation, each formulation was expelled from the syringe through the needle using an automated syringe pump at 150 mm/min and sample was collected in pre-rinsed polypropylene tubes and tested for sub-visible particle concentrations and for complement activation capacity.
Another set of siliconized glass and SiOPlas™ syringes were filled with 1mg/mL IVIg in PBS. These syringes were placed horizontally on an orbital shaker and shaken overnight at room temperature. Following shaking, the formulations were expelled from the syringes through the needle using an automated syringe pump at 150 mm/min, collected in pre-rinsed polypropylene tubes, and tested for sub-visible particle concentrations and complement activation capacity.
Accelerated stress testing of IVIg formulations using freeze thawing
Borosilicate vials (6mL) and SiOPlas™ vials (6mL) were filled with 4 mL of a formulation containing 1 mg/mL IVIg in PBS pH 7.4. The contents of the vials were subjected to 1 or 6 freeze-thaw cycles. In each freeze-thaw cycle, the vials were first dipped in liquid nitrogen for 2 minutes, then thawed in a water bath at 30 °C for 14.5 minutes. Each vial was swirled gently for mixing prior to next freeze-thaw cycle.
Analysis of sub-visible particle concentrations
Particle concentrations and images of particles in the various stressed formulations were obtained using flow imaging microscopy (FlowCAM®, Fluid Imaging Technologies, Scarborough, Maine), as previously described.15 Samples that had been subjected to 6 freeze-thaw cycles contained concentrations of particles that approached or exceeded the upper limit for the flow imaging microscopy instrument, and so these formulations were diluted 100-fold with phosphate buffered saline, pH 7.4 before analysis. Particle counts in the stressed formulations were measured in aliquots that were sampled from the same individual syringes and vials that were tested for complement activation. Each sample contained a distribution of particle sizes; no effort was made to fractionate the particles prior to testing the stressed formulation for their ability to activate complement.
Analysis of soluble protein fractions by size exclusion chromatography
Size exclusion chromatography was used to monitor the retention of monomeric protein and the appearance of any soluble aggregates in IVIG samples after application agitation or freeze-thawing stresses. A TSKgel G3000SWXL column (TOSOH Biosciences, Montgomeryville Pennsylvania) was used with an Agilent 1100 series system (Santa Clara, CA). The eluent was monitored at an absorbance of 280 nm in the Agilent ChemStation software. The mobile phase of 100 mM sodium sulfate, 100 mM sodium phosphate, and 0.05% (w/v) sodium azide at pH 6.7 was passed through the system at 0.6 mL/min. Peak areas in chromatograms were quantified in GRAMS/AI software version 9.1 (Thermo Fisher Scientific Inc., Waltham, Massachusetts). After application of accelerated stress conditions, the fractional loss of soluble protein from the (essentially) particle-free, centrifuged and filtered starting formulations was quantified using the ratio of the peak areas for stressed vs. unstressed formulations.
Complement activation in human serum samples
Samples pooled from three vials or syringes of the various stressed IVIg formulations as well as unstressed control samples of each formulation were delivered to Exsera Biolabs (Denver, CO) for analysis of their capacity to activate complement. Complement activation was measured in normal human serum pooled from three individual donors that had been previously screened for normal complement function. Test samples of stressed IVIg formulations were diluted tenfold into pooled human serum, mixed and allowed to incubate at 37 °C for 30 minutes. After incubation, samples were stored at −80 °C until further testing was conducted. For analysis of complement activation, concentrations of four complement cascade activation fragments, C3a, Bb, C4a and C5a, were measured by ELISA using kits purchased from Quidel Corporation (San Diego, CA). C4a was chosen because it is a marker of activation of the classical or lectin pathway, Bb as a distinct marker of the alternative pathway for complement activation, C3a as the central point of complement activation and C5a as a marker of terminal complement pathway activation.40 Triplicate ELISA measurements were conducted for each of the for complement cascade proteins. In addition to testing of stressed IVIG samples, several controls were analyzed. These control samples contained pooled serum alone, or samples of serum to which saline solution, phosphate buffered saline with zymosan or phosphate buffered saline with heat aggregated gammaglobulins were added in a 1:9 ratio. The average of the measurements were plotted versus particle sample counts as a fold increase compared to concentrations measured in the saline control.
Results
Particle formation during accelerated stress testing of IVIg formulations
As expected, each of the accelerated stress testing methods generated microparticles within the tested formulations (Table 1). Freeze thawing of IVIg in PBS pH 7.4 resulted in the highest number of sub-visible particles. A single freeze-thaw cycle produced 3.2 × 105 and 7.1 × 105 particles of size greater than 2 micron in borosilicate and SiOPlas™ vials, respectively. Particles larger than 10 microns were also produced, with 8×103 particles detected in borosilicate vials and 5.1×104 particles found in SiOPlas™ vials.
Table 1.
Concentrations of sub-visible particles in stressed samples submitted for complement activation testing. Samples were pooled from three samples from each stress/container combination. Container-to-container variation in particle concentrations in samples prior to pooling was <15%.
| Applied Stress | Container Type / Buffer Condition |
Particles of size >2 micron, #/mL |
Particles of size >10 micron, #/mL |
|---|---|---|---|
| Unstressed Control | Polypropylene / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 300 | 10 |
| Single Freeze Thaw Cycle | SiOPlas™ Vial / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 7.1×105 | 5.1×104 |
| Single Freeze Thaw Cycle | Borosilicate Glass Vial / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 3.2×105 | 8.0×104 |
| Six Freeze Thaw Cycles | SiOPlas™ Vial / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 2.1×106 | 3.6×105 |
| Six Freeze Thaw Cycles | Borosilicate Glass Vial / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 4.8×106 | 2.3×105 |
| Orbital Shaker, Overnight | SiOPlas™ Syringe / PBS pH 7.4 | 2.4×106 | 1.7 ×105 |
| Orbital Shaker, Overnight | Siliconized Glass Syringe / PBS pH 7.4 | 6.0×105 | 1.1 ×105 |
| End-over-end rotation, 10 days | SiOPlas™ Syringe / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 3×103 | 90 |
| End-over-end rotation, 10 days | Siliconized Glass Syringe / 250 mM glycine pH 4.25, 0.02% (v/v) polysorbate 20 | 1.0×105 | 2×103 |
Application of multiple freeze thaw cycles further increased particle numbers by about an order of magnitude (Table 1). After six cycles, particle concentrations in borosilicate vials and SiOPlas™ vials had increased to 4.8×106 and 2.1×106 particles/mL of size greater than 2 microns, respectively. Particles larger than 10 micron were also generated, with 2.3 ×105 and 3.6×105 in borosilicate and SiOPlas™ vials, respectively.
The number of particles produced as a result of agitation stresses depended on the container type and also on the type of agitation that was applied41. Overnight agitation of IVIg formulations in PBS pH 7.4 using an orbital shaker generated 2.4×106 particles of size greater than 2 microns in siliconized glass syringes, and 6.0×105 particles/mL in corresponding SiOPlas™ syringes (Table 1). As was the case with the freeze-thaw studies, roughly an order of magnitude fewer large particles (>10 micron) were formed in syringes of both types.
End-over end rotation for ten days was the gentlest accelerated stability test that was applied. After IVIg formulations in glycine pH 4.25 underwent end-over-end rotation for 10 days, only 3×103 and 1.03 ×105 particles/mL in the particle size range greater than 2 microns were detected in SiOPlas™ and siliconized glass syringes, respectively. Correspondingly few particles larger than 10 micron were also generated, with 2.0 × 103 and 90 particles/mL detected in siliconized glass and SiOPlas™ syringes, respectively.
Collections of typical flow imaging microscopy images of particles produced by the various accelerated stress methods are presented in Figure 1. Samples stressed by freeze-thawing in vials predominately contained aspherical microparticles characteristic of proteinaceous aggregates (Figure 1a), whereas samples stressed by agitation in siliconized glass syringes (Figure 1b) contained numerous spherical particles characteristic of droplets of lubricants. Agitation in the SiOPlas™ syringes (Figure 1c) produced particles of various morphologies that may have comprised both irregularly-shaped protein aggregates and spherical droplets of silicone oil.
Figure 1.

Flow imaging microscopy of stressed IVIg samples. Collages are randomly-selected set of images of particles present in IVIg samples that had been processed by a) overnight shaking in glass syringes on an orbital shaker; b) overnight shaking in SiOPlas™ syringes on an orbital shaker; c) 10 days of end-over-end rotation in silicone oil-lubricated glass syringes; d) 10 days of end-over-end rotation in SiOPlas™ syringes; e) freeze-thawed six times in glass vials; f) freeze-thawed six times in SiOPlas™ vials.
For all of the accelerated stress conditions tested, size exclusion chromatographic analysis showed that only minimal (<5%) amounts of insoluble protein were formed. No soluble aggregates were detected under any condition. Application of six freeze-thaw cycles to IVIg in borosilicate glass vials produced the highest numbers of particles per mL – nearly 5 million particles greater than 2 microns – but this represented a loss of only 3.7% of the original monomeric protein.
Complement activation in human serum in response to particles produced under accelerated stress conditions.
Particles of size greater than 2 microns within IVIg formulations that had been subjected to accelerated stress conditions activated complement in a linear, dose-dependent fashion when the IVIg formulations were diluted ten-fold into human serum (Figures 3-5). Levels of complement activation were generally higher in samples with larger numbers of particles of size greater than 10 microns, but these larger particles were present at low levels that did not allow any clear dose-dependence to be resolved (see Figure 6). .
Figure 3.
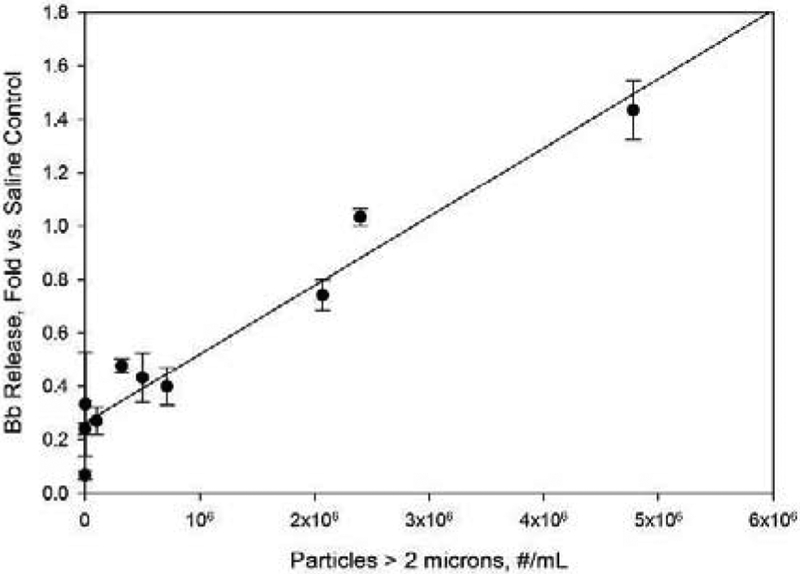
Effect of sample particle levels on Bb concentration in human serum samples. Concentrations are reported as fold increases vs. Bb levels for saline control samples. Particle concentrations refer to concentrations in IVIg formulations prior to 10-fold dilution into human serum. Line represents a least-squares linear fit, with correlation coefficient r2= 0.94.
Figure 5.
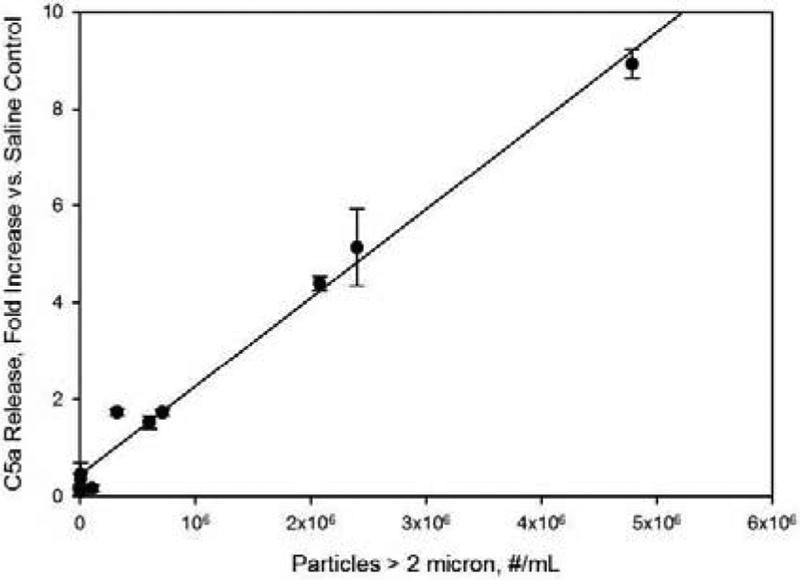
Effect of sample particle levels on C5a concentration in human serum samples. Concentrations are reported as fold increases vs. C5a levels for saline control samples. Particle concentrations refer to concentrations in IVIg formulations prior to 10-fold dilution into human serum. Line represents a least-squares linear fit, with correlation coefficient r2= 0.99.
Figure 6.

Effect of concentrations of particles of sizes larger than 10 microns on C5a (filled circles) and C3a (open circles) concentrations in human serum samples. Concentrations are reported as fold increases vs. C5a and C3a levels for saline control samples. Particle concentrations refer to concentrations in IVIg formulations prior to 10-fold dilution into human serum. Line represents a least-squares linear fit, with correlation coefficients r2= 0.60 and r2=0.47 for C5a and C3a, respectively.
The observed complement activation was consistent with activation through the alternative pathway. No increase over saline control levels was seen for C4a, which is a marker for the classical or lectin pathways (Figure 2). In contrast, Bb, a marker of the alternative pathway for complement activation, increased linearly (r2=0.94) as the concentrations of particles in the size range greater than 2 microns increased (Figure 3).
Figure 2.
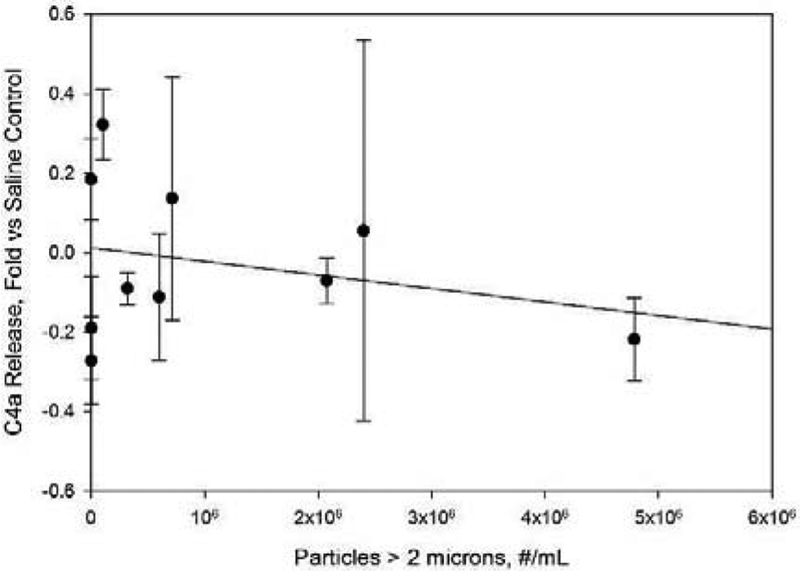
Protein particles within IVIg samples did not stimulate release of C4a concentration in human serum samples. Concentrations are reported as fold increases vs. C4a levels for saline control samples. Particle concentrations refer to concentrations in IVIg formulations prior to 10-fold dilution into human serum. Line represents a least-squares linear fit, with correlation coefficient r2= 0.005.
Increases in concentrations of the anaphylatoxins C3a and C5a were also observed when particles were diluted into serum (Figures 4 and 5). As with the Bb response, the fold increases vs. saline control were linearly dependent on particle dose, with correlation coefficients r2 of 0.85 and 0.99 for C3a and C5a, respectively. Responses to the IVIg formulations that were subjected to 6 freeze-thaw cycles in borosilicate glass vials produced increases in C3a and C5a concentrations that were 2.4 and 8.9 fold higher than those observed in saline controls.
Figure 4.
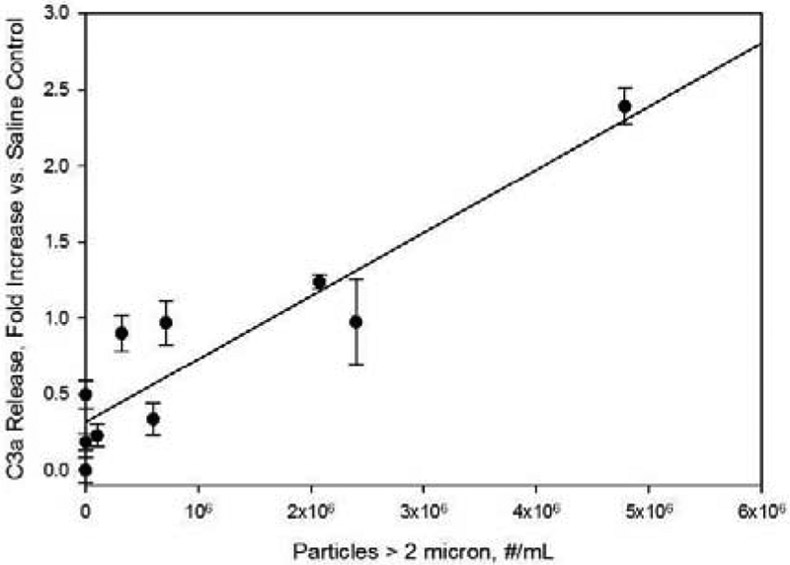
Effect of sample particle levels on C3a concentration in human serum samples. Concentrations are reported as fold increases vs. C3a levels for saline control samples. Particle concentrations refer to concentrations in IVIg formulations prior to 10-fold dilution into human serum. Line represents a least-squares linear fit, with correlation coefficient r2= 0.85.
For comparison, positive controls containing 1 mg/mL zymosan or 1 mg/L heat aggregated gamma globulins stimulated ca. 11-fold increases (vs. saline) of C3a, Bb, and C4a, and ca 32-fold increases (vs. saline) in C5a levels.
Discussion
Sub-visible particles are ubiquitous in formulations of therapeutic proteins, but their levels can be greatly increased by stresses encountered during shipping, handling and storage42-44. Agitation stresses are incurred as a result of vibration during shipping, and during routine handling of liquids. Although liquid formulations of therapeutic proteins are typically intended to remain in liquid form at controlled temperature, accidental freezing can occur, especially during shipping in containers cooled with gel packs38, or in formulations intended for home administration45. In the polyclonal antibody formulations tested here, increased levels of particles were seen when IVIg formulations in prefilled syringes were subjected to agitation, or when formulations were freeze-thawed in vials. The current study was designed to probe whether sub-visible particles in protein formulations are capable of complement activation, and so we used a variety of formulations and accelerated stress conditions to create samples with a range of particle sizes and concentrations, but we did not attempt to elucidate the detailed mechanisms by which each stress fostered particle formation.
The particle concentrations that were observed after freeze-thaw testing depended on both the vial type and on the number of freeze thaw cycles to which the formulations were subjected. After six freeze-thawing cycles, millions of particles of size greater than 2 μm and hundreds of thousands of particles of size greater than 10 μm were produced per mL of sample. Interestingly, samples in glass vials that underwent multiple freeze-thawing cycles produced more particles than samples frozen in the polymeric SiOPlas™ vials. We speculate that the higher numbers of particles in glass vials resulted from higher mechanical stresses as ice expanded during freezing in rigid glass vials compared to those experienced in the more flexible cyclic polyolefin vials.
The number of particles generated in response to agitation of IVIg formulations in syringes depended on the intensity of the applied agitation stress, buffer conditions, and on the syringe type. High-intensity, overnight agitation of syringes on a plate shaker generated more particles than were created after 10 days of low-intensity, end-over-end rotation of syringes. Agitation of silicone oil-lubricated prefilled syringes resulted in the production of spherical particles characteristic of silicone oil droplets, as well as smaller numbers of aspherical, nearly translucent particles typical of protein aggregates, and particles that appear to be composed of both silicone oil droplets and aggregated protein46. Fewer particles were produced in the SiOPlas™ syringes, where the plasma-deposited lubricating layer was likely less prone than liquid silicone oil lubricating layers to be removed from the surface by fluid shear and interfacial tension forces46.
Complement activation is an ADR that has been associated with the presence of nanoparticles in a number of nanoparticulate drugs.17, 19, 23, 26, 27, 47-52 Although many types of nanoparticles have been shown to activate complement53, there are other nanoparticles that exhibit minimal complement activation54. Nanoparticle-induced complement activation can occur through the classical, lectin, or alternative pathways53. Complement activation through the classical pathway is characteristic of activation by antigen-bound IgG. This activation occurs when Fc regions of antigen-bound IgG’s assemble to form hexameric complexes.55 In contrast, in the present case the increases in Bb levels in response to particles in IVIg formulations indicated that complement was activated via the alternative pathway, likely through non-specific binding that is different from the Fc-mediated activation characteristic of native IgG function. The structural basis for protein particle-induced complement activation via the alternative pathway is not completely understood, but for other nanoparticles alternative pathway activation results from non-covalent deposition of the complement cascade protein C3 on the particle surfaces56.
The complement activation in response to handling-induced particles in IVIg formulations was quite strong. Even under the freeze-thawing conditions that produced the highest numbers of particles that we tested, most of the protein remained soluble, in monomeric form. For example, after 6 freeze-thaw cycles in glass vials, based on monomer loss determined from HPLC analysis, only 3.7% of the original 1 mg/mL monomeric protein had aggregated, representing on 37 micrograms/mL particles. Yet this small amount of aggregate elicited release of Bb, C3a and C5a at a level that was approximately one fourth of the levels generated by 1 mg/mL levels of the positive controls zymosan and heat-aggregated gamma globulins.
Freeze thawing and agitation stresses applied to IVIg formulations in glass and SiOPlas™ containers yielded particle numbers that spanned several orders of magnitude, and different characteristic morphologies were observed when the particles were analyzed by flow imaging microscopy. However, when examined on a per-particle basis, the propensity of particles to activate complement did not depend on particle morphology. It should be noted that flow imaging microscopy is sensitive to particle morphological characteristics with micron-scale features. It is possible that at a finer scale, protein structures within the various particle types might have been similar, thus yielding similar levels of complement activation per particle.
Micelles of surfactants such as the polysorbate 20 and polysorbate 80 have been shown to be capable of activating complement57. But in the present case, complement activation did not appear to be strongly dependent on surfactant concentration, since the particle dose dependency was indistinguishable between formulations with and without added polysorbate 20.
The complement activation that we observed depended linearly on the concentrations of particles of size smaller than 10 microns. Complement activation was also generally higher in samples that contained the largest numbers of particles of size greater than 10 microns (see Figure 6), but these larger particles were found in the various stressed IVIg samples at concentrations roughly an order of magnitude lower than the corresponding concentrations for smaller, 2-10 micron-sized particles. Thus, our experiments were inconclusive as to whether complement activation depended on the doses of particles of size greater than 10 microns.. It is interesting to note that the smaller submicron particles are not currently regulated in protein products for parenteral administration; limits on particle content described in United States Pharmacopoeia <USP 787/788> are only for particles of size greater than 10 microns and greater than 25 microns. Monitoring particles below <USP 787/788> size ranges of less than 10 micron may provide insight into potential immunogenicity and complement activation capacity of therapeutic protein formulations.
Caution should be used in extending these results to the prediction of complement activation-related ADRs in clinical settings. Thresholds and extents of complement activation may vary from patient to patient due to genetic and acquired factors,21, 58-60 making direct prediction of in vivo responses to particles problematic.61 Furthermore, the use of cell-free serum in our in vitro test may underestimate the capacity for particles to activate complement; in whole blood, the presence of B cells increases sensitivity to complement activation by rituximab in patients.62 Finally, complement activation by nanoparticles can be highly material-specific, and particles formed from other protein drugs, e.g., monoclonal antibodies, may not behave in a fashion similar to particles found in the polyclonal mixture of antibodies present in IVIg.
Conclusions
We observed that as little as a single unintended freeze-thawing of an IVIg formulation creates a sufficient number of particles to activate complement in an in vitro serum-based assay. Complement activation was directly proportional to particle concentration for 2-10 micron sized particles in stressed samples, whereas no clear correlation was observed for >10 micron particles in the same samples. The level of activated complement was not dependent on the specific particle morphologies generated by multiple types of stresses, or on the presence of non-ionic surfactants in the formulations. Although these results may not be easily extrapolated to quantitatively predict risk of ADRs in patients infused with other monoclonal antibody drug products, it is clearly prudent to take steps in handling, storage and administration of therapeutic protein products to minimize generation of particles in infused therapeutic protein products, and it may be important to monitor particles below 10 microns in size for insight into immunogenicity.
Acknowledgements
Funding for this work was provided by the NIH under RO1 EB006006, and by SiO2 Medical Products, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bertolotto A; Malucchi S; Sala A; Orefice G; Carrieri PB; Capobianco M; Milano E; Melis F; Giordana MT, Differential effects of three interferon betas on neutralising antibodies in patients with multiple sclerosis: a follow up study in an independent laboratory. Journal Of Neurology Neurosurgery And Psychiatry 2002, 73 (2), 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deisenhammer F; Schellekens H; Bertolotto A, Measurement of neutralizing antibodies to interferon beta in patients with multiple sclerosis. Journal of Neurology 2004, 251, 31–39. [DOI] [PubMed] [Google Scholar]
- 3.Goodin DS; Frohman EM; Hurwitz B; O'Connor PW; Oger JJ; Reder AT; Stevens JC, Neutralizing antibodies to interferon beta: Assessment of their clinical and radiographic impact: An evidence report - Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. NEUROLOGY 2007, 68 (13), 977–984. [DOI] [PubMed] [Google Scholar]
- 4.Hartung HP; Schellekens H; Munschauer FE, Neutralizing antibodies to interferon beta in patients with multiple sclerosis: scientific background and clinical implications - Introduction. Journal of Neurology 2004, 251, 1–3.14999483 [Google Scholar]
- 5.Hartung HP; Munschauer F; Schellekens H, Significance of neutralizing antibodies to interferon beta during treatment of multiple sclerosis: expert opinions based on the Proceedings of an International Consensus Conference. European Journal of Neurology 2005, 12 (8), 588–601. [DOI] [PubMed] [Google Scholar]
- 6.Hemmer B; Stuve O; Kieseier B; Schellekens H; Hartung HP, Immune response to immunotherapy: the role of neutralising antibodies to interferon beta in the treatment of multiple sclerosis. Lancet Neurology 2005, 4 (7), 403–412. [DOI] [PubMed] [Google Scholar]
- 7.Malucchi S; Sala A; Gilli F; Bottero R; Di Sapio A; Capobianco M; Bertolotto A, Neutralizing antibodies reduce the efficacy of beta IFN during treatment of multiple sclerosis. Neurology 2004, 62 (11), 2031–2037. [DOI] [PubMed] [Google Scholar]
- 8.Yanai H; Hanauer SB, Assessing Response and Loss of Response to Biological Therapies in IBD. American Journal of Gastroenterology 2011, 106 (4), 685–698. [DOI] [PubMed] [Google Scholar]
- 9.Nanda KS; Cheifetz AS; Moss AC, Impact of antibodies to infliximab on clinical outcomes and serum infliximab levels in patients with inflammatory bowel disease (IBD): a meta-analysis. Am J Gastroenterol 2013, 108 (1), 40–7; quiz 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ordás I; Feagan BG; Sandborn WJ, Therapeutic drug monitoring of tumor necrosis factor antagonists in inflammatory bowel disease. Clin Gastroenterol Hepatol 2012, 10 (10), 1079–87; quiz e85-6. [DOI] [PubMed] [Google Scholar]
- 11.O'Meara S; Nanda KS; Moss AC, Antibodies to infliximab and risk of infusion reactions in patients with inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis 2014, 20 (1), 1–6. [DOI] [PubMed] [Google Scholar]
- 12.Vennegoor A; Rispens T; Strijbis EM; Seewann A; Uitdehaag BM; Balk LJ; Barkhof F; Polman CH; Wolbink G; Killestein J, Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult Scler 2013, 19 (5), 593–600. [DOI] [PubMed] [Google Scholar]
- 13.Kotarek J; Stuart C; De Paoli SH; Simak J; Lin TL; Gao Y; Ovanesov M; Liang Y; Scott D; Brown J; Bai Y; Metcalfe DD; Marszal E; Ragheb JA, Subvisible Particle Content, Formulation, and Dose of an Erythropoietin Peptide Mimetic Product Are Associated With Severe Adverse Postmarketing Events. J Pharm Sci 2016, 105 (3), 1023–7. [DOI] [PubMed] [Google Scholar]
- 14.Picard M; Galvao VR, Current Knowledge and Management of Hypersensitivity Reactions to Monoclonal Antibodies. Journal of Allergy and Clinical Immunology-in Practice 2017, 5 (3), 600–609. [DOI] [PubMed] [Google Scholar]
- 15.Atwal D; Safar AM; Govindarajan R; Makhoul I, Severe first infusion reaction related to cetuximab in cancer patients in Arkansas. Journal of Oncology Pharmacy Practice 2019, 25 (5), 1130–1134. [DOI] [PubMed] [Google Scholar]
- 16.Choquette D; Faraawi R; Chow A; Rodrigues J; Bensen WJ; Nantel F, Incidence and Management of Infusion Reactions to Infliximab in a Prospective Real-world Community Registry. Journal of Rheumatology 2015, 42 (7), 1105–1111. [DOI] [PubMed] [Google Scholar]
- 17.Szebeni J; Alving CR; Savay S; Barenholz Y; Priev A; Danino D; Talmon Y, Formation of complement-activating particles in aqueous solutions of Taxol: possible role in hypersensitivity reactions. Int Immunopharmacol 2001, 1 (4), 721–35. [DOI] [PubMed] [Google Scholar]
- 18.Szebeni J, Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology 2005, 216 (2-3), 106–21. [DOI] [PubMed] [Google Scholar]
- 19.Andersen AJ; Hashemi SH; Andresen TL; Hunter AC; Moghimi SM, Complement: alive and kicking nanomedicines. J Biomed Nanotechnol 2009, 5 (4), 364–72. [DOI] [PubMed] [Google Scholar]
- 20.Moghimi SM; Andersen AJ; Ahmadvand D; Wibroe PP; Andresen TL; Hunter AC, Material properties in complement activation. Adv Drug Deliv Rev 2011, 63 (12), 1000–7. [DOI] [PubMed] [Google Scholar]
- 21.Moghimi SM; Farhangrazi ZS, Nanomedicine and the complement paradigm. Nanomedicine 2013, 9 (4), 458–60. [DOI] [PubMed] [Google Scholar]
- 22.Szebeni J, Complement activation-related pseudoallergy: a stress reaction in blood triggered by nanomedicines and biologicals. Mol Immunol 2014, 61 (2), 163–73. [DOI] [PubMed] [Google Scholar]
- 23.Boraschi D; Italiani P; Palomba R; Decuzzi P; Duschl A; Fadeel B; Moghimi SM, Nanoparticles and innate immunity: new perspectives on host defence. Seminars in Immunology 2017, 34 (C), 33–51. [DOI] [PubMed] [Google Scholar]
- 24.Sacks SH, Complement fragments C3a and C5a: the salt and pepper of the immune response. Eur J Immunol 2010, 40 (3), 668–70. [DOI] [PubMed] [Google Scholar]
- 25.Klos A; Tenner AJ; Johswich KO; Ager RR; Reis ES; Köhl J, The role of the anaphylatoxins in health and disease. Mol Immunol 2009, 46 (14), 2753–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szebeni J; Baranyi L; Savay S; Milosevits J; Bunger R; Laverman P; Metselaar JM; Storm G; Chanan-Khan A; Liebes L; Muggia FM; Cohen R; Barenholz Y; Alving CR, Role of complement activation in hypersensitivity reactions to doxil and hynic PEG liposomes: experimental and clinical studies. J Liposome Res 2002, 12 (1-2), 165–72. [DOI] [PubMed] [Google Scholar]
- 27.Chanan-Khan A; Szebeni J; Savay S; Liebes L; Rafique NM; Alving CR; Muggia FM, Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil): possible role in hypersensitivity reactions. Ann Oncol 2003, 14 (9), 1430–7. [DOI] [PubMed] [Google Scholar]
- 28.Meszaros T; Csincsi AI; Uzonyi B; Hebecker M; Fulop TG; Erdei A; Szebeni J; Jozsi M, Factor H inhibits complement activation induced by liposomal and micellar drugs and the therapeutic antibody rituximab in vitro. Nanomedicine-Nanotechnology Biology and Medicine 2016, 12 (4), 1023–1031. [DOI] [PubMed] [Google Scholar]
- 29.Fulop T; Meszaros T; Kozma GT; Szebeni J; Jozsi M, Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H. Antibodies 2018, 7 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Kolk LE; Grillo-López AJ; Baars JW; Hack CE; van Oers MH, Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol 2001, 115 (4), 807–11. [DOI] [PubMed] [Google Scholar]
- 31.Randolph TW; Carpenter JF, Engineering challenges of protein formulations. Aiche Journal 2007, 53 (8), 1902–1907. [Google Scholar]
- 32.Rosenberg A In FDA Perspective on Immunogenicity Testing- A Risk Based Analysis, Bethesda, MD, Bethesda, MD, 2003. [Google Scholar]
- 33.Rosenberg AS, Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev Biol (Basel) 2003, 112, 15–21. [PubMed] [Google Scholar]
- 34.Rosenberg AS, Effects of protein aggregates: An immunologic perspective. Aaps Journal 2006, 8 (3), E501–E507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thirumangalathu R; Krishnan S; Ricci MS; Brems DN; Randolph TW; Carpenter JF, Silicone Oil- and Agitation-Induced Aggregation of a Monoclonal Antibody in Aqueous Solution. JOURNAL OF PHARMACEUTICAL SCIENCES 2009, 98 (9), 3167–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerhardt A; McGraw NR; Schwartz DK; Bee JS; Carpenter JF; Randolph TW, Protein Aggregation and Particle Formation in Prefilled Glass Syringes. Journal of Pharmaceutical Sciences 2014, 103 (6), 1601–1612. [DOI] [PubMed] [Google Scholar]
- 37.Pardeshi NN; Zhou C; Randolph TW; Carpenter JF, Protein nanoparticles promote microparticle formation in intravenous immunoglobulin solutions during freeze-thawing and agitation stresses. J Pharm Sci 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L; Ammar DA; Ross LA; Mandava N; Kahook MY; Carpenter JF, Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: effects of long-term storage and product mishandling. Invest Ophthalmol Vis Sci 2011, 52 (2), 1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weikart CM; Pantano CG; Shallenberger JR, Performance Stability of Silicone Oxide-Coated Plastic Parenteral Vials. PDA J Pharm Sci Technol 2017, 71 (4), 317–327. [DOI] [PubMed] [Google Scholar]
- 40.Janeway Charles A., P. T., Walport Mark, Schlomchik Mark, Immunobiology: The Immune System in Health. 6 ed.; Garland Science Publishing: New York, 2005; p 37–101. [Google Scholar]
- 41.Kiese S; Papppenberger A; Friess W; Mahler HC, Shaken, not stirred: Mechanical stress testing of an IgG1 antibody. Journal Of Pharmaceutical Sciences 2008, 97 (10), 4347–4366. [DOI] [PubMed] [Google Scholar]
- 42.Nejadnik MR; Randolph TW; Volkin DB; Schöneich C; Carpenter JF; Crommelin DJA; Jiskoot W, Postproduction Handling and Administration of Protein Pharmaceuticals and Potential Instability Issues. J Pharm Sci 2018. [DOI] [PubMed] [Google Scholar]
- 43.Jiskoot W; Nejadnik MR; Sediq AS, Potential Issues With the Handling of Biologicals in a Hospital. J Pharm Sci 2017, 106 (6), 1688–1689. [DOI] [PubMed] [Google Scholar]
- 44.Vlieland ND; Nejadnik MR; Gardarsdottir H; Romeijn S; Sediq AS; Bouvy ML; Egberts ACG; van den Bemt BJF; Jiskoot W, The Impact of Inadequate Temperature Storage Conditions on Aggregate and Particle Formation in Drugs Containing Tumor Necrosis Factor-Alpha Inhibitors. Pharmaceutical Research 2018, 35 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vlieland ND; Gardarsdottir H; Bouvy ML; Egberts TCG; van den Bemt BJF, The majority of patients do not store their biologic disease-modifying antirheumatic drugs within the recommended temperature range. Rheumatology 2016, 55 (4), 704–709. [DOI] [PubMed] [Google Scholar]
- 46.Gerhardt A; McGraw NR; Schwartz DK; Bee JS; Carpenter JF; Randolph TW, Protein Aggregation and Particle Formation in Prefilled Glass Syringes. J Pharm Sci 2014. [DOI] [PubMed] [Google Scholar]
- 47.Savay S; Szebeni J; Baranyi L; Alving CR, Potentiation of liposome-induced complement activation by surface-bound albumin. Biochim Biophys Acta 2002, 1559 (1), 79–86. [DOI] [PubMed] [Google Scholar]
- 48.Szebeni J, Complement activation-related pseudoallergy caused by amphiphilic drug carriers: the role of lipoproteins. Curr Drug Deliv 2005, 2 (4), 443–9. [DOI] [PubMed] [Google Scholar]
- 49.Moghimi SM; Andersen AJ; Hashemi SH; Lettiero B; Ahmadvand D; Hunter AC; Andresen TL; Hamad I; Szebeni J, Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: the challenges ahead. J Control Release 2010, 146 (2), 175–81. [DOI] [PubMed] [Google Scholar]
- 50.Moghimi SM; Hunter AC, Complement monitoring of nanomedicines and implants. Adv Drug Deliv Rev 2011, 63 (12), 963–4. [DOI] [PubMed] [Google Scholar]
- 51.Boraschi D; Costantino L; Italiani P, Interaction of nanoparticles with immunocompetent cells: nanosafety considerations. Nanomedicine (Lond) 2012, 7 (1), 121–31. [DOI] [PubMed] [Google Scholar]
- 52.Szebeni J; Storm G, Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem Biophys Res Commun 2015, 468 (3), 490–7. [DOI] [PubMed] [Google Scholar]
- 53.Moghimi SM; Simberg D; Skotland T; Yaghmur A; Hunter C, The Interplay between Blood Proteins, Complement, and Macrophages on Nanomedicine Performance and Responses. J Pharmacol Exp Ther 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas DG; Chikkagoudar S; Heredia-Langer A; Tardiff MF; Xu Z; Hourcade DE; Pham CT; Lanza GM; Weinberger KQ; Baker NA, Physicochemical signatures of nanoparticle-dependent complement activation. Comput Sci Discov 2014, 7 (1), 015003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diebolder CA; Beurskens FJ; de Jong RN; Koning RI; Strumane K; Lindorfer MA; Voorhorst M; Ugurlar D; Rosati S; Heck AJ; van de Winkel JG; Wilson IA; Koster AJ; Taylor RP; Saphire EO; Burton DR; Schuurman J; Gros P; Parren PW, Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343 (6176), 1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klapper Y; Hamad OA; Teramura Y; Leneweit G; Nienhaus GU; Ricklin D; Lambris JD; Ekdahl KN; Nilsson B, Mediation of a non-proteolytic activation of complement component C3 by phospholipid vesicles. Biomaterials 2014, 35 (11), 3688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiszhár Z; Czúcz J; Révész C; Rosivall L; Szebeni J; Rozsnyay Z, Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur J Pharm Sci 2012, 45 (4), 492–8. [DOI] [PubMed] [Google Scholar]
- 58.Agbeko RS; Fidler KJ; Allen ML; Wilson P; Klein NJ; Peters MJ, Genetic variability in complement activation modulates the systemic inflammatory response syndrome in children. Pediatr Crit Care Med 2010, 11 (5), 561–7. [DOI] [PubMed] [Google Scholar]
- 59.Heurich M; Martínez-Barricarte R; Francis NJ; Roberts DL; Rodríguez de Córdoba S; Morgan BP; Harris CL, Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci U S A 2011, 108 (21), 8761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris CL; Heurich M; Rodriguez de Cordoba S; Morgan BP, The complotype: dictating risk for inflammation and infection. Trends Immunol 2012, 33 (10), 513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szebeni J; Muggia F; Gabizon A; Barenholz Y, Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv Drug Deliv Rev 2011, 63 (12), 1020–30. [DOI] [PubMed] [Google Scholar]
- 62.Kozma GT; Meszarosa T; Weiszhar Z; Schneider T; Rosta A; Urbanics R; Rosivall L; Szebeni J, Variable association of complement activation by rituximab and paclitaxel in cancer patients in vivo and in their screening serum in vitro with clinical manifestations of hypersensitivity: a pilot study. European Journal of Nanomedicine 2015, 7 (4), 289–301. [Google Scholar]