

SUMMARY
Oncogenic RAS mutations drive cancers at many sites. Recent reports suggest that RAS dimerization, multimerization, and clustering correlate strongly with activation of RAS signaling. We have found that re-expression of DIRAS3, a RAS-related small GTPase tumor suppressor that is downregulated in multiple cancers, inhibits RAS/mitogen-activated protein kinase (MAPK) signaling by interacting directly with RAS-forming heteromers, disrupting RAS clustering, inhibiting Raf kinase activation, and inhibiting transformation and growth of cancer cells and xenografts. Disruption of K-RAS cluster formation requires the N terminus of DIRAS3 and interaction of both DIRAS3 and K-RAS with the plasma membrane. Interaction of DIRAS3 with both K-RAS and H-RAS suggests a strategy for inhibiting oncogenic RAS function.
Graphical Abstract
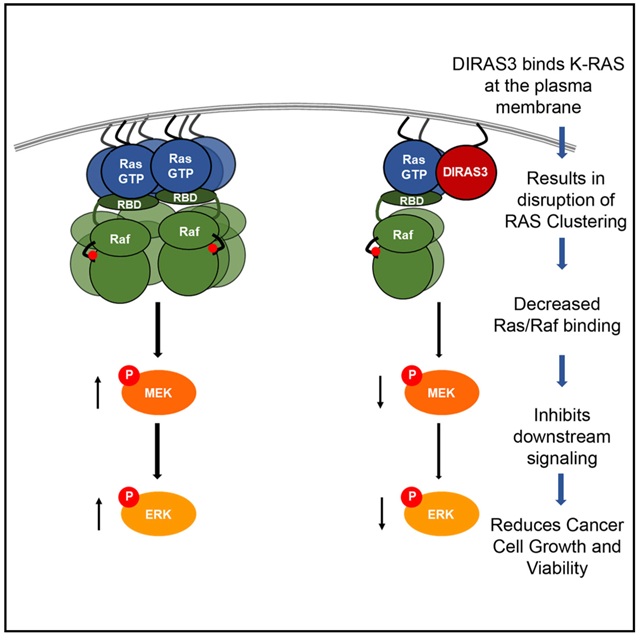
In Brief
Sutton et. al. show that re-expression of DIRAS3 can inhibit the growth of multiple cancer types driven by K-RAS mutations by a direct interaction and disruption of K-RAS higher ordered clusters. This phenotype is driven by an N-terminal extension, which distinguishes DIRAS3 from other RAS-related small GTPases.
INTRODUCTION
Oncogenic RAS mutations drive many tumor types, including approximately 90% of pancreatic adenocarcinomas, 30% of lung cancers, and up to 60% of low-grade ovarian cancers (Prior et al., 2012; Singer et al., 2003). Despite decades of research, targeting constitutively active RAS has remained elusive. Recent reports suggest that RAS dimerization, multimerization, or clustering correlate strongly with activation of RAS signaling (Muratcioglu et al., 2015; Nan et al., 2015). The specific mechanism(s) by which RAS activity is regulated have not been fully elucidated and, aside from wtRAS (Ambrogio et al., 2018), no physiologic inhibitors of oncogenic RAS clustering have previously been identified.
DIRAS3 (ARHI) is a maternally imprinted gene that was discovered in the context of ovarian cancer, as it was the most transcriptionally downregulated gene when compared to expression in normal ovarian epithelial cells (Yu et al., 1999). A 26-kDa small GTPase, DIRAS3, shares 50%–60% homology to the oncogene H/N/K-RAS, differing primarily by the addition of a 34-amino-acid N-terminal extension, which is essential for most DIRAS3 functions (Luo et al., 2003). To function, DIRAS3, like RAS, requires an association with the plasma membrane (PM) through a CAAX-box membrane-anchoring domain, and the loss of this membrane association reduces tumor suppressive function. DIRAS3 is downregulated transcriptionally and post-transcriptionally by several different mechanisms in ovarian cancers, including loss of heterozygosity, hypermethylation of the non-imprinted allele, transcriptional and microRNA (miRNA) regulation, and decreased DIRAS3 RNA binding to HuR (Feng et al., 2008; Lu et al., 2006; Luo et al., 2003). Downregulation of DIRAS3 has also been demonstrated in pancreatic, lung, breast, thyroid, prostate, and hepatocellular cancers (Dalai et al., 2007; Huang et al., 2009; Lin et al., 2011; Lu et al., 2006; Weber et al., 2005; Wu et al., 2013). Re-expression of DIRAS3 induces cytotoxicity and inhibits clonogenic growth, motility, and migration, while inducing autophagy and tumor dormancy (Badgwell et al., 2012; Lu et al., 2008; Luo et al., 2003; Yu et al., 1999). Moreover, re-expression of DIRAS3 can inhibit both the phosphatidylinositol 3-kinase (PI3K) and RAS/ mitogen-activated protein kinase (MAPK) signaling pathways (Lu et al., 2008; Lu et al., 2014).
Although many of the previously reported functions of DIRAS3 support its role as a tumor suppressor gene, classical experiments to demonstrate loss of tumor suppressor activity in genetically engineered mouse models have not been possible, due to the loss of DIRAS3 from the murine genome when mice and humans diverged evolutionarily some 60 million years ago (Fitzgerald and Bateman, 2004). In this study, we have demonstrated the ability of DIRAS3 to suppress H-RAS- and K-RAS-mediated transformation of murine and human cells. Both the N-terminal extension of DIRAS3 and its membrane association through the CAAX-C-terminal membrane-anchoring domain are required to suppress transformation. Re-expression of DIRAS3 downregulates the growth of transformed ovarian, pancreatic, and lung cancer cells. The clinical relevance and importance of DRAS3 expression are suggested by improved survival in patients with K-RAS-driven pancreatic cancers that express DIRAS3. In elucidating the mechanism(s) by which DIRAS3 inhibits RAS/Raf/MAPK signaling, we found that DIRAS3 binds directly with RAS through its A5 helical domain to form DIRAS3-RAS heteromers and disrupts RAS clustering, inhibiting Raf kinase and downstream MAPK signaling. Disruption of K-RAS cluster formation, like inhibition of RAS transformation, requires the N terminus of DIRAS3 and interaction of DIRAS3 with the PM. Thus, DIRAS3 regulates oncogenic RAS activity by inhibiting RAS clustering and multimerization.
RESULTS
DIRAS3 Inhibits RAS-Induced Malignant Transformation of Murine Fibroblasts and RAS-Induced Anchorage-Independent Growth of Partially Transformed Human Breast Epithelial Cells
As DIRAS3 can suppress the growth of transformed cancer cells (Luo et al., 2003; Yu et al., 1999), we asked whether DIRAS3 expression also inhibits RAS-driven transformation of murine fibroblasts (NIH 3T3) and partially transformed human breast epithelial cells (MCF10a). Cells in both models were transfected with plasmid cloning DNA (pcDNA) vectors containing mutant RAS (H- or K-) and wild-type (WT) DIRAS3 or DIRAS3 mutants that lacked the 34-amino-acid N-terminal extension (ΔNT) or had lost function of the cell-membrane-anchoring domain (C226S) and transformation was compared to mutant RAS co-transfected with an empty vector control (Figure 1A). Non-transfected cells, WT RAS, or vector-only transfected cells were used as negative controls (Figure S1). Using a classical transformation assay (Figure 1B) to study the effect of mutant K-RAS on murine fibroblasts (NIH 3T3), we found that co-transfection of WT DIRAS3 with constitutively active (G12V) mutant K-RAS and H-RAS constructs inhibited focus formation when compared to co-transfection of an empty vector with mutant K-RAS or H-RAS (Figures 1C and 1D). Co-transfection of an N-terminal-deleted construct of DIRAS3 (ΔNT DIRAS3) did not inhibit focus formation (Figures 1C and 1D), suggesting that the 34-amino-acid N terminus is required for inhibiting K-RAS-induced transformation, as it is for other DIRAS3-mediated suppressor functions (Luo et al., 2003). Although co-transfection of WT DIRAS3 and mutant RAS suppressed focus formation, co-transfection of mutant RAS with a DIRAS3 construct containing a single point mutation in the CAAX membrane-anchoring domain (C226S) substantially reduced inhibition of RAS-driven transformation, suggesting that membrane localization of DIRAS3 contributes to its tumor suppressive function. Results were confirmed using the partially transformed breast epithelial cell line MCF10a (Figure 1B), where co-transfection of DIRAS3 significantly inhibited clonogenic growth in soft agar induced by RAS (G12V) (Figures 1E and 1F). Co-transfection of the ΔNT DIRAS3 or C226S constructs failed to inhibit RAS (G12V)-induced anchorage-independent clonogenic growth of MCF10a cells (Figure 1E). Similar results were obtained for H-RAS (G12V) (Figure 1F). Thus, both an intact DIRAS3 N terminus and localization of DIRAS3 to the membrane are critical to antagonize mutant K- and H-RAS-mediated transformation.
Figure 1. DIRAS3 Inhibits RAS-Induced Malignant Transformation of Murine Fibroblasts and RAS-Induced Anchorage-Independent Growth of Partially Transformed Human Breast Epithelial Cells.

(A) Illustration of constructs in pcDNA3.1 vectors that were used for the following experiments.
(B) Experimental schema for the assays performed.
(C and D) DIRAS3 inhibits K-RAS- (C) and H-RAS-induced (D) transformation of murine NIH 3T3 cells. Cells were plated in 60-mm dishes and transfected with 10 μg of each DNA plasmid for 24 h prior to being separated into dishes for focus formation and clonogenic selection by G418. Transformed foci were counted at 10× magnification as they appeared within two 10 × 10-mm areas per plate. The columns indicate the mean, and the error bars indicate the SD (**p < 0.01; *p < 0.05). Clonogenic selection by G418 was used to ensure equal transfection efficiency.
(E and F) DIRAS3 inhibits anchorage-independent growth of MCF10a breast epithelial cells transformed with K-RAS (E) and H-RAS (F). Cells were plated in 6-well plates and transfected with 3 μg of each DNA plasmid for 24 h to being re-seeded at a cell density of 1.0 × 105 in soft agar. Cells were grown for 2 weeks and colonies were counted. The assay was performed three times with technical triplicates for each experiment. The columns indicate the mean, and the error bars indicate the SD (**p < 0.01).
Re-expression of DIRAS3 Inhibits RAS-Driven Anchorage-Dependent Short-Term Growth, Long-Term Clonogenic Growth, and Downstream Signaling in K-RAS-Mutated Cancers from Multiple Sites
Re-expression of DIRAS3 at physiologic levels in ovarian cancer cell lines attenuates RAS/Raf/MAP signaling, decreases p-ERK, and inhibits clonogenic growth (Lu et al., 2008). We demonstrated that several pancreatic, lung, and ovarian cancer cell lines with activating K-RAS mutations were dependent upon mutant K-RAS for viability and growth using knockdown of K-RAS with pooled small interfering RNAs (siRNAs) (Figure S2A). Compared to a non-targeted siRNA control, knockdown of K-RAS in AsPc-1 pancreatic cancer cells and HeyA8 ovarian cancer cells significantly impaired cell viability in a 5-day sulforhodamine B (SRB) cytotoxicity assay (Vichai and Kirtikara, 2006). Re-expression of DIRAS3, at near physiologic levels using a tet-ON inducible system in AsPc-1, Capan-2, HeyA8, and OVCAR8 cancer cell lines inhibited clonogenic growth and downstream p-ERK signaling driven by oncogenic K-RAS (Figures 2A-2D). Four additional pancreatic and lung cancer cell lines were evaluated that depended upon K-RAS for growth and viability (Panc-1, MiaPaca2, H358, and H441). In all four cell lines, transient expression of DIRAS3 inhibited long-term clonogenic growth of G418-resistant colonies, whereas transfection of ΔNT DIRAS3 did not (Figure S3). Importantly, the transient transfection of DIRAS3 decreased MAPK signaling (Figure S4). Re-expression of DIRAS3, but not ΔNT DIRAS3, decreased clonogenic growth of BxPc3 pancreatic cancer cells that harbor an oncogenic B-RAF mutation rather than K-RAS (Figure S3C). Re-expression of DIRAS3 also suppressed the clonogenic growth of mouse embryonic fibroblasts engineered to express only WT RAS or a single oncogenic K-RAS mutant (Figure S3D). Taken together, these data demonstrate that transient re-expression of the tumor suppressor DIRAS3 completely arrests long-term cell viability of cancer cells harboring both oncogenic RAS mutations or increased RAS/MAPK signaling, and this inhibition requires the full-length DIRAS3 protein containing the 34-amino-acid N-terminal extension.
Figure 2. Re-expression of DIRAS3 Inhibits RAS-Driven Growth and Downstream Signaling in Ovarian and Pancreatic Cancer Cells.

(A) Re-expression of DIRAS3 inhibits colony formation in Hey-A8-DIRAS3- and AsPc-1-DIRAS3-inducible cell lines. Cells were seeded in triplicate in a 6-well plate with or without DOX for 2 weeks. Colonies were stained with Coomassie blue (top) and counted (bottom).
(B) DIRAS3 re-expression inhibits MAPK signaling. Hey-A8-DIRAS3 and AsPc-1-DIRAS3 cells were seeded at 0.3 × 106 cells/well and treated with DOX for 24 h before cell lysate was collected and western blot analysis performed. Densitometry was measured with ImageJ (imagej.nih.gov/ij/docs/guide/), and experiments were performed at least three times. The ratio of p-ERK to t-ERK was calculated and normalized to the DOX− control. The columns indicate the mean and the bars indicate the SD (*p < 0.05).
(C) Expression of DIRAS3 inhibits colony formation in OVCAR8-DIRAS3- and Capan-2-DIRAS3-inducible cell lines. Cells were seeded in triplicate in a 6-well plate with or without DOX for 2 weeks. Colonies were stained with Coomassie blue and counted.
(D) DIRAS3 re-expression inhibits MAPK signaling. OVCAR8-DIRAS3 and Capan-2-DIRAS3 cells were seeded at 0.3 × 106 cells/well and treated with DOX for 24 h before the cell lysate was collected and western blot analysis performed. Densitometry was measured with ImageJ and experiments were performed at least three times. The ratio of p-ERK to t-ERK was calculated and normalized to the DOX− control. The columns indicate the mean, and the bars indicate the SD (*p < 0.05).
(E) DIRAS3 expression inhibits tumor growth in mouse orthotropic xenografts. Mice were injected intraperitoneally (i.p.) with five million OVCAR8-DIRAS3 cells or the parental control cells and were immediately fed with sucrose water with or without the addition of doxycycline for the duration of the experiment. Six weeks following the injection, the mice were sacrificed and the tumors were extracted upon necropsy. The tumor burden was determined by mass, and each dot represents a single mouse. Asterisk denotes significant difference (*p < 0.05).
(F) GEPIA overall survival analysis of pancreatic adenocarcinoma (PAAD) patients based on DIRAS3 mRNA expression (solid high = red; solid low = blue). Dashed lines indicate the co-proportional hazard ratio and the 95% confidence interval. Log rank, p = 0.033.
Silencing of DIRAS3 Accelerates Growth and Enhances p-ERK Signaling in SU86.86 Pancreatic Cancer Cells and Normal Epithelial Pancreatic Cells (HPNEs)
When gene expression in normal pancreatic and pancreatic cancer cell lines was compared, 21 of 23 (91%) of the pancreatic cancer cell lines exhibited reduced DIRAS3 mRNA expression when compared to DIRAS3 expression in normal pancreas. One pancreatic cancer cell line, namely, SU86.86, maintained high levels of DIRAS3 mRNA (Figure S5A). Knockdown of DIRAS3 with siRNA accelerated short-term growth of SU86.86 and increased p-ERK signaling (Figures S5B-S5D) when compared to cells treated with control siRNA. Similarly, HPNE normal pancreatic cells and CaOv3 ovarian cancer cells that express higher levels of DIRAS3 showed increased p-ERK signaling and enhanced proliferation upon knockdown of endogenous DIRAS3 with siRNA (Figures S5E-S5G).
Re-expression of DIRAS3 Inhibits Growth of a RAS-Mutated Ovarian Cancer Xenograft
To determine whether growth inhibition was observed in vivo, OVCAR8-DIRAS3-inducible cells and their parental controls (5 million) were injected intraperitoneally. Doxycycline was added to the drinking water to induce DIRAS3 gene expression in some groups of xenografts but not in others, and tumor burden was assessed 6 weeks post-injection. Re-expression of DIRAS3 inhibited tumor burden, but xenografts of parental non-inducible OVCAR8 cells were not affected by the addition of doxycycline to the drinking water (Figure 2E).
DIRAS3 Expression Impacts Survival in Pancreatic Cancer, which Often Has Oncogenic RAS Mutations
Given the impact of DIRAS3 expression on cancer growth of RAS-driven cancer cells and xenografts, we asked whether DIRAS3 expression might impact prognosis in patients with pancreatic cancer where more than 90% of cases are driven by mutated RAS. Using GEPIA (Tang et al., 2017), a web server for cancer and normal gene expression profiling analysis, Kaplan-Meyer survival analysis was performed to assess the overall survival of pancreatic adenocarcinoma patients with different levels of DIRAS3 mRNA expression. Using a log-rank test (Mantel-Cox test), we found that higher DIRAS3 expression (top 75%) showed a significant increase in overall survival (p = 0.033) compared to lower DIRAS3 expression (Figure 2F). The favorable prognosis of all groups in this study reflects the profiling of resectable early-stage pancreatic cancers to obtain The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) datasets.
DIRAS3 Forms a Heteromer with H-RAS and K-RAS
To determine how DIRAS3 inhibits RAS function, we first asked whether DIRAS3 binds directly to RAS. We used the HeyA8-DIRAS3 ovarian cancer cell line that harbors a K-RAS-activating mutation and that has been engineered to express DIRAS3 at near physiologic levels from a doxycycline-inducible promoter and observed co-immunoprecipitation of DIRAS3 with endogenous K-RAS (Figure 3A). Similarly, re-expression of DIRAS3 in AsPc-1 pancreatic cancer cells also documented co-immunoprecipitation with endogenous K-RAS (Figure 3B). We used a DuoLink proximity ligation assay and confirmed co-localization between K-RAS and DIRAS3 in these cell lines (Figure 3C). Although these data demonstrate cellular co-localization of DIRAS3 and K-RAS, sub-cellular localization of this interaction cannot be defined precisely by this approach.
Figure 3. DIRAS3 Co-localizes with K-RAS.

(A and B) Co-immunoprecipitation of DIRAS3 and K-RAS. Hey-A8-DIRAS3- (A) and AsPc-1-DIRAS3- (B) inducible cells at 70% confluency in 60-mm dishes were treated with or without DOX for 48 h. Cell lysate was collected, and 1.5 ug of lysate was used for immunoprecipitation with a rabbit immunoglobulin G (IgG) control or anti-K-RAS antibody. Western blotting was performed and probed with anti-DIRAS3 antibody before stripping and probing with anti-K-RAS.
(C) DIRAS3 and K-RAS formed heterodimers in Hey-A8-DIRAS3 cells. DIRAS3 and K-RAS complexes were analyzed with an in situ PLA assay. Scale bars represent 20 μm. Data were obtained from two independent experiments performed in duplicate. Columns indicate the mean, and the bars indicate the SD (**p < 0.01).
Co-localization of DIRAS3 and RAS Occurs at the PM
We used the Recombinase-enhanced BiLC (ReBiL) system to further analyze requirements for the DIRAS3-RAS interaction. This system uses Cre to insert a bi-directional, doxycycline-inducible expression module into a single pre-determined chromosomal location (Wong et al., 2005), enabling each of two proteins of interest to be appended by an N- or C-terminal split luciferase. Doxycycline induces each of the proteins, and their interaction is measured by an increase in luciferase signal (Li et al., 2014). This system has recently been used to evaluate homo- and hetero-meric interaction of K/N/H-RAS proteins (Li et al., 2018). Using the well-established protein-protein interaction of p53 and Mdm2 as a positive control and nLuciferase and cLuciferase as a negative control, we investigated the interactions of K-RAS with K-RAS, K-RAS with K-RASG12D, K-RAS with DIRAS3, and K-RAS with ΔNT DIRAS3 (Figure 4A). Importantly, ReBiL enabled near-physiologic level expression of DIRAS3 and K-RAS and monitoring of their interaction in living cells in real time. Interactions of K-RAS with K-RAS and K-RAS with DIRAS3 were readily detected (Figure 4A). A loss of the DIRAS3 N-terminal extension had no measurable effect (Figure 4A). Using confocal microscopy, we confirmed the plamsa membrane-dependent co-localization of DIRAS3 and K-RAS (Figure 4B). Importantly, when comparing the Pearson correlation coefficient to quantify the degree of co-localization between fluorophores, K-RAS and DIRAS3 were significantly correlated (R2 = 0.86) when imaged on the PM, compared to imaging the cytoplasm (R2 = 0.54). Although the expression and cellular localization of DIRAS3 within the cytoplasm are likely biologically relevant to the essential role that DIRAS3 plays in autophagy (Lu et al., 2008), it is not a major site of interaction with K-RAS, consistent with the possibility that the anchored orientation of the two proteins in the PM may facilitate their interaction. Additionally, stochastic optical reconstruction microscopy (STORM) (Bates et al., 2013) imaging performed at total internal reflection (TIRF) (Fish, 2009) confirmed the interaction between DIRAS3 and K-RAS on the membrane (Figure 4C). Finally, using TEM of the inner leaflet of PM sheets (Plowman et al., 2005; Prior et al., 2003a, 2003b), we documented membrane-associated co-localization between K-RAS and DIRAS3 (Figure 4D). Different antibody-conjugated gold nanoparticles were used to document K-RAS (4.5 nm) and DIRAS3 (2 nm) on the PM sheets, allowing for bi-variate K-function analysis to determine their co-localization (Figure 4E). In addition, we found that DIRAS3 co-localized with Sos-1, a well-established K-RAS-plasma-membrane-interacting protein by using immunofluorescent staining in U2OS cells (Figure S6A).
Figure 4. DIRAS3 Co-localizes with RAS at the Plasma Membrane (PM).

(A) Luminescent signals were determined for several ReBiL cell lines to detect low- affinity protein-protein interactions that were normalized to that of p53-MDM2. nLuc-cLuc was used as a negative control. K-RAS and K-RASG12D interaction was nearly 400% of p53-MDM2, and K-RAS-DIRAS3 or K-RAS-NTD was slightly less. K-RAS and DIRAS3 C226S did not show a robust luminescence signal. Data were obtained from three independent experiments performed in triplicate. Columns indicate the mean, and the bars indicate the SD (**p < 0.01).
(B) DIRAS3 co-localizes with K-RAS. U2OS-701 cells were treated with DOX for 24 h. Immunofluorescence staining of expressed DIRAS3 and K-RAS was analyzed by confocal microscopy. Scale bars represent 30 μM.
(C) U2OS TR2 cells were treated with doxycycline for 24 h to induce K-RAS expression and simultaneously transfected with DIRAS3 pcDNA plasmid. STORM imaging at TIRF confirmed the interaction between DIRAS3 and K-RAS on the membrane. Scale bars represent 2 μm and 1 μm for the higher magnification. DIRAS3 is labeled in red and K-RAS is labeled in green.
(D) TEM of DIRAS3- and K-RAS-labeled gold nanoparticles on the inner leaflet of the PM of Hey-A8-DIRAS3 ovarian cancer cells following induction of DIRAS3 by doxycycline for 24 h. GFP-tagged K-RASG12V, which was expressed by lentiviral infection, was immunolabeled with 6-nm gold nanoparticles linked to anti-GFP antibody, and endogenous DIRAS3 was immunolabeled with 2-nm gold particles linked to anti-DIRAS3 (ID8). Gold particles were then imaged by TEM at 100,000× magnification. Scale bars represent 100 nm and 20 nm for the higher magnification. DIRAS3 is labeled in red (2nm gold) and K-RAS is labeled in green (6 nm gold).
(E) Bivariate K-function analysis of TEM documenting the extent of clustering and size of the clusters observed between K-RAS and DIRAS3 on the surface of the PM. Spatial co-clustering between 6-nm and 2-nm gold particles was assessed using the bivariate K-function where values above 1 indicate statistically significant co-localization between DIRAS3 and K-RAS on the PM. At least 15 PM sheets were imaged, analyzed, and pooled for each condition.
DIRAS3 Interacts Directly with RAS at the A5 Helical Domain
To identify the specific regions required for K-RAS-DIRAS3 interaction, we used peptide arrays (with 15-mer polypeptide sequences spanning the entire length of the protein shifting by 1–3 amino acids at each spot) that identified the A5 helical domain of both H- and K-RAS as the primary region of DIRAS3 binding (Figure 5A). This sequence was further confirmed using a single amino acid shift peptide array spanning from amino acids 151 to 171. The binding pattern between full-length DIRAS3 and ΔNT DIRAS3 was nearly identical (Figure S5C). To confirm the A5 interaction site, K-RASG12V constructs harboring three point mutations (D153A, Y157A, and R161A) or deletion of the A5 helical domain (Δ153–161) were used for immunoprecipitation and split-luciferase complementation assays. The interaction between DIRAS3 and K-RASG12V was interrupted by mutation of the A5 helical domain (Figures 5B-5D). ReBiL analysis was performed with WT K-RAS and WT DIRAS3, compared to K-RAS C185S or K-RAS with mutations to the A5 helical domain, including the D154 deletion, which is inferred to be clinically relevant as it is present in the COSMIC database. K-RAS_A5_3A and K-RAS_A5del mutant proteins localized to the membrane fraction, indicating that the loss of interaction is not due to protein mislocalization to the cytoplasm (Figure 5C). We further confirmed that DIRAS3 and K-RAS directly interact by using co-immunoprecipitation of purified recombinant proteins, where the extent of interaction was modulated in a concentration-dependent manner (Figure 5E). Using crystal structures of H-RAS and B-Raf (PDB: 4G0N) (Fetics et al., 2015), we modeled the binding of DIRAS3 to RAS and Raf. This model suggested that DIRAS3 would be unlikely to interfere directly with the Raf-binding domain on RAS. However, these analyses do not eliminate the possibility that, by interacting with DIRAS3 at the A5 helical-domain, additional steric/conformational changes in RAS could occur to prevent or alter Raf-RAS binding (Figure 5F). We then used surface plasmon resonance (SPR) to determine the relative binding affinity between the two proteins. When different concentrations of K-RAS recombinant protein (810 to 10 nM) were injected onto a DIRAS3 sensor surface, we measured a dissociation constant KD value of 12.1 ± 0.3 nM (Figure 5G). Additional SPR studies were performed to determine the specificity of the K-RAS interaction with both WT DIRAS3 and ΔNT DIRAS3. Different recombinant RAS family proteins (K-RAS, N-RAS, and Rap1A) were titrated sequentially onto DIRAS3 or ΔNT DIRAS3 tethered by their C-terminals. K-RAS, but not N-RAS or Rap1A, bound to DIRAS3 and ΔNT DIRAS3 (Figure 5H). This is consistent with reports of NS1, a monobody that binds to K-RAS and H-RAS α4-α5 helical domains, but not N-RAS (Spencer-Smith et al., 2017). Compared to DIRAS3, ΔNT DIRAS3 showed faster kinetics but similar affinity for K-RAS. ΔNT DIRAS3 was co-expressed in murine 3T3 cells with WT and mutant (G12V) H-RAS or K-RAS and co-immunoprecipitation confirmed co-localization (Figures S6B and S6C). Interestingly, peptide array analysis of H-RAS and K-RAS showed similar binding patterns between WT DIRAS3 and ΔNT DIRAS3 (Figure S6D), suggesting that the interaction with the α5 helical domain is similar but that this alone is not sufficient enough to inhibit oncogenic function.
Figure 5. DIRAS3 Interacts with RAS at the A5 Helical Domain.

(A) The 15-mer peptide arrays generated for DIRAS3 and K-RAS were probed as described to determine possible sites of interaction. A confirmatory array of the A5 helical domain of K-RAS was also generated and probed with DIRAS3 recombinant protein.
(B) ReBiL split luciferase complementation of DIRAS3 with K-RAS A5 helical domain mutants document a decrease in luminescence following mutations to the A5 helical domain. Split luciferase constructs were induced with 40 ng/mL of doxycycline for 24 h prior to luminescence being measured. Western blot analysis was also performed, and complementation was normalized to the level of protein expression. Data were obtained from three independent experiments performed in triplicate. Columns indicate the mean, and the bars indicate the SD (**p < 0.01).
(C) ReBiL cell lysate was fractionated between the cytoplasm and membrane to determine the sub-cellular localization of DIRAS3 and K-RAS. The ReBiL line containing K-RAS C185S and WT DIRAS3 was used as a positive control for fractionation, as the point mutation to the CAAX domain of K-RAS impairs post-translational modifications and membrane localization.
(D) Immunoprecipitation of NIH 3T3 cells co-transfected with 10 μg of DIRAS3 and K-RASG12V constructs with mutations or deletion of the A5 helical domain. Western blot analysis was completed.
(E) DIRAS3 interacts with K-RAS in vitro. Purified recombinant DIRAS3 and K-RAS proteins were incubated together at different ratios to allow complex formation. The DIRAS3:K-RAS complexes that formed were immunoprecipitated with anti-K-RAS antibody and analyzed by western blotting.
(F) A model of DIRAS3, H-RAS, and B-Raf was developed based on the interactions predicted by peptide array analysis. The crystal structure of DIRAS3 has not been solved, but DIRAS3 33-229 was predicted based on the known crystal structures of the DIRAS family homologs, DIRAS1 and DIRAS2.
(G) SPR binding analysis. Three-fold serial dilutions of K-RAS (from 810 to 10 nM) were injected onto a DIRAS3 sensor surface. SPR sensorgrams were fitted to a 1:1 Langmuir binding model curve shown in black. The derived dissociation constant KD value of 12.1 ± 0.3 nM was calculated from the rate constants kon = (2.22 ± 0.68) × 104 M−1 s−1 and koff = (2.78 ± 1.25) × 10−4 s−1 (n = 3).
(H) SPR was used to determine the binding specificity between RAS family members and DIRAS3 or ΔNT DIRAS3. DIRAS3 (590 RU) and ΔNT DIRAS3 (520 RU) were tethered specifically through their C-terminus with Twin-Strep tag onto the immobilized Strep-TactinXT on a CM5 sensor chip. Recombinant RAS proteins (0.33, 1, and 3μM) were titrated sequentially onto the DIRAS3 sensor and the sensogram obtained.
DIRAS3 Inhibits RAS Clustering
Recent studies indicate that multimerization enhances RAS signaling (Nan et al., 2015). Using the ReBiL system to detect RAS-RAS interactions, we found that re-expression of DIRAS3, but not ΔNT DIRAS3, decreased luminescence produced by K-RAS multimers (Figure 6A). These results were confirmed by immunoprecipitation of the K-RAS multimer, where K-RAS was labeled with either an hemagglutinin (HA)-tag or a FLAG-tag and each was equally expressed using a bidirectional promoter following the addition of doxycycline. When DIRAS3 was introduced prior to immunoprecipitation, less interaction was observed between α-FLAG-K-RAS and HA-K-RAS, relative to empty vector controls or ΔNT DIRAS3 (Figure 6B). DIRAS3 disruption of RAS multimerization was associated with decreased p-ERK activation (Figure 6B). Transmission electron microscopy (TEM) detection of K-RASG12V clustering on the membrane of Hey-A8-DIRAS3 ovarian cancer cells documents a striking difference in the spatial distribution with and without DIRAS3 expression (Figures 6C and 6D). The presence of DIRAS3 markedly narrowed the peak of the K-function curve of GFP-K-RASG12V (Figure 6C); and increasing DIRAS3 expression shifts the peak of the K-function curves to the left (Figure 6C) and the maximal radius of K-RAS nanoclusters was decreased by ~50% (Figure 6D) and correlated with a decrease in p-ERK signaling (Figures 6E-6G). Taken together, these data indicate that the N terminus of DIRAS3 plays a critical role in inhibiting K-RAS multimer formation and downstream function but is not required for interaction with the α5 helical domain of K-RAS.
Figure 6. DIRAS3 but Not ΔNT DIRAS3 Reduces RAS Clustering and Downstream Signaling.

(A) DIRAS3 inhibits RAS multimerization. (A) Cells were plated in a 6-well plate and transfected with 3 μg of vector, DIRAS3, or ΔNT DIRAS3 plasmid DNA. Twenty four h post-transfection, cells were re-seeded into 96-well plates at a density of 5,000 cells/well. Doxycycline was added to induce the split luciferase K-RAS constructs, and 24 h later, the luminescent signals were determined for the K-RAS:K-RASG12D interaction. Data were obtained from three independent experiments performed in triplicate. Columns indicate the mean, and the bars indicate the SD (**p < 0.01).
(B) DIRAS3 reduced K-RAS co-immunoprecipitation. U2OS-605 cells were seeded in a 60-mm dish at 70% confluency prior to being transfected with 20 μg of vector, DIRAS3, or ΔNT DIRAS3 plasmid DNA. DOX was added to induce K-RAS and K-RASG12D for 24 h prior to collecting the cell lysate. A total of 1.5 mg of lysate was used for co-immunoprecipitation with anti-FLAG antibody or mouse IgG. Blots were probed as indicated.
(C) Cells lacking DIRAS3 form higher order clusters. TEM of GFP-K-RASG12V-labeled gold nanoparticles on the inner leaflet of the PM of Hey-A8-DIRAS3 ovarian cancer cells following induction of DIRAS3 by DOX for 24 h. GFP-K-RASG12V, expressed by lentiviral infection, was immunolabeled with 4.5-nm gold nanoparticles coupled to anti-GFP antibody. TEM was performed at 100,000× magnification, and the spatial distribution of the gold particles was analyzed using univariate K-function to assess K-RAS cluster frequency and size. At least 15 PM sheets were imaged, analyzed, and pooled for each condition.
(D) Analysis of the TEM data collected was used to determine the average cluster size (nm). Re-expression of DIRAS3 reduced the cluster size by approximately 6-fold, as compared to the Dox− sample or those expressing ΔNT DIRAS3. When the extent of nanoclustering L(r)-r was plotted against the length scale r, L(r)-r values above 1 indicate statistically significant clustering. Columns indicate the mean; bars indicate the SE (**p < 0.01).
(E) Lysate collected under the same experimental conditions used for TEM were assessed by western blot analysis and probed as indicated. Densitometry was measured with ImageJ, and experiments were performed in triplicate.
(F) Model demonstrating three-tiered process by which DIRAS3 regulates RAS/MAPK signaling through the direct interaction and clustering regulation of K-RAS at the PM.
DIRAS3 Inhibits RAS/Raf Interaction
Despite previous efforts to determine whether guanosine triphosphate (GTP) activation of RAS enhances RAS clustering, there has not been clear evidence that GTP binding is a critical step prior to multimerization or whether or not GTP activation can occur following homodimerization/multimerization. As demonstrated by Muratcioglu et al. (2015), simulations of GTP-bound K-Ras4B homodimerization was predicted at greater affinity than guanosine diphosphate (GDP)-bound K-Ras4B; however, only one predicted GTP-bound K-RAS4B dimer interface (α-helix) model permitted effector binding. To determine whether DIRAS3 altered GTP activation of RAS, we performed immunoprecipitation of the RAS binding domain (RBD) of Raf and measured levels of GTP-bound endogenous K-RAS following re-expression of DIRAS3. Re-expression of DIRAS3 reduced GTP-bound RAS, as determined by RBD Raf immunoprecipitation (Figures S7A and S7B). Using the ReBiL system to detect K-RASG12D:B-Raf interaction, we found that re-expression of DIRAS3 significantly reduced the K-RASG12D:B-Raf interaction, whereas ΔNT DIRAS3 and DIRAS3 C226S constructs did not (Figure S7C).
DISCUSSION
We used genetic and biochemical strategies to demonstrate that the small GTPase tumor suppressor protein DIRAS3 inhibits RAS-induced transformation as an endogenous antagonist of K-RAS, binding directly to the K-RAS protein and inhibiting multimer formation, clustering, and RAS/MAPK signaling. Direct interaction of DIRAS3 with K-RAS to form heteromers depends upon localized membrane interactions mediated by both C-terminal membrane binding and an interaction domain located in the A5 helical region of K-RAS. Although many tumor suppressors have been found to inhibit oncogenesis through cell cycle arrest, inducing programmed cell death, repairing DNA damage, and blocking metastasis, DIRAS3 antagonism occurs through a direct interaction and disruption of the cluster size of an oncogene, specifically K-RAS. DIRAS3 reduces or prevents mutant K-RAS-stimulated proliferation and clonogenic growth of cancer cell lines from multiple organ sites, including ovary, pancreas, and lung. As DIRAS3 is downregulated by multiple mechanisms in carcinomas from the ovary, pancreas, lung, breast, prostate, and thyroid (Dalai et al., 2007; Feng et al., 2008; Huang et al., 2009; Lin et al., 2011; Lu et al., 2006; Weber et al., 2005; Wu et al., 2013; Yu et al., 1999), the interaction of DIRAS3 and RAS is likely broadly relevant to oncogenesis, in both mutant K-RAS-dependent and -independent settings. This broad down-regulation of DIRAS3 provided a challenge in our study to fully examine the function of DIRAS3 at an endogenous level. Our inducible cell lines were titrated to re-express DIRAS3 at a similar level to the transformed HPNEs (Figure S8).
We showed that DIRAS3 binds to RAS at the PM and that RAS clustering and RAS-B-Raf association (which are required for downstream signaling) are decreased by full-length DIRAS3. Growth inhibition is associated with decreased p-ERK signaling, and knockdown of endogenous DIRAS3 can reverse this phenotype. Although ΔNT DIRAS3 can still interact with K-RAS, the 34-amino-acid N terminus of WT DIRAS3 is critical for inhibiting transformation, disrupting RAS clusters, and inhibiting RAS oncogenic functions, including the association of RAS and B-Raf. Consistent with the importance of the N terminus for suppressor function, the DIRAS3 N terminus fused to mutant RAS can block RAS-driven transformation (Figure S9). Consequently, we propose a model in which DIRAS3 regulation of RAS signaling and clustering follows a three-tiered process in which membrane localization is determined by specific C-terminal-directed co-localization that allows for proximity-induced interactions. These interactions can then be strengthened by a secondary interacting domain specific to each protein-protein interactant, which alters signaling and functional output. The third component is the N terminus of DIRAS3, which provides an additional nano domain regulating clustering and/or alternative protein-protein interaction partnerships (Figure 6F). Our work highlights the importance of using non-tagged constructs, as both the N terminus and C terminus of the DIRAS3 protein are important for its biological functions.
Spencer-Smith et al. (2017) recently demonstrated that NS1, a monobody that binds to the A4-A5 dimer interface of active H-RAS, inhibits RAS clustering and signaling. Interestingly, point mutations in the specific dimer interface residues (R135, D154, and R161) were not sufficient to disrupt RAS signaling, suggesting that this interface alone is not the only factor driving RAS clustering and activation (Spencer-Smith et al., 2017). Prakash et al. (2017) also documented the importance of the A4-A5 domain in forming K-RAS dimers and higher order oligomers. Based on computational and biochemical approaches, they determined that two partially overlapping domains (A3-A4 and A4-A5) were responsible for the weak but direct protein-protein interactions that occur between K-RAS molecules at the PM (Prakash et al., 2017). As DIRAS3 and ΔNT DIRAS3 interact similarly with K-RAS at the A5 helical domain, we were initially surprised to find that ΔNT DIRAS3 does not suppress RAS-driven transformation, signaling, and clustering. This led us to hypothesize that the leucine-rich hydrophobic region of the N terminus of DIRAS3 can alter the interfacial contacts or PM environment, leading to the observed reduction in K-RAS clustering and function; however, the specific mechanism remains to be elucidated.
Recent studies suggest that WT K-RAS can also serve as a tumor suppressor of constitutively active mutant K-RAS (Lin and Haigis, 2018; Nussinov et al., 2019). When clusters of mutant RAS are alloyed with the inactive WT isoform, downstream signaling is attenuated, suppressing transformation (Nussinov et al., 2019). Thus, interference with clustering by DIRAS3 or wtRAS can decrease mutant RAS function. Ambrogio et al. (2018) have documented that KRAS dimerization mediates the fitness of human and murine KRAS-mutant cancer cells and underlies resistance to MEK inhibition. These effects are abrogated when WT KRAS is replaced by KRASD154Q, a mutant that disrupts dimerization at the α4-α5 KRAS dimer interface without changing other fundamental biochemical properties of KRAS (Ambrogio et al., 2018). Our data suggest that both ΔNT DIRAS3 and WT DIRAS3 interact with the α5 helical domain of K-RAS, but only WT DIRAS3 is capable of inhibiting K-RAS multimerization and downstream signaling, suggesting that both the α5 helical region and the N-terminal region of DIRAS3 are important for tumor suppressor function. Thus, several factors likely cooperate to enable intact RAS dimers/multimers to more efficiently activate downstream signaling.
Our goal has been to identify the mechanism(s) by which DIRAS3 inhibits the RAS/MAPK signaling pathway and to determine whether DIRAS3-mediated inhibition of the MAPK signaling pathway occurs downstream of oncogenic RAS mutations. Using several cell lines that contain BRAF mutations (alone or in combination with K-RAS mutations), we found that re-expression of DIRAS3 decreased long-term clonogenic growth. Our results suggest that even in the presence of activating BRAF mutations, upstream clustering of RAS may still play a critical role in enabling heterodimerization of RAF to promote the signaling cascade, in that re-expression of DIRAS3 that disrupts clustering can affect growth mediated by downstream signaling. Taken together, we hypothesize that DIRAS3 plays a critical role in regulating RAS/MAPK signaling through altering the membrane-clustering dynamics of RAS and that this inhibition is one mechanism by which DIRAS3 suppresses tumorigenesis across multiple cancer types, especially considering the upregulation of the MAPK signaling cascade with or without oncogenic mutations in RAS (H-, N-, or K-RAS) itself.
The DIRAS family of tumor suppressor genes includes DIRAS1, DIRAS2, and DIRAS3. We have recently documented the tumor suppressive role of DIRAS1 and DIRAS2 in ovarian cancer (Sutton et al., 2018) and found that re-expression of DIRAS1 or DIRAS2 can also inhibit RAS multimerization (Sutton, 2017). These data support the hypothesis that regulation of RAS multimerization/clustering is critical to regulating activation of the RAS/MAPK signaling pathway and cell proliferation. Interestingly, DIRAS1 and DIRAS2 are found in the murine genome and may mediate many of the functions performed by DIRAS3 in humans (Sutton et al., 2018).
Mutant RAS proteins contribute to the malignant transformation of approximately one-third of human cancers. Distinguishing effectors that regulate RAS-driven signaling in tumors has remained a great challenge. In this report, we show that re-expression of DIRAS3 can inhibit the growth of multiple types of cancer driven by K-RAS mutations by a direct interaction and disruption of RAS higher ordered clusters. The importance of RAS dimerization/multimerization has only recently been appreciated. These studies are fundamentally important, as they demonstrate how members of an oncogenic superfamily can exhibit tumor suppressive function, and they establish DIRAS3 as a natural antagonist of oncogenic RAS clustering and signaling. The interaction between DIRAS3 and RAS provides an approach to target this RAS/MAP signaling axis, which drives multiple malignancies. Future work to determine the mechanisms by which DIRAS3 regulates RAS clustering on the PM will be critical for the clinical translation of these findings. Peptides or small molecules that mimic DIRAS3 activity could target the function of mutant RAS, providing one possible approach that is currently being pursued.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Bast Jr., M.D. (rbast@mdanderson.org)
All other unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell culture and transfection conditions
MCF10a and HeyA8 cells were provided by Dr. Gordon Mills, UT M.D. Anderson Cancer Center (Houston, TX); NIH 3T3, AsPc-1, Panc-1, MiaPaca-2, H358, Bx-PC3 and H441 cells were obtained from ATCC; U2OS cell lines were generated and obtained from Dr. Geoffrey Wahl, The Salk Institute for Biological Studies (La Jolla, California); U2OS TR2 cells expressing PAmCherry1-K-RAS G12D under doxycycline induction were obtained from Dr. Xiaolin Nan, OHSU (Portland, Oregon). Isogenic RAS-dependent cells were obtained from Frederick National Laboratory and were maintained in DMEM High Glucose medium with 10% FBS. NIH 3T3 cells were routinely grown in Dulbecco’s modified minimal essential medium (Cellgrow, VA) supplemented with 2 mM L-glutamine, nonessential amino acids, 100 U of penicillin, 100 μg of streptomycin and 10% fetal bovine serum. MCF10a cells were routinely grown in Dulbecco’s modified minimal essential medium (DMEM): nutrient mixture-F12 supplemented with 5% fetal bovine serum, 20 ng/mL EGF (Peprotech), 0.5 mg/mL Hydrocortisone (Sigma H-0888), 100 ng/mL cholera toxin (Sigma C-8052), 10 μg/mL insulin (Sigma I-1882), and 100 U of penicillin, 100 μg of streptomycin. AsPc-1, Hey-A8, Panc-1, MiaPaca-2, H358 and H441 cells were routinely grown in RPMI supplemented with 2 mM L- glutamine, nonessential amino acids, and 10% fetal bovine serum. DIRAS3 inducible cell lines were maintained as previously specified using tetracycline free fetal bovine serum and G418 to maintain clonal selection. DIRAS3 expression was induced by adding 1 μg/mL doxycycline (Dox). U2OS ReBiL cell lines were routinely grown in DMEM (Cellgrow, VA) supplemented with 2mM L-glutamine, nonessential amino acids, and 10% tetracycline-free fetal bovine serum. Gene expression was induced by adding 0.1 μg/mL doxycycline (Dox). All cells were maintained in a humidified atmosphere of 5% CO2.
Orthotopic Mouse Xenograft
Five million DIRAS3-inducible cells and the parental OVCAR8 cell lines were injected intraperitoneally into six-week old female athymic nu/nu mice were purchased from Taconic Biosciences. Each group contained 8-10 mice. 2 mg/mL DOX in 5% sucrose or sucrose alone water starting the day of injection, and supplemented every two days throughout the duration of the study. The tumor mass (g) was determined 4 weeks post injection after euthanizing mice with CO2 and excising and pooling each intraperitoneal tumor nodule. All procedures were carried out according to an animal protocol approved by the IACUC of The University of Texas MD Anderson Cancer Center.
METHOD DETAILS
Plasmids
The DIRAS3 plasmid was generated in our laboratory. H-RAS and H-RASGV12 plasmids were purchased from Clontech (631924). ΔNT DIRAS3, K-RAS, K-RASG12V, NT-K-RAS and 34S-K-RASG12V were custom synthesized by Blue Heron. DIRAS3 C226S, and NT-H-RASG12V were constructed in our laboratory. Plasmids synthesized by Blue Heron were re-cloned into plasmid containing a pCMV promoter. Viral GFP-K-RASG12V was provided by Dr. John Hancock (McGovern Medical School UT Health).
Antibodies
Anti-DIRAS3 (15E11 and 1D8) mouse monoclonal antibodies were developed in our laboratory. Other antibodies were obtained commercially: Anti-K-RAS antibodies were purchased from ThermoScientific (PA5-27234) and Abcam (Ab172949); anti-Sos-1 antibody (Cell Signaling Technologies D3T7T); anti-Flag antibody (Sigma F1804); anti-HA antibody (Cell Signaling Technologies 3724S); anti-H-RAS antibody (Abcam Ab96548). Antibodies against pERK (4370S), tERK (4695S), pAKT (9271S) and tAKT (9272S) were purchased from Cell Signaling Technologies. K-Ras(Ab172949), H-Ras (Abcam Ab96548) and DIRAS3 (15E11 and 1D8) antibodies were validated for specificity using recombinant purified protein, and very little cross reactivity was observed by western blot analysis.
Purified recombinant protein
His-K-RAS (1-185) (ab96817), H-RAS (1-186) (ab93949), and H-RASG12V (2-186) (ab140571) was purchased from Abcam. Flag-K-RAS (1-188) K-RAS (1-189) and DIRAS3 (1-226) were purified from BL21DE3 cells in our laboratory using a BioRad NGC Chromatography System.
Transient Transfection
Cells were transfected with MegaTran 1.0 (Origene) or XtremeGeneHP (Roche) for plasmid DNA or DharmaFECT I (ThermoScientific) for siRNA, using manufacturer’s protocols. The predesigned siRNAs were purchased from Dharmacon (ThermoScientific).
Surface plasmon resonance (SPR)
Direct binding of K-RAS to DIRAS3 was measured by SPR using a Biacore T200 optical biosensor (GE Healthcare) at 25°C. To capture GST fusion protein, approximately 12000 RU of goat anti-GST antibody (GE Healthcare) was immobilized on a CM5 chip (GE Healthcare) and GST-tagged DIRAS3 (40 μg/ml) was injected for 1 min at a flow rate of 10 μl/min, resulting in a DIRAS3 (250 −300 RU) ligand surface. Another flow cell with immobilized anti-GST antibody and captured 20 μg/ml of GST (150-200 RU) was used as a reference surface. Binding experiments were performed using a flow rate of 30 μl/min in TBST (50 mM Tris, pH7.5, 100 mM NaCl, 0.01% Tween 20). K-RAS (ab96817, abcam) was freshly buffer-exchanged in TBST using Zeba desalting spin columns (Thermo). To regenerate the antibody surface, captured molecules (GST and GST-tagged DIRAS3) were removed by 120 s injection of 10 mM glycine (pH2.2), then 60 s injection of 20 mM Tris (pH7.5) containing 1 M NaCl and 0.015% DDM. The SPR sensorgrams were reference and buffer subtracted, and evaluated using the Biacore T200 Evaluation Software (version 3.1).
Focus Formation/Transformation assay
Transformation of NIH 3T3 cells was performed following standard protocols (Clark et al., 1995). Low passage NIH 3T3 cells were plated in 60 mm dishes at a density of ~1.2 × 106 cells/plate. Transfection of single plasmids (20 μg) or co-transfection of differing plasmids (10 μg each) were completed 24 hours post seeding using MegaTran 1.0 (Origene) following manufacturers protocol. 24 hours post transfection, cells were trypsinized and plated onto 60 mm dishes at a ratio of 1:4 and kept in DMEM supplemented with dexamethasone (Sigma) or G418 to determine transfection efficiency for 14 days, with media changed every 3-4 days. Cells treated with DMEM supplemented with G418 media were stained by Coomassie blue and colonies were counted to ensure equal transfection efficiency. Those plates treated with DMEM supplemented with dexamethasone were examined microscopically for signs of contact-uninhibited growth and the appearance of morphologically transformed foci. Transformed foci were counted at 10x magnification as they appeared within two 10x10 mm areas per plate. The assay was performed three times with technical duplicates of each transfection group.
Anchorage independent growth
MCF10a cells were transfected with single or double DNA plasmids using MegaTran 1.0 according to the manufacturer’s protocol. 24 hours following transfection, MCF10a cells were trypsinized and resuspended in RMPI containing 10% fetal bovine serum (FBS) and 0.35% noble agar, and plated on top of a layer of RPMI containing 10% FBS and 0.5% noble agar in a 35-mm dish. Media without epidermal growth factor (EGF) supplement was changed every 3 days and colonies were allowed to grow until they reached more than 50 cells/colony (~2-3 weeks). Colonies were visualized and quantified microscopically.
Cell proliferation assay
Cell proliferation was determined using clonogenic and sulforhodamine B (SRB) assays. Clonogenic assays were performed using DIRAS3-inducible cell lines where 400-800 cells/well were cultured with or without Doxycycline for 72 hours. Alternatively, cells were transfected with specified plasmids and 24-hours post transfection were trypsinized and re-seeded (1:10-40) and selection with G418 was used until only single cells remained in the non-transfected control group. Cells were then allowed to grow in a clonogenic fashion for two weeks prior to staining and fixation with Coomassie blue. Clonogenic growth was quantified by manual analysis of colonies containing at least 50 cells. SRB short term viability assays were performed following siRNA knockdown of K-RAS in both AsPc-1 and HeyA8 cell lines. Cells were plated at 1x104 cell density and transfected with siRNA as described above. 72 hours post transfection with siRNA cells were fixed with 30% TCA at 4°C for 1 hour, following by washing. Cells were incubated with 0.4% SRB in for 30 min at room temperature before being washed with 1% acetic acid. Tris base was used to solubilize the SRB dye prior to reading the plates with a microplate-reader (Tecan) at 510 nm.
qPCR analysis of mRNA expression
SU86.86 pancreatic cancer cells were treated as described and RNA was extracted with Trizol (Invitrogen 11596026) following the manufacturers protocol. cDNA was synthesized from 1 μg of RNA using the Superscript II First Strand Synthesis Kit (Invitrogen, #11904-018). SYBR green based quantitative PCR was used to measure RNA levels. Relative expression was calculated by the 2−ΔΔCT method using glyceraldehydes-3-phosphatase dehydrogenase (GAPDH) as the reference gene. The experiments were repeated at least three times, and samples were measured in technical duplicate. DIRAS3 primer 5′ → 3′ Forward: CTACCTGCCGACCATTGAAAA Reverse: GGGTTTCCTTCTTGGTGACTG
RAS activity Raf-RBD Pull-down assay
H-RAS and K-RAS specific activity assays were purchased and performed following the manufacturers protocol from CellBioLabs (STA-400-Kand STA-400-H). NIH 3T3 cells were plated in 100 mm dish at ~80% confluence. Twenty-four hours post seeding, cells were transfected with 10 μg of DNA using MegaTran 1.0 (Origene) following manufacturers protocol. 24 hours post transfection, cells were lysed as suggested following the CellBioLabs protocol and the assay was carried out according to the manufacturer’s protocol.
ReBiL analysis of protein-protein interactions
Cells were seeded in 96-well or 384-well black wall, clear bottom plates and treated with doxycycline (100 ng/mL) to induce gene expression. 24-48 hours following gene expression the cells were washed briefly with 1X phosphate buffered saline (PBS) and D-luciferin (200μM) was added. Luminescence was determined within 15 minutes using a Tecan plate reader. Each experiment was performed at least three times with technical triplicates. Untreated cells were used for background subtraction.
Immunoprecipitation and Immunoblotting
NIH 3T3 cells were co-transfected with 10μg of each DNA plasmid for 24 hours using MegaTran 1.0 (Origene) transfection reagent prior to incubation in lysis buffer (50 mM HEPES, pH 7.0, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10 mM NaF, 10 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100) plus protease and phosphatase inhibitors (1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin and 1 mM Na3VO4). Cells were lysed for 30 min on ice, and then centrifuged at 17,000 x g for 30 min at 4°C. The protein concentration was assessed using a bicinchoninic acid (BCA) protein assay (Thermo, Waltham, MA). Lysates (0.8-1 mg protein) were diluted with lysis buffer to 1 mL. Immune complexes were incubated overnight with 2 μg of the antibody and precipitated with protein A/G-agarose beads for 90 min. Complexes were washed in lysis buffer (3 × 5 min) and in PBS (3 × 5 min). Immunoprecipitated proteins were separated by SDS-PAGE and transferred to PVDF membranes. Immunoblot analysis was performed with the indicated antibodies and visualized with an ECL enhanced chemiluminescence detection kit (GE Healthcare). Band intensity from western blots was quantified using the ImageJ program (https://imagej.nih.gov/ij/docs/guide/). Purified recombinant protein was also used to determine the specific protein-protein interactions. Magnetic beads (40 μL) were incubated with 1 μL anti-K-RAS antibody (Sigma) for 1 hour at room temperature with gentle rotation. Beads were then mixed with K-RAS and DIRAS3 purified proteins at a specific concentration in 1 mL lysis buffer and incubated with rotation overnight at 4°C. Beads were collected using the magnetic stand and washed (6 × 5 min) with wash buffer before elution with 1X SB at 70°C for 5 minutes.
Peptide array analysis
Peptide arrays were made using the MultiPep RS robot (Intavis, Bergisch Gladbach, Germany) according to the SPOT synthesis technique described by Frank, 2002. Arrays were developed by soaking membranes in 100% methanol for 10 minutes at room temperature followed by washes with PBS (3x 10 min). Membranes were then blocked overnight at 4°C in 5% BSA/PBS. Recombinant proteins were added to the membrane at a final concentration of 1 μg/mL in 1% BSA/PBS and shaken gently at room temperature for 2 hours. Membranes were washed 3x with 1% BSA/PBS for 10 minutes each prior to the addition of primary antibody diluted in wash buffer for 1 hour at room temperature. The membrane was washed 3x for 10 minutes and a diluted secondary antibody (1:10,000) was added for 45 minutes at room temperature, with gentle shaking. The membrane was washed 3x with wash buffer for 10 minutes, then followed with 3 washes with PBS-T for 10 minutes each. Membranes were developed with HRP substrates and exposed to X-Ray film. K-RAS4B Arrays (residues 1-189) used a 12-mer walk stepping by 3 amino acids; H-RAS Array (residues 1-186) a 12-mer walk stepping by 3 amino acids; and DIRAS3 Array (residues 1-229) used a 15-mer walk stepping by 3 amino acids.
Cytoplasm/membrane extraction
U2OS-ReBiL cells were seeded at 5 × 106 cells/100 mm dish and treated with doxycycline for 24 hours. Cells were collected and fractionated lysates were generated using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo-Fisher #89842) using the manufacturers recommended protocol. Added washes (3X) were performed between the cytoplasm extraction and membrane extraction to reduce contamination between the fractions.
DUOLINK in situ assay
Duolink in situ PLA probes and Duolink in situ detection reagents were purchased from Sigma. K-RAS/DIRAS3 heterodimer formation was studied in Hey-A8-DIRAS3 ovarian cancer cells and AsPc-1-DIRAS3 pancreatic cancer cells. Procedures were performed following the manufacturer’s instructions. Briefly, cells were seeded on chamber slides, fixed, blocked and incubated with primary antibodies and then with secondary antibodies conjugated with oligonucleotides (PLA probe MINUS and PLA probe PLUS). Finally, cells were incubated sequentially with ligation solution and amplification solution followed by analysis with fluorescence microscopy.
Confocal Microscopy
Immunofluorescence staining was performed using two well chamber slides, fixed with 4% Paraformaldehyde for 10 minutes prior to washing with PBS. Cells were permeabilized using 0.05% Triton X in PBS for 10 minutes at room temperature. Cells were washed 3X with PBS for 5 minutes each before being blocked with 0.5% BSA/PBS at room temperature for 1 hour. Primary antibodies were added at the dilution of 1:600 in 0.5% BSA/PBS for both 1D8 DIRAS3 and K-RAS and incubated overnight at 4°C. Cells were washed 3X with 0.5% BSA/PBS for 5 minutes each prior to the addition of secondary antibodies. Secondary antibodies were added at 1:250 dilution in 0.5% BSA/PBS and incubated at room temperature for 45 min. Cells were washed 3X with 0.5% BSA/PBS and DAPI staining at 1:10,000 dilution occurred at room temperature for 10 minutes. Slides were mounted, and images captured on an Olympus FV1000 Laser Confocal Microscope.
Super-resolution fluorescence microscopy
The custom multispectral super-resolution microscope (MSSRM) for multicolor super-resolution imaging was constructed as described elsewhere (T.H., unpublished data). Briefly, the MSSRM is housed on a Nikon Ti-U inverted microscope frame equipped with two lasers emitting at 405 and 638 nm, respectively, an oil immersion objective (60x, numerical aperture 1.49), and an EM-CCD (Andor iXon+). Signals from the sample is collected through the objective and split between a positional channel and a spectral channel to enable simultaneous imaging of multiple fluorophores without having to use emission filters. The MSSRM setups achieves ~10 nm spectral resolution and hence is able to distinguish single Alexa Fluor 647 and CF660C molecules reliably as previously demonstrated. MSSRM imaging of fluorescently stained cells were performed in PBS buffer supplemented with 1% beta-mercaptoethanol (βME), 5 μg/mL glucose oxidase (Sigma, G2133-50 kU), 0.4 μg/mL catalase (Sigma, C100-50 MG), and 10% glucose (w/v, Fisher Chemicals D16-500). The EM-CCD was operated in frame transfer mode at 15 ms per frame with a gain setting of 300. Acquisition of raw images was performed using the open source micromanager software suite (https://micro-manager.org/) (Stuurman et al., 2010). Image analyses for extracting single-molecule localization, spectra were all performed with custom MATLAB (Mathworks, MA) scripts as described previously (Nickerson et al., 2014). Coordinates of single molecules were grouped based on their peak emission wavelength. Images in each channel was then rendered separately and recombined in Fiji (https://imagej.net/Fiji/) (Schindelin et al., 2012) into a composite image.
EM Spatial Interaction and Cluster analysis
Using methods previously reported by Plowman et al. (2005) and Prior et al. (2003a, 2003b), we assessed the co-localization of K-RASG12V and DIRAS3 on the plasma membrane (PM) of Hey-A8-DIRAS3 ovarian cancer cells. HeyA8-DIRAS3 inducible cells were seeded at 1.0 × 105 cells/well in a six well plate onto gold EM grids with pioloform and poly-L-lysine coating. Expression of DIRAS3 was achieved by adding doxycycline to the media for 24 hours. Lentiviral K-RASG12V was then used to infect both doxycycline negative and positive samples for 18 hours prior to harvesting the EM grids and adherent cells for analysis. Briefly, the basolateral membrane was exposed and fixed using 4% paraformaldehyde (PFA) and 0.1% glutaraldehyde (GA). For the univariate experiments, 4.5-nm gold nanoparticles pre-coupled to anti-GFP antibody were used to immunolabel GFP-RAS on the PM and embedded in uranyl acetate. Gold particle distribution was visualized by transmission EM (TEM) using JEOL JEM-1400 transmission EM. ImageJ was then used to assign x and y coordinates of gold particles in a 1-μm2 area on intact and featureless PM region. Ripley’s K-Function was used to calculate the gold particle distribution and extent of nanoclustering (Plowman et al., 2008). (Eqs. A and B):
| (Eq. A) |
| (Eq. B) |
where K(r) is the univariate spatial distribution for a total of n labeled gold particles within a cell PM sheet area of A; r is length scale between 1 and 240 nm with 1nm increments; ∥ · ∥ is Euclidean distance. The indicator function of 1(·) is given a value of 1 if ∥xi-xj ≤ r and 1(·) has a value of 0 if ∥xi-xj∥ > r; and edge correction was accounted for by using Wij−1, which is the circumferential fraction of the circle defined with a center located at xi and with a radius ∥xi-xj∥. Monte Carlo simulations were then used to generate a 99% confidence interval (99% C.I.), which was employed to normalized K(r) to yield L(r) - r. For each condition, at least 15 basolateral PM sheets were pooled generate K-function curves. A 1000 bootstrap samples were constructed and pooled to examine the statistical significance between various conditions (Diggle et al., 2000; Plowman et al., 2008).
Bi-variate analysis of co-localization was performed in a similar fashion where the basolateral membrane was exposed and fixed with 4% PFA and 0.1% GA before sequential labeling with 6-nm gold nanoparticles pre-coupled to anti-GFP followed by 2-nm gold nanoparticles pre-coupled to anti-DIRAS3 (antibody ID8). Membranes were embedded in uranyl acetate and imaging performed (Prior et al., 2003b). At least 15 PM sheets were imaged, analyzed and pooled for each condition. Bivariate co-clustering was analyzed using the Ripley’s bivariate K-Function analysis (C-F):
| (Eq. C) |
| (Eq. D) |
| (Eq. E) |
| (Eq. F) |
where Kbs(r) describes the spatial distribution of 6nm (b stands for big) gold particles surrounding each 2nm (s stands for small) gold particle and vice versa Ksb(r) quantifies the distribution of small gold surrounding each big gold. Kbiv(r) is an estimator function that combines both Kbs(r) and Ksb(r) to yield a single equation, where nb = number of big (6nm) gold and ns = number of small (2nm) gold. Other symbols are the same as in Equation 1 and 2. Monte Carlo simulations were employed to generate a 95% confidence interval, which was then used to normalize the bivariate K function to yield Lbiv(r)-r. We used the same Bootstrap tests used in the univariate analysis to test the statistical significance between different sets of bivariate data.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism and Microsoft Excel were used for the statistical analyses performed in this manuscript. The statistical details for each experiment can be found in the figure legends, figures and results sections throughout the main text. All experiments were repeated independently at least three times and the data (bar graphs) expressed as mean ± s.e. Statistical analysis was performed using Student’s t test (two-sample assuming unequal variances). The criterion for statistical significance was taken as p < 0.05 (two-sided).
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-DIRAS3 (ID8 or 15E11) | Bast Lab | N/A |
| Anti-K-RAS | ThermoScientific | PA5-27234; RRID:AB_2544710 |
| Anti-K-RAS | Abcam | Ab172949 |
| Anti-Sos-1 | Cell Signaling Technologies (CST) | D3T7T; RRID:AB_2797902 |
| Anti-Flag | Sigma | F1804; RRID:AB_262044 |
| Anti-HA | CST | 3724; RRID:AB_1549585 |
| Anti-H-RAS | Abcam | Ab96548; RRID:AB_10862138 |
| Anti-pERK | CST | 4370; RRID:AB_2315112 |
| Anti-tERK | CST | 4695; RRID:AB_390779 |
| Anti-pAKT | CST | 9271; RRID:AB_329825 |
| Anti-tAKT | CST | 9272; RRID:AB_329827 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| His-K-RAS (1-185) | Abcam | Ab96817 |
| H-RAS (1-186) | Abcam | Ab93949 |
| H-RASG12V (2-186) | Abcam | Ab140571 |
| Flag-K-RAS (1-188) | Bast Lab | N/A |
| K-RAS (1-189) | Bast Lab | N/A |
| DIRAS3 (1-226) | Bast Lab | N/A |
| GST-DIRAS3 (1-226) | Bast Lab | N/A |
| Critical Commercial Assays | ||
| Ras Activity Raf-RBD Pull-Down Assay | CellBioLabs | STA-400-K STA-400-H |
| Cytoplasm/Membrane Extraction Mem-PER Plus | ThermoFisher | 89842 |
| DUOLINK in Situ Assay | Sigma | N/A |
| Experimental Models: Cell Lines | ||
| MCF10a | Gordon Mills Lab | N/A |
| HeyA8 | Gordon Mills Lab | N/A |
| NIH 3T3 | ATCC | CRL-6442 |
| AsPC-1 | ATCC | CRL-1682 |
| Panc-1 | ATCC | CRL-1469 |
| Capan-2 | ATCC | HTB-80 |
| MiaPaca2 | ATCC | CRM-CRL-1420 |
| H358 | ATCC | CRL-5807 |
| Bx-PC3 | ATCC | CRL-1687 |
| H441 | ATCC | HTB-174 |
| HPNE | ATCC | CRL-4023 |
| U2OS ReBiL | Geoffrey Wahl Lab | N/A |
| U2OS TR2 | Xiaolin Nan Lab | N/A |
| Isogenic RAS-dependent MEFs | Frederick National Lab | N/A |
| OVCAR8 | Bast Lab | N/A |
| Oligonucleotides | ||
| DIRAS3 primer 5′ → 3′ Forward: CTACCTGCCGACCATTGAAAA | IDT | Custom |
| DIRAS3 primer 5′ →3′ Reverse: GGGTTTCCTTCTTGGTGACTG | IDT | Custom |
| KRAS siRNA | Dharmacon | LQ-005-069-00-0005 |
| DIRAS3 siRNA | Dharmacon | LQ-008660-00-0005 |
| Non-targeting control siRNA | Dharmacon | D-001810-10-05 |
| Recombinant DNA | ||
| DIRAS and DIRAS3 mutant constructs | Bast Lab | N/A |
| H-RAS and H-RASG12V | Clonetech | N/A |
| ReBiL constructs | Bast and Wahl Labs | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| MATLAB | MathWorks | N/A |
| ImageJ-Fiji | N/A |
Highlights.
Re-expression of DIRAS3 (ARHI) inhibits ovarian, lung, and pancreatic cancer growth
DIRAS3 (ARHI) interacts directly with RAS at the plasma membrane
Re-expression of DIRAS3 inhibits RAS clustering and downstream MAPK signaling
The N-terminal extension of DIRAS3 is required for its tumor-suppressive function
ACKNOWLEDGMENTS
The authors would like to thank members of the Bast laboratory for their discussion and suggestions and Dr. Robert Langley for careful review of the manuscript. The Protein Production and Analysis Core at IBT of Texas A&M University provided services for molecular cloning and partial purification of the KRAS and DIRAS3 recombinant proteins, and Dr. Chun Li in the Department of Cancer Systems Imaging provided access to the Biacore 3000 instrument. This work was supported by the MD Anderson SPOREs in Ovarian Cancer grants NCI P50 CA 83639 and CA 217685; the National Cancer Institute grant R01 CA135354; a grant from the Cancer Prevention and Research Institute of Texas (RP140429); the Shared Resources of the MD Anderson CCSG grant NCI P30 CA16672; The National Foundation for Cancer Research; philanthropic support from the Anne and Henry Zarrow Foundation; and generous donations from Stuart and Gaye-Lynn Zarrow, the Mossy Foundation, and the Roberson endowment. M.N.S. was supported by a pre-doctoral fellowship from NIH TL1TR000369 and UL1TF000371 and a fellowship in cancer research from the American Legion Auxiliary as well as the Ann and Sol Schreiber Mentored Investigator Award from the Ovarian Cancer Research Fund Alliance. SPR studies were conducted in the Protein Production and Analysis Core at Texas A&M IBT. This work was performed in partial fulfillment of the requirements for a PhD degree from The University of Texas MD Anderson UTHeath GSBS, Houston, TX 77030, USA.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.045.
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
REFERENCES
- Ambrogio C, Kohler J, Zhou ZW, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al. (2018). KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 172, 857–868.e815. [DOI] [PubMed] [Google Scholar]
- Badgwell DB, Lu Z, Le K, Gao F, Yang M, Suh GK, Bao JJ, Das P, Andreeff M, Chen W, et al. (2012). The tumor-suppressor gene ARHI (DIRAS3) suppresses ovarian cancer cell migration through inhibition of the Stat3 and FAK/Rho signaling pathways. Oncogene 31, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Jones SA, and Zhuang X (2013). Stochastic optical reconstruction microscopy (STORM): a method for superresolution fluorescence imaging. Cold Spring Harb. Protoc 2013, 498–520. [DOI] [PubMed] [Google Scholar]
- Clark GJ, Cox AD, Graham SM, and Der CJ (1995). Biological assays for Ras transformation. Methods Enzymol. 255, 395–412. [DOI] [PubMed] [Google Scholar]
- Dalai I, Missiaglia E, Barbi S, Butturini G, Doglioni C, Falconi M, and Scarpa A (2007). Low expression of ARHI is associated with shorter progression-free survival in pancreatic endocrine tumors. Neoplasia 9, 181–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle PJ, Mateu J, and Clough HE (2000). A comparison between parametric and non-parametric approaches to the analysis of replicated spatial point patterns. Adv. Appl. Probab 32, 331–343. [Google Scholar]
- Feng W, Marquez RT, Lu Z, Liu J, Lu KH, Issa JP, Fishman DM, Yu Y, and Bast RC Jr. (2008). Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 112, 1489–1502. [DOI] [PubMed] [Google Scholar]
- Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, and Mattos C (2015). Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish KN (2009). Total internal reflection fluorescence (TIRF) microscopy. Curr. Protoc. Cytom Chapter 12, Unit12.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald J, and Bateman JF (2004). Why mice have lost genes for COL21A1, STK17A, GPR145 and AHRI: evidence for gene deletion at evolutionary breakpoints in the rodent lineage. Trends Genet. 20, 408–412. [DOI] [PubMed] [Google Scholar]
- Frank R (2002). The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports—principles and applications. J. Immunol. Methods 267, 13–26. [DOI] [PubMed] [Google Scholar]
- Huang J, Lin Y, Li L, Qing D, Teng XM, Zhang YL, Hu X, Hu Y, Yang P, and Han ZG (2009). ARHI, as a novel suppressor of cell growth and down-regulated in human hepatocellular carcinoma, could contribute to hepatocarcinogenesis. Mol. Carcinog 48, 130–140. [DOI] [PubMed] [Google Scholar]
- Li YC, Rodewald LW, Hoppmann C, Wong ET, Lebreton S, Safar P, Patek M, Wang L, Wertman KF, and Wahl GM (2014). A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep. 9, 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Wang L, Hayes TK, Sutton MN, Bast RC, McCormick F, Der CJ, and Wahl GM (2018). Facile real time detection of membrane colocalization of RAS superfamily GTPase proteins in living cells. bioRxiv. 10.1101/369587. [DOI] [Google Scholar]
- Lin D, Cui F, Bu Q, and Yan C (2011). The expression and clinical significance of GTP-binding RAS-like 3 (ARHI) and microRNA 221 and 222 in prostate cancer. J. Int. Med. Res 39, 1870–1875. [DOI] [PubMed] [Google Scholar]
- Lin YJ, and Haigis KM (2018). Brother’s Keeper: Wild-Type Mutant K-Ras Dimers Limit Oncogenesis. Cell 172, 645–647. [DOI] [PubMed] [Google Scholar]
- Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB, et al. (2008). The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest 118, 3917–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Luo RZ, Peng H, Rosen DG, Atkinson EN, Warneke C, Huang M, Nishmoto A, Liu J, Liao WS, et al. (2006). Transcriptional and posttranscriptional down-regulation of the imprinted tumor suppressor gene ARHI (DRAS3) in ovarian cancer. Clin. Cancer Res 12, 2404–2413. [DOI] [PubMed] [Google Scholar]
- Lu Z, Yang H, Sutton MN, Yang M, Clarke CH, Liao WS, and Bast RC Jr. (2014). ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ. 21, 1275–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo RZ, Fang X, Marquez R, Liu SY, Mills GB, Liao WS, Yu Y, and Bast RC (2003). ARHI is a Ras-related small G-protein with a novel N-terminal extension that inhibits growth of ovarian and breast cancers. Oncogene 22, 2897–2909. [DOI] [PubMed] [Google Scholar]
- Muratcioglu S, Chavan TS, Freed BC, Jang H, Khavrutskii L, Freed RN, Dyba MA, Stefanisko K, Tarasov SG, Gursoy A, et al. (2015). GTP-Dependent K-Ras Dimerization. Structure 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Tamgüney TM, Collisson EA, Lin LJ, Pitt C, Galeas J, Lewis S, Gray JW, McCormick F, and Chu S (2015). Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 112, 7996–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson A, Huang T, Lin LJ, and Nan X (2014). Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells. PLoS One 9, e100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019). Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin. Cancer Biol 54, 114–120. [DOI] [PubMed] [Google Scholar]
- Plowman SJ, Ariotti N, Goodall A, Parton RG, and Hancock JF (2008). Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol. Cell. Biol 28, 4377–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowman SJ, Muncke C, Parton RG, and Hancock JF (2005). H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 102, 15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Sayyed-Ahmad A, Cho KJ, Dolino DM, Chen W, Li H, Grant BJ, Hancock JF, and Gorfe AA (2017). Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci. Rep 7, 40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Muncke C, Parton RG, and Hancock JF (2003a). Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol 160, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Parton RG, and Hancock JF (2003b). Observing cell surface signaling domains using electron microscopy. Sci. STKE 2003, PL9. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, and Shih IeM. (2003). Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst 95, 484–486. [DOI] [PubMed] [Google Scholar]
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, et al. (2017). Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuurman N, Edelstein AD, Amodaj N, Hoover KH, and Vale RD (2010). Computer Control of Microscopes using μManager. Curr. Protoc. Mol. Biol Chapter 14, Unit14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MN (2017). The Role of the DIRAS Family Members in Regulating Ras Function, Cancer Growth and Autophagy (The University of Texas; ), PhD thesis. [Google Scholar]
- Sutton MN, Yang H, Huang GY, Fu C, Pontikos M, Wang Y, Mao W, Pang L, Yang M, Liu J, et al. (2018). RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 14, 637–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V, and Kirtikara K (2006). Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Weber F, Aldred MA, Morrison CD, Plass C, Frilling A, Broelsch CE, Waite KA, and Eng C (2005). Silencing of the maternally imprinted tumor suppressor ARHI contributes to follicular thyroid carcinogenesis. J. Clin. Endocrinol. Metab 90, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Wong ET, Kolman JL, Li YC, Mesner LD, Hillen W, Berens C, and Wahl GM (2005). Reproducible doxycycline-inducible transgene expression at specific loci generated by Cre-recombinase mediated cassette exchange. Nucleic Acids Res. 33, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liang L, Dong L, Yu Z, and Fu X (2013). Effect of ARHI on lung cancer cell proliferation, apoptosis and invasion in vitro. Mol. Biol. Rep 40, 2671–2678. [DOI] [PubMed] [Google Scholar]
- Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y, Cuevas B, Kuo WL, Gray JW, Siciliano M, et al. (1999). NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc. Natl. Acad. Sci. USA 96, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.