

SUMMARY
High-salt diets inhibit the suppressive function of thymus-derived natural regulatory T cells (tTreg). Transforming growth factor β (TGF-β)-induced ex vivo regulatory T cells (iTreg) comprise another Treg subset that exhibits similarities and differences with tTreg. Here, we demonstrate that iTregs are completely stable and fully functional under high salt conditions. High salt does not influence the development, differentiation, and functional activities of iTreg but affects Foxp3 stability and function of tTreg in vitro and in vivo. In addition, high salt does not significantly change the transcription profiles of the iTreg signature or pro-inflammatory genes. Therefore, we conclude that iTreg, unlike tTreg, are stable and functional in the presence of high salt. Our findings provide additional evidence that iTreg may have different biological features from tTreg and suggest a greater potential for clinical utility in patients with autoimmune diseases, in which the complicated role of environmental factors, including diet, must be considered.
In Brief
Luo et al. show that high salt does not affect the characteristics of TGF-β-induced regulatory T cells (iTregs). iTregs are stable and functional in the presence of high salt during autoimmune responses.
Graphical Abstract
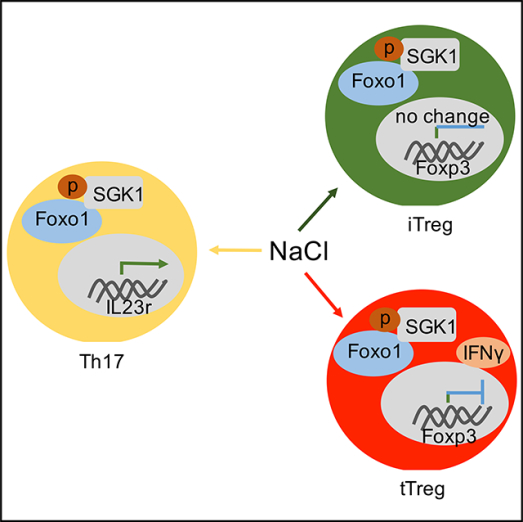
INTRODUCTION
It has been well recognized that high dietary salt intake impacts obesity and cardiovascular diseases (Farquhar et al., 2015; He and MacGregor, 2009). Hypertension is a predominant consequence of continuous high salt diet (Wang et al., 2012). Interestingly, recent studies have also demonstrated that high dietary salt intake promotes autoimmunity via pro-inflammatory responses (Appel et al., 2011; McGuire, 2010). Excessive NaCl intake can dampen the innate immune system (Kleinewietfeld et al., 2013; Machnik et al., 2009), as well as promote the differentiation of pathogenic Th17 cells in either in vitro (Kleinewietfeld et al., 2013; O’Shea and Jones, 2013; Wu et al., 2013) or in vivo in experimental autoimmune encephalomyelitis (EAE), a mouse model of the autoimmune disease multiple sclerosis (O’Shea and Jones, 2013; Wu et al., 2013). Clinical and epidemiological studies in accord with these findings indicate that high sodium intake presents a notably positive correlation with increased disease activity in multiple sclerosis and rheumatoid arthritis patients (Farez et al., 2015; Sundström et al., 2015).
Thymus-derived, naturally occurring CD4+Foxp3+ cells (tTreg) play an essential role in immunologic homeostasis and the prevention of autoimmune diseases (Liu et al., 2015a). Although tTreg cells have an ideally preventive role in controlling autoimmune diseases with numerous animal models, the therapeutic effects of tTreg on established autoimmune diseases are still unsatisfactory (Koenecke et al., 2009; Sakaguchi, 2005). One of the important reasons is that the phenotype and function of tTreg subset are not stable in the inflammation conditions (Zheng et al., 2008). We and others have previously reported that tTreg can convert into other T effector cell subsets, such as Th1, Th2, Th17, and Tfh cells, followed by Foxp3 downregulation and reduction of the immunosuppressive function in the presence of pro-inflammatory cytokines in vitro and in vivo (Ding et al., 2010; Lu et al., 2010a; Zheng et al., 2008). Hernandez et al. (2015) recently provided a new line of evidence that high salt also markedly diminishes the functional activities of tTreg cells, although it did not change Foxp3 expression.
In addition to the tTreg subset, CD4+ Treg cells can be induced in the periphery outside of thymus (pTreg) or from conventional non-Treg cells in the presence of transforming growth factor β (TGF-β) and interleukin (IL)-2 with appropriate antigens (iTreg) (Abbas et al., 2013; Zheng et al., 2002, 2007). Both pTreg and iTreg subsets also express Foxp3 and for the most part share the phenotypic and functional features of the tTreg subset (Abbas et al., 2013; Curotto de Lafaille and Lafaille, 2009; Laurence et al., 2012). However, the iTreg subset also displays substantial differences with tTreg subset (e.g., iTreg may be stable in the inflammatory conditions) (Zheng et al., 2008). It is unknown whether high levels of dietary salt also affect the phenotype and function of the iTreg subset.
Here, we report that neither the differentiation nor function of the iTreg subset was affected by physiologically elevated levels of sodium chloride in vitro. The iTreg subset that had differentiated in vitro was resistant to Th17 and Th1 cell conversion in the presence of high levels of sodium chloride although high salt significantly increased iTreg proliferation. High dietary salt intake also did not change the differentiation and function of pTreg in vivo, although it significantly promoted Th17 cell differentiation in both in vitro and in vivo systems. Conversely, high salt reduced the Foxp3 expression and functionality of tTreg subset in vitro and in vivo. RNA sequencing (RNA-seq) analyses showed that high levels of dietary salt did not significantly change the gene profiles related to Treg and inflammatory cell signatures. Thus, our data provide evidence for an additional difference between tTreg and iTreg or pTreg subsets, and implies the induced Treg subset can easily adapt to environmental factors. This subset may therefore have advantages in treating patients with autoimmune diseases in the future.
RESULTS
Neither Differentiation nor Stability of iTreg Subset Is Affected by High Salt In vitro
As recent studies have demonstrated that high salt conditions promoted Th17 cell differentiation (Kleinewietfeld et al., 2013), we sought to determine whether high salt also similarly affects the biological features of iTreg subset. The iTreg differentiation was conducted with a standard protocol as previously described in the presence of different concentrations of NaCl (20–40 mM) with different time points (Zheng et al., 2008). Foxp3-GFP knock in mice were used to facilitate the Foxp3 identification and expression on the iTreg subset by flow cytometry. The addition of TGF-β dramatically promoted Foxp3 induction, and around 90% of naive CD4+ cells became CD4+CD25+Foxp3+ cells, but high salt did not diminish the Foxp3 induction effect (Figures 1A and 1B). If any effect was measured, it was that high levels of sodium chloride slightly increased Foxp3 induction (Figures 1A and 1B). We observed that high salt expanded total numbers of CD4+Foxp3+ cells and elevated the mean fluorescence intensity (MFI) of CD25 (Figure S1). Additionally, we also induced Th17 cells and observed that the addition of high salt significantly increased Th17 cell differentiation (Figure S2), indicating the precursor cells we used responded to high salt.
Figure 1. High-Salt Condition Does Not Influence Foxp3 Expression and Conversion of iTregs In vitro.

Naive CD4+ T cells from C57BL/6 mice were stimulated with anti-CD3/28 microbeads in the presence of IL-2 ± TGF-β in standard media, or with additional 20 or 40 mM NaCl for 3 days. Some iTregs were generated normally, harvested, and thoroughly washed on day 3, then renewed either in normal media or media enriched with 20 or 40 mM NaCl for an additional 3 to 7 days with anti-CD3/28 microbeads (5 cells per bead) and IL-2 (50 U/mL).
(A) Naive CD4+ T cells were differentiated into iTregs in the presence or absence of additional 20 or 40 mM NaCl for 3 days and detected by FACS for GFP-Foxp3+.
(B) The line graph depicts a summary of experiments (n = 6).
(C) Cells were stimulated as in (A) and were re-stimulated under the indicated increased NaCl concentrations for another 3–7 days and analyzed by fluorescence-activated cell sorting (FACS) (one representative experiment of six is shown).
(D) The line graph depicts a summary of experiments.
(E) iTregs were re-stimulated under 40 mM NaCl for 3 days and analyzed by flow cytometry for IL-17A and IFNγ. The bar graph depicts a summary of independent experiments (n = 6). Statistical analyses were performed using one-way ANOVA. ns, not significant.
We then determined whether high salt affects the stability of iTregs that had been induced. We first induced the iTreg subset as shown in Figures 1A and 1B, and high salt was then added to cultures for an additional 3–7 days. Either untreated or NaCl-treated iTregs showed no Foxp3 reduction during these days (Figures 1C and 1D). As high salt promotes the tTreg subset to secrete interferon gamma (IFNγ) (Hernandez et al., 2015), we also analyzed whether the iTreg subset began to express IFNγ as for the tTreg subset when they encountered high salt. The iTreg subset did not produce relevant cytokines including IFN-γ and IL-17A and was almost completely resistant to Th1 and Th17 conversion even in the presence of high salt (Figure 1E).
High Salt Has Little Effect on Suppressive Function of iTregs In vitro
High salt does not reduce Foxp3 expression but diminishes tTreg functionality (Hernandez et al., 2015). We examined whether high salt also affects the functionality of iTreg subset. A standard in vitro co-culture suppression assay was used to study iTreg in vitro functionality in the presence or absence of high-salt conditions. Splenic enriched T cells from normal C57BL/6 mice labeled with carboxyfluorescein succinimidyl ester (CFSE) were cultured with γ-irradiated antigen-presenting cells (APC) and with anti-CD3 antibody in the absence or presence of iTregs with or without addition of sodium chloride. T cell proliferation was determined by the CFSE dilution rates after 3 days of culture. There was no obvious proliferation difference of T responders between normal media and with addition of physiologically relevant 20 mM or 40 mM NaCl (Figure 2A). When iTreg (with different ratios) were added to T responder cells, T cell proliferation was remarkably inhibited and exhibited a dose-dependent effect (Figures 2A and 2B).
Figure 2. Both Fresh iTreg and NaCl-Primed iTregs Possess Suppressive Function In vitro.

(A) Enriched T cells (Teff) were labeled with CFSE, stimulated with soluble anti-CD3, and cultured alone or cocultured with iTregs at various ratios as indicated. The culture system was either in normal media or with indicated increased NaCl concentrations. Baseline indicates no added conditioned iTregs. CFSE dilution was detected by FACS after 3 days. Histograms depict cellular proliferation and are gated on CD8+ cells.
(B) The line graph depicts a summary of experiments at iTreg to Teff ratios as indicated (n = 5).
(C) Naive CD4 cells were differentiated into Tregs as described in STAR Methods, then re-stimulated under the indicated increased NaCl concentrations for another 3 days. Following incubation, iTregs were washed, counted and plated in a suppression assay in normal media as in (A). CFSE dilution was detected by FACS after 3 days. Histograms depict cellular proliferation and are gated on CD8+ cells.
(D) The line graph depicts a summary of experiments at iTreg to Teff ratios as indicated (n = 5).
To exclude the possibility that the time of exposure was insufficient for priming iTreg subset with high salt, we pretreated iTreg with high salt for 3 days and then tested their functional activities as described in Figure 2A. The suppressive ability of iTreg pretreated with high salt was similar to the iTreg that had been cocultured with high salt (Figure 2C). This result was consistent under the conditions of different ratios of iTreg and T responder cells (Figure 2D).
High-Salt Primed iTregs Rescue T Cell-Induced Colitis In vivo
The different effects of high-salt on tTreg and iTreg subsets in vitro prompted us to further study whether the iTreg subset is still resistant to high salt in vivo. We used a colitis model to validate this possibility. As described previously (Chen et al., 2017), CD4+CD25−CD62L+CD44− naive T cells from C57BL/6 mice were transferred into Rag1−/− mice, developing a typical autoimmune intestinal inflammation. Mice gradually lost weight and showed lethargy, piloerection, hunching, and dehydration with a clinical score 2–3 (Figures 3A and 3B). However, when iTreg or high salt-primed iTreg were co-transferred into Rag1−/− mice, body weight improved dramatically; meanwhile, adoptive transfer of high salt-treated iTreg had the same effect on controlling the progress of colitis as normal iTreg (Figure 3A and 3B). Additionally, splenomegaly, intestine swelling colon length, and shaped stool are proven to be useful indicators of the severity of colitis (Chen et al., 2017; Hernandez et al., 2015). Our results showed that model mice had significantly swollen colons, obvious splenomegaly, and less shaped stool compared with iTreg-treated mice. Similarly, high salt-treated iTreg can also markedly alleviate these typical features as normal iTreg, and this was consistent with weight gain (Figures 3C and 3D). Histologically, colitis mice showed mucosal ulceration, with loss of the normal crypt structure and intense goblet cell depletion, together with obvious inflammatory cell infiltration and edema. Matching the above data, no matter whether modified by high salt or not, iTreg identically lowered the pathological index (Figures 3E and 3F). In an additional experiment, we also observed that the iTreg subset maintained a suppressive effect on colitis when mice were fed a high salt diet (HSD) (Figure S3A). Thus, we have further confirmed that iTreg can strongly inhibit T cell-induced colitis, and high salt does not compromise iTreg in vivo function.
Figure 3. Immunosuppressive Activity of NaCl-Primed or Normal iTregs In vivo Is Similar In vitro When Assessed in the Rag1−/− Mice Inflammatory Bowel Disease Model.

iTregs were prepared from CD90.1+ Foxp3−GFP knock-in mice. Naive CD4+ (CD90.2+CD4+CD25−CD62L+CD44−) cells from C57BL/6 mice alone or together with CD90.1+ iTregs were adoptively transferred into Rag1−/− mice intraperitoneally (i.p.). The mice were killed at 6 weeks after the cell transfer and analyzed for disease severity (6 mice in each group in one experiment).
(A) Body weight of the recipient mice was presented as a percentage of the initial weight.
(B) Average clinical scores of each group are shown.
(C) Gross morphology of colons and spleens are shown.
(D) Absolute numbers of total splenocytes were compared.
(E) H&E staining of colon sections. Scale bars, 100 μm. Representative results (mean ± SEM) from four independent experiments are shown.
(F) Histological score. Statistical analyses were performed using one-way ANOVA between respective dietary and adoptive transfer groups. *p < 0.05. **p < 0.01. ns, not significant.
iTreg Subset Primed with or without High Salt Similarly Restrict Inflammatory Responses and Prevent T Cell Migration to the Inflamed Colon
Given that Th17 and Th1 cells play an important role in the pathogenesis of colitis (Burstein and Fearon, 2008), we also analyzed the frequency of these pathogenic cells in lamina propria (LP), spleens (SP), and mesenteric lymph nodes (mLN) at 6 weeks after cell transfer under different interventions. In line with previous reports, substantial amounts of Th17 and Th1 cells were identified in spleens and mesenteric lymph nodes in Rag1−/− mice when naive CD4+ cells were transferred. Co-transfer of iTreg or iTreg that had been pretreated with high salt significantly but similarly inhibited the Th17 and Th1 production in spleens and Th17 production in mLN (Figures 4A and 4B). The percentages and total CD4+ cells in LP were lowered to a greater extent in both Treg infusion groups than in the colitis control group, suggesting that either iTreg or iTreg pretreated with high salt dramatically and similarly suppressed the migration of CD4+ pathogenic cells to the lamina propria (Figures 4C and 4D).
Figure 4. NaCl-Primed or Normal iTregs Inhibit Differentiation of Pathogenic Cells in Inflammatory Colitis and Prevent Pathogenic Cells from Infiltrating to Colon.

(A) Colitis mice were sacrificed at 6 weeks after cell administration, and CD4+ cells from the spleen (SP) and mesenteric lymph nodes (mLN) were examined. Flow cytometric analysis and frequencies of IL-17A+CD90.2+ and IFNγ+CD90.2+ cells were examined in the respective mouse groups (6 mice in each group in one experiment).
(B) The bar graph depicts a summary of the experiment (6 mice in each group in one experiment).
(C) The infiltrating cells in large intestines (LP) were analyzed on total CD4+ T cells.
(D) The bar graph depicts a summary of the experiment (6 mice in each group in one experiment).
(E) On day 35 and 45 after cells transfer, iTreg− Foxp3 loss were compared in SP and LN, cells were gated on CD90.1+. Representative results (mean ± SEM) from four independent experiments are shown.
(F) The line graph depicts a summary of the experiment (6 mice in each group in one experiment). Statistical analyses were performed using one-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
Treg subset functionality is usually associated with Foxp3 expression (Wu et al., 2006). We also studied the in vivo Foxp3 stability of these iTreg subsets pretreated with or without high salt in the colitis model. As shown in Figures 4E and 4F, about 35% of Foxp3 (range from 65% to 30%) was maintained at 6 weeks after iTreg transfer, the Foxp3 was similarly sustained on the iTreg subset that had been pretreated with high salt. High salt also did not promote their conversion to Th1 and/or Th17 cells (Figures S3B–S3D). Additionally, we also observed no difference of Foxp3 expression in colitis or in colitis mice fed with a high salt diet (Figures 4E and 4F), suggesting that high salt did not change the transferred iTreg subset stability in vivo.
In order to explore the possible mechanisms whereby high salt does not change iTreg subset function, we performed a RNA-seq analysis. Purified iTreg subset was restimulated with anti-CD3/CD28 microbeads in the presence of IL-2 with or without high salt for 72 h, and mRNA expression was determined using RNA-seq (Figure S4A). Apart from CCR-6, which was increased around 2.5-fold, almost all mRNA signatures related to immunoregulation and inflammation features of Treg cells are not changed. High salt slightly increased cd274, nt5e, il10, nrp1, and tigit that are related to Treg cell phenotype and function, although it slightly reduced Foxp3 and TGF-β (Figures S4A and S4B). High salt did not significantly change the levels of Th1, Th2, and Tfh transcripts but somewhat decreased IL-17a and IL-17f, consistent with the stability of the iTreg subset in vitro and in vivo, as shown in Figure 5B.
Figure 5. Sodium Chloride Reverts the Suppressive Function of Naturally Occurring Treg Cells but Not TGF-β-Induced Treg Cells In vitro and In vivo.

(A) tTregs isolated from thymus and TGF-β-induced iTregs were stimulated with anti-CD3/28 microbeads (5 cells per bead), IL-2 (50 U/mL) in standard media, or with additional 40 mM NaCl for 72 h, followed by co-cultured with enriched T cells labeled with CFSE in the presence of soluble anti-CD3 (0.025 μg/mL). Baseline indicates no addition of conditioned Treg subsets. CFSE dilution was detected by FACS after 3 days.
(B) Histograms depict cellular suppression and are gated on CD8+ T cells (n = 5).
(C) tTregs and iTregs were prepared from CD90.1+ Foxp3+ GFP knock-in mice and some of them were pre-treated by sodium chloride (40 mM). D90.2+CD4+CD25−CD62L+CD44− naive T cells alone or together with the various Treg subsets were adoptively transferred into Rag1−/− mice via i.p. injection. All mice were sacrificed at 35 days after the cell transfer, weight loss was analyzed for disease severity (6 mice in each group in one experiment). Body weight of the recipient mice was presented as a percentage of the initial weight.
(D) Gross morphology of colons was shown.
(E) The loss of Foxp3-GFP of the four sets were also compared in mLN, cells were gated on CD90.1+ cell. Representative results (mean ± SEM) from three independent experiments are shown.
(F) The bar graph depicts a summary of the experiment (6 mice in each group in one experiment). Statistical analyses were performed using Student’s t test and one-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
SGK1 serves as a critical node in high salt-driving Th17 induction due to prominent IL-17 production, which is also responsible for high salt-mediated loss of suppression in the tTreg subset. It is notable that NaCl-primed iTregs also had slightly elevated SGK1 expression (Figure S4B), and although SGK1 was upregulated in both tTreg and iTreg subsets following high salt priming, the functional consequences in both Treg subsets were different.
FOXO proteins are the chief downstream targets of SGK1, which functions in a Treg cell-intrinsic manner to regulate Foxp3 expression of T regulatory cells. SGK1 is known to inactivate both FOXO1 and FXO3a, two main members of the FOXO protein family, to indirectly reduce tTreg cell activities (Safa et al., 2015). In fact, a recent study has demonstrated an increase of phosphorylation of FOXO1/FOXO3a in tTreg subset under high salt conditions (Hernandez et al., 2015). Interestingly, we did not observe an increase but found a decrease in phosphorylation of FoxO1/FoxO3a in iTreg subset with the high-salt condition (Figures S4C and S4D).
Sodium Chloride Reverts the Suppressive Function of Naturally Occurring tTreg Cells but Not iTreg and pTreg Cells In vitro and In vivo
To exclude the possibility that the different stability and functionality of the tTreg subset reported by others could be attributed to subtle methodological differences, we designed a face-to-face comparison experiment where both tTreg and iTreg subsets were simultaneously analyzed for their phenotype and functionality under a high salt environment. An in vitro experiment was similarly conducted as in Figure 2A, and showed that both tTreg and iTreg cells similarly suppressed the proliferation of CD8+ T responder cells; tTreg pretreated with NaCl significantly diminished their inhibitory activity, while iTreg pretreated with high salt maintained their suppressive ability (Figures 5A and 5B). A similar result was also observed when high salt was added to cell cultures (data not shown). We then used colitis models to further confirm that high salt subverted the therapeutic effect of tTreg on colitis, while the effect of the iTreg subset on colitis was sustained even if they had been pretreated with high salt (Figures 5C and 5D). We further analyzed their Foxp3 expression during colitis. In these experiments, we co-transferred CD90.1+ tTreg or iTreg subsets and CD90.2+ naive CD4+ cells into Rag1−/− mice to identify the biological features of Treg subsets during colitis. At 5 weeks after cell transfer, we found similar levels of Foxp3+ donor cells in both tTreg and iTreg subsets in colitis. However, tTreg cells pretreated with high salt significantly reduced their Foxp3 level (Figures 5E and 5F). Conversely, the iTreg subset had no such change on the Foxp3 expression during colitis (Figures 5E and 5F). These results further validate that both Treg subsets have different biological characteristics in the high salt condition.
High-Salt Does Not Significantly Affect the Development and Function of iTreg Cells in Humans
To consider the clinical relevance of our findings, we extended this study from mouse cells to human cells. Although a difference between mouse and human iTreg cell development appears to exist, recent studies have demonstrated that the addition of all-trans retinoic acid permits induction of human iTreg in the presence of TGF-β (Lu et al., 2010b). Using this protocol, we found that the human naive CD4+ cells differentiated into Foxp3+ cells that have displayed potent in vitro suppression. Interestingly, neither the differentiation nor function of human iTreg cells was reduced by high salt exposure (Figure 6). Thus, the human iTreg subset may be also resistant to high salt.
Figure 6. High Salt Does Not Affect the Differentiation and Function of Human iTreg Cells.

Human naive CD4+CD25−CD45RA+CD45RO− cells were stimulated with anti-CD3/28 microbeads in the presence of rhIL-2 and TGF-β in standard media, or in the presence of an additional 20 or 40 mM NaCl for 5 days.
(A) Foxp3 expression was detected by FACS.
(B) The bar graph depicts a summary of independent experiments in A (n = 5).
(C) T responder cells labeled with CFSE were stimulated with anti-CD3 in the presence of γ-irradiated antigen presenting cells (APCs). iTreg cells or iTregs primed with NaCl were added to culture wells. CFSE dilution and suppressive rates were calculated. Histograms depict cellular proliferation and are gated on CD8+ cells.
(D) The bar graph depicts a summary of independent experiments in (C) (n = 5). Statistical analyses were performed using one-way ANOVA. ns, not significant.
DISCUSSION
Although sodium is an indispensable nutrient and is crucial for cellular function in appropriate amounts, high intake of sodium chloride has wide-ranging effects in renal, cardiovascular, and endocrine diseases (Appel et al., 2011). Interestingly, recent studies have also demonstrated that a high salt diet promotes the onset of multiple sclerosis and other related autoimmune diseases (Krementsov et al., 2015; Zostawa et al., 2017), raising the possibility that high salt affects the immune system.
Initial studies revealed that excessive sodium chloride uptake mainly affects the innate immune system. Machnik et al. (2009) revealed that a high salt diet stimulates macrophages to secrete vascular endothelial growth factor-C to promote inflammation and hypertension. Zhang et al. (2015) reported that high salt increased pro-inflammatory signatures and decreased anti-inflammatory and pro-endocytic molecules in both human and mouse macrophages, eventually leading to inflammation. Subsequent studies provided evidence that high salt also affects the adaptive immune system. Indeed, high salt promotes Th17 cells and aggravates Th17-mediated inflammation and diseases (Kleinewietfeld et al., 2013).
In addition to pro-inflammatory cells and T effector cells, T regulatory cells (Treg) are an integral component of the normal immune system and contribute to the maintenance of peripheral tolerance, which can downregulate immune responses and are essential for immune homeostasis (Horwitz et al., 2008; Lan et al., 2012). Although many Treg subsets have been identified, CD4+Foxp3+ Treg cells are the most important population. These are naturally occurring and develop in the thymus, and therefore are named natural Treg (tTreg) cells (Abbas et al., 2013; Sakaguchi et al., 1995; Shevach, 2009). Although the tTreg cell subset has a potent preventive role in inflammatory diseases, the therapeutic effect of tTreg on established disease is less satisfactory and related to their stability (Lu et al., 2014; Xu et al., 2007; Zheng et al., 2008; Zhou et al., 2010). When the diseases are established, pro-inflammatory cytokines can reduce Foxp3 expression and diminish Treg function. Interestingly, the high salt environment also dramatically diminishes the Treg functionality. Although high salt does not reduce Foxp3 expression and change epigenetic marks, high salt promotes tTreg to convert into aTh1-like phenotype with loss of suppressive activity (Hernandez et al., 2015). However, others have also reported that tTreg cells are notably stable under physiologic and inflammatory conditions in an animal model (Rubtsov et al., 2010). The possible explanation for this difference is that they have used a Th1 inflammation model, whereas our group and others have previously observed that IL-6 and IL-21 rather than IFNγ are crucial pro-inflammatory cytokines to make this conversion (Lu et al., 2014; Xu et al., 2007; Zheng et al., 2008; Zhou et al., 2010).
Our group first reported that CD4+ Treg cells can be induced from the conventional non-Treg cells and this subset has been named induced Treg (iTreg) (Zheng et al., 2002). iTreg subset shares phenotypic characteristics and suppressive activity of tTreg subset (Huter et al., 2008a; Zheng et al., 2004, 2006, 2007), however, some differences have been identified between both subsets. For example, the tTreg subset suppresses immune responses mainly through cell contact although they can also produce TGF-β and IL-10, while the iTreg suppresses in a “classical” contact-dependent manner, as well as by cytokines like TGF-β and IL-10 (Zheng et al., 2004, 2006). Both tTreg and iTreg subsets directly suppress B cell responses but their mechanisms of action are completely different (Iikuni et al., 2009; Xu et al., 2016; Zhao et al., 2006). While tTreg failed to suppress Th17-mediated disease, the iTreg subset can suppress Th17-mediated disease (Huter et al., 2008b). Indeed, inflammatory cytokines such as IL-6 and transforming growth factor α (TNF-α) reduce Foxp3 expression and the function of the tTreg subset (Gao et al., 2015; Xu et al., 2007; Zheng et al., 2008), nonetheless, the iTreg subset seems to be resistant to these pro-inflammatory cytokines (O’Connor et al., 2012; Zheng et al., 2008). In addition to the two Treg subsets described above, others also suggest existence of a third population of Treg cell differentiation in vivo and named them the pTreg subset (Abbas et al., 2013). As pTreg cells at least contaminate the tTreg subset that has immigrated from thymus, this classification needs further study (Josefowicz et al., 2012; Sakaguchi et al., 2008).
However, there is also a controversial argument on the stability of iTreg cells induced ex vivo. Several groups considered the iTreg subset to be unstable because they observed Foxp3 in conserved non-coding DNA sequence (CNS) elements is methylated in iTreg but not in tTreg subset (Floess et al., 2007; Hadaschik and Enk, 2015; Kanamori et al., 2016). There are some reasons that can explain this difference. First, it is not surprising the Foxp3 is turned off in iTreg subset in vitro because these cells are differentiated ex vivo with short-term exposure of TGF-β. However, continuous exposure of low doses of IL-2 and/or TGF-β can sustain Foxp3 expression (Zheng et al., 2007). Second, Foxp3 methylation in CNS is not the only factor that affects Treg function. Many studies have demonstrated iTreg subset can prevent and even treat autoimmune diseases although they could be methylated in Foxp3 CNS. Our previous study also demonstrated that all-trans retinoic acid (atRA) enhances the quantity and quality of development of iTregs, however, atRA significantly increased histone acetylation within the promoter and conserved non-coding DNA sequence (CNS) elements at the Foxp3 gene locus, while DNA methylation in the CNS was not significantly altered (Liu et al., 2015b; Lu et al., 2014). Third, iTreg stability mostly depends upon the methods by which they are induced. When high levels of TCR stimulation were used, Foxp3 expression occurs rapidly but is not easily sustained (Koenecke et al., 2009), however, suboptimal TCR stimulation and appropriate doses of IL-2 can overcome this problem (Gu et al., 2014; Zheng et al., 2007). Fourth, Foxp3 is not the only factor that contributes to Treg function. Some reports show that cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) plays an important role in T cell-mediated dominant immunologic self-tolerance (Takahashi et al., 2000; Wing et al., 2014), and CTLA-4 could function in the Foxp3-independent non-Treg compartment (Walker, 2013). Others have also observed protective human TGF-β-iTregs that exhibit methylated Foxp3 (Hippen et al., 2011). Moreover, CD103+ iTreg suppress immune responses independent of Foxp3 expression (Liu et al., 2014). Furthermore, high salt did not affect Foxp3 level but did reduce its function (Hernandez et al., 2015). Our previous work also demonstrated that iTreg exert their suppressive activity through releasing active TGF-β and IL-10 (Horwitz et al., 2003; Zheng et al., 2004).
Now, we provide an important line of evidence that the iTreg subset may be more adaptable to environmental change. Our results reveal that a high salt did not affect the development and function of iTreg cell in both mouse and human cells in vitro. High salt also does not reduce the differentiation and functionality of the iTreg subset in the mouse in vivo. Additionally, high salt does not promote the iTreg subset to convert into Th1- or Th17-like cells, displaying a complete difference between iTreg and tTreg subsets. This cannot be explained by the nonresponse of CD4+ cells to high salt, because high salt promoted Th17 cell differentiation, and RNA-seq displayed the changed RNA signatures of high salt-primed CD4+ cells.
SGK1 activation leads to downstream phosphorylation of FoxO1/FoxO3a, resulting in Foxp3 degradation and reduction of Treg stability (Hernandez et al., 2015). Interestingly, pFoxO1 expression by the iTreg subset is not increased as in the tTreg subset when exposed to high salt in this study. In fact, high salt even decreases pFoxO1, and thus high salt probably increases the functional activity of the iTreg subset in the high salt environment. The mechanisms of different responses of tTreg and iTreg subsets to high salt deserves further study. However, the high salt diet has significantly promoted disease activity in autoimmune and inflammatory conditions, so it is likely that high salt mainly promotes the activities of macrophages and T effector cells, including Th17 cells, and interferes with function of tTreg subsets (Wu et al., 2013; Zhang et al., 2015).
Taken together, our findings add further evidence to show that TGF-β-induced CD4+Foxp3+ regulatory T cells are somehow different from natural CD4+Foxp3+ regulatory T cells, not only in the pro-inflammatory milieu, but also in high salt conditions. Thus, manipulation of iTreg as therapy may provide a superior approach to combat autoimmune and inflammatory diseases in complicated environmental conditions.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be addressed to and will be fulfilled by the Lead Contact, Song Guo Zheng (szheng1@pennstatehealth.psu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and donors
C57BL/6 (B6), C57BL/6-Foxp3GFP and B6 Rag1−/− mice were purchased from Jackson Laboratory. All mice were maintained in a single specific pathogen-free room in the animal facility at the Sun Yat-sen University or the Penn State Hershey College of Medicine. Sex-matched 6- to 12-week-old mice were used in all experiments. All the experimental procedures were approved by the Sun Yat-sen University and the Penn State Hershey College of Medicine Institutional Animal Care and Use Committee. All protocols that involved healthy human blood donors were approved by the third affiliated hospital at Sun Yat-Sen University. Informed consent was obtained from recruited healthy volunteers.
METHOD DETAILS
In vitro polarization and proliferation
tTregs were sorted from thymus in C57BL/6 mice or Foxp3GFP C57BL/6 mice by gating on CD4+CD25high or CD4+GFP+ cells. tTregs cells were stimulated with anti-CD3/CD28 microbeads (1 bead: 5 cells; GIBCO) and IL-2 (50 U/ml; R&D) in the presence or absence of high salt (NaCl 40mM) for 3 to 7 days. IL-2 was renewed every other day. The percentage of Foxp3 was examined by flow cytometry. Naive CD4+CD25−CD62L+ CD44− T cells were isolated from spleen cells of C57BL/6 and C57BL/6 Foxp3GFP mice using naive CD4+ T cell isolation kit (Miltenyi Biotec, Auburn, CA). Naive cells were > 95% pure as verified by CD4-Percp-Cy5.5, CD25-PE and CD62L-APC. To generate CD4+ induced regulatory T cells (iTregs), naive CD4+ cells (3 × 105) were cultured in 96-well plates and stimulated with anti-CD3/CD28 microbeads (5 cells per bead, GIBCO), IL-2 (50 U/ml; R&D) ± TGF-β (2 ng/ml; R&D) for 3 days in X-VIVO 15 medium (LONZA) in the presence or absence of an additional 20 or 40 mM NaCl (Sigma-Aldrich). For iTreg cells proliferation, iTregs were differentiated from naive CD4+ T cells, then removed beads, washed the cells, and re-stimulated with anti-CD3/CD28 microbeads (5 cells per bead) in fresh standard media with or without additional of NaCl for another 3 days. Cells were harvested for intracellular staining of IL-17A and IFNγ.
Suppression assay in vitro
To examine the suppressive activity of iTregs and tTreg as described in Zheng et al. (2004), enriched T cells (3 × 105) were stained with carboxyfluorescein succinimidyl ester (CFSE) at 2 μM and cultured with Treg subsets at various ratios as indicated. Cells mixtures were stimulated with soluble anti-CD3 (0.025 μg/ml) in the presence of APCs with or without an additional 40 mM NaCl for 3 days and analyzed by FACS. In addition, both Treg subsets were pre-incubated with or without NaCl (40 mM) in the presence of anti-CD3/CD28 microbeads (5 cells per bead), IL-2 (50 U/ml) for 3 days and then washed the cells. These iTregs were re-cultured with CFSE-T cells for an additional three days. The proliferative levels of CFSE-CD8+ cells were judged by the rates and intensity of CFSE dilution measured with flow cytometry.
Generation of human iTregs ex vivo
Human PBMCs were prepared from heparinized venous blood by Ficoll- Hypaque density gradient centrifugation. CD4+CD25−CD45RA+CD45RO− T cells were sorted from PBMCs (> 98% purity). These cells (2×105) were cultured in 96-well plates and stimulated with anti-human CD3/CD28 beads (10 cells per bead), rhIL-2 (50 U/ml; R&D Systems) and rhTGF-β (5 ng/ml) in X-VIVO 15 medium (LONZA) in the presence or absence of an additional 20 or 40 mM NaCl (Sigma-Aldrich) for 5 days. Foxp3 expression was determined by flow cytometry assay. The human iTreg suppression assay was similarly assessed as described above.
Flow cytometry
The following fluorescence conjugated mouse antibodies were used for flow cytometric analysis from Biolegend (San Diego, CA): Percp-Cy5.5-CD4 (GK1.5), PE-CD25 (PC61), PE-CD62L (MEL-14). Cell subsets were stained with mAbs and isotype control indicated above, and analyzed on a FACS Calibur flow cytometer. For intracellular staining of cytokines, cells were stimulated with PMA (0.25 μg/ml), ionomycin (0.25 μg/ml) for five hours and brefeldinA (5 μg/ml) for four hours at 37°C, and then permeabilized and stained with IFNγ (XMG1.2) and IL-17A (TC11–18H10.1) antibodies. Data were acquired on FACS and analyzed with flowJo software.
Mice; adoptive transfer colitis
For T cell-induced colitis, methods were followed as outlined previously (Chen et al., 2017). iTreg and tTreg subsets were prepared from CD90.1+ Foxp3GFP mice. Naive CD4+ (CD90.2+CD4+CD25−CD62L+CD44−) cells from C57BL/6 mice alone or together with CD90.1+ iTregs or tTregs (with or without 40 mM NaCl pretreatment) were adoptively transferred into Rag1−/− mice, and weight loss was monitored. The mice were killed at 5 or 6 weeks after the cell transfer, and analyzed for disease severity as described (Hernandez et al., 2015).
RNA-seq
Total RNA was extracted with the RNeasy mini kit (QIAGEN). The RNA degradation and contamination was monitored on 1% agarose gels, and its quality were assessed using the NanoPhotometer spectrophotometer (IMPLEN) and RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies). cDNA library construction and Illumina sequencing were completed by Beijing Novogene Bioinformatics Technology Co., Ltd. Briefly, sequencing libraries were generated using NEBNext Ultra RNA Library Prep Kit following manufacturer’s recommendations. PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The library preparations were sequenced on Illumina Hiseq 2000 platform, and 125 bp/150 bp paired-end reads were generated.
Western Blotting
iTreg cells were re-stimulated with anti-CD3/CD28 microbeads (5 cells per bead) and IL-2 (50 U/ml) and culture for indicated times prior to being harvested. Anti-GAPDH (#5174) and anti-phospho-FoxO1(Thr24)/FoxO3a (Thr32) (#9464) were obtained from Cell Signaling Technology. Primary antibodies were detected by secondary anti-rabbit-HRP conjugated (#7074, Cell Signaling Technology) antibodies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism (GraphPad Software). Data were analyzed by Student’s t test in case of two groups and one-way ANOVA analysis was performed for three and more groups in mice studies. Data are presented if not indicated elsewhere as Mean ± SEM. A value of p < 0.05 was considered to be statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Percp/cy5.5 anti-mouse CD4 antibody | Biolegend | Cat# 100540; RRID: AB_893326 |
| PE anti-mouse CD25 antibody | Biolegend | Cat# 101904; RRID: AB_312847 |
| APC anti-mouse CD62L antibody | Biolegend | Cat# 104412; RRID: AB_313099 |
| PE/Cy7 anti-mouse/human CD44 antibody | Biolegend | Cat# 103030; RRID: AB_830787 |
| LEAF Purified anti-mouse CD3ε antibody | Biolegend | Cat# 100331; RRID: AB_1877073 |
| PE anti-mouse IL17A antibody | Biolegend | Cat# 506904; RRID: AB_315464 |
| APC anti-mouse IFNγ antibody | Biolegend | Cat# 505810; RRID: AB_315404 |
| APC anti-mouse CD8a antibody | Biolegend | Cat# 100712; RRID: AB_312751 |
| GAPDH (D16H11) Rabbit mAb | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| Phospho-FoxO1(Thr24)/FoxO3a(Thr32) Antibody | Cell Signaling Technology | Cat# 9464; RRID: AB_329842 |
| Anti-Rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IL-2 Protein | R&D | Cat# 202-IL-050 |
| Recombinant Human TGF-β1 protein | R&D | Cat# 240-B |
| Sodium chloride solution | Sigma Aldrich | Cat# S5150 |
| Brefeldin A | Sigma Aldrich | Cat# B7651–5MG |
| Ionomycin calcium salt | Sigma Aldrich | Cat# I0634–1MG |
| Phorbol 12-myristate 13-acetate | Sigma Aldrich | Cat# P8139–1MG |
| Dynabeads Mouse T-activator CD3/28 | Thermo Fisher | Cat# 11453D |
| Dynabeads Human T-activator CD3/28 | Thermo Fisher | Cat# 11132D |
| X-VIVO15 | LONZA | Cat# 04–744Q |
| Critical Commercial Assays | ||
| CellTrace CFSE Cell proliferation kit | Thermo Fisher | Cat# C34554 |
| Naive CD4+ T cell isolation kit, mouse | Miltenyi | Cat# 130–104–453 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse: B6-Foxp3 GFP | The Jackson Laboratory | Cat# 006772 |
| Mouse: Rag1 KO | The Jackson Laboratory | Cat# 002216 |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Sequence data, analyses. | This paper | N/A |
Highlights.
High salt restrains thymic regulatory T cell (tTreg) cell function
High salt does not affect induced Treg (iTreg) cell function
High salt promotes Th17 development via SGK1
ACKNOWLEDGMENTS
This study was supported by grants from the National Key R&D Program of China (2017YFA0105801 to Y.L.), NIH (R01 AR059103, R61 AR073 409, and NIH Star Award to S.G.Z.), the Zhujiang Innovative and Entrepreneurial Talent Team Award of Guangdong Province (2016 ZT 06S 252 to Y.L.), the National Natural Science Foundation of China (30972951 and 81671611 to Y.X.), the Natural Science Foundation of Guangdong Province (2014A030308005 to Q.F.), the Department of Science and Technology in Guangzhou City, and the Department of Science and Technology in Guangdong Province (to Q.F.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.066.
REFERENCES
- Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, Jiang S, Kuchroo VK, Mathis D, Roncarolo MG, et al. (2013). Regulatory T cells: recommendations to simplify the nomenclature. Nat. Immunol 14, 307–308. [DOI] [PubMed] [Google Scholar]
- Appel LJ, Frohlich ED, Hall JE, Pearson TA, Sacco RL, Seals DR, Sacks FM, Smith SC Jr., Vafiadis DK, and Van Horn LV (2011). The importance of population-wide sodium reduction as a means to prevent cardiovascular disease and stroke: a call to action from the American Heart Association. Circulation 123, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Burstein E, and Fearon ER (2008). Colitis and cancer: a tale of inflammatory cells and their cytokines. J. Clin. Invest 118, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Xu Z, Zheng Y, Wang J, Qian W, Olsen N, Brand D, Lin J, and Zheng SG (2017). A protocol to develop T helper and Treg cells in vivo. Cell. Mol. Immunol 14, 1013–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curotto de Lafaille MA, and Lafaille JJ (2009). Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30, 626–635. [DOI] [PubMed] [Google Scholar]
- Ding ZC, Blazar BR, Mellor AL, Munn DH, and Zhou G (2010). Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood 115, 2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farez MF, Fiol MP, Gaitán MI, Quintana FJ, and Correale J (2015). Sodium intake is associated with increased disease activity in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 86, 26–31. [DOI] [PubMed] [Google Scholar]
- Farquhar WB, Edwards DG, Jurkovitz CT, and Weintraub WS (2015). Dietary sodium and health: more than just blood pressure. J. Am. Coll. Cardiol 65, 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al. (2007). Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, Wu Q, Chen Z, Gao Z, Fan H, et al. (2015). Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc. Natl. Acad. Sci. USA 112, E3246–E3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu L, Chen M, Xu L, Lan Q, Li Q, Liu Z, Chen G, Wang P, Wang X, et al. (2014). TGF-β-induced CD4+Foxp3+ T cells attenuate acute graft-versus-host disease by suppressing expansion and killing of effector CD8+ cells. J. Immunol 193, 3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadaschik EN, and Enk AH (2015). TGF-b1-induced regulatory T cells. Hum. Immunol 76, 561–564. [DOI] [PubMed] [Google Scholar]
- He FJ, and MacGregor GA (2009). A comprehensive review on salt and health and current experience of worldwide salt reduction programmes. J. Hum. Hypertens 23, 363–384. [DOI] [PubMed] [Google Scholar]
- Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, and Hafler DA (2015). Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Invest 125, 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, June CH, Miller JS, Wagner JE, and Blazar BR (2011). Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant 11, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz DA, Zheng SG, and Gray JD (2003). The role of the combination of IL-2 and TGF-beta or IL-10 in the generation and function of CD4+ CD25+ and CD8+ regulatory T cell subsets. J. Leukoc. Biol 74, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz DA, Zheng SG, and Gray JD (2008). Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 29, 429–435. [DOI] [PubMed] [Google Scholar]
- Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, and Shevach EM (2008a). TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur. J. Immunol 38, 1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, and Shevach EM (2008b). Cutting edge: antigen-specific TGF beta-induced regulatory T cells suppress Th17-mediated autoimmune disease. J. Immunol 181, 8209–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iikuni N, Lourenço EV, Hahn BH, and La Cava A (2009). Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J. Immunol 183, 1518–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M, Nakatsukasa H, Okada M, Lu Q, and Yoshimura A (2016). Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 37, 803–811. [DOI] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, and Hafler DA (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, Huehn J, Ganser A, Förster R, and Prinz I (2009). Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur. J. Immunol 39, 3091–3096. [DOI] [PubMed] [Google Scholar]
- Krementsov DN, Case LK, Hickey WF, and Teuscher C (2015). Exacerbation of autoimmune neuroinflammation by dietary sodium is genetically controlled and sex specific. FASEB J. 29, 3446–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Q, Fan H, Quesniaux V, Ryffel B, Liu Z, and Zheng SG (2012). Induced Foxp3(+) regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J. Mol. Cell Biol 4, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, O’Shea JJ, and Fowler DH (2012). STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 37, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lan Q, Lu L, Chen M, Xia Z, Ma J, Wang J, Fan H, Shen Y, Ryffel B, et al. (2014). Phenotypic and functional characteristic of a newly identified CD8+Foxp3-CD103+ regulatory T cells. J. Mol. Cell Biol 6, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, and Germain RN (2015a). Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZM, Wang KP, Ma J, and Guo Zheng S (2015b). The role of all-trans retinoic acid in the biology of Foxp3+ regulatory T cells. Cell. Mol. Immunol 12, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Wang J, Zhang F, Chai Y, Brand D, Wang X, Horwitz DA, Shi W, and Zheng SG (2010a). Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J. Immunol 184, 4295–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Zhou X, Wang J, Zheng SG, and Horwitz DA (2010b). Characterization of protective human CD4CD25 FOXP3 regulatory T cells generated with IL-2, TGF-b and retinoic acid. PLoS ONE 5, e15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, Wang J, Chen M, Liu Y, Shen Y, et al. (2014). Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. USA 111, E3432–E3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, et al. (2009). Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med 15, 545–552. [DOI] [PubMed] [Google Scholar]
- McGuire S (2010). Institute of Medicine. 2010. Strategies to reduce sodium intake in the United States Washington, DC: The National Academies Press; Adv. Nutr 1, 49–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor RA, Floess S, Huehn J, Jones SA, and Anderton SM (2012). Foxp3+ Treg cells in the inflamed CNS are insensitive to IL-6-driven IL-17 production. Eur. J. Immunol 42, 1174–1179. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, and Jones RG (2013). Autoimmunity: rubbing salt in the wound. Nature 496, 437–439. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa K, Ohori S, Borges TJ, Uehara M, Batal I, Shimizu T, Magee CN, Belizaire R, Abdi R, Wu C, et al. (2015). Salt accelerates allograft rejection through serum- and glucocorticoid-regulated kinase-1-dependent inhibition of regulatory T cells. J. Am. Soc. Nephrol 26, 2341–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol 6, 345–352. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, and Toda M (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol 155, 1151–1164. [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Shevach EM (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645. [DOI] [PubMed] [Google Scholar]
- Sundström B, Johansson I, and Rantapää-Dahlqvist S (2015). Interaction between dietary sodium and smoking increases the risk for rheumatoid arthritis: results from a nested case-control study. Rheumatology (Oxford) 54, 487–493. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, and Sakaguchi S (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med 192, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LS (2013). Treg and CTLA-4: two intertwining pathways to immune tolerance. J. Autoimmun 45, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cui Y, Shen J, Jiang J, Chen S, Peng J, and Wu Q (2012). Salt-sensitive hypertension and cardiac hypertrophy in transgenic mice expressing a corin variant identified in blacks. Hypertension 60, 1352–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing JB, Ise W, Kurosaki T, and Sakaguchi S (2014). Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 41, 1013–1025. [DOI] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387. [DOI] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, and Kuchroo VK (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Fuss I, and Strober W (2007). Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol 178, 6725–6729. [DOI] [PubMed] [Google Scholar]
- Xu A, Liu Y, Chen W, Wang J, Xue Y, Huang F, Rong L, Lin J, Liu D, Yan M, et al. (2016). TGF-β-induced regulatory T cells directly suppress B cell responses through a noncytotoxic mechanism. J. Immunol 196, 3631–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, et al. (2015). High salt primes a specific activation state of macrophages, M(Na). Cell Res. 25, 893–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DM, Thornton AM, DiPaolo RJ, and Shevach EM (2006). Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 107, 3925–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, and Horwitz DA (2002). Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25-precursors. J. Immunol 169, 4183–4189. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang JH, Gray JD, Soucier H, and Horwitz DA (2004). Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J. Immunol 172, 5213–5221. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, and Horwitz DA (2006). TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J. Immunol 176, 3321–3329. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Wang P, Gray JD, and Horwitz DA (2007). IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol 178, 2018–2027. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang J, and Horwitz DA (2008). Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J. Immunol 180, 7112–7116. [DOI] [PubMed] [Google Scholar]
- Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, and Zheng SG (2010). Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J. Immunol 185, 2675–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zostawa J, Adamczyk J, Sowa P, and Adamczyk-Sowa M (2017). The influence of sodium on pathophysiology of multiple sclerosis. Neurol. Sci 38, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.