

ABSTRACT
The CRISPR-Cas9 system has become the most promising and versatile tool for genetic manipulation applications. Albeit the technology has been broadly adopted by both academic and pharmaceutic societies, the activity (on-target) and specificity (off-target) of CRISPR-Cas9 are decisive factors for any application of the technology. Several in silico gRNA activity and specificity predicting models and web tools have been developed, making it much more convenient and precise for conducting CRISPR gene editing studies. In this review, we present an overview and comparative analysis of machine and deep learning (MDL)-based algorithms, which are believed to be the most effective and reliable methods for the prediction of CRISPR gRNA on- and off-target activities. As an increasing number of sequence features and characteristics are discovered and are incorporated into the MDL models, the prediction outcome is getting closer to experimental observations. We also introduced the basic principle of CRISPR activity and specificity and summarized the challenges they faced, aiming to facilitate the CRISPR communities to develop more accurate models for applying.
KEYWORDS: CRISPR-Cas9, machine learning, on-target, off-target, predicting models, features
1. Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) is an adaptive immune system found in bacteria and archaea, which was harnessed for programmable and precise gene editing in 2012 [1,2]. It mediates cleavage the invading DNA by the RNA-guided DNA endonuclease Cas9 [3,4]. There are two key components in the CRISPR-Cas9 gene editing system: a small guide RNA (gRNA) and a Cas9 endonucleases [5, Martin 4]. The gRNA is a chimeric RNA consisting of a tracrRNA and a crRNA, of which the crRNA contains a guide (spacer) sequence [6] that precisely directs the Cas9 protein to the corresponding target site in the genome. Another important feature of the CRISPR-Cas9 system is the protospacer adjacent motif (PAM), which is a CRISPR-dependent and conserved DNA sequence motif adjacent to the target site (protospacer), and are used by the endogenous CRISPR in bacteria to distinguish self and invading DNAs [7–12].
The Cas9 protein has two nuclease domains: RUVC and HNH, which cleaves the non-complementary and complementary strand respectively upon CRISPR gene editing [14–16]. The resulting consequence is a double-stranded DNA break (DSB) that will be generated at the target site. DSBs are detrimental for cells if leaving unrepaired as this will lead to chromosomal abnormality. Mammalian cells have thus developed several DNA repair pathways: with nonhomologous-mediated end joining (NHEJ) and homology-directed repair (HDR) as the two major pathways for DSBs repair [17] (Figure 1). Although CRISPR-Cas9 gene editing is becoming one of the routinely used methods for genetic perturbation applications, one general question that almost all applications will encounter is that how to select the optimal gRNA with high activity and specificity. Briefly speaking, an effective CRISPR gene editing application depends on the choice of the best gRNA target site (or guide sequence), the best delivery method, and introducing the right genetic modification after DSB repair [18].
Figure 1.
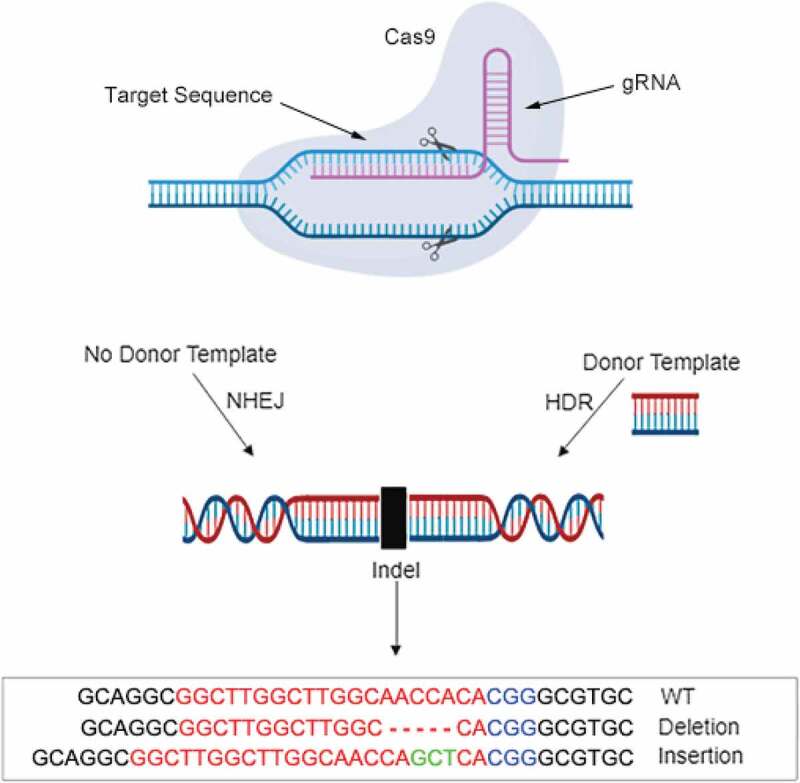
The mechanism of CRISPR-Cas9 genome editing system. Briefly, RNA-guide nuclease (RNG) is introduced into the organism, and the gRNAs are targeted to the target sequences after recognition by the PAM sequence. Two main repair methods are HDR and NHEJ, the application of which is depended on whether there is a donor sequence. After that, indels are induced for maintaining the liveness of cells. The regions complementary to the gRNA variable region are coloured in red in the bottom box. The insertion regions and PAM sequences are marked in green and blue, respectively. Short dash line represents the deletion region.
Over the past several years, several CRISPR activity (on-target) and specificity (off-target) scoring algorithms and in silico gRNA designing web tools have been developed to facilitate the design of CRISPR gRNAs and experiments [19]. All these in silico gRNA design and off-target prediction tools have dramatically facilitated the broad applications and success of CRISPR gene editing technologies. For a noncomprehensive overview of all these CRISPR designing tools, please refer to a recent review by Guo-hui Chuai et al [20]. In this review, we concentrate on algorithms which use machine and deep learning (MDL) methods for streamlining CRISPR design. We compared and evaluated the processing of data, algorithm characteristics, selection of features of all the MDL-based CRISPR designing tools. And finally, by analysing all the pros and cons of currently available MDL algorisms for CRISPR activity and specificity design, we suggested future improvements that should be taken into consideration to develop the next generation of MDL-based CRISPR designing tools.
2. The basic principle of CRISPR activity and specificity
To carry out a successful CRISPR gene editing study, optimal gRNAs should be firstly selected, which means choosing gRNAs with both high on-target efficiency and low (no) off-target activity [21]. The CRISPR/Cas9 system functions with a principle that once the gRNA forms a complementary base pairing (R loop) with the target site, the Cas9 endonuclease activity is activated and introduces a DSB to the target site [22–24]. The DSB is subsequently repaired by endogenous DNA repair mechanisms and the introduction of changes (or indels) at the DSB site can be captured by various methods, such as surrogate reporter vectors, T7E1 assay, TIDE, ICE, and deep sequencing [25,26]. Statistical quantification of the percentage of indels is the most broadly used measurement for the activity and specificity of CRISPR. Multiple studies have found that the CRISPR-Cas9 activity varies significantly among different gRNAs [4,27].Previously, we discovered that the gene editing activity was affected by several factors, such as the guide sequences secondly structure and chromatin accessibility [28]. Using a dual-fluorescence surrogate reporter system [29], we also discovered that through fine-tuning the DSB repair pathway, several variants of recombinant Cas9 proteins are generated to enhance DSB repair by MMEJ [30] or by HDR (SpCas9-KRAB, submitted for publication). The development of in silico gRNA designing web tools, such as CRISPOR [31], ChopChop [32], and Cas-Designer [33,34], as well as algorisms for prediction of gRNA activity, have greatly facilitated the application and improvement of CRISPR-Cas9 gene editing technologies.
Compared to CRISPR activity prediction, confidentially and precisely predicting the CRISPR gRNA off-target effect is more challenging. The potential and un-invertible off-target effect caused by CRISPR is the most frequently raised concern and impedes clinical applications of CRISPR [35]. Since it is the first invention of RNA-guide CRISPR gene editing technology, great efforts have been made to understand the mechanisms causing CRISPR off-targets and significant improvements have been achieved. One major cause of the CRISPR off-target is that the minimum mismatches (up to 3nt) between the gRNA spacer and the off-target site are tolerated [36]. Forming the R loop among the gRNA, Cas9 and the target site is essential for activating the nuclease activity [37]. From a molecular and physical energy point of view, the R loop requires to reach a minimum energy level (Nmin) to accomplish the activation of Cas9 nuclease activity and gene editing. The Nmin comes from the DNA:RNA Watson-Crick base-pairing between the target DNA and the gRNA spacer, binding of the Cas9 to DNA, binding of the Cas9 to the PAM, interaction between Cas9 and the gRNA scaffold, local chromatin status of the target site, and many unrevealed factors. Based on the Nmin theory, many improvements have been made to increase CRISPR specificity, such truncated gRNAs with shorter spacer sequences [38–41], Cas9 variants (eSpCas9, SpCas9-HF, SpCas9-HF1) with neutral amino acids to the DNA binding domain, modified gRNA scaffolds. Additionally, titrating the amount of Cas9 and gRNA delivered [6,42], combining catalytically inactive Cas9 with Fokl nuclease domain (fCas9) [43,44] together with combining a Cas9 nickase mutant with pair gRNAs [103] can also increase the CRISPR specificity but via other mechanisms than Nmin.
Although the PAM is highly conserved for each Cas9 ortholog, which means specific Cas9 protein will specifically targets to according sites, considerable but lower cleavage activity was observed for alternative PAMs though. For example, the sole PAM of SpCas9 protein is a 3ʹPAM (protospacer preceding NGG). However, the SpCas9 also shows significant but lower activity for NAG and NGA PAM in comparison to the NGG PAM [45]. The underlying mechanism is that the Cas9 protein contains a PAM interaction domain (PID) that is specifically selected to recognize one PAM sequence. However, to keep the possibility of adaptation to newly evolved phases (bacterial viruses), the PID still retains its evolution feature and amino acid changes to the PID for recognizing different PAMs. Taking advantages of this mechanism, several Cas9 variants, i.e., xCas9, Cas9-VQR/EQR, and Cas9-VRER, have been generated to broaden the PAM specificities [46,47].
3. An overview of CRISPR gRNA design tools
Currently, three types of CRISPR designing tools have been developed based on experimental and simulated data: (i) Alignment-based, of which the CRISPR guide sequences (spacers) are simply retreated based on mapping PAM sequences in the genome; (ii) Hypothesis-driven, of which the gRNA activity is predicted based on the specific features such as GC content; and (iii) Machine and Deep Learning (MDL)-based, of which the gRNA activity score is predicted with algorisms trained with big datasets of CRISPR KO experiments conducted in different cell types [20]. As increasing CRISPR gene editing datasets are generated by the global CRISPR community, data-driven MDL-based methods have become the key choice for predicting CRISPR gRNA activity and specificity. For instance, comparing to the experimentally off-target detect methods of GUIDE-seq [48], HTGTS [49], BLESS [50] or IDLV [51], the MDL-based prediction methods built on experimental datasets are more efficient and cost-effective.
However, all current prediction models have four major problems: (1) Data insufficiency. Machine learning models outperform other methods owing to the data-driven mechanism, but they cannot predict the unseen data efficiently unless there are enough data for extracting features thoroughly. (2) Unclear mechanism. The mechanism of the CRISPR-Cas9 gene system has not been comprehensively explored and restrict the features used in the current state-of-the-art algorithms. With features not well representing the mechanism of the CRISPR-Cas9 systems, MDL-based methods can hardly achieve ground-breaking improvements with sufficient data. Some crucial features are even lacking, such as the local chromatin state that affects Nmin in specific cell types. Although the deep neural network (DNN) may automatically extract features, it is still required to functionally validate these DNN-predicted features and their importance for CRISPR functions. (3) Data heterogeneity. Datasets generated from different platforms and cell types need to be integrated for data augment. (4) Last but not least, data imbalance. Most frequently, the number of off-target sites detected by whole-genome high throughput sequencing is significantly less than that identified by prediction software (like Cas-OFFinder).
4. On-target activity prediction
A number of MDL-based methods have been developed to predict CRISPR on-target activity (Table 1), which can be roughly classified into two categories, (1) Machine learning based, including sgRNA Designer [52], sgRNA Scorer [53], CRISPR-Scan [54], SSC [55], and CRISPRater [56]. However, most of these models cannot be intuitively explained. Theoretically, the computational processes an interpretable model could be repeated by other groups (‘simulatability’) with a full understanding of the algorithm (‘algorithmic transparency’). Furthermore, every part of the model should have an intuitive explanation (‘decomposability’) [57]. For instance, CRISPRater, CRISPRScan, SSC are trained by a simple linear model (Table1), Azimuth1.0 was trained by generalized linear models Logistic Regression. These models are the most easily interpretable models. These linear models can easily to be trained, and users can run the trained models rapidly, also, it is suitable to be applied to a large scale of sgRNA predictions, but it has limitations to process the non-linear relation of features. TUSCAN, an user-friendly model trained by random forest, is explainable and it does not need the normalization or parameter tuning steps, but it performs poorly when the features grow rapidly. Other models trained by SVM (Support Vector Machine), which works slow for data with large volume, and the GBRT (Gradient Boost Regression Tree), which can process different features naturally, cannot be interpreted as we do not know the training processes precisely. However, almost all these models are benefited from the large-scale library generation but modest performance for individual gRNA/target design [56]. Also, the process of feature selection is labour-intensive and acquires specialized validation for model developers. For instance, some features influent the sgRNA efficiency have been reported by other groups, including second structure, epigenetics, and physicochemical property of sequences, which could be regarded as an important feature added to the feature matrix. In fact, almost all of these features were manually curated rather than extracted automatically by machines. More importantly, generalization is a common drawback of these models, namely a model only performs well in a specific dataset (always the training dataset), but not in a new testing dataset [31]. (2) Deep learning based. Over the last several years, researchers have successfully applied deep learning techniques in the CRISPR design. DeepCas9 [58], DeepCRISPR [59], DeepCpf1 [60], and CRISPRCpf1 [61] using the CNN (convolution neural network) to predict the sRNA activity based on the automatic recognition of sequence features. The greatest strength of deep learning is that its complex structure of neural network allows identifying important features automatically. But the feature extraction step resembles a black box making it difficult to functionally validate the features [62]. In addition, current public datasets have only tens of thousands of human cells. Although we can adopt data amplification methods to artificially expand the data, the real information of the data may be masked as the granularity of the data is refined, and it is difficult to achieve millions of data as Google and other group did [63–67]. Hence, publicly available on-target data are still insufficient for building up a powerful deep learning model. Current on-target datasets can be accessed on their website, https://github.com/maximilianh/crisporPaper/tree/master/effData [31].
Table 1.
Publicly available tools for gRNA on-target prediction.
| On-target software | Model | Ref | PAM | URL |
|---|---|---|---|---|
| DeepCRISPR | CNN1 | [59] | NGG | http://www.deepcrispr.net/ |
| DeepCpf1 | CNN | [61] | TTTN | http://deepcrispr.info/ |
| DeepCas9 | CNN | [58] | NGG | https://github.com/lje00006/DeepCas9 |
| CRISPRater | L1-reg2 | [56] | NGG | https://crispr.cos.uni-heidelberg.de/ |
| WU-CRISPR | SVM3 | [96] | NGG | http://crispr.wustl.edu/ |
| SgRNAScorer | SVM (C)6 | [68] | NGG, NAG, NNAGAAW, NNNNGMTT |
https://crispr.med.harvard.edu/sgRNAScorerV2/ |
| TUSCAN | RF4 | [98] | NGG | https://github.com/BauerLab/TUSCAN |
| SSC | Elastic Net | [55] | NGG | http://crispr.dfci.harvard.edu/SSC/ |
| CRISPRScan | Linear reg | [54] | NGG | http://www.crisprscan.org/?page=welcome |
| TSAM | GBRT5+ SVM | [75] | NGG | http://www.aai-bioinfo.com/CRISPR |
| Azimuth1.0 | Logistic reg | [89] | NGG | no available |
| Azimuth2.0 | GBRT | [52] | NGG | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| CRISPRpred | SVM | [69] | NGG | https://github.com/khaled-buet/CRISPRpred |
| ge-CRISPR | SVM | [70] | NGG | http://bioinfo.imtech.res.in/manojk/gecrispr/ |
1. CNN: Convolution Neural Network.
2. L1-Reg: L1-Regression.
3. SVM: Support Vector Machine.
4. RF: Random Forest.
5. GBRT: Gradient Boost Regression Tree.
6. SVM (C): using SVM to classify (+1 represent high activity, −1 represent low-activity).
The accuracy of these gRNA activity prediction tools implemented in different cell types and different species is still not clear [20]. Because of the high variabilities among species, species-specific software has been developed, such as fryCRISPR for Drosophila [71], CRISPR-P for plant [72,73], CRISPRscan for zebrafish [54], and EuPaGDT for pathogens [74]. Among them, only CRISPRscan was developed based on machine learning, whereas the others are hypothetically driven software. Notably, most of these algorithms were designed with the rule sets derived from human and mouse datasets, which would result in severe overfitting problem [56].
5. Off-target prediction
Previous studies have found that the off-target sites of the CRISPR-Cas system are not random [52,13]. In this review, we used five sets of benchmarks to calculate the mutation frequency and their bases preference in each position among the spacer sequence of gRNAs. Similarly, we found that the mutation at the 5ʹ end was more likely to be active, while in the active off-target, the A to C mutation at the 8th position was more likely to occur (Figure 2). This observation partially explains that the off-target activity would be decreased when the 5ʹ-end of gRNA was truncated [76–78]. In other words, if we truncate the length of the gRNA, especially the 5ʹ-end, the off-target activity will be reduced.
Figure 2.

Heatmap of the percent activity value in each position. Darker grid indicates more frequent mismatch. The x-axis indicates nucleotide position while the y-axis shows all paired gRNA-DNA interactions with one nucleotide was removed from gRNA, producing a bulged DNA base.
Two major steps are generally adapted to further understand and most importantly quantify the CRISPR off-target effect: (1) Bioinformatically searching the off-target sites. There are a great number of tools for off-target sites searching, such as conventional alignment algorithms: bowtie [79], bowtie2 [80], bwa [81], TagScan [82], GPGPU-enabled CUSHAW [83]. All software above still have two limitations: a restricted number of mismatches and fixed PAM. Hence, new algorithms customized for CRISPR-Cas systems to predict off-target sites are developed such as CasOFFinder [84], FlashFry [85], dsNickFury [86], and CRISPOR [31]. (2) Scoring based on ranking and selection. In addition to alignment-based methods, it was initially incorporated with hypothesis-driven methods (evaluating off-target activity base on formula) and then developed to learning-based methods (see Table 2). For instance, the MIT server [42] evaluates the off-target score by hand craft, a formula based on the number of mismatch nucleotides and the distance between them. This was then used to classify whether the gRNA off-target score reaches the cut-off value of 66 [87]. Subsequently, a method called CFD [52] (cutting frequency determination) is used to predict off-target score by multiplying the frequency of bases in each position of the gRNA spacer sequence. Haeussler et al. [31] evaluated most of the current machine learning methods and integrated them into a gRNA designing tool CRISPOR [31]. The MIT score was recommended by CRISPOR as an off-target reference because it can get the aggregation score of a single gRNA which summarizes all influence the off-target sequences and high accuracy. It was not until 2018 that deep learning-based methods have been applied for CRISPR off-target scoring. Two models named CNN_std [88] and DeepCRISPR [59] used the CNN model to predict gRNA specificity score for CRISPR-Cas9 system. A group of scientists from the Microsoft and the Broad Institution developed a model named elevation [86], and integrated it with Azimuth [86] (an activity model that they developed previously) into a website, which provides great a convenient platform for further application and development. All the data used in these MDL-based models were shown in Supplementary Table 3.
Table 2.
Publicly available tools for gRNA off-target prediction based on machine learning.
| Off-target software | Model | Ref | PAM | URL |
|---|---|---|---|---|
| CNN_std | CNN2 | [88] | NGT,NAG,NGC,NTG, NGG,NGA,NCG,NAA |
https://github.com/MichaelLinn/off_target_prediction |
| DeepCRISPR | DCDNN1 | [59] | NGT,NAG,NGC,NTG, NGG,NGA,NCG,NAA |
http://www.deepcrispr.net/ |
| Elevation | BRT4+ L1-reg+GBRT3+ LR5 | [86] | NAG, NCG, NGA, NGC, NGG, NGT, NTG |
https://crispr.ml/ |
| CRISPOR | hand craft | [31] | Nearly all | http://crispor.tefor.net/ |
| SynergizingCRISPR | AdaBoost | [102] | NGG | https://github.com/Alexzsx/CRISPR |
| CRISTA | RF6 | [103] | NGG | http://crista.tau.ac.il/pair_score.html |
| Predict CRISPR | ensemble SVM classifier |
[13] | NGG | https://github.com/penn-hui/OfftargetPredict |
1. DCDNN: Deep Convolutional Denoising Neural Network
2. CNN: Convolution Neural Network.
3. GBRT: Gradient Boost Regression Tree.
4. BRT: Boost Regression Tree
5. LR: Logistic Regression
6. RF: Random Forest.
In this review, we did not evaluate DeepCRISPR because it is not user-friendly, no encode source code provided, and running too slow on website. So, only one deep-learning based software, CNN_std, was included here. SynergizingCRISPR, which integrated the prediction result of five other models (CFD, MIT Website, MIT, Cropit, and CCTop) as input features, is running extremely slow and it was filtered out, too. As a consequence, we comprehensively compared six methods in this review, i.e., CFD, CCtop, preCRISPR, CNN_std, CRISTA, and elevation, using five benchmarks (Figure 3). We used the weighted Spearman correlation to minimized the false negative of gRNAs. The two methods built based on hypothesis and statistic (CFD and CCTOP) always obtain poor performance among all evaluated benchmarks (Figure 3). CNNstd and CRISPRpred are comparable in overall rankings across all the benchmark. It is noted that no significant advantage was found between deep learning and machine learning software. elevation, constructed as multi-level model, performs the best across all the weights. CRISTA produces random results that may not predict precisely. It performs the worst for all the datasets and therefore this software is not recommended (Figure 3). On the contrary, our reanalysis results show that the elevation model consistently outperforms the others in all evaluation dataset. The comparison detail was attached in the supplementary text, and corresponding test data are available in supplementary Table 4. This result can facilitate the CRISPR community to use at least by far the most powerful and accurate tool to select gRNAs with low off-target effects.
Figure 3.
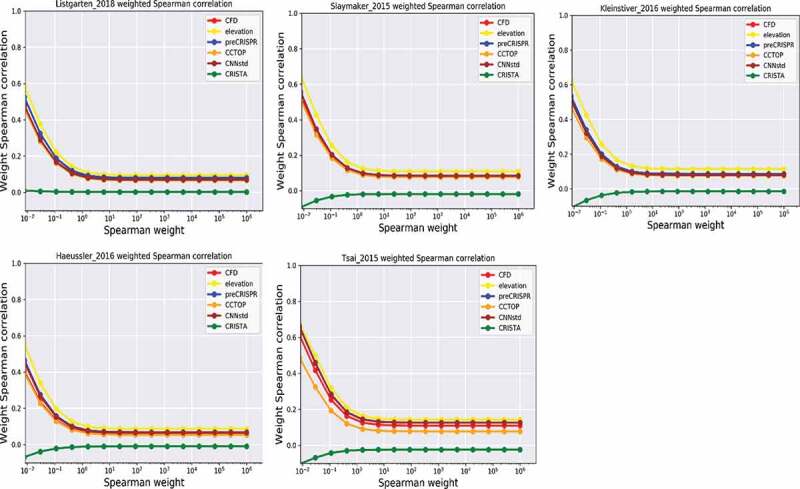
The comparison of different off-target prediction algorithms. Y-axis represents the weighted Spearman correlation determined by the weight of the X-axis counterpart. The weight ranges from 10−2 to 106. High weight indicates high normalized activity value of the positive off-target gRNA. The five independent datasets were tested separately.
6. Challenges in CRISPR activity and specificity prediction
6.1. Data heterogeneity
CRISPR datasets from different cell types, gRNA libraries, and organisms are heterogeneous and could not be simply combined. Current gRNA design rules are likely incomplete and biased due to the small number of gRNAs studied [20]. For instance, the SSC model used a mixed dataset from mouse mESC and human HL60 for model training [55], which may cause bias in this combination since the sequence features and the epigenetic states were from distinct cell types and species. Moreover, different methods used in quantifying CRISPR activity may cause batch-effect and heterogeneity among different experiments. Currently, nearly ten methods have been designed for gRNA on-target activity detection [26]. Two of them were wildly used as training data of gRNA efficiency predicting model. Firstly, measure gRNA-mediated CRISPR-Cas9 activity by capturing the phenotypic outcome. The gene functional knockout (KO) is used to quantify the gRNA activity by measuring the intensity of a green fluorescent protein (GFP) [89]. As the GFP-based method depends on intensive fluorescence-activated cell sorting (FACS) analysis, Doench et al. also used a drug-resistant assay to measure the gRNA efficiency [52]. However, these methods usually underestimate the actual CRISPR gRNA activity and cause false-negatives as frameshift deletion/insertion could potentially not cause a change in GFP intensity. Secondly, the most broadly used method of CRISPR gRNA activity measurement is based on deep sequencing of indels introduced at the target site [61], which directly measure the presence of indels introduced by CRISPR-Cas function. The endogenous DNA repair machinery might affect the readout of these CRISPR activity detection methods. Thus, instead of merging CRISPR activity datasets measured by different methods, large-scale CRISPR activity measurement experiments should be carried out on identical detection method to reduce the data heterogeneity as much as possible.
6.2. Data imbalance
Data imbalance is a common issue in the off-target prediction. A majority of the available gRNA off-target data are measured based on high-throughput sequencing, like Guide_seq by Tsai et al. [48] and Kleinstiver et al. [76], Listgarten et al. [86], HTGTS [49], Digenome_seq [90], CIRCLE_Seq [91], and low-throughput techniques like target PCR and flanking PCR [38,42,50,51,90,92]. For each target site, the homologous off-target sequences with cleavage activity can be genome-wide detected. These homologous gRNAs detected by different methods are defined as positive sets, however, more negative sets are arranged among the genome. The homologous gRNA target sites with undetected cleavage are much more than that of the detective ones. This issue will cause the data deviated to the negative group, as the true positive sRNA off-target sites account for an extremely low proportion. To date, the evaluating method (PRC curve) and bootstrapping sampling could solve this problem. The latter can sample the positive and negative samples into the same size. It should be noted that nearly all the exiting tools tend to avoid missing any true off-target cleavage site by weighting more on the true positive inputs. As for the CRISPR gene therapy purpose, the cost of false negative was much higher than that of false positive. Therefore, Listgarten et al. [86] proposed a weight Spearman correlation to address this problem, where the weight was added to the activity score of the sgRNAs ranged from 10−2 and 106 to reduce the false discoveries (Supplementary Text). Furthermore, Lin et at [88] used the stratify cross-validation model, which samples the positive class to be the same scale as a negative class. When the data augmentation is a concern, bootstrapping has been applied in DeepCRISRP [59] and Predict CRISPR [13] 75.
6.3. On-target data featurization
Featurization has been commonly used to improve the performance of in silico methods for gRNA activity prediction, despite this procedure is labour-intensive and needs strong knowledge about the CRISPR gene editing mechanism. Several features have been proven essential for gRNA on-target activity, including sequence compositions, thermodynamics, secondary structure, and physiochemical properties [93]. The conventional features used in the gRNA Designer (rule set II) [52], which was regarded as the state-of-the-art tool before 2017, can be classified into four types: 1) Sequence composition. The nucleotide composition of the gRNA spacer sequence has a preference. For instance, cytosine is predominant in the upstream of the PAM [89]. These features can be encoded into the single nucleotide and dinucleotide with position-dependent and position-independent using binary features. What is more, the flanking bases of PAM should also be considered. 2) GC content. Doench et al. found that gRNAs with low or high GC content tend to be less active [89]. And the most active gRNAs are those with approximately 50% GC content. Besides, this feature type also includes GC count, GC content, the latter means the percent of GC in spacer sequences. 3) Physicochemical features. Biochemical and structural studies have suggested that the thermodynamic of gRNA may influence the binding of gRNA to the target DNA. Doench et al. split the thermodynamic of sequences into melting temperature (Tm) of the spacer, 5mer Tm in the left side (5ʹ) of the spacer, 8mer Tm in the middle of the spacer sequence and also 5mer Tm in 3ʹ of the spacer [52]. 4) Cutting position. In addition to the four types of features mentioned above, Doench et al. pioneered in adding the cutting information in features, such as the amino acid cut position, in which the DSB occurred in the peptide of target sequences.
Besides, more features have been implemented to facilitate the model construction. 1) Secondary structure of spacer. Higher Gibbs free energy decides the higher self-folding ability of the gRNA spacer sequences. However, this folding ability should not be too high to achieve gRNAs with high activity. This will prevent the binding of gRNA to the target [28]. Moreover, the length of the gRNA scaffold should be considered, too [42]. Experiments have demonstrated that the gRNA scaffold with a length of 67nt and 85nt may have higher efficiency when compared to the original size [42]. 2) Epigenetic features. Chromosome accessibility influents the combination of gRNAs and the target sites [28]. For instance, H3K4me3 and chromosome accessibility, RRBS, CTCF have been applied in algorithms like DeepCRISPR [59] and DeepCpf1 [61]. However, Listgarten et al. failed to improve the model performance after adding the chromosome in the feature [86]. On the other hand, the epigenetic features are different cross-species, restricting the application of these algorithms for cross-species prediction.
It is noted that the number of features in each category may be insufficient, because the mechanism of CRISPR on-target has not been fully resolved. Although the gRNA Designer (rule set II) resulted in an excellent performance, there is still much room for further improvement. Several features have been added for possible performance improvement (supplementary Table1). For example, Hui Peng et al. extended the features from cutting position in spacer sequences of gRNA to protein, transcriptome, genome. Extending the thermodynamic to the context sequence in the flanking of the spacer region also improve prediction outcome [75]. Besides, previous studies indicated that structure accessibility also played an essential role in the recognized of miRNA and microRNA [94,95]. Hence, Wong et at [96] excavated the other types of second structure features, such as accessibility of individual nucleotide and stability of gRNA, and apply those in the CRISPR-Cas gRNA efficiency prediction. Khaledur Rahman et al. [97] first utilized the feature of the second structure, the specific heat of the corresponding 30-mer (4bp+23bp+3bp) of gRNAs, to train the SVM model called CRISPRpred (supplementary Table1). Nevertheless, these features cannot effectively represent and affect the activity of gRNAs, as the mechanism of the CIRSPR/Cas system has not been fully figured out.
An increasing number of features were developed to assess gRNA performance, but the importance of these features has not been fully evaluated, given that feature selection is an essential step to prioritize the features and eliminate features of less importance. Previously, Doench et al. [52] did not include the feature selection step. Increasing studies tend to discard several unimportant features by various feature selection methods. For example, Labuhn et al. [56] ranked the RMSE of the 1024 features using the linear regression. Similarly, Wilson et al. [98] used a strategy of forward-selection by incrementally inputting to select important features. Nevertheless, none of them evaluated the performance between feature selection and non-selection. Moreover, the selected features would just with the best representation in specific datasets, which is still needed to be evaluated by independent datasets. Deep learning method based features extraction such as DeepCpf1 [61], DeepCRISPR [59], DeepCas9 [58] appears successively. Automatic feature extraction may be the most advantageous characteristic of deep learning. These deep learning models do not require intense attention to the featurization of the gRNA sequences. Based on account of the auto feature extraction, the algorithms of deep learning can identify the sequences deeper and deeper. However, deep learning works slowly and it is difficult to be interpreted.
6.4. Off-target data featurization
The major features of off-target prediction are the number, composition, and combination of mismatches Initially, only sequence mismatches were considered. The MIT server considered the mean distance between two bases of mismatch and the number of mismatches, together with an experimentally-determined position-specific mismatch penalty matrixes to calculate the off-target efficiency [42]. After that, they also developed a formula for sequence off-target score calculation, which was applied in the CRISPRseek program [99] and CRISPOR [31] to facilitate the design of gRNAs. A similar method of activity evaluation was implemented by CCTOP [100] and CROPIT [101]. However, CCTOP and MIT score consider all off-target sites to calculate an aggregation score for one target site by a hypothetic formula, which was convenient for the gRNA library building and screening the optimal gRNA for a gene of interest. The CCTOP focuses more on the position of mismatches in the target site, while the CROPIT [101] weights more on the number of mismatches of three segments in the seed region of spacers and uses the whole-genome chromatin state information (DNase I HS data) as the features. The CFD [52], a Naive Bayes model [86], calculates the frequency of each type of mismatches in each site of the gRNA spacer region. Hence, the efficiency of gRNA specificity in CFD dependents on the position, number, and composition of mismatches between the gRNA and target DNA sequences [52]. In addition to sequences related features, SynergizingCRISPR [102] used the prediction score generated by other methods as features. Although CRISTA [103] uses the most extensive features to train the Random Forestmodel, only the top 30 most related features were integrated into the model.
Epigenetic features are highly variable and stochastically depend on the cell types and cell state. Hence, models developed by epigenetic features are usually limited by cell types, such as DeepCRISPR [59]. In contrast, CROPIT summarized the chromatin accessibility from 200 cells and took the parameters of the overlap sites and the number of different types of cells. Therefore, the CROPIT model can be applied to different cell types in principle. More information was listed in Supplementary Table2.
7. Conclusion
In conclusion, the CRISPR-Cas9 technology has rapidly emerged as a facile and efficient platform for gene editing. Because of its simplicity, efficacy, specificity, and programmability, this technology has tremendous advantages compared to other gene-editing technologies. Machine learning has been integrated into multiple bioinformatics fields, such as predicting the splicing site in genome [104,105] and long non-coding RNA region identification [106,107] . The work of designing gRNA is a typical interdisciplinary task. It needs to fully understand the biological mechanism of action of CRISPR and the algorithmic features of machine learning. The machine learning gRNA design tools serve as an important platform for the efficient application and development of the CRISPR system. However, the existing models still have some flaws, such as unclear mechanism, data imbalance, data heterogeneity, insufficient training dataset, lacking generalization ability, and inefficiency of cross-species. In the near future, it is expected that comprehensive, consistent, and sequencing-based datasets with high efficiency and specificity will be continuously generated. Hence, continuous efforts are required to further improve the accuracy and design gRNAs with high on-target activity and low (no) off-target effects. With the increase of the data volume from the application of CRISPR and more deeper mechanisms of CRISPR to be found, learning-based gRNA design tools will improve the prediction effect and aid designing gRNAs with the least off-target effects and high activity to meet the requirement in clinical applications.
Funding Statement
This work was supported by the Danish Research Council for Independent Research [DFF–1337–00128, and 9041-00317B]; Guangdong Provincial Key Laboratory of Genome Read and Write [No. 2017B030301011]; Lundbeck Foundation [(R219–2016-1375, R173–2014-1105)]; Sapere Aude Young Research Talent Prize [DFF-1335–00763A].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary materials
The supplemenatry data for this article can be accessed here.
References
- [1].Koonin EV, Makarova KS.. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol Rep. 2009;1:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. [DOI] [PubMed] [Google Scholar]
- [3].Gasiunas G, Barrangou R, Horvath P, et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109(39):E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ran FA, Hsu PD, Wright J, et al . Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Le Rhun A, Escalera-Maurer A, Bratovič M, et al. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019;16(4):380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mojica FJ, Díez-Villaseñor C, García-Martínez J, et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174–182. [DOI] [PubMed] [Google Scholar]
- [10].Mojica FJ, Díez-Villaseñor C, García-Martínez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(Pt 3):733–740. [DOI] [PubMed] [Google Scholar]
- [11].Tamulaitis G, Venclovas C, Siksnys V. Type III CRISPR-Cas immunity: major differences brushed aside. Trends Microbiol. 2017;25(1):49–61. [DOI] [PubMed] [Google Scholar]
- [12].Abudayyeh OO, Page RA, Geipel I, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peng H, Zheng Y, Zhao Z, et al. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics. 2018;34(17):i757–i765. [DOI] [PubMed] [Google Scholar]
- [14].Anders C, Niewoehner O, Duerst A, et al. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513(7519):569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yamano T, Nishimasu H, Zetsche B, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165(4):949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343(6176):1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shan Q, Wang Y, Li J, et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol. 2013;31(8):686 %@ 1546–1696. [DOI] [PubMed] [Google Scholar]
- [18].Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Henry VJ, Bandrowski AE, Pepin A-S, et al. OMICtools: an informative directory for multi-omic data analysis. Database (Oxford). 2014;2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chuai G-H, Wang Q-L, Liu Q. In silico meets In Vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol. 2017;35(1):12–21. [DOI] [PubMed] [Google Scholar]
- [21].Lee CM, Cradick TJ, Fine EJ, et al. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol Ther. 2016;24(3):475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jiang F, Taylor DW, Chen JS, et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016;351(6275):867–871%@ 0036–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Szczelkun MD, Tikhomirova MS, Sinkunas T, et al. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Nat Acad Sci. 2014;111(27):9798–9803%@ 0027–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin L, Luo Y. Tracking CRISPR’s Footprints. Methods Mol Biol. 2019;1961:13–28. [DOI] [PubMed] [Google Scholar]
- [26].Zischewski J, Fischer R, Bortesi L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv. 2017;35(1):95–104. [DOI] [PubMed] [Google Scholar]
- [27].Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jensen KT, Fløe L, Petersen TS, et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. [DOI] [PubMed] [Google Scholar]
- [29].Zhou Y, Liu Y, Hussmann D, et al. Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system. Cell Mol Life Sci. 2016;73(13):2543–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin L, Petersen TS, Jensen KT, et al. Fusion of SpCas9 to E. coli Rec A protein enhances CRISPR-Cas9 mediated gene knockout in mammalian cells. J Biotechnol. 2017;247:42–49. [DOI] [PubMed] [Google Scholar]
- [31].Haeussler M, Schönig K, Eckert H, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Montague TG, Cruz JM, Gagnon JA, et al. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42(WebServer issue):W401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Park J, Bae S, Kim JS. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31(24):4014–4016. [DOI] [PubMed] [Google Scholar]
- [34].Aach J, Mali P, Church GM. Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. bioinformatics. 2014;31(24):4014–4016. [DOI] [PubMed] [Google Scholar]
- [35].Zhang XH, Tee LY, Wang X-G, et al. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin Y, Cradick TJ, Brown MT, et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42(11):7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lim Y, Bak SY, Sung K, et al. Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat Commun. 2016;7:13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cho SW, Kim S, Kim Y, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fu Y, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pattanayak V, Lin S, Guilinger JP, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32(6):577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tsai SQ, Wyvekens N, Khayter C, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32(6):569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523(7561):481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Anders C, Bargsten K, Jinek M. Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol Cell. 2016;61(6):895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hu JH, Miller SM, Geurts MH, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tsai SQ, Zheng Z, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Frock RL, Hu J, Meyers RM, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33(2):179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ran FA, Cong L, Yan WX, et al. In vivo genome editing using staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang X, Wang Y, Wu X, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. 2015;33(2):175–178. [DOI] [PubMed] [Google Scholar]
- [52].Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chari R, Mali P, Moosburner M, et al. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12(9):823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12(10):982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xu H, Xiao T, Chen C-H, et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25(8):1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Labuhn M, Adams FF, Ng M, et al. Refined sgRNA efficacy prediction improves large-and small-scale CRISPR–cas9 applications. Nucleic Acids Res. 2017;46(3):1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lipton ZC. The mythos of model interpretability. arXiv preprint arXiv:1606.03490 2016.
- [58].Xue L, Tang B, Chen W, et al. Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J Chem Inf Model. 2018;59(1):615–624. [DOI] [PubMed] [Google Scholar]
- [59].Chuai G, Ma H, Yan J, et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018;19(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Luo J, Chen W, Xue L, et al. Prediction of activity and specificity of CRISPR-Cpf1 using convolutional deep learning neural networks. BMC Bioinformatics. 2019;20(1):332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kim HK, Min S, Song M, et al. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat Biotechnol. 2018;36(3):239–241. [DOI] [PubMed] [Google Scholar]
- [62].Angermueller C, Pärnamaa T, Parts L, et al. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Raina R, Madhavan A, Ng AY.. Large-scale deep unsupervised learning using graphics processors. In: Proceedings of the 26th annual international conference on machine learning. ACM; 2009. p. 873–880. [Google Scholar]
- [64].Coates A, Huval B, Wang T, et al. Deep learning with COTS HPC systems. In: International conference on machine learning; 2013. p. 1337–1345.
- [65].Dean J, Corrado G, Monga R, et al. Large scale distributed deep networks. In: Advances in neural information processing systems; 2012. p. 1223–1231. [Google Scholar]
- [66].Le QV, Marc’Aurelio R, Rajat M, et al. Building high-level features using large scale unsupervised learning. arXiv preprint arXiv:1112.6209 2011. In: 2013 IEEE international conference on acoustics, speech and signal processing. IEEE; 2013. p. 8595–8598.
- [67].Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. In. 2012;1097–1105. [Google Scholar]
- [68].Chari R, Yeo NC, Chavez A, et al. sgRNA scorer 2.0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth Biol. 2017;6(5):902–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tang H, Rahman MK, Rahman MS. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. Plos One. 2017;12(8):e0181943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kaur K, Gupta AK, Rajput A, et al. ge-CRISPR - An integrated pipeline for the prediction and analysis of sgRNAs genome editing efficiency for CRISPR/Cas system. Sci Rep. 2016;6:30870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gratz SJ, Ukken FP, Rubinstein CD, et al. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lei Y, Lu L, Liu H-Y, et al. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant. 2014;7(9):1494–1496. [DOI] [PubMed] [Google Scholar]
- [73].Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Peng D, Tarleton R. EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb Genom. 2015;1(4):e000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Peng H, Zheng Y, Blumenstein M, et al. CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics. 2018;34(18):3069–3077. [DOI] [PubMed] [Google Scholar]
- [76].Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ren X, Yang Z, Xu J, et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014;9(3):1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sternberg SH, Doudna JA. Expanding the biologist’s toolkit with CRISPR-Cas9. Mol Cell. 2015;58(4):568–574. [DOI] [PubMed] [Google Scholar]
- [79].Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25 %@ 1474–1760X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Iseli C, Ambrosini G, Bucher P, et al. Indexing strategies for rapid searches of short words in genome sequences. PLoS One. 2007;2(6):e579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu Y, Schmidt B, Maskell DL. CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows–wheeler transform. Bioinformatics. 2012;28(14):1830–1837%@ 1460–2059. [DOI] [PubMed] [Google Scholar]
- [84].Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30(10):1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].McKenna A, Shendure J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018;16(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Listgarten J, Weinstein M, Kleinstiver BP, et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng. 2018;2(1):38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Anderson KR, Haeussler M, Watanabe C, et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat Methods. 2018;15(7):512–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lin J, Wong K-C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics. 2018;34(17):i656–i663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Doench JG, Hartenian E, Graham DB, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12(3): 237–243, 231. following 243. [DOI] [PubMed] [Google Scholar]
- [91].Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–cas9 nuclease off-targets. Nat Methods. 2017;14(6):607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kim D, Kim S, Kim S, et al. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016;26(3):406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kuan PF, Powers S, He S, et al. A systematic evaluation of nucleotide properties for CRISPR sgRNA design. BMC Bioinformatics. 2017;18(1):297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Robins H, Li Y, Padgett RW. Incorporating structure to predict microRNA targets. Proc Nat Acad Sci. 2005;102(11):4006–4009%@ 0027–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang X, Wang X, Varma RK, et al. Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic Acids Res. 2009;37(22). e152-e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wong N, Liu W, Wang X. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015;16:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rahman MK, Rahman MS, Tang H. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PloS One. 2017;12(8):e0181943%@ 0181932–0186203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wilson LOW, Reti D, O’Brien AR, et al. High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. Crispr J. 2018;1(2):182–190. [DOI] [PubMed] [Google Scholar]
- [99].Zhu LJ, Holmes BR, Aronin N, et al. CRISPRseek: a bioconductor package to identify target-specific guide RNAs for CRISPR-Cas9 genome-editing systems. PLoS One. 2014;9(9):e108424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dobson L, Remenyi I, Tusnady GE. CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res. 2015;43(W1):W408–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Singh R, Kuscu C, Quinlan A, et al. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015;43(18):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhang S, Li X, Lin Q, et al. Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics. 2018;35(7):1108–1115. [DOI] [PubMed] [Google Scholar]
- [103].Abadi S, Doherty N, Page AML, et al. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13(10):e1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Cawley SL, Pachter L. HMM sampling and applications to gene finding and alternative splicing. Bioinformatics. 2003;19(Suppl 2):ii36–41. [DOI] [PubMed] [Google Scholar]
- [105].Oubounyt M, Louadi Z, Tayara H, et al. Deep learning models based on distributed feature representations for alternative splicing prediction. IEEE Access. 2018;6:58826–58834. [Google Scholar]
- [106].Tripathi R, Patel S, Kumari V, et al. DeepLNC, a long non-coding RNA prediction tool using deep neural network. Network Model Anal Health Inf Bioinf. 2016;5:1. [Google Scholar]
- [107].Han S, Wang Z, Wang R, et al. Long noncoding RNA identification: comparing machine learning based tools for long noncoding transcripts discrimination. Biomed Res Int. 2016;2016:8496165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.