

ABSTRACT
A sequencing-based profiling method (RiboMeth-seq) for ribose methylations was used to study methylation patterns in mouse adult tissues and during development. In contrast to previous reports based on studies of human cancer cell lines, almost all methylation sites were close to fully methylated in adult tissues. A subset of sites was differentially modified in developing tissues compared to their adult counterparts and showed clear developmental dynamics. This provides the first evidence for ribosome heterogeneity at the level of rRNA modifications during mouse development. In a prominent example, the expression levels of SNORD78 during development appeared to be regulated by alternative splicing of the Gas5 host-gene and to correlate with the methylation level of its target site at LSU-G4593. The results are discussed in the context of the specialized ribosome hypothesis.
KEYWORDS: Ribose methylation, rRNA, mouse development, Gas5, SNORD78, alternative splicing, specialized ribosomes
Introduction
Eukaryotic ribosomes are composed of a small (SSU) and a large (LSU) ribosomal subunit that are formed by assembly of four ribosomal RNA (rRNA) species (18S, 5.8S, 28S, and 5S) and ~80 ribosomal proteins (RPs). For decades, ribosomes were perceived as interchangeable and invariant ribonucleoprotein particles (RNPs) that convey, in the cytoplasm, the genetic information embedded within mRNAs into proteins. However, a bulk of recent findings suggests that ribosomes can be heterogeneous both in terms of their protein and RNA components. It is argued that changes in the composition of ribosomes may influence their structure and activity and confer an additional regulatory layer in the control of gene expression either by modulating translation speed and fidelity, or by influencing mRNA selectivity. This appealing hypothesis of ‘specialized ribosomes’ has the potential to considerably change our view of the regulation of the flow of genetic information [1–5].
An enormous source of ribosome heterogeneity can theoretically originate from changes in the rRNA content of post-transcriptionally modified nucleotides. A recent inventory based on quantitative mass spectrometry of a human cell line listed 14 types at 228 sites [6]. Among these were 112 ribose methylations (2´-O-Me) that constitute one of the most studied rRNA modifications. Almost all of these are introduced co-transcriptionally in the nucleolus, concomitantly with pre-rRNA processing and pre-ribosomal subunit assembly [7]. Their synthesis involves a specific family of antisense box C/D small nucleolar RNAs: the so-called SNORDs that form RNPs composed of four stably-bound proteins: Nhpx/Snu13, Nop56, Nop58, and Fibrillarin/Nop1 [8,9]. Indeed, the vast majority of SNORDs form transient, perfect ~9–20 bp-long double-stranded RNA structures with pre-rRNA in which the nucleotide substrate for 2´-O-methylation occupies a fixed position: it always faces the fifth nucleotide upstream of the D (or D´) box, a conserved sequence motif found in all SNORDs [10,11]. Thus, most, if not all, human sites of rRNA ribose methylation are guided by a tailored SNORD bearing an appropriated antisense element [12]. Finally, there are also numerous SNORDs lacking obvious base-pairing potential with other RNAs [13]. Among these so-called orphan SNORDs, many display preferential or even exclusive expression in brain. Whether these highly-expressed, still enigmatic SNORDs impact – directly or indirectly – rRNA modification profiles in brain remains an open issue [14].
An essential role of rRNA ribose methylations is evidenced by loss of function studies of the rRNA methyltransferase gene. In mice, genetic ablation of the Fibrillarin gene results in 100% lethality occurring very early in development [15] while in S. cerevisiae, the thermosensitive nop1.3 mutant, which is unable to 2´-O-methylate pre-rRNAs, does not grow at the non-permissive temperature [16]. More recently, it was also shown that Fibrillarin depletion in the zebrafish impacts early development, particularly neuronal differentiation [17]. At the molecular level, the prevailing view is that ribose methylations fine-tune the fidelity and/or efficiency of translation [18–21]. This is very likely achieved by altering chemical and structural properties of the modified nucleotides which, in turn, optimize rRNA folding, conformational flexibility and/or reactivity with protein factors. Of relevance, many of these chemical modifications are clustered in conserved and functional rRNA segments, i.e. the decoding centre region (DCR), the peptidyl transferase centre (PTC), the peptide tunnel and the inter-subunit domain [22,23]. However, the precise regulatory functions of the vast majority of rRNA methylation remain to be fully appreciated. Indeed, the lack of most individual rRNA 2´-O-methylations in genetically-tractable model, such as yeast, is often dispensable for survival, yet high-resolution phenotyping unveil subtle defects under challenging growth conditions [24]. In this situation, an alternative approach would be to study patterns of modification changes in biological settings, e.g. during development. Indeed, altered methylation levels at some rRNA sites have been associated with impaired development in the zebrafish [25].
The study of ribose methylations in rRNA has recently been boosted by developments in RP-HPLC [26], quantitative mass spectrometry (qMS) [6] and sequencing-based methods [12,27] (reviewed in [28]). Although most rRNA 2´-O-methylated sites were found to be close to fully modified (90–100%), an important finding from human cancer cell lines was that one-third of rRNA methylation sites were found to be fractionally modified ranging from ~56–90% [6,12]. The implication of this is ribosome heterogeneity at the level of nucleotide modifications. Interestingly, these so-called fractional rRNA modifications are more sensitive to the dosage of the Fibrillarin and they are preferentially found externally relative to the 3D structure of the ribosome [29,30]. In contrast, methylation levels at fully modified sites, particularly those located at the DCR and PTC, remain unchanged in Fibrillarin-depleted cells. Altogether, it has been surmised that fractionally methylated positions are bona fide candidates to physiological regulations, possibly in a cell-type and/or developmental-specific manner [12,29,30].
Here, we apply a sequencing-based method (RiboMeth-seq; Fig. 1(A)) for profiling of 2´-O-Me sites in rRNA to adult tissues from the mouse and their developing (E16.5) counterparts. In contrast to previous studies of human cancer cell lines, we find by far the majority of sites to be fully methylated in adult tissues. Strikingly, RNA from developing tissues is considerably less methylated at a discrete set of sites, suggesting a ‘ribosomal code’ to development. Furthermore, we provide a detailed analysis of a subset of 2´-O-methylations guided by SNORDs encoded within the growth arrest-specific 5 (Gas5) host gene. Of particular relevance, our study points to a developmentally regulated post-transcriptional mechanism that specifically prevents the release of SNORD78 in adult tissues and leaves the expression of other Gas5-encoded SNORDs mostly unchanged. Together our study provides a framework for the study of ribose methylations in rRNA during development and draws a more realistic picture of these modifications than that derived from studies of cultured cells.
Figure 1.
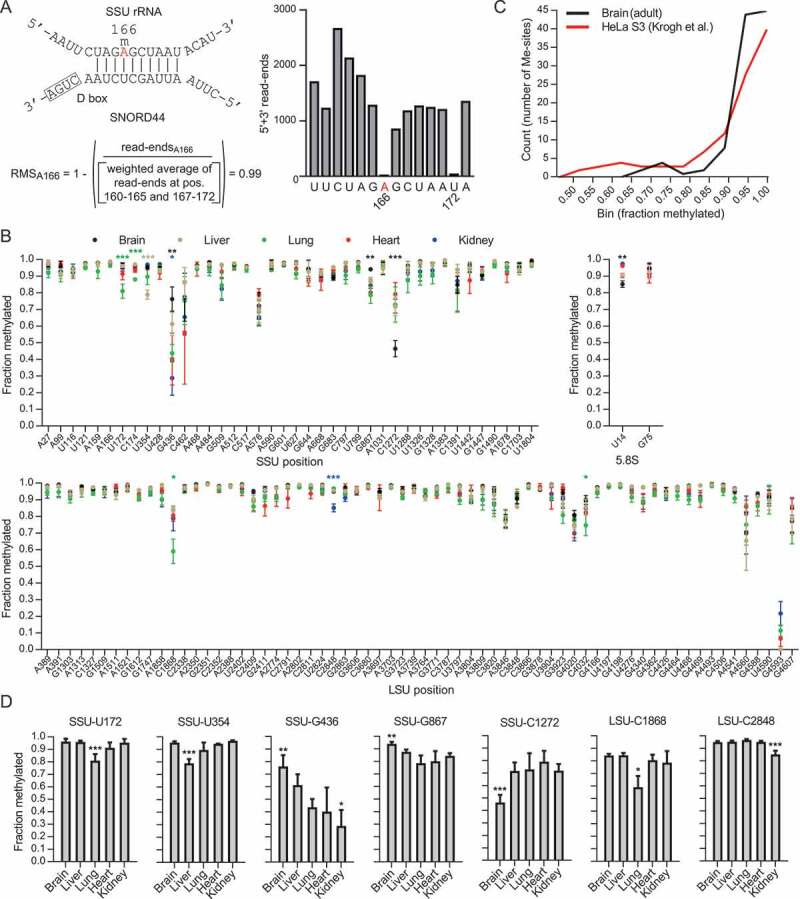
(A) The principle of RiboMeth-seq analysis and calculation of RiboMeth-seq score (RMS) illustrated by analysis of SSU-Am166 (labelled in red). The modification is guided by SNORD44. The RNA is partially fragmented by alkaline that results in cleavage at all positions except those that are resistant to activation of the 2ʹOH due to methylation. RNA fragments are cloned and sequenced and the number of read-ends recorded. Based on this, a score is calculated by comparison of the read-ends at the queried position and that of a weighted average of six flanking positions at either side. The score describes the fraction of molecules methylated at the queried position. Note that the flanking sequence includes SSU-U172 that is also methylated. (B) Graph showing methylation scores at all methylated positions in mouse rRNA in five adult tissues. Sites are numbered according to human rRNA to allow comparisons. The mouse positions can be found in Table S3. (C) Comparison of the distributions of methylation scores in HeLa cells and mouse adult brain. (D) Examples of differentially methylated sites between adult tissues. Asterisks indicate levels of statistical significance when comparing the tissue in question to all other tissues.
Results
rRNA in adult tissues is highly methylated
We first examined rRNA profiles by RiboMeth-seq analysis in whole-cell RNA from adult mouse tissues, including brain, liver, heart, lung, and kidney. 104/106 of the sites previously reported from human cancer cell lines [12] were detected (Fig. 1(B) and Table S3). SSU-U1668 is generally found to be methylated at very low levels (0.08 in TK6 cells [6]) that escape detection by RiboMeth-seq. Thus, the notable exception was LSU-G4593 that is fully methylated in human cancer cell lines, but scored at background levels in the present analysis. In addition, we detected methylations at SSU-U354 and LSU-G3606 that both were added to the list of human methylation sites subsequent to the original profiling [12]. In contrast, we did not find evidence for methylation at LSU-A1310 that is well documented in human cell lines or at SSU-C621 and LSU-U1760 that were suggested as new sites in the qMS study of TK6 cells [6], but currently have not been ascribed plausible SNORD guide RNAs. Thus, RiboMeth-seq analyses did not reveal any novel mouse- or tissue-specific rRNA 2´-O-Me sites. There are two reservations to this claim. First, by analysing tissues, the score reflects an average of all cell types. Since most sites are close to fully methylated, this applies to all major cell types within the tissue, but does not exclude a different methylation pattern in a minor cell type within the tissue. Second, due to the relatively high background of the method, sites with low modification stoichiometry might have escaped detection. However, due to the limited number of SNORDs that potentially target rRNA, we nevertheless conclude that the methylation sites found in adult mouse tissues is very similar to those described for human cell lines. Obviously, this claim does not exclude that a few rRNA methylation sites may be discovered as more cell types or experimental conditions are examined.
The methylation scores varied little between the tissues analysed (Fig. 1(B)). Almost all sites scored above 0.9 in all tissues with only 31 sites scoring below this value in all tissues combined and only 11 of these consistently scoring below 0.9. Thus, the score distribution was distinctly different from previous analyses of human cancer cell lines, e.g. HeLa cells [12], with a larger fraction of sites that were fully or close to fully methylated clearly separated from a subset of fractional sites (Fig. 1(C)). Among the fractional sites, less than 10 sites were single outliers (Fig. 1(D)), but no trend in relation to tissues or ribosomal location was observed. Thus, we did not find evidence for substantial tissue-specific variation in methylation patterns in adult tissues. We conclude that adult tissues are fully or close to fully methylated at almost all sites with a distinct subset of sites showing fractional methylation in different tissues.
Loss of SNORD126 and the corresponding LSU-Am1310 in the mouse
To understand the absence of a RiboMeth-seq signal at LSU-A1310 in the mouse, we conducted a focused study of this site. In parallel, we studied the expression of SNORD126 that has the potential to target LSU-A1310 for 2´-O-methylation via a conserved, 12 nt-long antisense element (http://snoatlas.bioinf.uni-leipzig.de/index.php; see also Fig. 2(A)). SNORD126 was previously listed as an orphan SNORD with significant expression in rat spleen and thymus [31]. Furthermore, LSU-A1310 is methylated in human cell lines; albeit at low levels (e.g. 0.44 in TK6 cells [6]) and, accordingly, SNORD126 is expressed, e.g. in HeLa and Jurkat cells (Fig. 2(B); top panel). Remarkably, the SNORD126 gene is not present in the mouse genome and, accordingly, northern blotting analysis failed to detect SNORD126 in several mouse tissues (Fig. 2(B); top panel). As expected from previous studies [31], SNORD126 was readily detected in rat spleen or thymus (Fig. 2(B); bottom panel). To obtain evidence independent of RiboMeth-seq profiling that have problems detecting low stoichiometry sites, we then tested whether LSU-A1310 is methylated as predicted by the SNORD126:rRNA duplex. This was achieved through the use of primer extension assay that relies on the fact that most 2´-O-Me cause pausing of reverse transcriptase (RT) at low, but not high, dNTP concentrations [32]. As shown in Fig. 2(C), we detected a dNTP-dependent pause positioned one nucleotide upstream of LSU-A1310 in rat spleen and thymus, but not in liver. In contrast, no dNTP-dependent pause was observed when the same mouse tissues were examined. Altogether, this provides strong support for the involvement of SNORD126 in guiding a rat, tissue-specific 2´-O-methylation at LSU-A1310. Note that the stoichiometry of modification could not be inferred because the primer extension assay is not quantitative. This is a rare example of tissue-specificity of rRNA ribose methylation.
Figure 2.
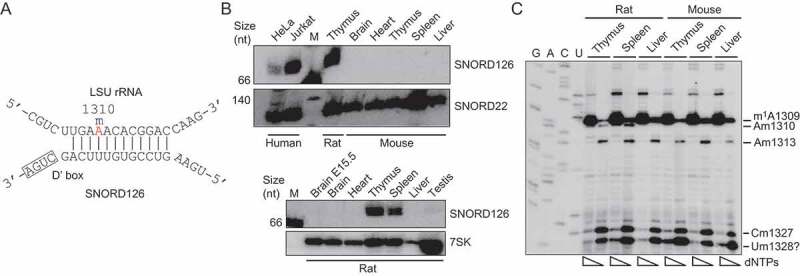
(A) Base pairing between the antisense element associated with box D´ in SNORD126 and the target sequence in rat LSU rRNA that result in methylation of Am1310 (labelled in red). (B) Northern blotting analysis of SNORD126 expression in mouse (upper panel) and rat (lower panel) tissues. SNORD22 and 7SK RNA were hybridized in parallel to serve as loading controls. (C) Primer extension analysis of ribose methylation at LSU-Am1310 and flanking modification sites in mouse and rat tissues. For each sample, primer extension was carried out at high and low dNTP concentration and run in parallel to reveal dNTP-dependent RT-pausing signals. A dideoxy-sequencing ladder was generated using the same primer and run in parallel to achieve mapping at sequencing resolution. Note that the sequencing lanes are labelled according to the RNA-like sequence. The pause signal labelled ‘Um1328?’ could represent a novel 2´-O-Me site with a RiboMeth-seq score below the threshold.
Developing tissues show distinct patterns of hypomethylated 2´-O-Me sites
We next assayed rRNA methylation profiles in developing (E16.5) tissues, including whole (E9.5) head (Data S1-2). No additional 2´-O-Me sites compared to adult tissues were revealed. Unsupervised clustering analysis showed that developing and adult tissues clustered separately (with the exception of the brain E16.5 SSU sample) (Fig. 3(A)), with rRNA in developing tissues being less methylated than their adults counterparts (Fig. 3(B)). Indeed, we detected 59 sites below a score of 0.9 in at least one tissue and with a subset of these being consistently lower in E16.5 tissues (Fig. S1-S3). Interestingly, two rRNA sites (SSU-A576 and LSU-G4593, Fig. 3(B)) showed higher degrees of methylation in developing tissues. In fact, methylation at LSU-G4593 was not detected above background by RiboMeth-seq analyses in any adult tissue but was clearly methylated at E16.5. A closer examination revealed that, upon brain development, the most variable rRNA sites showed distinct trajectories in terms of methylation dynamics. As illustrated in Fig. 3(C–E), they could be roughly ranked into three main classes: (i) rRNA sites with sharp increased methylation (SSU-U354, SSU-G436, SSU-G867); (ii) rRNA sites with decreased methylation (SSU-A576, LSU-G4593) and (iii) rRNA sites for which methylation levels slightly increased (SSU-G509, SSU-A590, SSU-U1442, SSU-G1447, LSU-C1868, LSU-G2411, LSU-G3923, LSU-C4032). Surprisingly, many more developmentally-regulated rRNA sites were found in SSU than LSU rRNA (Fig. 3(B), Figure S1 and S2). In conclusion, we have uncovered a developmental change in ribosome methylation patterns that affects a subset of sites only and by definition contributes to ribosome heterogeneity.
Figure 3.
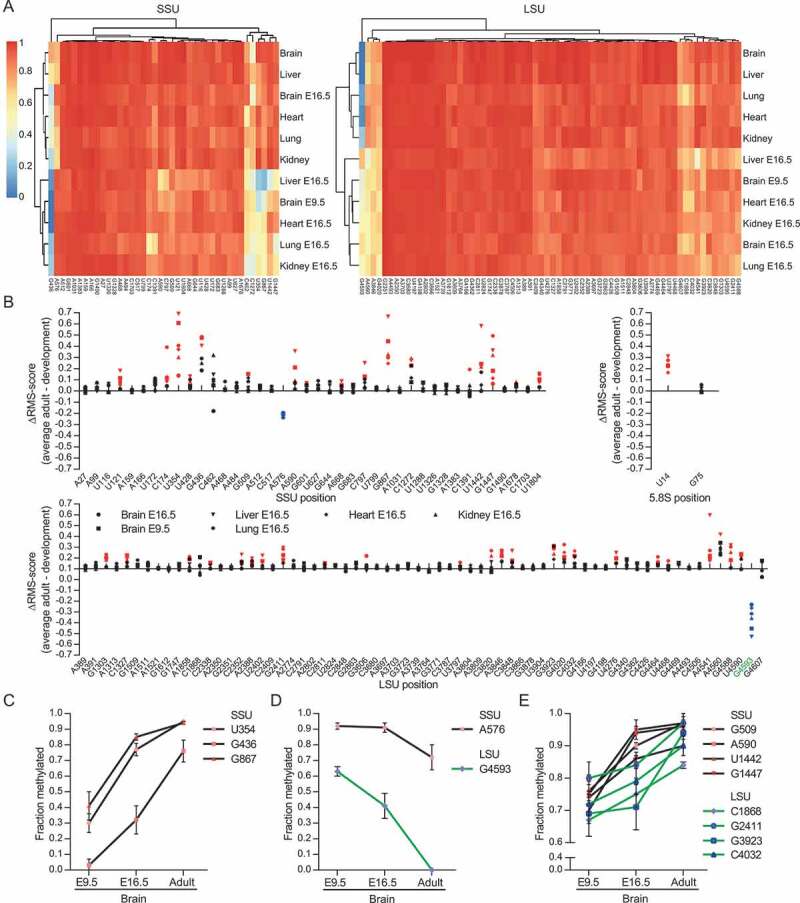
(A) Heat-map and cluster analysis of RiboMeth-seq scores of rRNA from adult and developing tissues. (B) Graph showing the differences in RiboMeth-seq scores between the average score for all adult tissues and average scores from each developmental tissues (n = 3). Positions showing statistically significant higher or lower scores are labelled in red and blue, respectively. Position LSU-Gm4593 is discussed extensively in the text and is labelled in green. (C–E) Graphs showing changes in RiboMeth-seq score for those positions that show variation during brain development sorted into three main types; sites that are significantly up-regulated >0.25 (C), down-regulated >0.10 (D), and up-regulated between 0.15 and 0.25 (E).
Differential expression of SNORDs from within the same host-gene during development
In a first attempt to address the potential significance of modulating 2´-O-methylations at selective rRNA sites during mouse development, we decided to concentrate on LSU-Gm4593. This site stands out for several reasons. First, it was the only site that was barely detected in adult mouse tissues. Second, it decreases dramatically during development. Third, methylation at LSU-G4593 is predicted to be guided by the evolutionarily conserved SNORD78 which is processed from intron 6 of the Gas5 non-coding RNA gene (Fig. 4(A)). Remarkably, eight additional SNORDs are also generated from the same Gas5 transcript and levels of their associated 2´-O-methylation sites do not change substantially during development and in adult tissues (Fig. 4(A)). It, therefore, suggests that a developmentally-regulated mechanism specifically influence the methylation guiding activity of SNORD78. Fourth, an important biological role for SNORD78, particularly in brain development, was inferred from its knock-down in zebrafish one-cell stage embryos [25]. Finally, several studies have implicated SNORD78 and/or Gas5 in cancer [33–36].
Figure 4.

(A) Map of the mouse Gas5 pre-RNA (Mouse mm10 assembly) showing exons (light grey boxes) and introns and the locations of intron-encoded SNORDs (dark grey boxes). Red lines indicate alternative splicing events. The numbering of exons deviates from standard to emphasize similarities with human Gas5. Below the map, the 2´-O-Me sites guided by the SNORDs are indicated together with RiboMeth-seq scores for brain E9.5 (whole head) and adult, respectively. (B) Primer extension analysis of LSU-Gm4593 and flanking positions conducted as described in the legend to Fig. 2(C). (C) Northern blotting analysis of SNORD78 expression in adult and E16.5 tissues. SNORD22 was hybridized in parallel to serve as a loading control. (D) Northern blotting analysis of all Gas5 encoded SNORDs during brain development. Note that whole head was used to represent brain samples in E9.5 and E12.5 embryos. M: Molecular weight marker. (E) Quantitation of signals from biological replicates of northern blots comprising RNA from E9.5, E16.5, and adult. Signals were normalized against SNORD22 and E9.5 was set to 1. Statistical significance was calculated by comparing E16.5 and adult to E9.5 (n = 3). (F) RT-qPCR analysis of Gas5 expression during brain development using two different primer sets (n = 4–5).
By using a method independent of RiboMeth-seq, namely primer extension assay, we first confirmed differential methylation levels at LSU-G4593 in developing and adult tissues. As reported in Fig. 4(B) (left panel), we readily detected dNTP-dependent reverse transcription pauses positioned one nucleotide upstream of LSU-G4593 and, consistent with RiboMeth-seq data, their signal intensities decreased in adult brain as compared to developing E16.5 brain or E9.5 head (Fig. 4(B); left panel). The same trend holds true for the other tissues we examined (Fig. 4(B); right panel). Remarkably, reduced methylation levels at LSU-G4593 were accompanied by a decreased expression of SNORD78 in the adulthood, ranging from 60% to 90% depending on the tissues (Fig. 4(C)). This rather unexpected observation prompted us to systematically monitor the expression of all Gas5-encoded SNORDs. Using developing brain tissues as a model, we found that only SNORD78 expression levels displayed a dramatic decrease during brain development from E9.5 to adult (Fig. 4(D–E)), yet a slight decrease was also apparent for SNORD74, SNORD76, and SNORD44. During the same time course, expression level of the spliced Gas5 transcript, as judged by RT-qPCR with two different pairs of primers, remained mostly unchanged (Fig. 4(F)). Hence, developmentally-regulated post-transcriptional regulations preferentially dampen SNORD78 production. This represents one of the first, if not the only, example demonstrating that the release of distinct SNORDs from the same host-gene transcript can be differentially regulated.
Splicing of the SNORD78-coding intron 6 changes during development
In mammals, intron-encoded SNORDs are preferentially positioned 70–80 nt upstream of the 3´ splice site, thus pointing to a critical distance between SNORD sequences and branch point as experimentally documented for the two Gas5-encoded SNORD75 and SNORD76 [37]. The SNORD78-coding intron 6 of the Gas5 transcript is known to be alternatively spliced (Fig. 4(A)) through skipping of exon 7 [38]. This results in an excised intron in which SNORD78 is placed further upstream in relation to the branch site which may impede SNORD78 processing. The specific details of the mechanism behind alternative splicing of the Gas5 transcript have not been worked out. The splice sites of all introns comply with the consensus rules in particular at the intron side of the splice sites (Fig. 5(A)). This is particularly clear from comparative analysis of Gas5 genes from different rodents as exemplified by introns 6 and 7 (Fig. 5(B)). These two introns differ additionally from the other introns in the Gas5 transcript by having a conserved sequence element in the branch site region. The element in intron 7 is not found in human Gas5 which may be related to a different type of alternative splicing based on alternative 5´ splice sites in exon 7 compared to rodents rather than skipping of exon 7 (Figure 5(A)).
Figure 5.
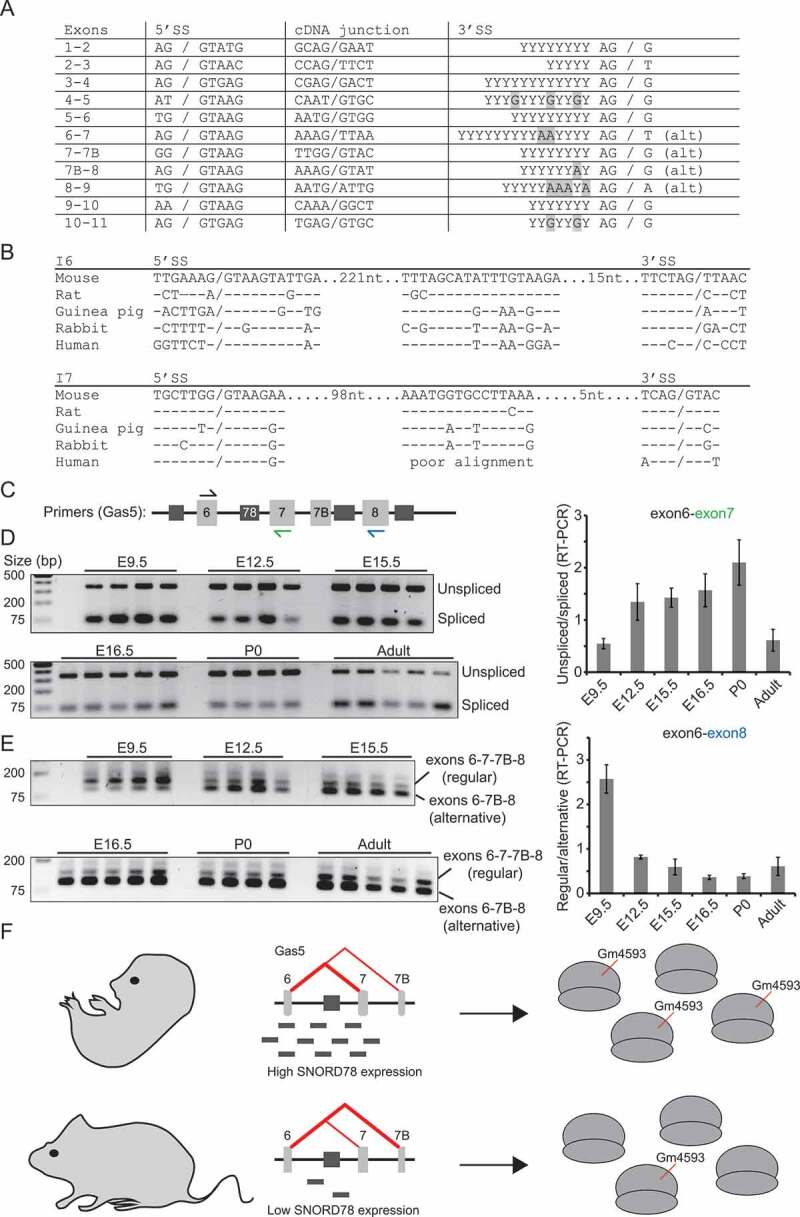
(A) Splice-site sequences in the mouse Gas5 transcript. For exon numbering, refer to Fig. 4(A). (B) Alignment of splice-site sequences and conserved intronic sequence elements in exons 6 and 7 of rodents and human Gas5. (C) Diagram showing placement of primers for RT-PCR experiments. Primers coloured in blue and green were used as the downstream primers in (D) and (E), respectively. (D) Gel picture (left) and quantitation of gels (right) of an RT-PCR analysis of spliced and unspliced transcripts containing exons 6 and 7 during brain development (n = 4–5). (E) Similar analysis as in (C) using primers located in exons 6 and 9 to discriminate between two main types of splicing to reveal exon 7 skipping (n = 4–5). (F) Model describing that alternative splicing of intron 6 in the Gas5 transcript, results in high SNORD78 levels and many ribosomes methylated at LSU-G4593 in the early embryo, and, conversely, in low SNORD78 levels and few ribosomes methylated at LSU-G4593 in the adult mouse.
To address a possible role of alternative splicing in SNORD78 expression, we conducted two RT-PCR experiments. First, we measured the ratio of unspliced and regularly spliced intron 6 during brain development using primers placed in the flanking exons 6 and 7, respectively (Fig. 5(C–D)). Here, we observed a decrease in splicing of intron 6 correlating with a decrease in SNORD78 levels. Note that this experiment does not detect alternatively spliced transcripts that skip exon 7. Then, we compared regular splicing and alternative splicing by using primers in exons 6 and 8, respectively. We observed a sharp decline in regular splicing after E9.5 (Fig. 5(C–E)). Both of these observations support our idea of specific regulation of SNORD78 expression through splicing of the Gas5 transcript during development. At E9.5, transcripts are spliced relatively efficiently and splicing efficiency then decreases during development (Fig. 5(D)). The spliced transcripts are predominantly spliced by regular splicing but this is shifted during development towards skipping of exon 7 (Fig. 5(E)). In the adult, splicing is efficient (Fig. 5(D)) and splicing predominantly involves skipping of exon 7 (Fig. 5(E)). Our analysis does not rule out other contributing factors, e.g. relating to the stability of SNORD78. It should also be kept in mind that regulation of Gas5 expression in developing tissue is markedly different from adult tissues (transcriptional compared to post-transcriptional regulation [39]) suggesting that the two may be difficult to compare. Based on the sequence structure and the splicing analysis, we propose that SNORD78 expression is regulated during development, mostly through regulation of splicing of intron 6 in the Gas5 transcript. This results in low methylation level at LSU-G4593 in adult mouse tissues (Fig. 5(F)).
CRISPR-Cas9 mediated knock-out of SNORD78
In order to unveil potential regulatory roles of SNORD78 and/or LSU-Gm4593, we constitutively inactivated the SNORD78 gene. For these experiments, we switched to the human embryonic kidney 293T (HEK293T) cell line that provided a more clear experimental situation by having close to full methylation of the site (RMS = 0.93 [40]). Inactivation was achieved through the use of the CRISPR-Cas9 system via two sgRNAs designed to tether Cas9 endonuclease within and upstream of the SNORD78 sequence (Fig. 6(A)). Two independent SNORD78-deficient clones (KO1 and KO2) were then selected for further analyses and two wild-type clones (WT1 and WT2) that followed the same transfection and cloning selection procedures but without any genomic alteration at the SNORD78 gene were selected as controls (Fig. 6(B)). Note that the two sets of KO and WT cell lines were generated using two different approaches. KO1 and WT1 were selected based on puromycin resistance and limited dilution, whereas KO2 and WT2 were selected by single-cell sorting based on GFP expression. As expected, SNORD78 was detected in WT1 and WT2, but not in KO1 and KO2 cells (Fig. 6(C)). KO1 has a large insertion in the SNORD78 sequence and thus generates longer and aberrant SNORD78 transcripts denoted as SNORD78* (Fig. 6(C), upper panel, Figure S5 for sequences of the clones). Constitutive deletion of the SNORD78 gene did not impact strongly on the expression of the two neighbouring SNORDs, even though expression levels of spliced Gas5 transcripts appeared slightly increased in SNORD78-deficient cells (Fig. 6(D)). More importantly, LSU-G4593 was not methylated in KO1 and KO2 cells (Fig. 6(E)). Finally, cell counting (Fig. 6(F)), flow cytometry analysis using propidium iodide DNA staining (Fig. 6(G)) and flow cytometry analysis using propidium iodide and Annexin V staining (Fig. 6(H)), did not reveal any impairments in cell growth, cell cycle profiles and apoptotic pathways, respectively. Hence, the lack of SNORD78 and/or the disappearance of LSU-Gm4593 are unlikely to elicit any strong nucleolar stress response. Consistent with this assumption, pre-rRNA processing pathways remained unaltered in SNORD78-KO cells (Figure S4).
Figure 6.

(A) Map of the human Gas5 pre-RNA (human hg38 assembly) showing exons (light grey boxes) and introns and the locations of intron-encoded SNORDs (dark grey boxes). Red lines indicate alternative splicing events. The numbering of exons deviates from standard to emphasize similarities with mouse Gas5 in Fig. 4(A). The lower part of the figure shows an enlargement of the intron 6 region with detailed annotation of SNORD78 and the relative location of the two DNA sequences targeted by sgRNAs for CRISPR-Cas9 knock-out. (B) Agarose gel of PCR analysis confirming changes at the SNORD78 locus in CRISPR-Cas9 KO clones. (C) Northern blotting analyses of a subset of SNORDs showing specific changes (aberrant migration in KO1 (SNORD78*) and absence in KO2) relating to SNORD78 compared to several other SNORDs encoded by the Gas5 transcript. (D) Gas5 expression in SNORD78-KO cell lines compared to WT as revealed by RT-qPCR (n = 7). (E) Primer extension analyses of SNORD78-KO cell lines showing absence of the SNORD78-guided Gm4593 compared to flanking 2´-O-Me sites. (F) Growth curves (n = 4), (G) flow cytometry using propidium iodide DNA staining (n = 2), (H) and flow cytometry using propidium and Annexin V staining of SNORD78-KO cell lines compared to WT (n = 2). (I) Luciferase reporter assays of stop-codon read-through and arginine to serine misincorporation (n = 5) as well as (J) IRES-elements in SNORD78-KO cell lines compared to WT (n = 12).
To demonstrate a potential regulatory role of LSU-Gm4593 in modulating translation, WT and SNORD78-KO cells were transiently transfected by sensitive luciferase reporter genes, classically used to monitor translation fidelity. These reporters contain in-frame stop codons or mutations so that firefly luciferase activities can be measured as a read-out of codon stop read-through or amino acid mis-incorporation. As shown in (Fig. 6(I)), we found that the SNORD78-KO2 clone exhibited a slight increase in recoding at UAG stop-codon and also to some extent at UGA while accuracy of translation at the other stop-codons was normal. These defects in translation fidelity were less apparent with the other SNORD78-KO1 clone, which only showed a tendency of increased read-through UAG stop codons (P = 0.0636). In any event, amino acid mis-incorporation was also unaffected for the two SNORD78-KO clones when a luciferase reporter gene with mutations in amino acid residues (R to S) involved in enzymatic activities was tested. Abrogation of Fibrillarin or inhibition of rRNA methylation can impact cap-independent translation [29,35,41,42]. This prompted us to interrogate the efficiency of internal ribosome entry site (IRES)-mediated translation in SNORD78-KO cells. As indicated in (Fig. 6(J)), we did not uncover any deficiencies in cap-independent translation when IRESs from FGF2 and FGF1A transcripts were tested in the Renilla-IRES-Firefly di-cistronic constructs. Finally, we measured the protein synthesis rates by incorporation of puromycin and detection using an anti-puromycin antibody (SUnSET assay [43]) without finding significant changes in SNORD78-KO clones compared to WT (data not shown). Overall, SNORD78 and the highly variable LSU-Gm4593 appear not to be essential and have little impact, if any, on translation in the unperturbed cell model system used in the present study.
Discussion
We have used a sequencing-based profiling method (RiboMeth-seq) to study ribose methylation sites in mouse rRNA from adult and developing tissues. Several general observations were made. The repertoire of 2´-O-Me sites in mouse was very similar to human and we found no evidence for substantial variation in the methylation patterns in five adult tissues. This further implies that no tissue-specific methylations were found in the mouse. Strikingly, we found 2´-O-Me sites to be highly methylated in adult tissues compared to what was previously described for rRNA in human-cultured cancer cells. Together, these observations paint a conservative picture of ribose methylation in rRNA and serves as a corrective to publications based on cultured cells, some of which claim considerable variation in methylation patterns. Of significance, we found developing tissues to show hypomethylation at a discrete subset of sites that showed developmental dynamics. The variable sites were not linked in a trivial manner, e.g. by shared host-genes of the guide RNAs. By focusing on the lncRNA host-gene Gas5 that harbours nine methylation guiding SNORDs, we found support for a model where alternative splicing specifically regulates the expression of SNORD78 and thereby its cognate modification at LSU-G4593 during mouse development without significantly affecting methylations guided by other Gas5 SNORDs.
Adult tissues show close to full methylation at the majority of 2´-O-Me sites
RiboMeth-seq analyses revealed that all samples of adult mouse tissues showed the same repertoire of rRNA ribose methylations. Although it is impossible to claim that the analyses were comprehensive, a comparison between previous RiboMeth-seq analyses and other methods suggest that only few additional sites will be found [28]. Specifically, a quantitative mass spectrometry of a human lymphoblast cell line uncovered only two additional sites (SSU-C621 and LSU-U1760), both methylated at relatively low stoichiometry [6]. The proposed guide RNAs for these sites does not comply with the consensus of interaction with their targets and no other supporting evidence for these sites was provided. Furthermore, we found no evidence for these sites in the mouse in the present study. On the other hand, a limitation of the RiboMeth-seq is in detection of sites with low stoichiometry. As an example, LSU-Gm4593 was not detectable in several adult tissues by RiboMeth-seq, but a signal was observed by primer extension of the same samples (Fig. 4(A)). Thus, it is not possible to exclude the existence of additional sites with low stoichiometry or with high stoichiometry restricted to less abundant cell types in the sample. One example of an unexpected difference in methylation patterns was the absence of LSU-Am1310 in the mouse. This site is found as one of few tissue-specific methylations in the rat guided by SNORD126 and is located as a neighbour to LSU-m1A1309 in H26 in LSU Domain II. The loss of the SNORD126 gene appears to have taken place more than once during evolution of the vertebrates and may be related to changes in transcription start sites of the host gene encoding Cyclin B1 Interacting Protein 1.
A key observation was the much higher methylation stoichiometry in adult tissues compared to the previously reported cancer cell lines [12]. This conclusion must apply to all abundant cell types in the tissue sample and is supported by similar analyses of four human tissues (brain, skin, liver, and skeletal muscle) each pooled from five donors (Krogh et al., unpublished). Consistent with this notion, U14 of 5.8S rRNA in HeLa or Novikoff hepatoma cells was already reported to be less methylated as compared to normal tissues [44]. Furthermore, some 2´-O-methylation sites in spliceosomal small RNAs in human tissues were also recently found to display higher methylation levels as compared to immortalized line of human T lymphocyte (Jurkat) cells [45]. Cancer cell lines are characterized by considerably more fractional sites and a much wider distribution of scores as shown in a comparison of the scores distribution of tissue (exemplified by brain) and cell lines (exemplified by HeLa) in (Fig. 1(C)). Furthermore, cancer cell lines show more variation in methylation pattern ([12], and unpublished) than tissues. In light of this, we speculate that the methylation pattern in cancer cell lines is due to large-scale changes in gene expression, including SNORD host-genes, and rapid growth rates. Ribosome production may be limiting the growth rate in cancer cells [46,47] and the RNA-guided rRNA modification systems may be unable to keep up with the increased rates of RNA polymerase I transcription and pre-ribosome assembly. Thus, we propose that the present description of the ribose methylation pattern based on analyses of adult tissues provides a better baseline than our previous analyses of cultured cancer cell lines.
Developmental tissues
Another key observation in our study was the identification of several rRNA sites whose stoichiometry of methylation appears developmentally-regulated (Fig. 3). Although some of these overlap with sites that were described as substoichiometric in cultured cells, some sites were not observed as such previously. Thus, our study emphasizes a subset of sites showing specific changes in a biological setting, namely during development. Interestingly, most fractional sites at E16.5 are preferentially located on SSU rRNA (Fig. 3(B)) and the changes that occur in SSU rRNA appear to occur earlier than the changes in LSU rRNA (Fig. 3(C–E)). This may, in fact, drive the unexpected clustering of the brain E16.5 SSU rRNA sample in (Fig. 3(A)) with adult tissues. The reason for the observed subunit bias is not known. In principle, it could be related to changes in transcription rate and/or ribosome biogenesis. SSU rRNA is transcribed first and it is known from studies in yeast, that there is more co-transcriptional modification and pre-rRNA processing in the SSU compared to the LSU rRNA [27,48]. Establishment of rRNA 2´-O-methylation patterns involves a complex series of interlaced base pairing interactions between SNORDs and nascent pre-rRNA. It is therefore more than likely that kinetic features governing snoRNA:rRNA duplex formation and dissociation as well as the accessibility of the target nucleotide in the context of the folded rRNA impose tight temporal and spatial constraints [12,29,30]. One can intuitively reason that increased rDNA transcription rates, as expected in developing cells or exponentially growing human cancer cells in vitro, might limit the window of opportunity for a subset of SNORD base-pairing, thus reducing the probability of their methylation sites to be heavily modified. This point of view may imply that decreased methylation at some rRNA sites simply accompanies cellular growth and/or cell differentiation and could be interpreted as biological noise or ‘bystander effect’ without necessarily functional consequences. Another possibility is changes in availability of active SNORDs, although the differences are less likely to derive from changes in Fibrillarin levels that have been shown to preferentially affect sites in LSU rRNA [29,30].
A more interesting possibility is that the methylation changes affect the functional properties of the ribosome. In the context of the specialized ribosome hypothesis [1–5], we have demonstrated that ribosome heterogeneity exist in the mouse and varies during development, but the functional implications of this were not directly addressed. The variable SSU rRNA sites do not display any obvious structural clustering and most sites are buried in the mature ribosome with SSU-A590 as the exception. However, the SSU serves dual functions in mRNA recruitment and translation. An attractive hypothesis is that the methylation changes could be part of developmentally-regulated reprogramming, possibly by favouring translation of a subset of embryonic mRNAs. As a proof of principle, altered rRNA methylation profiles, as observed in Fibrillarin-depleted cells, affect IRES-dependent translation both in vivo and in hybrid in vitro translation assays [29].
Splicing as a regulator of SNORD expression
In mammals, all methylation guiding SNORDs are intron-encoded and processed from precursor transcripts by exonucleolytic trimming of the spliced and debranched, linearized host-intron. This implies that SNORD expression is sensitive to alternative splicing, as this dictates both the introns available for processing and the structure of the substrate to be processed, e.g. the distance of the SNORD from the branch site that appears to be a factor in expression [38]. Moreover, alternative splicing is coupled to non-sense mediated decay (NMD) and a model has been put forward that suggest uncoupling of the expression of host-gene exon- and intron-encoded transcripts by alternative splicing and NMD [49]. Usually, NMD is elicited by illegitimate stop codons. The Gas5 transcript may encode short peptides but does not encode a conserved protein. Frame-shift mutations in the longest open reading frame (ORF) occur naturally in inbred mouse strains without obvious deleterious phenotypes [50]. However, it is frequently observed that lncRNA are associated with ribosomes [51] and thus may be subject to NMD, possibly related to the existence of short ORFs. In this context, it is interesting to note that the longest ORF in regularly spliced mouse Gas5 is 72 amino acids, the last codon of which is generated by splicing of intron 6 and directly followed by a stop codon. The predominant alternative splicing in the mouse involves skipping of exon 7 resulting in replacement of the terminal serine with an arginine and extension of the putative peptide by two additional amino acids before a stop codon is encountered. Thus, the stop codon in relation to splice junctions differs in the two transcripts which could impact their relative sensitivity to NMD that depends on the location of stop codons in relation to downstream splice junctions. In a broader context, a multitude of different Gas5 spliced expressed sequence tags (EST) have been described as well as five main mRNA isoforms that all have a 5´ splice site at the exon 6 – intron 6 junction in common and differ in having 3´ splice sites at intron 6 (the predominant form), 7 (the main splice variant), 8 or 10 (two variants that differ at their 3´ end). In terms of overall Gas5 levels, transcriptional mechanisms combined with NMD are responsible for low Gas5 levels during differentiation, whereas decreased NMD underlie the increased Gas5 levels associated with growth arrest due to, e.g. increased cell density [39]. Thus, the complex and diverse regulatory mechanisms that govern Gas5 expression provide an ideal framework for regulating expression of intron-encoded SNORDs through alternative splicing and NMD. Consistent with this notion, we observed that the overall levels of Gas5 expression was quite constant during brain development (Fig. 4(F)) while the alternative splicing related to alternative splicing of exon 7 varied considerably (Fig. 5(D–E)) and correlated with the expression levels of SNORD78 (Fig. 4(D–E)) and the cognate methylation at LSU-G4593 (Fig. 3(D)). This is unusual as the correlation between expression levels of SNORDs or other features of SNORDs and methylation of their cognate target sites in previous reports were weak, at best [12,29,30]. Processing of SNORD78 into smaller fragments [35] as an alternative explanation for the observed down-regulation was unlikely because such fragments were absent in northern blots and in the low-coverage RNA-seq that is part of the RiboMeth-seq analysis. In conclusion, we propose that SNORD78 is specifically down-regulated early during development by alternative splicing and processing of Gas5 transcripts which in turn results in very low levels of the cognate ribose methylation at LSU-G4593 in differentiated adult tissues (Fig. 5(E)).
CRISPR-Cas9 mediated knock-out of SNORD78 and the cognate LSU-Gm4593
LSU-Gm4593 is located at the base of Helix 95 close to the four-way junction in Domain VI of the large ribosomal subunit. Helix 95 presents the sarcin-ricin loop for interaction with translational GTPases [52]. We thus speculated that ribose methylation, by stabilizing base-paired segments, may modulate the conformational flexibility of Helix 95 which could, in turn, fine-tune translation. Further indications of the importance of this modification come from the association of Gas5 and SNORD78 with cancer. The Gas5 gene is frequently reported to be misexpressed in cancers, with its down-regulation associated with poor prognosis [53,54]. Conversely, up-regulation of SNORD78 correlates with prostate cancer [33], hepatocellular carcinoma [34] or non-small cell lung cancer [36,55]. As a first attempt to establish an experimental system to assess the cellular function, if any, of LSU-Gm4593, we used the CRISPR-Cas9 system to knock-out SNORD78 from HEK293T cells that normally has almost full methylation of the site. The constitutive lack of LSU-Gm4593 in exponentially growing HEK293 cell did not yield any overt cellular phenotypes, even though translation fidelity was slightly affected in one of two SNORD78-KO clones. Strikingly, our cellular phenotypic analysis does not corroborate with conclusions drawn from another study where stably shRNA-mediated knock-down of SNORD78 in H1975 cells was accompanied by decreased proliferation, G0/G1 arrest and increased apoptosis [36]. The reason of such discrepancies remains to be further clarified. Yeast entirely lacks ribose methylations in all of Domain VI, so a strong effect on basic properties of translation or cell growth was not to be expected in a cell culture setting. In our opinion, the approach of deleting individual modifications and scoring the effect in cultured cells may turn out to be naïve and the effects of SNORD78-KO will most likely only be revealed when ribosome functions are challenged in more biologically relevant settings. As a prelude to such studies, we have here demonstrated the full pattern of changes, including that of LSU-Gm4593/SNORD78 in a biological context.
Materials and methods
Cell culture
HEK293T cells (a gift from Dr. L. Haren, CBI-Toulouse) were grown at 37°C with 5% CO2 in Dulbecco’s Modifier Eagle Medium (Gibco; 4.5 g/L glucose) supplemented with 10% fetal bovine serum (PAN biotech), 1 mM Sodium Pyruvate (Gibco) and 1% penicillin-streptomycin (Sigma-Aldrich).
Isolation of whole-cell RNA from cells and mouse tissues
Whole-cell RNA was isolated from cells using Tri-Reagent (Euromedex) followed by RQ1 RNAse-free DNAse (Promega) and proteinase K (Sigma-Aldrich) treatments. RNA was then extracted with phenol-chloroform (saturated with water) and ethanol precipitated before storage at −20°C in RNAse-free water. Whole head samples were used to represent brain in E9.5 and E12.5 embryos. E16.5 and adult mouse tissues (C57BL/6J obtained from Charles River) were manually dissected according to French institutional guidelines and quickly frozen in liquid nitrogen before storage at −80°C. Frozen tissues were homogenized in Tri-Reagent (Euromedex) using an Ultra-Turrax homogenizer and whole-cell RNA was isolated according to the manufacturer. RNA was subsequently treated with DNase, proteinase K, and extracted and precipitated as described above.
Ribometh-seq analysis
Five micrograms of whole-cell RNA was degraded by alkaline hydrolysis, size fractionated and ligated to adaptors as previously described [27,56]. In brief, a 20–40 nucleotide fraction of alkaline degraded RNA was excised and purified from 10% polyacrylamide/7 M urea denaturing gels. Adaptors were ligated to the RNA using a tRNA ligase and cDNA was synthesized using SuperScript IV reverse transcriptase (Thermo Fisher Scientific). Libraries (cDNA) were sequenced using the Ion Proton sequencing platform, reads were mapped against a curated mouse rDNA sequence (Table S1 and Data S1) and RiboMeth-seq scores (RMS) representing ‘fraction methylated’ were calculated as described previously (‘score C’) in [27]. In a few cases a barcode correction was applied when calculating the RMS [45]. Sites that have been barcode corrected are indicated in the excel sheet with RiboMeth-seq data (Data S2) and at NCBI Gene Expression Omnibus: GSE128947. All analyses were conducted in biological triplicates.
Northern blotting analysis
Ten micrograms of whole-cell RNA was fractionated by electrophoresis on a 6% polyacrylamide/7 M urea denaturing gel. Electro transferred onto a nylon membrane (Amersham Hybond-N, GE Healthcare) followed by UV crosslinking (Stratalinker). Hybridizations were carried out with 5′-end 32P-labelled-DNA oligonucleotide probes (Table S2). Membranes were incubated overnight at 50°C in 5× SSPE, 5× Denhardt’s, 1% SDS, 150 μg/mL yeast tRNA and washed twice in 0.1% SSPE, 0.1% SDS for 15 min at room temperature. Radioactive signals from the probes were revealed using a Typhoon™ Biomolecular Imager (Amersham) and intensities were quantified using Multi Gauge V3.0 software.
Detection of ribose methylation by primer extension
The presence of 2´-O-methylation was tested by primer extension using low dNTP concentration [57]. Five micrograms of whole-cell RNA was reverse transcribed in 25 µl 1× RT buffer at 42°C for 60 min using AMV (Promega, 20 U) at two dNTP concentrations (1 mM and 0.01 mM, respectively). Primer sequences can be found in Table S2. cDNAs were fractionated by electrophoresis on a 8% polyacrylamide/7 M urea denaturing gel, the gel was dried and radioactive signals from the probes were revealed using a Typhoon™ Biomolecular Imager (Amersham).
Analysis of Gas5 expression by RT-qPCR
One microgram of whole-cell RNA was reverse transcribed with random hexamer primers using Go Script™ Reverse Transcriptase kit (Promega) at 42°C for 60 min. 1/20 of the RT reaction was amplified by PCR using GoTaq polymerase (Promega). mRNA expression was performed using the IQ™ Custom SYBR® Green Supermix (Bio-Rad) qPCR on a CFX96TM Bio-Rad Real-Time Thermal Cycler. The relative quantification of gene expression was performed using the standard curve method with triplicates for each datapoint. Primer sequences can be found in Table S2.
Analysis of Gas5 splicing
RNA splicing at Gas5 pre-mRNA were assayed by RT-PCR, as described above. DNA products were then fractionated by electrophoresis on a 2% agarose gel and stained by ethidium bromide. The identity of the RT-PCR products was further confirmed by DNA sequencing and the relative intensities of PCR products were quantified using Image J software. Primer sequences can be found in Table S2.
Generation and characterization of SNORD78-deficient HEK293 cells
The human SNORD78 gene was disrupted via CRISPR/Cas9-mediated deletion. Two 20-bp guide sequences, sgRNA1 and sgRNA2 (Table S2) were selected using ATUM’s design tool (https://www.atum.bio/products/crispr) and cloned into pX458 (Addgene #48138) and pX459 V2.0 (Addgene #62988) plasmids which contains a GFP marker and a puromycin resistance cassette, respectively. HEK293T cells seeded in six-well dishes (70–80% confluency) were transfected using lipofectamine 2000 (Thermo Fisher Scientific) with pX459_sgRNA1 and pX459_sgRNA2 (500 ng each). Forty-eight hours post-transfection, cells were transiently treated with puromycin (2 µg/mL; 36–48 h) before being subjected to clonal selection by limiting dilution. Expected deletion events were validated by PCR (primer sequences are listed in Table S2). Two clones (WT1 and KO1) were randomly chosen for further analyses. HEK293Tcells were also transfected with pX458_sgRNA1 and pX458_sgRNA2 as described above. Forty-eight hours post-transfection, cells were suspended in PBS, 2 mM EDTA, 0.5% BSA, filtered (40 µM) and single cells were sorted in 96-well plates using fluorescence-activated cell sorting (BD INFLUX). Single cells were expanded and screened for the targeted deletion as described above. Two clones (WT2 and KO2) were randomly chosen for further analyses. The rate of cell growth was assayed using Beckman Coulter Z1 particle counter. Cells were seeded into a 24-well plate (50,000 cells per well) and counted in quadruplicate at each time points. Four independent proliferation assays were performed. Cell cycle profiles were monitored using propidium iodide DNA staining. Cells were harvested and fixed in ice-cold 70% ethanol, washed in PBS + 0.5% Tween and suspended in PI/RNase staining buffer (BD Pharmingen). Data were collected on a FACSCalibur cytometer (Becton Dickinson) and analysed using FlowJo software (FlowJo, LCC). Two independent experiments were performed. Apoptosis was detected using Annexin V-FITC/propidium iodide staining according to the manufacturer’s instructions (BioLegend). Cells were analysed using FacsVerse (Becton Dickinson) with acquisition of 20,000 total events (BD FacsSuite). Two independent experiments were performed.
Dual luciferase reporter assay
100,000 cells were seeded into a 24-well plate for 24 h. Cells were transfected using lipofectamine 2000 (Invitrogen) with 0.06 ng Renilla vector (internal control) and 500 ng of reporter Firefly vectors (a gift of Drs. Anne-Catherine Prats and Frederic Catez). Twenty-four hours post-transfection, cells were harvested, lysed and luciferase activity was monitored using the Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer. Luciferase detection was assayed on Centro LB 960 Microplate Luminometer (Mikrowin 2000 Software). Six independent transfections were performed with each measurement made in triplicate.
Statistical methods
Results are expressed as mean ± standard error of the mean (SEM) except for RiboMeth-seq, cell cycle profiling, and apoptosis assay results which were presented as mean ± standard deviation (SD). Comparison of two groups was analysed by two-tailed Student’s unpaired t-test. Statistically significant differences between groups are indicated as *P < 0.05, **P < 0.01, and ***P < 0.001. Hierarchical cluster analysis and heatmap were generated in R using the pheatmap package (https://CRAN.R-project.org/package=pheatmap). A number of different clustering methods separated robustly embryonic and adult tissues. In the figure, the result using default settings and the complete linkage method was used.
Funding Statement
The work was supported by the Lundbeck Foundation under grant [R198-2015-174; to HN], the Danish Cancer Society under grant [R167-A10943-17-S2; to NK], and the Agence Nationale de la Recherche under grant [ANR-18-CE12-0008-01; to JC].
Acknowledgments
We are grateful to A. Zakaroff-Girard and E. Riant (Cytometry Core Facility, INSERM U1048, part of TRI Imaging Platform, Genotoul) and V. Daburon (CBI-LBCMCP, Toulouse) for cytometry analysis, advice and technical assistance. We thank Sowmya Ramachandran for critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability
RiboMeth-seq data have been deposited at GEO under accession number GSE128947.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Gilbert WV. Functional specialization of ribosomes? Trends Biochem Sci. 2011;36:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Guo H. Specialized ribosomes and the control of translation. Biochem Soc Trans. 2018;46:855–869. [DOI] [PubMed] [Google Scholar]
- [3].Shi Z, Barna M. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol. 2015;31:31–54. [DOI] [PubMed] [Google Scholar]
- [4].Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ferretti MB, Karbstein K. Does functional specialization of ribosomes really exist? RNA. 2019;25:521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Taoka M, Nobe Y, Yamaki Y, et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018;46:9289–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lafontaine DL. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat Struct Mol Biol. 2015;22:11–19. [DOI] [PubMed] [Google Scholar]
- [8].Reichow SL, Hamma T, Ferre-D’Amare AR, et al. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. [DOI] [PubMed] [Google Scholar]
- [10].Cavaille J, Nicoloso M, Bachellerie JP. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383:732–735. [DOI] [PubMed] [Google Scholar]
- [11].Kiss-Laszlo Z, Henry Y, Bachellerie JP, et al. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell. 1996;85:1077–1088. [DOI] [PubMed] [Google Scholar]
- [12].Krogh N, Jansson MD, Hafner SJ, et al. Profiling of 2ʹ-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016;44:7884–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dupuis-Sandoval F, Poirier M, Scott MS. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA. 2015;6:381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cavaille J. Box C/D small nucleolar RNA genes and the Prader-Willi syndrome: a complex interplay. Wiley Interdiscip Rev RNA. 2017;8(4):e1417. [DOI] [PubMed] [Google Scholar]
- [15].Newton K, Petfalski E, Tollervey D, et al. Fibrillarin is essential for early development and required for accumulation of an intron-encoded small nucleolar RNA in the mouse. Mol Cell Biol. 2003;23:8519–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tollervey D, Lehtonen H, Jansen R, et al. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. [DOI] [PubMed] [Google Scholar]
- [17].Bouffard S, Dambroise E, Brombin A, et al. Fibrillarin is essential for S-phase progression and neuronal differentiation in zebrafish dorsal midbrain and retina. Dev Biol. 2018;437:1–16. [DOI] [PubMed] [Google Scholar]
- [18].Baudin-Baillieu A, Fabret C, Liang XH, et al. Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 2009;37:7665–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liang XH, Liu Q, Fournier MJ. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA. 2009;15:1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu B, Liang XH, Piekna-Przybylska D, et al. Mis-targeted methylation in rRNA can severely impair ribosome synthesis and activity. RNA Biol. 2008;5:249–254. [DOI] [PubMed] [Google Scholar]
- [21].Baxter-Roshek JL, Petrov AN, Dinman JD. Optimization of ribosome structure and function by rRNA base modification. PloS One. 2007;2:e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sharma S, Lafontaine DLJ. ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem Sci. 2015;40:560–575. [DOI] [PubMed] [Google Scholar]
- [23].Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Esguerra J, Warringer J, Blomberg A. Functional importance of individual rRNA 2ʹ-O-ribose methylations revealed by high-resolution phenotyping. RNA. 2008;14:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Higa-Nakamine S, Suzuki T, Uechi T, et al. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res. 2012;40:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang J, Sharma S, Watzinger P, et al. Mapping of complete set of ribose and base modifications of yeast rRNA by RP-HPLC and mung bean nuclease assay. PloS One. 2016;11:e0168873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Birkedal U, Christensen-Dalsgaard M, Krogh N, et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew Chem. 2015;54:451–455. [DOI] [PubMed] [Google Scholar]
- [28].Krogh N, Nielsen H. Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods. 2019;156:5–15. [DOI] [PubMed] [Google Scholar]
- [29].Erales J, Marchand V, Panthu B, et al. Evidence for rRNA 2ʹ-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci U S A. 2017;114:12934–12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sharma S, Marchand V, Motorin Y, et al. Identification of sites of 2ʹ-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci Rep. 2017;7:11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang JH, Zhang XC, Huang ZP, et al. snoSeeker: an advanced computational package for screening of guide and orphan snoRNA genes in the human genome. Nucleic Acids Res. 2006;34:5112–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Maden BE, Corbett ME, Heeney PA, et al. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie. 1995;77:22–29. [DOI] [PubMed] [Google Scholar]
- [33].Martens-Uzunova ES, Hoogstrate Y, Kalsbeek A, et al. C/D-box snoRNA-derived RNA production is associated with malignant transformation and metastatic progression in prostate cancer. Oncotarget. 2015;6:17430–17444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ma P, Wang H, Han L, et al. Up-regulation of small nucleolar RNA 78 is correlated with aggressive phenotype and poor prognosis of hepatocellular carcinoma. Tumour Biol. 2016;37:15753–15761. [DOI] [PubMed] [Google Scholar]
- [35].Su H, Xu T, Ganapathy S, et al. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene. 2014;33:1348–1358. [DOI] [PubMed] [Google Scholar]
- [36].Zheng D, Zhang J, Ni J, et al. Small nucleolar RNA 78 promotes the tumorigenesis in non-small cell lung cancer. J Exp Clin Cancer Res. 2015;34:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hirose T, Steitz JA. Position within the host intron is critical for efficient processing of box C/D snoRNAs in mammalian cells. Proc Natl Acad Sci U S A. 2001;98:12914–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Smith CM, Steitz JA. Classification of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5ʹ-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol Cell Biol. 1998;18:6897–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pickard MR, Williams GT. Molecular and cellular mechanisms of action of tumour suppressor GAS5 LncRNA. Genes (Basel). 2015;6:484–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Iyer-Bierhoff A, Krogh N, Tessarz P, et al. SIRT7-dependent deacetylation of fibrillarin controls histone H2A methylation and rRNA synthesis during the cell cycle. Cell Rep. 2018;25:2946–2954 e2945. [DOI] [PubMed] [Google Scholar]
- [41].Basu A, Das P, Chaudhuri S, et al. Requirement of rRNA methylation for 80S ribosome assembly on a cohort of cellular internal ribosome entry sites. Mol Cell Biol. 2011;31:4482–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Marcel V, Ghayad SE, Belin S, et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013;24:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schmidt EK, Clavarino G, Ceppi M, et al. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. [DOI] [PubMed] [Google Scholar]
- [44].Nazar RN, Sitz TO, Busch H. Tissue specific differences in the 2ʹ-O-methylation of eukaryotic 5.8S ribosomal RNA. FEBS Lett. 1975;59:83–87. [DOI] [PubMed] [Google Scholar]
- [45].Krogh N, Kongsbak-Wismann M, Geisler C, et al. Substoichiometric ribose methylations in spliceosomal snRNAs. Org Biomol Chem. 2017;15:8872–8876. [DOI] [PubMed] [Google Scholar]
- [46].Ruggero D. Revisiting the nucleolus: from marker to dynamic integrator of cancer signaling. Sci Signal. 2012;5:pe38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2010;50:131–156. [DOI] [PubMed] [Google Scholar]
- [48].Kos M, Tollervey D. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell. 2010;37:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lykke-Andersen S, Chen Y, Ardal BR, et al. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014;28:2498–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Muller AJ, Chatterjee S, Teresky A, et al. The gas5 gene is disrupted by a frameshift mutation within its longest open reading frame in several inbred mouse strains and maps to murine chromosome 1. Mamm Genome. 1998;9:773–774. [DOI] [PubMed] [Google Scholar]
- [51].Zeng C, Fukunaga T, Hamada M. Identification and analysis of ribosome-associated lncRNAs using ribosome profiling data. BMC Genomics. 2018;19:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Maracci C, Rodnina MV. Review: translational GTPases. Biopolymers. 2016;105:463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gao Q, Xie H, Zhan H, et al. Prognostic values of long noncoding RNA GAS5 in various carcinomas: an updated systematic review and meta-analysis. Front Physiol. 2017;8:814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li W, Li N, Shi K, et al. Systematic review and meta-analysis of the utility of long non-coding RNA GAS5 as a diagnostic and prognostic cancer biomarker. Oncotarget. 2017;8:66414–66425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Su J, Liao J, Gao L, et al. Analysis of small nucleolar RNAs in sputum for lung cancer diagnosis. Oncotarget. 2016;7:5131–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Krogh N, Birkedal U, Nielsen H. RiboMeth-seq: profiling of 2ʹ-O-Me in RNA. Methods Mol Biol. 2017;1562:189–209. [DOI] [PubMed] [Google Scholar]
- [57].Maden BE. Mapping 2ʹ-O-methyl groups in ribosomal RNA. Methods. 2001;25:374–382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RiboMeth-seq data have been deposited at GEO under accession number GSE128947.