

ABSTRACT
Once believed to be unique features of neoplasia, chimeric RNAs are now being discovered in normal physiology. We speculated that some chimeric RNAs may be functional precursors of genes, and that forming chimeric RNA at the transcriptional level may be a ‘trial’ mechanism before the functional element is fixed into the genome. Supporting this idea, we identified a chimeric RNA, HNRNPA1L2-SUGT1 (H-S), whose sequence is highly similar to that of a ‘pseudogene’ MRPS31P5. Sequence analysis revealed that MRPS31P5 transcript is more similar to H-S chimeric RNA than its ‘parent’ gene, MRPS31. Evolutionarily, H-S precedes MRPS31P5, as it can be detected bioinformatically and experimentally in marmosets, which do not yet possess MRPS31P5 in their genome. Conversely, H-S is minimally expressed in humans, while instead, MRPS31P5 is abundantly expressed. Silencing H-S in marmoset cells resulted in similar phenotype as silencing MRPS31P5 in human cells. In addition, whole transcriptome analysis and candidate downstream target validation revealed common signalling pathways shared by the two transcripts. Interestingly, H-S failed to rescue the phenotype caused by silencing MPRS31P5 in human and rhesus cells, whereas MRPS31P5 can at least partially rescue the phenotype caused by silencing H-S in marmoset cells, suggesting that MRPS31P5 may have further evolved into a distinct entity. Thus, multiple lines of evidence support that MRPS31P5 is not truly a pseudogene of MRPS31, but a likely functional descendent of H-S chimera. Instead being a gene fusion product, H-S is a product of cis-splicing between adjacent genes, while MRPS31P5 is likely produced by genome rearrangement.
KEYWORDS: Chimeric RNA, gene evolution, HNRNPA1L2, SUGT1, MRPS31P5, RNA-Seq
Introduction
How does a novel gene evolve is a fundamental question in biology. Gene duplication [1], genomic rearrangement [2], transposable elements domestication [3], and lateral gene transfer [4] are known mechanisms of novel gene formation. Among them, gene duplication is considered to be a dominant mechanism in the creation of thousands of new genes [1]. Genomic rearrangements, including inversions, translocations, and deletions, have the possibility of combining sequences from different genes to make a new gene [2]. Transposable elements (TE) are DNA sequences which can move within a genome and consist of two classes: retrotransposons and transposons. Retrotransposons are first transcribed into RNA and then inserted into the genome by reverse transcription, while DNA transposons utilize transposase enzyme to move around in the genome [3]. Lateral gene transfer refers to the transfer of DNA sequences between different organisms, including prokaryotes to eukaryotes [4].
A Chimeric RNA is defined as a transcript consisting of combined nucleotide sequences from different genes [5]. The first well known chimeric RNA was the transcriptional product of the fusion gene BCR-ABL, resulting from the ‘Philadelphia Chromosome’ [6]. Although traditionally thought to be unique features of cancer cells, chimeric RNAs are also widely present in normal physiology [7–14], and several chimeric RNAs have also been demonstrated to play critical roles in cell maintenance, motility, and stemness [15–18]. As more functional chimeric RNAs are being identified, we speculate that they may serve as precursors of more stable hereditary elements, i.e. genes. We reason that during evolution, a chimeric RNA may have been produced at the transcriptional level and been beneficial to the species; through evolutionary selection, the chimeric RNA can be fixed into the genome as a new gene. If this hypothesis is true, we expect that the chimeric RNA occurs first in evolution, while its descendant gene follows. The RNA should play similar functional roles with the gene. It may also be true that once the gene is formed in evolution and takes over the functional role the chimeric RNA plays, the chimeric RNA is repressed.
In this study, we developed a pipeline to discover such chimeric RNAs. By analysing RNA-Seq data from the TCGA bladder cancer study [19], we successfully identified six chimeric RNAs with highly similar junction sequences matching to six intact gene transcripts. Among them, one chimeric RNA was found to exist in marmosets, rhesus monkeys, and humans, while the corresponding gene is found in rhesus monkeys and humans, but not in marmosets. Intriguingly, silencing the gene in human cells and silencing the chimera in marmoset cells resulted in a similar cellular phenotype. RNA-sequencing analysis also revealed highly similar whole transcriptome changes associated with these perturbations in the two species, although rescue experiments suggested that they have distinct function. We further investigated the mechanism for chimeric RNA formation in marmosets and found that it is a product of cis-splicing between adjacent genes.
Results
The discovery of chimeric RNAs and matched genes in human
In order to identify candidate genes derived from chimeric RNAs, we developed a pipeline outlined in Fig. 1(A). From 433 RNA-Sequencing datasets from the TCGA bladder cancer study [20], we identified 778 unique chimeric RNAs [19]. We then selected 50 bp upstream and 50 bp downstream of the junction as input, and blasted it against the Refseq_RNA database. Through this procedure, we identified 356 chimeric RNAs that matched to the transcript of another intact gene with greater than 90% similarity in sequence. We then verified the alignment between the candidate chimeric RNAs and genes individually and found that the majority of these matches are due to incorrect annotation of the chimeric RNAs or repeat sequences (SINE, LINE, LTR etc.). After filtering out these false positives, six candidates of chimeric RNA and gene pairs remained (Table S1).
Figure 1.
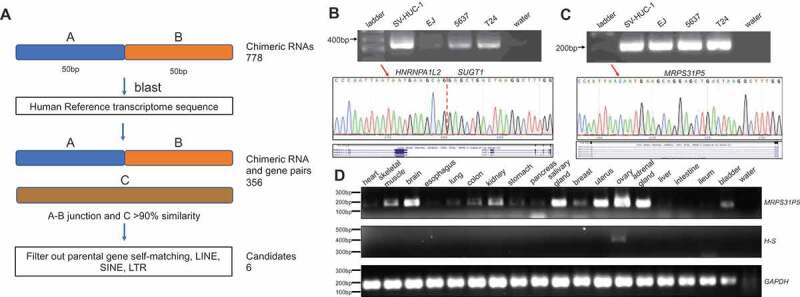
Discovery of chimeric RNAs and their potential descendent genes.
(A) the pipeline for discovering potential genes derived from chimeric RNAs. The junction sequence of 778 chimeric RNAs with 50bp on each side was used to blast against reference RNA. Candidates were identified when over 90% similarity was found from the transcript of an intact gene. After filtering out false matches and repeat sequences, six candidates remained. (B,C) upper panels, RT-PCR of HNRNPA1L2-SUGT1 and MRPS31P5 in four different bladder cancer cell lines; middle panels, Sanger sequencing of chimeric RNA HNRNPA1L2-SUGT1 and MRPS31P5, with red arrows highlighting the nucleotide difference between HNRNPA1L2-SUGT1 and MRPS31P5; bottom panel, the sequence alignment in UCSC Genome Browser. (D) RT-PCR of MRPS31P5 and HNRNPA1L2-SUGT1 in normal human tissues.
Primers were then designed to amplify the chimeric RNAs and genes in human bladder cancer cells. PCR products were validated through Sanger sequencing. We were able to detect the HNRNPA1L2 (NM_001011725.1)-SUGT1 (NM_006704.4) (H-S) chimeric RNA and MRPS31P5 (NR_051963.1) (Fig. 1(B,C)). We could successfully amplify the other five pseudogenes and validated by Sanger sequencing, but failed to specifically amplify the corresponding five chimeric RNA (data not shown).
We then examined the expression of HNRNPA1L2, SUGT1, and MRPS31P5 in The Genotype-Tissue Expression (GTEx) database, which contains RNA sequencing data for a large number of normal human tissues (Fig. S1). MRPS31P5 exhibited comparable expression to both HNRNPA1L2 and SUGT1 in a wide range of human tissues, whereas the chimeric RNA was not found in the GTEx RNA-Sequencing analysis, suggesting that the chimera may be minimally expressed in normal physiology. Experimentally, MRPS31P5 was detected in a wide range of human tissue samples, while H-S was only detectable in human ovary tissue samples (Fig. 1(D), and Fig. S2).
MRPS31P5 has higher sequence similarity to the fusion than to MRPS31
MRPS31P5 is annotated as Mitochondrial Ribosomal Protein S31 pseudogene. We first compared the full length transcript sequence of MRPS31P5 to MRPS31 (NM_005830.4) and then to H-S chimera. Both transcripts have similar sequences to the fragment of MRPS31P5 containing the 5ʹ end to around 1000 bp. However, the scores and percent identities showed higher similarity of MRPS31P5 to H-S chimeric RNA, then to MRPS31 (the alignment showed that the similarity of MRPS31P5 to MRPS31 was 81% while its similarity to the H-S chimera was 95%) (Fig. S3). We then examined the whole genomic locus of MRPS31P5, ad compared it to the sequencing covering HNRNPA1L2 and SUGT1 (HNRNPA1L2 and SUGT1 are two neighbouring genes), as well as the genetic sequence of MRPS31. Analysis of these whole gene locus sequences including introns, the percent identities between MRPS31P5 and MRPS31 was found to be 88%, while its similarity to the fragment containing the fusion parental genes HNRNPA1L2 and SUGT1 was 93%. These findings support that MRPS31P5 is more closely related to the HNRNPA1L2 and SUGT1 fusion than to MRPS31. MRPS31P5 was likely wrongly annotated as the pseudogene of MRPS31, due to the unawareness of the existence of the H-S chimera.
H-S and MRPS31P5 in different species
H-S chimeric RNA consists of the sequence of the first five exons from HNRNPA1L2 joined to the final twelve exons of SUGT1. MRPS31P5 has a similar sequence to the first five exons of HNRNPA1L2 and the third and fourth exons of SUGT1 (Fig. S4) The rest of SUGT1 exons do not match to MRPS31P5. In comparison, MRPS31 does not have similar sequence to MRPS31P5 after its exon6. To investigate whether the chimeric RNA exists in other species, we downloaded several RNA-Seq datasets to search for the presence of H-S junction sequence (14 nt on each side) in other primates. We found that H-S exists in rhesus macaques (SRR832944), crab-eating macaques (SRR832917, SRR832918), and orangutans (ERR247256, ERR247255). The MRPS31P5 gene can also be found by searching the genomic sequence using UCSC blat in rhesus macaques, crab-eating macaques, orangutans, gorillas, bonobos, and chimpanzees (Table S2). Interestingly, even though the chimeric RNA sequence was found in the RNA-Seq data of marmosets (SRR850169), MRPS31P5 was not found in the marmoset genome.
Experimentally, we designed primers to specifically amplify the chimeric RNA, but not MRPS31P5, in marmosets and rhesus monkeys. Sanger sequencing results showed the correct sequence of the chimeric RNAs (Fig. 2(A,B)). The configuration of the chimeric RNA is also identical to that in humans, suggesting that the chimera is conserved from marmoset to human. We also designed consensus primers to amplify the fragment of MRPS31P5 transcript. Even though the transcript was readily amplified and confirmed by Sanger Sequencing in rhesus monkeys and humans (Fig. 2(C), Fig. S5A, and 5B), it was not detected in marmosets. Consistently, the emergence of MRPS31P5 was predicted after the separation of marmoset and rhesus, 29.6 Mya (million years ago) by Gentree [21] (Fig. 2(D)), while the flanking genes, NEK3 and THSD1 are conserved in humans, rhesus monkeys and marmosets (Fig. 2(E) and Fig. S5C). HNRNPA1L2 and SUGT1 are neighbouring genes starting from the Platyrrhini in evolution, suggesting that it may be formed via transcriptional readthrough (addressed later). Consistently, the H-S chimeric transcript was detected in marmoset. Thus, we placed the appearance of the chimera before the divergence of Platyrrhini and Catarrhini (44.2 Mya) (Fig. 2(D)).
Figure 2.
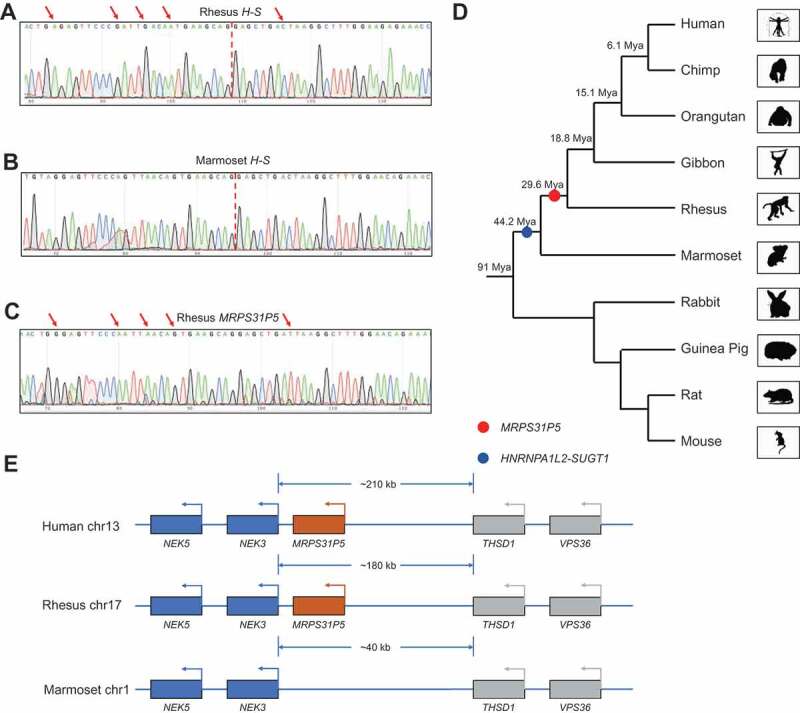
HNRNPA1L2-SUGT1 and MRPS31P5 in evolution.
(A) Sanger sequencing of HNRNPA1L2-SUGT1 in rhesus monkey. (B) Sanger sequencing of HNRNPA1L2-SUGT1 in marmoset. (C) Sanger sequencing of MRPS31P5 in rhesus monkey, with red arrows pointing to the difference between HNRNPA1L2-SUGT1 and MRPS31P5. (D) Phylogenetic tree view of the origination of HNRNPA1L2-SUGT1 (blue dot) and MRPS31P5 (red dot). The evolutionary age data of MRPS31P5 was mined from GenTree. Mya stands for million years ago. (E) Genomic region flanking MRPS31P5 in human, rhesus and marmoset. MRPS31P5 is absent in marmosets
Functional study of Chimeric RNA H-S and MRPS31P5
The chimeric RNA H-S and pseudogene MRPS31P5 are both predicted to be long non-coding RNAs based on sequence analysis. First, we compared the expression of the chimeric RNA between human and marmoset. To do so, we used conserved primers for H-S and selected fibroblast cells of both species to avoid the effect of tissue specificity. qPCR results showed much higher expression levels of H-S in the marmoset fibroblast cell line, CJ-157, than in the human fibroblast cell line HFF. We then used qPCR to evaluate the expression of MRPS31P5 and found that it is abundantly expressed in HFF. Consistent with sequence analysis, no expression of MRPS31P5 could be detected in CJ-157 (Fig. 3(A)).
Figure 3.
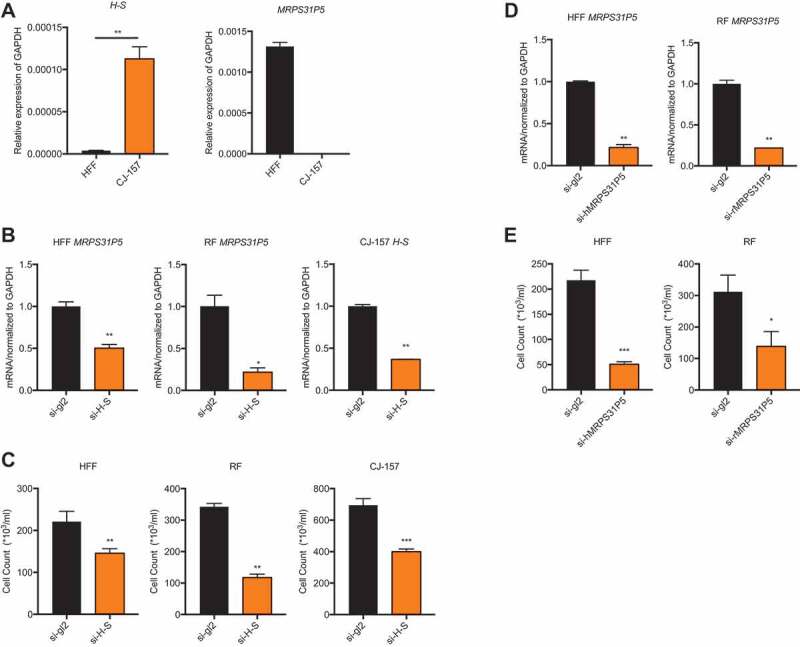
Silencing MRPS31P5 in human and rhesus fibroblasts as well as silencing H-S in marmoset fibroblasts reduced cell proliferation.
(A) left panel, expression level of the chimeric RNA HNRNPA1L2-SGUT1 in the human fibroblast cell line (HFF), and marmoset fibroblast cell line (CJ-157); right panel, MRPS31P5 is expressed in HFF, while the it does not exist in marmoset. (B) With siRNAs targeting the junction sequence (si-H-S), qRT-PCR showed significantly knocking down of MRPS31P5 in HFF and RF, H-S in CJ-157. (C) With transfection of si-H-S, cell number in HFF, RF and CJ-157 were significantly reduced in compared with si-gl2 group. (d) with siRNAs specific for human MRPS31P5 (si-hMRPS31P5) and rhesus monkey MRPS31P5 (si-rMRPS31P5), qRT-PCR showed significant knocking down of MRPS31P5 in both HFF and RF cells. (e) With transfection of si-hMRPS31P5 and si-rMRPS31P5, the cell numbers in HFF and RF were significantly reduced compared with si-gl2 controls.
Sequence similarities between the H-S chimeric RNA and MRPS31P5, coupled with a decrease in expression levels of H-S in normal human tissues, invited speculation that they play similar roles, and that during evolution, the function of H-S was taken over by MRPS31P5. To investigate this possibility, we first designed siRNAs targeting the junction sequence of the H-S chimera in various species. Because H-S and MRPS31P5 share the same junction sequence, the siRNAs targeting H-S also target MRPS31P5 in the RF and HFF cell lines. Consistently, the expression levels of MRPS31P5 was reduced upon transfecting the siRNAs in the HFF and RF cell lines (Fig. 3(B)). We noticed significantly reduced cell numbers in the cells transfected with these siRNAs in comparison to the cells transfected with the negative control si-gl2 (Fig. 3(C)). To determine if this proliferation inhibition was indeed due to the silencing of MRPS31P5 and not the H-S chimera in human and rhesus, we designed specific siRNAs to target the non-homologous sequence part of MRPS31P5 (Fig. S4), which only affected MRPS31P5 transcripts (Fig. 3(D)). Similarly to si-H-S, si-hMRPS31P5 and si-rMRPS31P5 transfection resulted in significantly reduced cell proliferation (Fig. 3(E)). We observed no effect on cell count following transfection of these two siRNAs into marmoset CJ-157 cells, ruling out potential off-target effects on cell proliferation (Fig. S6).
Human and rhesus cells transfected with siRNAs targeting MRPS31P5 and marmoset cells transfected with si-H-S had similar morphological changes: an increase in size and a more spread-out configuration (Fig. S7A), although these morphological changes may be due to more open space caused by slowed proliferation. To further investigate the mechanism for this reduced cell proliferation, we performed propidium iodide (PI) staining and FACS analysis. Even though we detected very few cells in G2 phase, we noticed an increased proportion of cells in G0/G1 phase (Fig. S7B), suggesting a G1/S arrest with the knockdown of H-S or MRPS31P5 in the fibroblast cell lines.
MRPS31P5 in HFF share similar functions with H-S in CJ-157
The phenotype following MRPS31P5 knockdown in human cells strongly argues against its annotation as a pseudogene. We then performed RNA-sequencing comparing whole-transcriptome changes in HFF cells transfected with si-MRPS31P5 versus control si-gl2. Using a fold change of 2 as the cut-off, we found 986 downregulated, and 373 upregulated genes. Gene Ontology analysis [22] (http://cbl-gorilla.cs.technion.ac.il) revealed that almost all top GO terms are related to cell cycle and division in the downregulated genes. For instance, the top three enriched terms were mitotic cell cycle, cell cycle, and cell division processes in down-regulated genes (Fig. 4(A)), consistent with the phenotype we observed. A few terms related to cell response were enriched in upregulated genes, albeit with much less significant p values.
Figure 4.
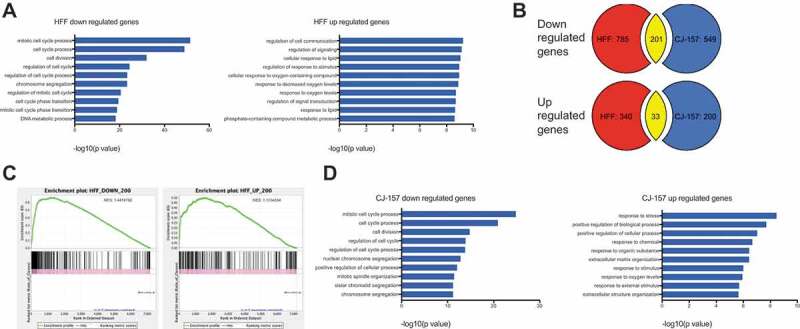
Whole transcriptome analysis of MRPS31P5 silenced HFF cells and H-S silenced CJ-157 cells.
(A) left panel, Gene Ontology analysis of si-hMRPS31P5 downregulated genes in HFF; right panel, GO term analysis of upregulated genes. (B) Venn diagrams illustrating the overlap between common upregulated and downregulated genes in HFF with si-hMRPS31P5 and CJ-157 with si-H-S. There are 201 shared downregulated genes between HFF si-hMRPS31P5 and CJ-157 si-H-S, while 33 genes are commonly upregulated genes between HFF si-hMRPS31P5 and CJ-157 si-H-S. (C) GSEA of CJ-157 si-H-S against the top 200 downregulated or upregulated genes in MRPS31P5-silenced human HFF. (D) left, Gene Ontology analysis of CJ-157 si-H-S downregulated genes. right, Gene Ontology analysis of CJ-157 si-H-S upregulated genes.
We then performed RNA-Seq, comparing marmoset CJ-157 cells transfected with si-H-S versus si-gl2. We noticed a large portion of genes that are downregulated after transfection with si-H-S in marmoset CJ-157 cells are the same genes downregulated in human HFF after si-MRPS31P5 transfection (201 out of 750). In comparison, the commonly upregulated genes are much fewer in number, with only 33 common genes between the two RNA-Seq datasets. We then performed Gene Set Enrichment Analysis (GSEA) [23] using the top 200 downregulated and 200 upregulated genes in human cells as reference. When we used up- and downregulated marmoset genes as queries, significant enrichment was observed in both analyses, with NES scores of 1.4 and 1.1 respectively (Fig. 4(C)). Impressively, GO term analysis for downregulated marmoset genes revealed the exact same terms as in humans, and in a similar successive order (Fig. 4(D)). Though less significant, many GO terms associated with upregulated genes in humans are also seen enriched in the upregulated marmoset genes.
For validation, we first picked the top ten down-regulated genes related to the cell cycle and designed conserved primers for human, rhesus, and marmoset. qPCR results showed that all ten genes were significantly downregulated in the HFF cell line transfected with si-hMRPS31P5. They were also all significantly downregulated in si-rMRPS31P5 transfected rhesus RF cells and si-H-S transfected marmoset CJ-157 cells (Fig. 5(A)). We then randomly picked another ten down-regulated genes in HFF with si-hMRPS31P5. Again, the majority of these genes were also significantly downregulated in RF cells with si-rMRPS31P5 and CJ-157 cells with si-H-S (Fig. 5(B)).
Figure 5.
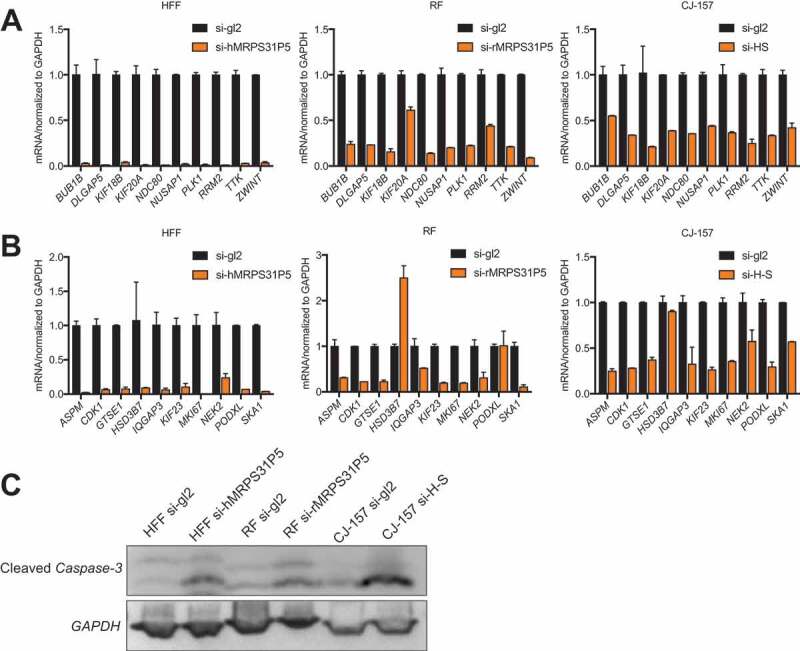
Downstream targets are shared in MRPS31P5-silenced HFF, MRPS31P5-silenced RF cells, and H-S-silenced CJ-157.
(A) qRT-PCR validation of the top ten target genes of si-MRPS31P5 from cell cycle list in HFF, RF and CJ-157 cell lines. (B) ten additional genes from were also tested in HFF, RF and CJ-157 cell line. (C) Western blot measuring cleaved Caspase 3. Cleaved Caspase-3 was increased in si-MRPS31P5 transfected HFF and RF cells, and si-H-S transfected CJ-157 cell line.
As prolonged cell cycle arrest will result in apoptosis, we evaluated cleaved Caspase-3 as an indicator for apoptosis. As shown in Fig. 5(C), we observed a higher level of cleaved Caspase-3 in all the knockdown groups compared to the control group.
H-S chimeras and MRSP31P5 are not functionally equivalent
The above results strongly support that H-S and MRPS31P5 are involved in similar signling pathways. To further investigate whether MRPS31P5 in humans is the functional descendent of H-S chimeric RNA in marmoset, we performed the rescue experiments. First we cloned H-S chimera from marmoset CJ-157, then overexpressed the chimera ino HFF cells, which were transfected with si-hMRPS31P5. If the chimera and MRPS31P5 are functionally equivalent, we expect to observe the rescue of the phenotype caused by silencing MRPS31P5. However, no obvious difference in cell number was observed in HFF cells transfected with empty vector or H-S chimera (Fig. 6(A)). Consistently, no rescue effect was seen in rhesus fibroblast cells as well.
Figure 6.
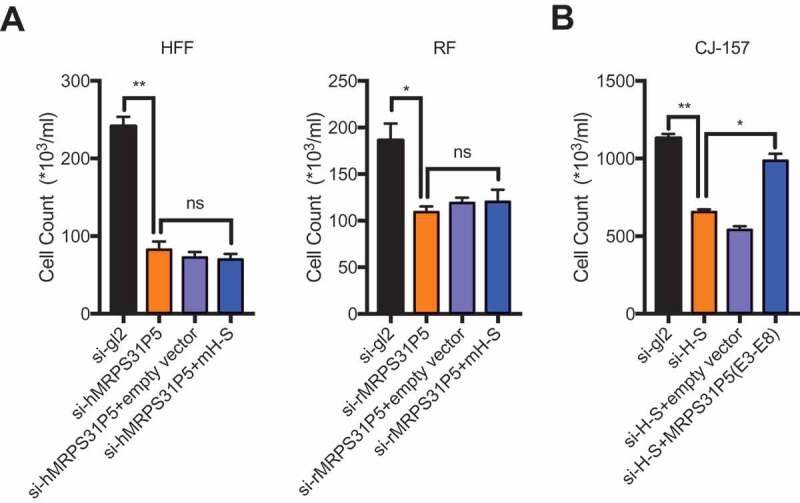
MRPS31P5 and H-S chimera are not functionally equivalent.
(A) In HFF and RF cells, marmoset H-S failed to rescue the reduced cell proliferation, caused by knocking down of hMRPS31P5 or rMRPS31P5. (B) In contrast, in CJ-157 cells, the phenotype caused by knocking down of H-S could partially be rescued by human MRPS31P5.
However, we observed significant rescue effect when we introduced human MRPS31P5 into CJ1-57 cells, which were transfected with si-H-S (Fig. 6(B)), suggesting that human MRPS31P5 can rescue the reduced cell proliferation caused by silencing of H-S in marmosets. These results are consistent with the model that H-S and MRPS31P5 are functional homologues in evolution; and that while MRPS31P5 is the descendent of H-S, it has acquired additional function, presumably due to the additional sequence in exon9.
Chimeric RNA H-S is a product of transcriptional readthrough
Chimeric RNAs can be generated through gene fusion, trans-splicing, and cis-splicing of adjacent genes (cis-SAGe) [24,25]. HNRNPA1L2 and SUGT1 are neighbouring genes located on chromosome 13q14.3 separated by approximately 9 kb and are transcribed in the same direction, making the chimera a candidate for cis-SAGe. We then performed the transcriptional readthrough assay to validate if this chimeric RNA is indeed generated by cis-SAGe in human bladder cancer cell lines. A 3ʹ reverse primer annealing to the SUGT1 exon6 was used to convert RNA to cDNA. A pair of primers annealing to the exon5 and intron5 of HNRNPA1L2 was then used to perform PCR to detect the upstream transcript from the same cDNA (Fig. 7(A)). DNase-I was used to eliminate potential DNA contamination. In both SV-HUC1 and T24 cell lines, we observed the correct size band (Fig. 7(B)), which was confirmed by Sanger sequencing (Fig. S8A). No band was observed in the control group without avian myeloblastosis virus (AMV) reverse transcriptase, suggesting the absence of DNA contamination. Similarly, we observed the transcriptional readthrough signal in marmoset CJ-157 cells (Fig. 7(C)), which was confirmed by Sanger sequencing (Fig. S8b).
Figure 7.
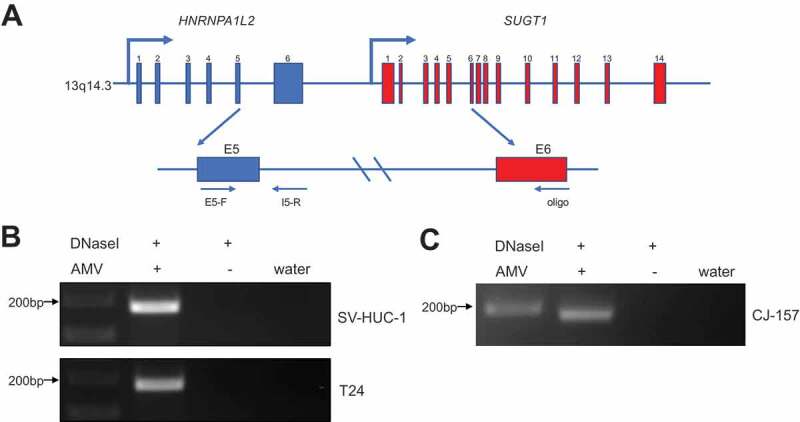
H-S chimeric RNA is a product of cis-SAGe.
(A) The schematic diagram of cis-SAGe assay. RNAs were treated with DNaseI. With a downstream oligo annealing to the exon6 of SUGT1, RNA was reverse transcribed into cDNA. PCR was then performed with a primer pair annealing to the exon5 and intron5 regions of HNRNPA1L2. (B) In bladder cancer cell lines SV-HUC-1 and T24, correct bands were only detected in AMV + group, but not in the group omitting AMV, suggesting the complete removal of DNA template. (C) Same experiment was performed using RNAs extracted from marmoset cell line CJ-157. Same result was observed.
Discussion
The chimeric RNA, H-S precedes the actual gene MRPS31P5. This type of phenomenon is not unprecedented. One prominent example is the jingwei gene, which contains a fragment of a retrotransposed region from Adh [26]. The other human example is PIPSL, which was found to be the product of retrotransposed chimeric RNA, PIP5K1A-PSD4 [27]. Babushok and colleagues confirmed that PIP5K1A-PSD4 was reverse transcribed and integrated into the genome by the L1 retrotransposon [28]. However, several lines of evidence made us conclude that MRPS31P5 is not a product of L1-mediated retrotransposition: 1) MRPS31P5 contains similar intron sequences to HNRNPA1L2 and SUGT1; 2) we failed to detect 7-bp to 20-bp target site duplications, a signature of L1 mediated retrotransposition [29]; and 3) no poly-A tail was found at the end of MRPS31P5 gene. Based on sequence analysis, we propose that MRPS31P5 is a product of duplication involving a fragment covering HNRNPA1L2 and SUGT1, inverted and inserted into the close chromosomal region 424kb away. A fragment covering exon 6 of HNRNPA1L2, exon 1 and 2 of SUGT1, and intergenic region is deleted to connect the first exons of HNRNPA1L2 and exon 3 and 4 of SUGT1. Subsequently, the rest of SUGT1 is replaced with another fragment (Fig. S9). Interestingly, with the junction sequence of PIP5K1A-PSD4, our pipeline can re-discover PIPSL as a descendent gene, proving its capability to also identify retrotransposition-mediated gene evolution.
We have accumulated multiple lines of evidence to support that MRPS31P5 is not a pseudogene of MRPS31, but rather a functional descendent of H-S chimeric RNA: 1) the sequence of the MRPS31P5 transcript is more similar to the H-S chimera than to that of MRPS31; 2) the DNA sequence of MRPS31P5 is more similar to the genomic fragment of HNRNPA1L2 and SUGT1 than to MRPS31; 3) the appearance of H-S chimeric RNA precedes that of MRPS31P5; 4) H-S chimera expression is diminished in human when compared to marmoset, transitioning to MRPS31P5 expression; 5) silencing MRPS31P5 resulted in cell cycle arrest and apoptosis; 6) silencing MRPS31P5 in human cells phenocopied H-S in marmoset cells; 7) silencing MRPS31P5 in human cells and silencing H-S in marmoset cells share similar genome-wide transcriptome changes; and 8) H-S failed to rescue the phenotype caused by silencing MRPS31P5 in human cells, whereas MRPS31P5 at least partially rescued the phenotype caused by silencing H-S in marmoset cells.
An interesting phenomenon we observed is that H-S in normal human tissues is minimally expressed. Consistently, we failed to identify the chimera in GTEx analysis. However, the chimera is readily detectable in cancer cell lines. It may be because the epigenetic factors that normally suppress the H-S chimera become misregulated in cancer. In addition, it may be beneficial for the cancer cells to make use the chimeric RNA to perform some ancient function.
We do not have a definitive mechanism for how the H-S chimeric RNA becomes the MRPS31P5 gene. However, there are several possibilities that chimeric RNA may facilitate novel gene formation. 1) Retrotransposition may occur, as in the case of PIPSL and jingwei genes. As explained earlier, MRPS31P5 does not fit this scheme. 2) The chimeric RNA may function as a guide RNA, bringing the parental genes in close proximity, which facilitates genome rearrangement. This possibility is indicated by a recently published RNA-poise model in which RNA-DNA interaction may constrain the genomic loci to a spatial proximity to facilitate genome rearrangement [30]. 3) Chimeric RNA invades chromosomal DNA to stabilize a transient RNA/DNA duplex using DNA sequences located in two distant genes [31]. DNA repair mechanisms might yield the final gene fusion through recombination in regions prone to DNA breaks. This possibility is supported by evidence that RNA can facilitate DNA repair in both human and yeast cells [32], and further substantiated by the recent discovery that a chimeric RNA mimicking a fusion RNA can drive the formation of common gene fusions in prostate cancer [31]. At the same time, we must also consider the possibility that chimeric RNA does not directly contribute to the formation of the gene. Rather, the same factors such as chromatin structures, close proximity, etc., that facilitate the expression of the chimeric RNA may also facilitate genomic rearrangement for novel gene formation.
Methods and materials
Bioinformatic pipeline for initial Chimeric RNA and matched gene discovery
From the RNA-Seq data of the TCGA bladder cancer study, we identified 778 chimeric RNAs [19]. We then used 50bp on each side of the junction as input to the blat software to compare against the Human Reference transcriptome sequence to confirm parental gene matching. We identified matches of this same sequence to transcripts of other genes using cut-offs of ≥90% sequence identity over 80bp of the junction sequence. 356 hits were identified. Finally, we manually curated these alignments to filter out likely false-positives due to inconsistencies in annotation and repeat elements.
Clinical samples
18 different normal human samples from various tissues were obtained from the Department of Pathology at the University of Virginia. The IRB committee of the University of Virginia approved the use of human clinical samples. All samples were de-identified.
RNA extraction and qRT-PCR
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR) were performed as described previously [33]. Primers are listed in Table S3.
Plasmid construction
pBABE-puro was used for construction of human MRPS31P5 gene and marmoset H-S full length. HFF or CJ-157 cDNA were used for PCR, primers were listed in Table S3.
siRNA transfection
Custom siRNAs were ordered from Invitrogen, and transfection was performed using Lipofectamine® RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. siRNA targeting sequences are listed in Table S4. For rescue experiment, empty vector or overexpression retrovirus were used to infect cells on day 1, followed by transfection with siRNAs one day later.
Cell culture
Human foreskin fibroblast (HFF) and Rhesus fibroblast (RF) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose (Gibco), 10% Foetal Bovine Serum (FBS) (Invitrogen, Gaithersburg, MD) and 1% Pen/Strep solution (Hyclone). The marmoset fibroblasts from the CJ-157 cell line were received from Dr. Behr’s lab, and were cultured in fresh M10 medium [DMEM (Gibco), 10% FBS, 1% Pen/Strep solution (Hyclone), 0.25 μg/mL Amphotericin B (Sigma), 1% MEM Non-Essential Amino Acids Solution (Gibco), and 2 mM GlutaMAX (Gibco)]. Cells were cultivated at 37°C in 5% CO2 humidity.
Propidium iodide (PI) staining and FACS
HFF, RF or CJ-157 cells were trypsinized 72 hours after siRNA transfection, then washed thrice with PBS and fixed with 80% ethanol (overnight in −20 °C). Cells were rehydrated in PBS for 15 minutes then stained by propidium iodide for 6 hours. Finally, cells were subjected to cell cycle analysis using the FACSCalibur flow cytometer. The results were analysed with FlowJo.
RNA sequence
HFF and CJ-157 cells transfected with si-hMRPS31P5, si-H-S, or si-gl2 (control) were harvested 72 hours after transfection. RNA was extracted and purified with QIAGEN RNeasy Mini Kit according to the manufacturer’s instructions. The quality of RNA-Seq raw reads was evaluated using NGSQC Toolkit software with default parameters, and only high quality reads were retained. We then used the featureCounts script from the Subread software package (http://subread.sourceforge.net/) to calculate count data for genes in human and marmoset using their reference annotations from Ensembl version 89. Next, we calculated the FPKM value from the counts data by normalizing it to the length of the gene and by the total number of high quality RNA-Seq reads from the sample. Common genes (using the gene name) between humans and marmosets were extracted. If the FPKM of any gene from all the human samples was less than 1, the entry was discarded. Similarly, if the FPKM of any gene from all the marmoset samples was less than 1, it was discarded. In the end, we were left with 7174 common genes between the two species. To find differentially expressed genes in humans, we calculated the ratio of sample ‘si-gl2_FPKM’ and sample ‘si-hMRPS31P5_FPKM’ and used a cut-off of ≥2 to identify upregulated genes, and a cut-off of ≤0.5 to identify downregulated genes. Similarly, differentially expressed genes were found in marmosets.
Western blotting
Western blotting was conducted according to our previously published procedure [34]. Antibodies for Western blot were anti-Cleaved Caspase-3 (CST, #9661, 1:1000 dilution) and anti-GAPDH (Proteintech, 60004-1-Ig, 1:2000 dilution).
Statistics
Quantitative results were presented as the mean ± standard error of the mean (SEM). Two-tailed t-tests were used for expression and cell number comparisons. GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA, USA) was used for statistical analyses. For all analyses, p < 0.05 was considered statistically significant.
Data access
The RNA-Seq data has been deposited into Gene Expression Omnibus (GEO), with accession number: GSE130594.
Funding Statement
This work was supported by NIH (GM132138) (HL) and China Scholarship Council (CSC, No. 201706370109) (HW).
Acknowledgments
Hui LI was supported by NIH GM132138. Hao Wu was supported by China Scholarship Council (CSC, No. 201706370109). We thank Dr. Rüdiger Behr for providing the marmoset fibroblast cell lines. We thank Justin Elfman and Aadi Sharma for their help with English editing. We thank the Biorepository and Tissue Research Facility (BTRF) at the University of Virginia for providing the clinical samples.
References
- [1].Long MY, VanKuren NW, Chen SD, et al. New gene evolution: little did we know. Annu Rev Genet. 2013;47:307–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rogers RL, Bedford T, Hardl DL.. Formation and longevity of chimeric and duplicate genes in drosophila melanogaster. Genetics. 2009;181(1):313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alzohairy AM, Gyulai G, Jansen RK, et al. Transposable elements domesticated and neofunctionalized by eukaryotic genomes. Plasmid. 2013;69(1):1–15. [DOI] [PubMed] [Google Scholar]
- [4].Sieber KB, Bromley RE, Hotopp JCD. Lateral gene transfer between prokaryotes and eukaryotes. Exp Cell Res. 2017;358(2):421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li Z, Qin FJ, Li H. Chimeric RNAs and their implications in cancer. Curr Opin Genet Dev. 2018;48:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nowell PC. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut. 1962;8:65–66. [DOI] [PubMed] [Google Scholar]
- [7].Li H, Wang JL, Mor G, et al. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321(5894):1357–1361. [DOI] [PubMed] [Google Scholar]
- [8].Yuan HL, Qin F, Movassagh M, et al. A Chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013;3(12):1394–1403. [DOI] [PubMed] [Google Scholar]
- [9].Gorohovski A. ChiTaRS-3.1-the enhanced chimeric transcripts and RNA-seq database matched with protein-protein interactions. Nucleic Acids Res. 2017;45(D1):D790–D795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jividen K, Li H. Chimeric RNAs generated by intergenic splicing in normal and cancer cells. Gene Chromosome Canc. 2014;53(12):963–971. [DOI] [PubMed] [Google Scholar]
- [11].Babiceanu M, Qin F, Xie X, et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016;44(6):2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chwalenia K, Facemire L, Li H. Chimeric RNAs in cancer and normal physiology. Wires Rna. 2017;8:6. [DOI] [PubMed] [Google Scholar]
- [13].Finta C, Zaphiropoulos PG. Intergenic mRNA molecules resulting from trans-splicing. J Biol Chem. 2002;277(8):5882–5890. [DOI] [PubMed] [Google Scholar]
- [14].Ren GP, Zhang Y, Mao X, et al. Transcription-mediated chimeric rnas in prostate cancer: time to revisit old hypothesis? Omics. 2014;18(10):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wu CS, Yu CY, Chuang CY, et al. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24(1):25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xie ZQ, Babiceanu M, Kumar S, et al. Fusion transcriptome profiling provides insights into alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A. 2016;113(46):13126–13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Finckenstein FG, Shahbazian V, Davicioni E, et al. PAX-FKHR function as pangenes by simultaneously inducing and inhibiting myogenesis. Oncogene. 2008;27(14):2004–2014. [DOI] [PubMed] [Google Scholar]
- [18].Tang Y, Qin F, Liu A, et al. Recurrent fusion RNA DUS4L-BCAP29 in non-cancer human tissues and cells. Oncotarget. 2017;8(19):31415–31423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu D, Singh S, Chen X, et al. The landscape of chimeric RNAs in bladder urothelial carcinoma. Int J Biochem Cell Biol. 2019;110:50–58. [DOI] [PubMed] [Google Scholar]
- [20].Robertson AG, Kim J, Al-Ahmadie H. et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171(3):540–556 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shao Y, Chen C, Shen H, et al. GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res. 2019;29(4):682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eden E, Navon R, Steinfeld I, et al. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Subramanian A, Tamayo P, VK Mootha, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Gong M, Yuan H, et al. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012;2(7):598–607. [DOI] [PubMed] [Google Scholar]
- [25].Elfman J, Li H. Chimeric RNA in cancer and stem cell differentiation. Stem Cells Int. 2018;2018:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Long MY, Langley CH. Natural-selection and the origin of Jingwei, a Chimeric processed functional gene in Drosophila. Science. 1993;260(5104):91–95. [DOI] [PubMed] [Google Scholar]
- [27].Akiva P, Toporik A, Edelheit S, et al. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16(1):30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Babushok DV, Ohshima K, EM Ostertag, et al. A novel testis ubiquitin-binding protein gene arose by exon shuffling in hominoids. Genome Res. 2007;17(8):1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ostertag EM, Kazazian HH. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. [DOI] [PubMed] [Google Scholar]
- [30].Yan ZM, Huang N, Wu W, et al. Genome-wide colocalization of RNA-DNA interactions and fusion RNA pairs. Proc Natl Acad Sci U S A. 2019;116(8):3328–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gupta SK, Luo LM, Yen LS. RNA-mediated gene fusion in mammalian cells. Proc Natl Acad Sci U S A. 2018;115(52):E12295–E12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Storici F, Bebenek K, Kunkel TA, et al. RNA-templated DNA repair. Nature. 2007;447(7142):338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chwalenia K, Qin F, Singh S, et al. A cell-based splicing reporter system to identify regulators of cis-splicing between adjacent genes. Nucleic Acids Res. 2019;47(4):e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xie Z, Tang Y, Su X, et al. PAX3-FOXO1 escapes miR-495 regulation during muscle differentiation. RNA Biol. 2019;16(1):144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]