

We have identified small molecules that potentiate aspects of mitochondrial dynamics and function in primary neurons.
Abstract
Impaired mitochondrial dynamics and function are hallmarks of many neurological and psychiatric disorders, but direct screens for mitotherapeutics using neurons have not been reported. We developed a multiplexed and high-content screening assay using primary neurons and identified 67 small-molecule modulators of neuronal mitostasis (MnMs). Most MnMs that increased mitochondrial content, length, and/or health also increased mitochondrial function without altering neurite outgrowth. A subset of MnMs protected mitochondria in primary neurons from Aβ(1–42) toxicity, glutamate toxicity, and increased oxidative stress. Some MnMs were shown to directly target mitochondria. The top MnM also increased the synaptic activity of hippocampal neurons and proved to be potent in vivo, increasing the respiration rate of brain mitochondria after administering the compound to mice. Our results offer a platform that directly queries mitostasis processes in neurons, a collection of small-molecule modulators of mitochondrial dynamics and function, and candidate molecules for mitotherapeutics.
INTRODUCTION
The unique architecture of post-mitotic neurons with their elaborate dendritic trees and far-reaching axons imposes extraordinary demands on the mitochondrial system for satisfying the neuron’s need for energy, calcium buffering, neurotransmitter metabolism, and other physiological processes (1). These demands are largely met by neuronal mitostasis, which includes the combined processes that maintain mitochondrial number and quality in the various compartments of the neuron across its lifetime. These processes include the regulation of the cell’s content of mitochondria through a balance between biogenesis and turnover, the length of each mitochondrion from a balance between fusion and fission, and other processes such as transport across the length of axons and dendrites. Biogenesis is largely controlled by the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), which interacts with multiple transcription factors and nuclear hormone receptors to regulate ~1500 genes in the nucleus that code for mitochondrial proteins and other factors that coordinate mitochondrial biogenesis and oxidative phosphorylation (2). The dynamic processes of fission, mediated by dynamin-related protein 1 (DRP1), and fusion, mediated by mitofusins and other proteins, provide for quality control over the system. Mitochondria can mix their contents upon fusion, spreading metabolites, enzymes, and other constituents throughout the fused mitochondrion. Damaged parts of a mitochondrion can be segregated into a daughter mitochondrion destined for destruction through fission. However, excessive fission leads to mitochondrial fragmentation and cell death. Increases in mitochondrial length, or elongation, caused by enhanced fusion or decreased fission can be beneficial in some situations, increasing the efficiency of the mitochondrial system and offering protection during stressed conditions (3, 4). Dysfunctions of neuronal mitostasis are found in most neuropsychiatric disorders including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, mood disorders, schizophrenia, and others (5–10). Mitochondrial failure leads to increased oxidative stress and damage to lipids, proteins, and nucleic acids from the generated reactive oxygen species (ROS) (11). Toxic Aβ species are reported to accumulate intraneuronally, and within mitochondria, during the development of Alzheimer’s disease, impairing energy metabolism, increasing ROS, and decreasing adenosine triphosphate (ATP) production (12).
The importance of mitostasis to the function of healthy neurons commands a deeper knowledge of each of its processes and ways to rectify mitostatic dysfunction in disease. Targeting mitochondria in screens for compounds that may protect them from disease or other insults is viewed as one promising strategy toward this goal (3). Unexpectedly, very little effort has been made in identifying compounds that modulate neuronal mitostasis. A few small-molecule screens for enhanced mitochondrial function or dynamics have been performed using yeast, human umbilical vein endothelial cells, or other mammalian cell lines (13–15), but no phenotypic screens for processes of mitostasis have been reported using neurons. Given the unique physiology and architecture of neurons, cell-based screens that use neurons are predicted to be more successful in identifying potential mitotherapeutics for brain disorders compared to traditional screens using immortalized, non-neuronal cell lines.
Here, we describe an assay platform for multiplexed, high-throughput, and phenotypic screening of small molecules for modulatory effects on neuronal mitostasis, focusing on the neuron’s content of mitochondria, mitochondrial elongation, and health. The latter parameter, health, can be assayed using an imaging function of “circularity.” When mitochondria become defective, they assume a small and rounded morphology. We used the platform to screen 2400 compounds and identified 67 modulators of neuronal mitostasis, abbreviated as “MnMs,” that modulate mitochondrial content, elongation, and health. More than 90% of the MnMs also increased mitochondrial function as measured by the potential across the inner mitochondrial membrane (IMM) and ATP generation by the cells, suggesting an extremely high correlation between mitochondrial morphology and function. Some of the MnMs proved to be protective to cultured neurons against insults that are associated with neuropsychiatric disorders, including increased oxidative stress, excess glutamate, or Aβ(1–42)–induced mitotoxicity and neurotoxicity. Using isolated mitochondria, we found that some MnMs that fully protected damaged mitochondria from these insults directly target mitochondria. One promising compound, dyclonine, was explored in more detail and shown to enhance synaptic activity in cultured hippocampal neurons and increase respiration in brain mitochondria isolated from mice treated with the compound.
RESULTS
Developing a multiplexed, high-throughput, and high-content phenotypic screen for MnMs using primary neurons
To simultaneously assay multiple processes of neuronal mitostasis in primary neurons, we generated a ROSA26 knock-in mouse line carrying a floxed tagGFP2 expression cassette with an N-terminal mitochondrial targeting sequence. This strain was generated in a C57Bl/6J background (Fig. 1A; referred to as Mt mice). Dissociated cells from the forebrains of P0 pups of Mt mice were mixed with replication-deficient AAV9 carrying a calcium/calmodulin-dependent protein kinase II (CaMKII) promoter–driven iCre recombinase gene (fig. S1A) and plated onto 384-well plates. Using Cre-dependent, mitochondrial green fluorescent protein (GFP) expression offers experimenter control over the number of GFP-labeled mitochondria in each imaging field so they are not too dense or too sparse. The floxed Mt expression cassette also included a nuclear-localized tagBFP2, which was used for the visualization of the neuronal nuclei. GFP-labeled mitochondria [mitochondrial GFP (Mt-GFP)] began to appear ~DIV8 (8 days in vitro), and by DIV14, the signal was sufficiently strong to be imaged and analyzed (fig. S1B). Compound treatment was performed 24 hours before imaging, on DIV13. Correct subcellular targeting of the Mt-GFP reporter was validated by costaining the Mt-GFP with tetramethylrhodamine methyl ester (TMRM) and MitoTracker Orange, two well-characterized, cell-permeable fluorescent probes known to label mitochondria (fig. S1C).
Fig. 1. Assaying mitochondrial morphology in primary neurons.
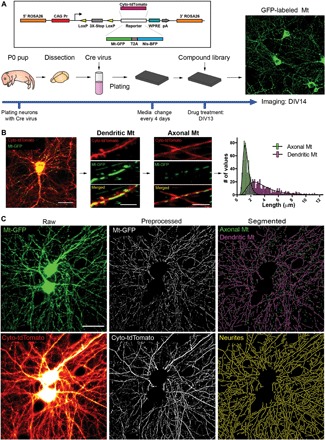
(A) Mt mice, work flow, and timeline of the screen. Mitochondria were visualized in primary mouse neurons by the Cre-dependent expression of mitochondria-targeted GFP (Mt-GFP). The image shows the transduced neurons in the field expressing GFP in somatic and neuritic mitochondria (maximum-projection image of confocal z-stacks, three slices, z = 0.7 μm with a 60× objective). (B) Classification of axonal and dendritic mitochondria. Representative images of a neuron (left) expressing Cyto-tdTomato (red) and Mt-GFP (green) and zoomed-in segments of a dendrite and two axons. The frequency distributions of mitochondrial length, located in either dendrites or axons, are plotted in the right panel. Best-fit lognormal distributions (black lines) show that 15% of the mitochondrial lengths can be found in both axons and dendrites (overlap). naxonal = 517, ndendritic = 525. Scale bars, 25 μm (left image) and 5 μm (middle and right images). (C) Image analysis. Maximum z-projection of the green channel (Mt-GFP) containing mitochondria and wide-field image of neurites (Cyto-tdTomato). After image preprocessing (somatic mitochondria removal from both channels, background subtraction and median filtering of mitochondria, and tubeness filtering of neurites), axonal and dendritic mitochondria and neurites were segmented (axonal, 0.5 to 1.4 μm; dendritic, >2.4 μm) by top-hat filtering followed by adaptive thresholding.
Mitochondria in neurons are morphologically heterogeneous. In the soma, they form an irregularly shaped interconnected network, while in neurites, they exist as rod-like structures of various lengths. We focused our efforts on discovering compounds that increase mitochondrial content, promote elongation, or induce healthier, less round mitochondria in the axons and/or dendrites. We first classified Mt-GFP into three classes: somatic, axonal, and dendritic mitochondria. The separation of axonal and dendritic mitochondria was achieved by their length differences. To measure the length differences between axonal and dendritic mitochondria, we used neurons from pups of Mt mice crossed with a ROSA26 knock-in for expressing cytoplasmic tdTomato (Cyto-tdTomato, Fig. 1A), which provided a strong signal in the neurites. We then measured the length of ~500 mitochondria in axons (axonal mitochondria) and ~500 in dendrites (dendritic mitochondria) (Fig. 1B). We considered mitochondria to be axonal if located in a process that was thin with a presynaptic bouton and dendritic if located in a thick process populated with dendritic spines. By plotting the frequency distribution of the mitochondrial length values, we found that mitochondria with lengths between 0.5 and 1.4 μm are predominantly axonal, while mitochondria with lengths longer than 2.4 μm are predominantly dendritic; therefore, we excluded the range 1.4 to 2.4 μm from the analysis of axonal and dendritic mitochondria. This resulted in eliminating 4.5% of axonal mitochondria (0.4 to 1.4 μm) and 3% of dendritic mitochondria (>2.4 μm). To identify somatic mitochondria, we took advantage of their tightly packed, bright and circular shape and analyzed them as one mass. Since extracting individual mitochondrial features from somatic mitochondria was not feasible, we excluded them from further analysis.
We considered the possibility that compounds might increase mitochondrial content by promoting neurite sprouting. This hypothesis envisions that the compounds might increase neurite sprouting with mitochondrial biogenesis being spurred to fill the new neurites with mitochondria. Alternatively, increased biogenesis of mitochondria might drive neurite sprouting to accommodate the additional organelles. It was also possible that compounds might increase the biogenesis-turnover balance without altering neurite sprouting, which would increase the density of the mitochondrial mass in the processes. To distinguish between these possibilities, the Cyto-TdTomato signal was used in our rescreen to probe for altered neurite area.
To collect high-quality data with sufficient replication for the screen, we used automated confocal imaging and captured four z-stacks in each well across four replicate plates (Fig. 1C). We then preprocessed and segmented the raw images to identify dendritic and axonal mitochondria and measured their area, length, and circularity. The morphological data of the mitochondria of every field were aggregated to a population sum (of individual mitochondrial area) or median (of the population of individual mitochondrial circularity and length) values.
Defining image-based parameters for mitochondrial content, elongation, and health
There are currently no small-molecule probes reported to have a robust beneficial effect on mitostatic processes of primary neurons. As a first step, we selected and tested 23 compounds with reported effects on mitochondrial dynamics in other cell types, attempting to find positive controls for a screen for mitochondrial content, elongation, or health in neurons (fig. S1D). Unexpectedly, none of the tested compounds had marked effects on the measured morphological features of mitochondria in primary forebrain neurons. In contrast, toxic compounds causing mitochondrial loss, fragmentation, and swelling were readily available (16). We chose to use the uncoupling agent carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) to perturb mitochondrial function and morphology to help identify and validate the image-based parameters chosen as measures of mitostatic processes. FCCP treatment induced marked mitochondrial loss and fragmentation (Fig. 2A). To quantify the mitochondrial content in the images, we defined the cumulative area of a field (CA) as the sum of the areas occupied by individual mitochondria. FCCP treatment caused a large decrease in the number of dendritic mitochondria and their average area (Fig. 2B, left) and a ~90% reduction in the average dendritic mitochondrial CA per well (Fig. 2B, right). The area distribution pattern for average axonal mitochondria did not change, but the number of mitochondria classified as axonal increased (Fig. 2C, left). This resulted in an increase of axonal mitochondrial CA per well upon FCCP treatment (Fig. 2C, right). The average sums of the dendritic and axonal mitochondrial CAs upon dimethyl sulfoxide (DMSO) or FCCP treatment altogether indicate a neuron-wide mitochondrial loss due to FCCP treatment (CADend+Ax,DMSO = 1292 + 404 = 1696 μm2, CADend+Ax,FCCP = 123 + 629 = 754 μm2). Although the observed increase in axonal mitochondrial CA (225 μm2) due to FCCP treatment probably arises from excessive fragmentation of dendritic mitochondria yielding short objects that become classified as axonal mitochondria, this increase accounts for only ~20% of the decrease of the dendritic CA. For this reason, we chose the parameter of dendritic mitochondrial CA > 2.5 robust Z-scores for identifying compounds that increase mitochondrial content.
Fig. 2. Hit parameters for the mitochondrial dynamics screen.

(A) Representative images showing the effects of FCCP on mitochondrial morphology. FCCP (12.5 μM) for 24 hours induced fragmentation of dendritic mitochondria compared to DMSO treatment. Scale bar, 20 μm. (B to E) Mitochondrial morphology of DMSO- and FCCP-treated neurons. Frequency distribution of the area occupied by individual dendritic (B) and axonal (C) mitochondria. FCCP treatment reduced the average area of dendritic mitochondria (meanDMSO = 1.44 μm2, meanFCCP = 1.06 μm2) and decreased the number of dendritic mitochondria (nDMSO = 28,718, nFCCP = 3713). FCCP increased the number of axonal mitochondria (nDMSO = 33,457, nFCCP = 51,767), while it did not change the average area of axonal mitochondria (meanDMSO = 0.39 μm2, meanFCCP = 0.39 μm2). Average well CA differed significantly between DMSO and FCCP treatment for dendritic (meanDMSO,Dend CA = 1292 ± 213, meanFCCP,Dend CA = 123 ± 44) (B, right) and axonal mitochondria (meanDMSO,Ax CA = 404 ± 79, meanFCCP,Ax CA = 629 ± 83 (C, right). FCCP treatment produced a significant decrease in dendritic length (D), with average medianFCCP = 2.77 ± 0.07 μm compared to medianDMSO = 3.35 ± 0.08 μm (D, right). FCCP treatment also produced a significant increase in the median axonal mitochondrial circularity compared to DMSO treatment (average medianDMSO = 0.83 ± 0.007 and medianFCCP = 0.94 ± 0.004; E, right), also illustrated by their frequency distribution (E, left). Bar graphs represent mean ± SD, nwells = 8 per treatment. Unpaired t tests, ****P < 0.0001, nDMSO,axonal = 33,457 and nFCCP, axonal = 51,767, nDMSO, dendritic = 28,718 and nFCCP, dendritic = 3713, 32 fields, eight wells.
To identify compounds that elongate mitochondria, we focused on the feature of dendritic length. FCCP treatment of neurons produced shorter dendritic mitochondria (Fig. 2D, left). Since individual dendritic mitochondrial length measures did not follow a normal distribution, we chose the median of the population within a field to obtain field values. Moreover, the median is less sensitive to outliers than the mean of the population and is thus more reliable for a screen. FCCP treatment produced a significant decrease in the median of dendritic mitochondrial length (Fig. 2D, right). Thus, we chose median dendritic mitochondrial length as the measure of elongation and defined hits that increase mitochondrial length as those that increase this parameter by more than 2.5 robust Z-scores.
We did not observe a significant decrease in average axonal mitochondrial area with FCCP treatment, but the axonal mitochondria became more rounded compared to their usual oblong shape. This increase in roundness of the axonal class can be measured by circularity (4πarea/perimeter2), a measure that correlates well with mitotoxicity and neurotoxicity (fig. S2). The ratio of highly circular mitochondria increased in the axonal mitochondrial population with FCCP treatment (Fig. 2E, left), and this was reflected in the average of axonal circularity median as well (Fig. 2E, right). Highly circular mitochondria are well documented to occur in many pathological conditions (17, 18). Therefore, we chose a decrease in the median of axonal mitochondrial circularity by >2.5 robust Z-score for identifying a third group of compounds—enhancers of mitochondrial health. In summary, FCCP treatment of neurons produced marked mitochondrial loss, dendritic mitochondrial fragmentation, and more circular axonal mitochondria (Fig. 2), as also described by others (16). Given this relationship, we concluded that small molecules that increase mitochondrial content or elongation, and those that decrease circularity, offer a starting point in identifying probes that improve the mitochondrial system in neurons.
We constructed an automated data analysis pipeline for our screen, where the input values were the medians (circularity and length) or sums (CA) of the individual mitochondrial measures of the fields. This data analysis pipeline is described in detail in Materials and Methods. A control experiment performed using this image and data analysis pipeline and vehicle-treated plates failed to identify any false positives in any of our hit categories (fig. S3).
Small-molecule screen and rescreen for MnMs
We screened a chemical library consisting of 2400 compounds (Spectrum Collection from MicroSource) for MnMs using the screening platform and data analysis pipeline described above. We used FCCP as a negative control and neutral wells from the plate interior for calculating robust Z-scores (see Materials and Methods).
In the primary screen, we selected 149 putative hits (6.2%) that met our threshold for modulating mitochondrial content (>2.5 robust Z-scores), elongation (>2.5 robust Z-scores), and/or health (<−2.5 robust Z-scores) (Fig. 3, A to C). Many compounds in the library proved to be neurotoxic, causing mitochondrial loss, fragmentation, and increased circularity similar to FCCP treatment. The putative hits were rescreened using 42 DMSO-treated wells scattered randomly across the plate for the calculation of robust Z-scores. As expected, the population of wells treated with putative hits yielded mitochondrial features that were shifted from the population of wells treated with DMSO (Fig. 3, D to F), indicating a hit enrichment. Close examination of selected hit compounds on mitochondrial features (Fig. 3, G to K) revealed that they had an opposite effect of FCCP treatment as predicted. Elongation hits led to an increased median of dendritic mitochondrial length (Fig. 3, G and K). Mitochondrial content enhancers increased the count and area occupied (CA) by dendritic mitochondria (Fig. 3, H and K). Health enhancers decreased the number of rounded axonal mitochondria and increased the number of elongated ones, thereby decreasing the median of axonal mitochondrial circularity (Fig. 3, I and J).
Fig. 3. Small-molecule modulators increasing mitochondrial elongation, content, and health.
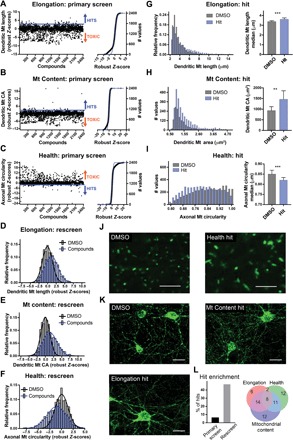
(A to C) Primary screen. Average robust Z-scores (N = 4 replicate plates) (left) and the cumulative distributions (right) for median dendritic mitochondrial length (A), dendritic mitochondrial CA (B), and axonal mitochondrial circularity (C). Populations followed a normal distribution (R2Elongation = 0.986, R2Mt Content = 0.987, R2Health = 0.987; cumulative Gaussian fits, blue lines; right). Hits: robust Z-scores > 2.5 (A and B) or < 2.5 (C). Toxic: median axonal circularity > 3 robust Z-scores and median dendritic length < −3 robust Z-scores. (D to F) Rescreen of primary hits. Relative frequency distributions of average robust Z-scores (N = 10 replicate plates) for median dendritic mitochondrial length (D), dendritic mitochondrial CA (E), and axonal mitochondrial circularity (F). Gaussian fits to frequency histograms of compound (blue, n = 149 compounds) and DMSO-treated (gray, n = 42 wells) neurons yielded population means: MDMSO = −0.05 and MCompounds = 1.07 for median dendritic mitochondrial length (D), MDMSO = −0.015 and MCompounds = 1.36 for dendritic mitochondrial content (E), and MDMSO = 0.046 and MCompounds = −1.01 for median axonal mitochondrial circularity (F). (G) Elongation hits increase dendritic mitochondrial length. Relative frequency distributions of dendritic mitochondrial length (left) and the average of their median values (right, mediansDMSO = 3.39 ± 0.08 μm, mediansHit = 3.62 ± 0.14 μm). (H) Mitochondrial content hits increase dendritic mitochondrial CA. Frequency distribution histograms of individual mitochondrial areas (left) and average CA values (right, meanDMSO = 936 ± 183 μm2, meanHit = 1467 ± 397 μm2). (I) Health hits reduce axonal mitochondrial circularity. Frequency distribution histograms of axonal mitochondrial circularities (left) and averages of their median values (right, mediansDMSO = 0.85 ± 0.0185, mediansHit = 0.82 ± 0.01). (G to I) Bar graphs are means ± SD of 10 well values from the 10 replicate plates (ncompound = 1 well per plate, nDMSO = 42 wells per plate); frequency distribution data were collected from four fields per treatment, total mitochondria nDMSO,Elongation = 1741, nHit,Elongation = 3074, nDMSO,Mt Content = 1365, nHit,Mt Content = 2782, nDMSO,Health = 4262, nHit,Health = 4779. Unpaired t test, **P < 0.01, ***P < 0.001. (J and K) Representative images of compounds that increase elongation and mitochondrial content or improve health. Z-projections of fluorescent images of primary neurons expressing Mt-GFP were collected using a 60× objective. Scale bars, 5 μm (J) and 20 μm (K). (L) Hit enrichment during the screen and hit distribution among mitochondrial parameters. The primary screen of 2400 compounds produced 149 potential hits (6.2%), of which 67 were confirmed in the rescreen (45%). Thirty-five of the confirmed hits were hits for more than one parameter.
We confirmed 67 of the initial 149 compounds in the rescreen (45%; Fig. 3L) using 10 replicate plates. Notably, some molecules both present in the Spectrum Collection and in the candidate molecule screen were confirmed in the rescreen as weak hits (daidzein, memantine, and the 4′-methyl ether derivative of resveratrol). Of the confirmed hits, 32 increased elongation, 45 increased mitochondrial content, and 33 increased mitochondria health (Fig. 3L). Thirty-five of the compounds altered more than one mitochondrial parameter. Analysis of the compounds based on structural fingerprint similarities revealed that those modulating the three hit categories fell into multiple structural clusters (fig. S4), and none of the hit categories enrich structurally similar compounds. This suggests that multiple and diverse mechanisms of action can lead to similar effects on aspects of mitochondrial dynamics. The classification itself revealed three primary clusters. Two of the clusters contained compounds that are structurally distinct. Most members from the third cluster are from the flavonoid family (flavones, isoflavones, and chalcones), implying common targets.
Mitostasis processes and function dissected with small-molecule probes
To distinguish compounds that increase mitochondrial content with or without neurite sprouting, we measured the total area covered by neurites across the fields imaged for each well in the rescreen (Figs. 1C and 4A, left). Eleven of the 53 compounds that significantly increased mitochondrial content (>2 robust Z-scores) also increased neurite area by more than 2 robust Z-scores (table S1), although there was no correlation between dendritic mitochondrial CA increase and neurite area across this population (Fig. 4A, right). The lack of correlation between these two parameters unexpectedly suggests that they are not coupled: Mechanisms that increase mitochondrial content are not triggered in response to increased sprouting, nor is increased neurite sprouting triggered by events that enhance mitochondrial content. A complete understanding of how neurons provide a sufficient supply of mitochondria to distal neurites has not been achieved, although biogenesis in the soma and anterograde transport along with de novo synthesis in the neurites offer partial explanations (1, 19).
Fig. 4. Mitochondrial dynamics and function.

(A) Uncorrelated mitochondrial content and neurite sprouting. Rescreen data of dendritic mitochondrial CA and neurite area of the compounds that significantly increased mitochondrial content (>2 robust Z-scores, dashed line). There was no correlation between increased neurite area and increased dendritic mitochondrial CA (Pearson’s r = 0.13, right). Eleven compounds that increased mitochondrial content (gray) increased neurite sprouting (pink) by >2 robust Z-scores. See table S1 for names of numbered compounds. (B) No correlation between dendritic and axonal mitochondrial content. There was no correlation between axonal and dendritic CA (Pearson’s r = 0.22). (C) Coupling of dendritic mitochondrial content and length. Dendritic mitochondrial length correlated with dendritic mitochondrial content (left, Pearson’s r = 0.76) and dendritic mitochondrial count (right, Pearson’s r = 0.6) after compound treatment. The correlation lines are bounded by a 95% confidence interval (gray shaded area). (D) Representative images showing the coupling of mitochondrial content and elongation. Mt-GFP expressing neurons were treated with a hit compound (12.5 μM) that increased both content and elongation by 12 hours after treatment. White arrows indicate the sites of mitochondrial growth. Scale bar, 20 μm. (E) Axonal mitochondrial length is uncoupled with content and count. There was no correlation between axonal mitochondrial length and axonal CA (left, Pearson’s r = −0.22) or count (right, Pearson’s r = −0.23). (F) Mitochondrial dynamics hits increase mitochondrial function. TMRM signal and ATP production of the rescreened mitochondrial dynamics hits (confirmed hits, n = 67) were measured, and a functional increase was determined as robust Z-scores >2 for TMRM signal or ATP production. Sixty-one increased (91%) one or both functional readouts. (G) Mitochondrial dynamics hit parameters differentiate compounds with increased mitochondrial function. Decreased axonal circularity, increased dendritic length, together with increased dendritic CA identified a population of compounds showing increased mitochondrial function measured by ATP production, TMRM signal, or both (functional increase, blue points, >2 robust Z-scores, average from four replicate plates) among the rescreened primary hits (n = 149). (H) Distribution of the 67 rescreened compounds across content, health, elongation, and function. Venn diagram of compounds that significantly increased ATP production or TMRM signal (n = 61), mitochondrial content (n = 53), elongation (n = 50), and/or health (n = 42). Significance threshold, 2 robust Z-scores.
Our rescreen data using compounds that modulate neuronal mitostasis along with neutral compounds offered a unique opportunity to explore the relationship between several mitochondrial properties in neurons. Plotting dendritic mitochondrial CA as a function of axonal mitochondrial CA showed that none of the 53 compounds that significantly increased dendritic mitochondrial CA (>2 robust Z-scores) increased axonal mitochondrial CA (Fig. 4B, Pearson’s r = 0.22). Given the large number of compounds with effects on dendritic mitochondrial CA, it seems unlikely that this results from the simple failure to find a compound that alters both. Rather, the result is more consistent with the possibility that increases in mitochondrial content can occur solely in dendrites and/or that axonal mitochondrial content is more tightly regulated and is harder to perturb with small molecules. Nevertheless, compound-induced dendritic mitochondrial content seems to be uncoupled from axonal mitochondrial content. However, we did observe a correlation between dendritic mitochondrial CA and dendritic mitochondrial length as expected (Fig. 4C, Pearson’s r = 0.76). Moreover, dendritic mitochondrial count and dendritic mitochondrial length also correlated positively (Fig. 4C, Pearson’s r = 0.6), indicating that compounds that increase mitochondrial content often do so by increasing both count and length.
To explore the possible connection between mitochondrial content and elongation, we collected time-lapse images of primary neurons for 12 hours after treatment with content and elongation hits along with untreated controls. As reported by others (20), we observed fast-moving short mitochondria that continuously underwent fusion and fission events or nearly stationary longer mitochondria that did not. However, in neurons treated with compounds, the stationary long mitochondria sometimes fused together by apparent growth toward each other (Fig. 4D and movie S1). This observation explains the correlation between dendritic mitochondrial content and elongation. Compounds that increased axonal mitochondrial length did not significantly influence axonal mitochondrial CA or count (Fig. 4E).
Do the MnMs alter mitochondrial function? To address this issue, we tested the 149 rescreened compounds for effects on mitochondrial function by measuring two parameters, the IMM potential and ATP generation. We used the well-characterized rhodamine derivative, TMRM, to measure the IMM potential and a commercial kit for measuring ATP generation. Notably, 61 of the 67 confirmed hits (91%) also significantly increased ATP production, TMRM signal, or both (>2 robust Z-scores), showing that the vast majority of MnMs also increase mitochondrial function (Fig. 4F). This indicates a tight link between mitochondrial dynamics and function. Next, we probed the correlation between individual mitochondrial morphological features and function using the rescreened 149 compounds. This revealed only weak correlations between the individual mitochondrial features (area, length, count, CA, and circularity) and function, with Pearson correlations (r) ranging from ~0.4 to 0.5. This included the three hit parameters of mitochondrial content, elongation, and health: dendritic mitochondrial CA (rATP = 0.5, rTMRM = 0.47), dendritic mitochondrial length (rATP = 0.41, rTMRM = 0.53), and axonal mitochondrial circularity (rATP = −0.44, rTMRM = −0.41). However, plotting the rescreened compounds in three-dimensional space defined by the three hit parameters and separately marking whether they increase function or not revealed that compounds boosting mitochondrial function form a distinct cluster from those that do not (Fig. 4G). This indicates that increased mitochondrial function is generally accompanied by a combination of phenotypic changes that can be described by mitochondrial content, length, and health.
The functional increase of mitochondria induced by 61 different MnMs can occur through at least three mechanisms: (i) an increase in mitochondrial mass present in the neurons (mitochondrial content), (ii) no change in mass but an increase in the efficiency of individual mitochondria, and (iii) a combination of (i) and (ii). To help distinguish between these possible mechanisms, we constructed a four-way Venn diagram of the compounds that increased function, content, health, or length (Fig. 4H). Most of the compounds that increased mitochondrial function (n = 61) also increased mitochondrial content (n = 49), revealing an expected relationship between increased mitochondrial mass and measured function. However, the screen also identified compounds that only increased ATP production and/or membrane potential change without increasing content (n = 12), suggesting the second mechanism of increasing the output efficiency of the existing mitochondria. All of these compounds improved health and/or induced elongation while increasing function. The Venn diagram further supports that all three parameters drive functional increases with a similar contribution (number of compounds increasing the hit parameter and function/number of compounds increasing hit parameter): biogenesis, 92% (49 of 53); health, 93% (39 of 42); and elongation, 90% (45 of 50).
Modulators of mitochondrial dynamics protect mitochondria against insults associated with neurodegenerative disorders
Altered mitochondrial dynamics and function have been identified to be part of the neuropathology found in neuropsychiatric disorders along with increased oxidative stress and glutamate toxicity (5–10). We hypothesized that the MnMs identified using wild-type neurons might correct the disease-related pathology. We thus challenged primary neurons with increasing concentrations of toxic Aβ(1–42) oligomers, peroxide (tert-butyl hydroperoxide), and excess glutamate to obtain a deeper understanding of the mitochondrial phenotypes that emerge (fig. S5). We observed severe fragmentation of both axonal and dendritic mitochondria with all three treatments, identified by a decrease of axonal mitochondrial area, a decrease in dendritic mitochondrial length, and an increase of axonal mitochondrial circularity. Treatment of neurons with 10 μM Aβ(1–42) oligomers, 75 μM peroxide, or 25 μM glutamate for 48 hours produced reproducible and sufficient effect sizes for our assays of mitotoxicity while keeping a significant fraction of neurons alive.
We then treated neuronal cultures with the same three toxic substances, in the absence or in the presence of the 12 most reliable MnMs chosen by effect sizes for mitochondrial function and mitostasis parameters, to determine whether the modulators offered protection against the toxic agents assayed 48 hours later. The MnMs by themselves had robust effects on mitochondrial health, elongation, or content (Fig. 5A). Five of the MnMs induced neuronal sprouting (increased neurite area), and nearly all increased ATP production and/or mitochondrial membrane potential. Most of the MnMs exhibited relatively high potency and dose-dependent effects on processes of mitostasis and neurite sprouting, with half-maximal effective concentration (EC50) values in the low to submicromolar range (table S2).
Fig. 5. MnMs protect from oligomeric Aβ(1–42)–, peroxide-, and glutamate-induced mitochondrial damage in primary neurons.
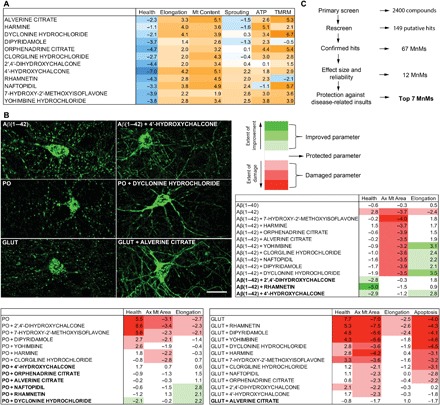
(A) Effects of the selected MnMs. Parameters were measured 24 hours after treatment with 12.5 μM of compounds. Data are mean robust Z-scores from Fig. 4. (B) Effects of selected MnMs in the presence of disease-related insults. Representative images of mitochondrial fragmentation induced by 10 μM Aβ(1–42), 75 μM peroxide (PO), or 25 μM glutamate (GLUT) in Mt-GFP–expressing neurons, and the mitochondrial protection by 12.5 μM of compounds after 48 hours of cotreatment (top left; scale bar, 20 μm). After cotreatment, axonal mitochondrial circularity (health), axonal mitochondrial average area, dendritic mitochondrial length (elongation), and live-dead cell ratio (survival) were either protected (within 2 Z-scores of vehicle-treated control), improved (>2 Z-scores for axonal mitochondrial area, elongation, and survival and <−2 Z-scores for health), or damaged (<−2 Z-scores for axonal mitochondrial area, elongation and survival and > 2 Z-scores for health) compared to the vehicle-treated control (top right and bottom). Compounds fully protective (all parameters are protected or improved) against an insult are bolded. Color bar represents absolute Z-score values toward either improvement (green) or damage (red) of the parameters. Data are mean Z-scores (n = 12 to 18 wells, four fields per well, two independent experiments). (C) Selection of the top seven MnMs. Summary of the steps for the selection of the top seven compounds.
We categorized the effects of MnMs in the presence of the toxic substances into three groups: (i) full protection: all three parameters measuring fragmentation (axonal mitochondrial area, dendritic mitochondrial length, axonal mitochondrial health) were normalized to within control values or improved, (ii) partial protection: at least one of the three parameters was within control values or improved, and (iii) no protection: none of the parameters were within control values or improved (Fig. 5B). The compounds exhibited different protective power against the three insults, suggestive of different mechanisms of action (Fig. 5B). Rhamnetin, 4′-hydroxychalcone, and 2′,4′-dihydroxychalcone conferred complete protection from Aβ(1–42)–induced mitochondrial fragmentation. The nine other compounds provided only partial protection, failing to protect against the decreased area of individual axonal mitochondria caused by the toxic peptide. As a control, we treated the cultures with the less toxic peptide oligomeric Aβ(1–40). This oligomeric peptide failed to cause mitochondrial fragmentation under the conditions used.
Mitochondrial fragmentation caused by treatment with peroxide was fully protected by six compounds—4′-hydroxychalcone, orphenadrine, alverine, naftopidil, rhamnetin, and dyclonine—and partially protected by four others. Glutamate-induced mitochondrial fragmentation was fully protected by one compound, alverine. We also measured the live-dead cell ratio in the presence of toxic concentrations of glutamate and the selected MnMs and found that alverine, along with 4′-hydroxychalcone and 2′,4′-dihydroxychalcone, offered protection. In summary, seven of the MnMs offered complete protection against Aβ(1–42) oligomer–, peroxide-, or glutamate-induced mitochondrial fragmentation in addition to their robust and reliable improvement of mitostatic processes using wild-type neurons (Fig. 5C).
Dyclonine directly enhances mitochondrial function, potentiates basal synaptic activity, and increases the respiration of brain mitochondria in treated mice
To determine whether select compounds directly alter mitochondrial function, isolated forebrain mitochondria of P0 mouse pups were treated with the compounds, and ATP production was measured using a luciferase-based kinetic assay (Fig. 6, A and B). We observed a significant direct effect on mitochondria for six from the top seven MnMs that offered complete protection against cell-based insults. Dyclonine, alverine, and naftopidil significantly increased ATP production, while rhamnetin, 4′-hydroxychalcone, and 2′,4′-dihydroxychalcone unexpectedly decreased it within the 45-min measurement period. After 24 hours of treatment with the latter compounds, ATP production in neurons showed no significant change (with the two chalcones) or increase (rhamnetin, Fig. 5A), suggesting that the direct inhibition of ATP production by mitochondria may have been normalized via longer-term compensatory mechanisms. Dyclonine and alverine increased ATP production of mitochondria in primary neurons after 24 hours of incubation as well.
Fig. 6. MnMs enhance ATP production from isolated mitochondria, potentiate basal synaptic activity, and increase respiration of mitochondria in vivo.
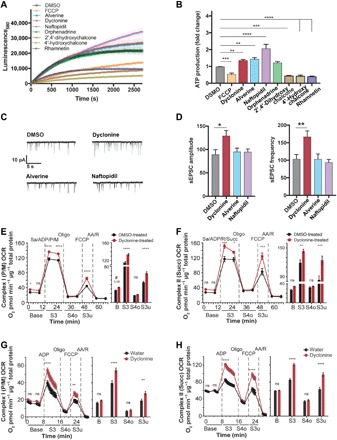
(A) Kinetics of ATP production of isolated mitochondria. Baseline-corrected average curves of the ATP production of isolated forebrain mitochondria of newborn C57BL/6J mice starting 20 min after the addition of vehicle (DMSO), 12 μM FCCP, and the top seven MnMs. Data are means ± SD of four runs from one experiment. Single exponential fits yielded amplitudes of 22,498 ± 358 (DMSO, R2 = 0.9), 17,958 ± 216 (FCCP, R2 = 0.98), 28,383 ± 278 (alverine, R2 = 0.96), 28,882 ± 673 (dyclonine, R2 = 0.91), 49,917 ± 838 (naftopidil, R2 = 0.98), 22,764 ± 315 (orphenadrine, R2 = 0.91), 9853 ± 132 (2′,4′-dihydroxychalcone, R2 = 0.92), 6121 ± 99 (4′-hydroxychalcone, R2 = 0.9), and 10,059 ± 166 (rhamnetin, R2 = 0.9). (B) ATP production of isolated mitochondria of the top seven MnMs. Average ATP production of forebrain mitochondria of newborn mice in the presence of DMSO, 12.5 μM FCCP, or 12.5 μM of the top seven MnMs, normalized to average DMSO treatment. Data are means ± SEM of three independent experiments (four runs per experiment) compared to DMSO by one-way analysis of variance (ANOVA) with Dunnett’s post hoc test, **P < 0.01, ***P < 0.001, ****P < 0.0001. (C and D) Synaptic activity of hippocampal neurons. Example traces (C) and quantified sEPSC amplitude/frequency (D) of cultured hippocampal neurons at DIV8 after 24 hours of incubation with DMSO, 12.5 μM dyclonine, alverine, or naftopidil using whole-cell patch-clamping techniques. Data are means ± SEM (n = 33 to 36 cells) compared to DMSO by one-way ANOVA with Dunnett’s post hoc test, *P < 0.05, **P < 0.01. (E and F) Mitochondrial respiration in primary neurons exposed to dyclonine for 24 hours. Before OCR measurements, DIV13 neurons were treated for 24 hours with DMSO (0.1%, black) or dyclonine (10 μM, red). OCRs were measured at baseline respiration (Base) and after addition of mitochondrial substrates pyruvate (P) and malate (M) or succinate (Succ) along with ADP to the permeabilized (saponin, Sa) neurons (first dashed line, S3). Oligomycin addition (second dashed line) blocked proton re-entry through ATP synthase, slowing ETC function to levels necessary to maintain Δψm (S4o; o = oligomycin induced). Addition of FCCP (third dashed line) short-circuited proton influx to bypass ATP synthase, revealing maximal, uncoupled OCR (S3u; u = uncoupler induced). Addition of rotenone (R)/antimycin A (AA), complex I and complex III inhibitors, respectively, were added to block oxidative phosphorylation and measure nonmitochondrial respiration (fourth dashed line). ns, not significant. (G and H) Respiration of isolated mitochondria from dyclonine-treated mice. OCR of whole brain mitochondria of 9-month-old C57BL/6J mice kept on either dyclonine-supplemented (25 mg/kg) or standard water for 7 months was measured in the presence of complex I (G) or complex II (H) substrates preincubated with mitochondria, yielding results similar to those in (E) and (F). Bar graphs [right panels of (E) to (H)] compare the OCR value of the first measured point in each condition between the water- and dyclonine-treated groups (mean ± SEM, n = 11 to 12 wells). Total protein was used to normalize OCR into pmol O2 min−1 μg−1 total protein. Data in (E) to (H) were analyzed using two-way ANOVA with repeated measures. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We hypothesized that an increased energetic state of neurons may be accompanied by altered neurophysiological properties, such as enhancement of synaptic activity measured by spontaneous excitatory postsynaptic currents (sEPSCs). To test this hypothesis, primary hippocampal neurons were treated for 24 hours with the MnMs that increased mitochondrial function, and sEPSCs were measured using patch-clamp techniques. Dyclonine was the only compound of the three that directly increased mitochondrial ATP production and significantly altered sEPSCs, increasing both frequency and amplitude (Fig. 6, C and D). Thus, increased spontaneous activity is not a universal feature of compounds that increase mitochondrial ATP synthesis.
We selected dyclonine for subsequent tests, first to measure the rate of respiration in cultured neurons treated with dyclonine and, second, to measure respiration in mitochondria isolated from mice treated chronically with dyclonine. The latter experiment was designed to test whether dyclonine, as a representative MnM, alters mitochondrial function in an in vivo environment. Establishing in vivo efficacy is critical given that cultured cells may or may not reflect an in vivo environment. We measured the absolute oxygen consumption rate (OCR) at baseline and in response to excess adenosine diphosphate (ADP)/substrate (State3/3u) as normalized to protein content in primary neurons exposed to dyclonine for 24 hours. We observed a significant increase of State 3 and 3u respiration stimulated by either complex I or complex II substrates in mitochondria from the treated cultured neurons (Fig. 6, E and F). A similar respiratory rate increase was observed in mitochondria isolated from the whole brains of mice treated chronically for 7 months with dyclonine laced into their water supply (Fig. 6, G and H). The latter results demonstrate that dyclonine increases the capacity of substrate oxidation of mitochondria in vivo when administered chronically.
DISCUSSION
Abundant evidence indicates that deficits in neuronal mitostasis contribute in a major way to the pathophysiology of neuropsychiatric disorders. Beyond the perspective of human health, the process of mitostasis in neurons is extraordinarily complex and has kept important biology hidden (1). Despite these facts, no systematic and large-scale method of interrogating the mitochondrial system in neurons has been developed. We provide such methodology here with a high-throughput, high-content, and multiplexed assay system for processes included in neuronal mitostasis. This assay system offers an effective way forward for discovering novel mitotherapeutics and new biology about the mitochondrial system in neurons and other cell types.
Our assay system and analysis pipeline offer several advantages over other options, including (i) live-cell imaging that circumvents fixation artifacts such as fragmentation, shrinkage, or swelling; (ii) the use of primary neurons, which provides confidence that newly discovered probes will be effective in neurons; (iii) the ability to obtain image data at multiple points across time rather than end point only; (iv) the ability to simultaneously query mitochondrial content, health, elongation, and neurite branching using different fluorescent reporters and mice heterozygous for two tandem, ROSA26 knock-in constructs; (v) the capability of separately addressing neuronal mitostasis issues in axons and dendrites; and (vi) the opportunity to assay or screen in the genetic background of mice engineered to model human disease states. Despite all the advantages of our assay system, there are some limitations. First, our mitochondrial classification is specific to cultured mouse neurons, since it is based on the observed length distribution of the validated axonal and dendritic mitochondrial pool in P0 cultured neurons. Changing the species and/or the cell type may require changing the predefined length thresholds. In addition, the assay is image-based and morphological in nature, and functional readouts must be obtained in independent assays. Nevertheless, we demonstrate that functional properties are correlated with morphological parameters.
We draw several important conclusions about neuronal mitostasis from the effects of 67 compounds identified from our primary screen and rescreen. First, most of the compounds modulated several aspects of mitostasis, including mitochondrial content, elongation, and health. This indicates that these mitochondrial parameters are generally coupled, at least as revealed by perturbations with random collections of small molecules. Second, despite this general coupling, there exists an interesting uncoupling of mitostasis processes in axons and dendrites. The compounds that promoted dendritic mitochondrial content (CA) failed to increase the axonal mitochondrial CA. Although there is some possibility that the assay is more sensitive in dendrites than axons that could account for this observation, it seems more likely that dendritic and axonal mitochondrial content is separately regulated. Third, our results show that dendritic mitochondrial elongation is coupled with mitochondrial content. The compounds that promoted mitochondrial elongation did not decrease the number of mitochondria, indicating that elongation did not occur by simple fusion of preexisting dendritic mitochondria. Rather, elongation was associated with an increase in count and CA. From another perspective, it appears that increased mitochondrial content as modulated by small molecules occurs by simultaneously increasing the count and length of dendritic mitochondria. It may be that dendritic mitochondrial elongation occurs from increased biogenesis in the soma, transport to dendrites, and subsequent fusion of mitochondria. Alternatively, there may exist an unrecognized growth process that occurs in individual mitochondria. Fourth, mechanisms that stimulate neurite sprouting seem largely distinct from those that stimulate increased mitochondrial content. Fifth, modulating the processes of mitostasis generally leads to increased function as measured by IMM potential and ATP generation. Finally, and most importantly, modulators of mitostasis generally provide protection against insults associated with neuropsychiatric disorders in the cell culture models tested here.
We identified seven promising MnMs from our orthogonal assays for mitochondrial function, the magnitude and reliability of the modulatory effect on mitostasis processes, and the extent of protection offered in three different disease models. Three of these MnMs—rhamnetin, 4′-hydroxychalcone, and 2′,4′-dihydroxychalcone—are from the same structural cluster that includes flavones, isoflavones, and chalcones (fig. S4). Their effects on processes of mitostasis and protection against Aβ(1–42) oligomers are similar (Fig. 5). Members of the flavone family, but not the chalcones, have been reported to be positive modulators of mitochondrial biogenesis and function, although the mechanism of action remains unknown (21). Both classes of molecules can inhibit phospholipase A2 (PLA2) enzymes (22, 23). Since increased PLA2 activity has been associated with neuropsychiatric disorders and inhibitors of PLA2 have been shown to be effective against amyloid beta peptide–induced apoptosis in primary neuronal cultures (24), it may be that PLA2 inhibition is part of the mechanism of action of these three MnMs in providing protection in the cellular disease models tested here. 4′-Hydroxychalcone is also a monoamine oxidase (MAO) inhibitor (25), which may help to preserve mitochondrial health under disease conditions. MAO resides in the mitochondrial outer membrane and produces hydrogen peroxide by oxidation of monoamine substrates to increase oxidative stress. Prior studies have shown that inhibitors of type B MAO alleviate this stress and have neuroprotective effects (26). Notably, all three molecules from this group inhibited the ATP production of isolated forebrain mitochondria, which is most likely due to the known inhibitory effect of isoflavones, flavones, and chalcones on F0F1-adenosine triphosphatase/ATP synthase (complex 5) (27). It is possible that their observed long-term enhancement on mitostasis and ATP production are due to some hitherto unknown cellular compensatory mechanism triggered by the mild inhibition of complex 5.
The remaining four compounds of the selected seven MnMs (dyclonine, alverine, orphenadrine, and naftopidil) are all drugs approved by the U.S. Food and Drug Administration, and three of them directly increased mitochondrial ATP production. Dyclonine is used as a local oral anesthetic, found in throat lozenges. Besides its most widely reported inhibition of voltage-dependent Na channels, it binds to NRF2 (nuclear respiratory factor 2) transcription factor (PubChem AID 743219) and activates the NRF2-ARE (antioxidant response element) signaling pathway, which constitutes a major mechanism in the cellular defense against oxidative stress by controlling the expression of genes involved in detoxication and cellular protection (28). Increased NRF2 activity defends against mitochondrial toxins and counterbalances mitochondrial ROS production. NRF2 function has been shown to be suppressed in mitochondria-related disorders, including Parkinson’s disease and Friedrich’s ataxia (29). Dyclonine also increased both the frequency and the amplitude of sEPSCs (Fig. 6D) and, most importantly, enhanced the function of neuronal mitochondria in vivo (Fig. 6, G and H). However, its exact mechanism of action needs to be investigated in further studies.
Both dyclonine and naftopidil target estrogen receptors (PubChem AID 743079 and 1259378), which are known to localize within mitochondria. In addition to their well-known roles in regulating gene expression, they have also been implicated in mitochondrial biogenesis through NRF1, which is a nuclear-encoded transcription factor that regulates the expression of multiple nuclear-encoded genes regulating mitochondrial function (30). Naftopidil, which is used in benign prostatic hypertrophy through its selective α1-adrenergic receptor antagonism, also stimulates estrogen-related receptor α (ESRRA) (PubChem AID 1224848). ESRRA is a PGC1α-activated transcription factor involved in numerous aspects of energy homoeostasis including mitochondrial biogenesis and function (31).
Proteins associated with mitochondrial elongation, such as thyroid hormone receptor β (THRB) and the androgen receptor (AR), are targeted by several top MnMs: THRB by dyclonine (32), orphenadrine (PubChem AID 743065), and rhamnetin (PubChem AID 1479), and AR by dyclonine (PubChem AID 1259243) and naftopidil (PubChem AID 1259247). Activation of these receptors promotes mitochondrial fission by increasing the expression of DRP1 (through AR) (33) or increasing the activity of DRP1 through phosphorylation at Ser616 by ULK1 (recruited to the mitochondria through THRB signaling) (34). Thus, the MnMs that antagonize these receptors are predicted to decrease DRP1 expression or activity, which would lead to mitochondrial elongation. Orphenadrine, besides targeting THRB, is an anticholinergic drug of the ethanolamine antihistamine class and used as a muscle relaxant treatment for Parkinson’s disease. It is also found to be protective against glutamate-induced neurotoxicity and mitotoxicity (35). Alverine is a smooth muscle relaxant used for the treatment of gastrointestinal disorders and irritable bowel syndrome. It also targets dopamine D2-like receptors (PubChem AID 485344), together with naftopidil (PubChem AID 485344), dyclonine, and orphenadrine (PubChem AID 652054). DRD2 antagonists can influence protein kinase A (PKA) activation by elevating intracellular cyclic adenosine monophosphate concentration and PKA phosphorylation of DRP1 at Ser637 that decreases DRP1 activity (36). Therefore, DRD2 antagonists may produce longer neuronal mitochondria by reducing DRP1 activity. Thus, it is possible that mitochondrial elongation occurs by the MnMs influencing three distinct receptor pathways—THRB, AR, and DRD2.
In summary, our assay system identified a set of MnMs with direct mitochondrial actions and/or indirect actions by targeting upstream modulatory elements of mitostasis and different cellular protection pathways, providing a valuable source for future central nervous system active mitotherapeutics. Future studies will include identifying molecular targets for this important set of small molecules and testing their efficacy in vivo and in animal models of disease.
MATERIALS AND METHODS
Mouse lines
The Mt mouse line was generated at the ROSA26 locus in a C57BL/6J (The Jackson Laboratory) background using standard knock-in methodology. The targeting vector contained a CAG promoter, a loxP-flanked STOP cassette (STOP codons with all three reading frames), and a mitochondrial-targeted GFP2 (Mt-GFP) and nuclearly targeted tagBFP2 transgenes (nls-BFP) separated by T2A peptide sequences (37). A Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE) at the 3′ end was included to enhance mRNA stability. The Nls-BFP signal was not used in this screen. A similarly engineered ROSA26 knock-in (Ai14 strain; 007908, The Jackson Laboratory), which contains a Cre-dependent cassette for the expression of cytosolic tdTomato (Cyto-tdTomato) was a gift from D. Page (The Scripps Research Institute, Florida). Mt mice were used in the primary screen. Progeny of Mt mice crossed with Ai14 mice that carried both fluorophores were used in the rescreen.
For respiration experiments using isolated mitochondria, C57BL/6J mice were grouped to receive dyclonine (25 mg/kg)–supplemented or standard water for 7 months starting at 2 months of age. The water supply was refreshed weekly. Mice were housed on a 12-hour light/12-hour dark cycle with ad libitum access to food and water. All experiments were performed during the light part of the diurnal cycle. Housing, animal care, and experimental procedures were consistent with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Virus
A replication-deficient AAV9 virus was packaged with a custom-designed plasmid containing a CaMKII promoter to confer neuron-specific expression followed by iCre and tdTomato sequences in tandem but separated by T2A peptide sequences. The expression of tdTomato was at very low levels and not used here.
Primary forebrain cell culture and maintenance
Forebrains of P0 Mt or Mt/Ai14 pups without olfactory bulbs and meninges were collected and placed in ice-cold Ca2+- and Mg2+-free Hanks’ balanced salt solution (HBSS) supplemented with gentamicin (2 μg/ml), 25 mM d-glucose, 1 mM pyruvate, and 20 mM Hepes. Brain tissue was digested with papain (0.6 mg/ml) at 37°C for 20 min. Forebrains were washed with plating media [Neurobasal (Gibco), 5% fetal bovine serum, 4 mM GlutaMAX (Gibco), and gentamicin (2 μg/ml)]. The tissue was gently triturated and allowed to settle, and the supernatant was filtered through a 40-μm mesh-sized cell strainer. Cells were centrifuged at 420g for 4 min and resuspended in plating media. Fifteen thousand cells per well were plated with 2 × 108 viral particles/ml onto black Greiner μClear (catalog no. 781946) or Ultra-Low Base Aurora with evaporation barrier (catalog no. ABE201201B) 384-well plates (80 μl per well) precoated with poly-d-lysine (PDL) for the primary screen and rescreen, respectively. Plating and media changes were performed using an automated Biomek FXP liquid handling workstation. Four hours after plating, 75% of the plating media was replaced by feeding media [Neurobasal-A (Gibco), 4 mM GlutaMAX (Gibco), gentamicin (2 μg/ml), and 2% B27 (Gibco)]. At DIV4, 50% of the media was exchanged with fresh feeding media supplemented with 16 μM 5-fluoro-2′-deoxyuridine (FUdR) to prevent glial cell overgrowth. At DIV8 and DIV12, additional 50% media changes were made without FUdR. Cells were maintained at 37°C and 5% CO2; plates were topped with Breathe-Easy adhesive membranes (Greiner plates) to minimize evaporation.
Compounds and compound transfer
The Spectrum Collection (Microsource) consists of structurally diverse compounds covering a large chemical space with 1600 compounds currently marketed or in clinical trials, 300 natural products and their derivatives, and 500 bioactive compounds not yet in clinical trials. For the primary screen and rescreen, 100 nl of 10 mM compounds (in DMSO) was pintooled into each well on DIV13 using the Biomek FXp robot, resulting in 12.5 μM final concentration of compound and 0.125% final DMSO concentration per well. FCCP (100 nl in DMSO) was pintooled into some wells at a 12.5 μM final concentration as a negative control. The primary screen and rescreen were performed with 4 and 10 replicate plates, respectively. Repurchased compounds and peroxide (Luperox, tert-butyl hydroperoxide) for all other experiments were dissolved in DMSO at the desired concentrations and pintooled similarly, keeping the final 0.125% DMSO concentrations in each well.
Glutamate (monosodium glutamate) was dissolved in 50:50% DMSO/water and pintooled as described above. Glutamate and peroxide stocks were made fresh. FCCP was protected from light and kept at −20°C as a 10 mM stock solution dissolved in DMSO to avoid freeze-thaw cycles. Compound plates for all assays were kept at −80°C and thawed just before use.
Amyloid-β peptide treatment
Aβ(1–42) and Aβ(1–40) peptide fragments were prepared fresh as described previously (5). Briefly, hexafluoroisopropanol-treated peptide (BACHEM) was solubilized in water-free DMSO to 2.2 mM and sonicated in a bath sonicator for 30 s. Cold feeding media without B27 was added to achieve a concentration of 125 μM peptide. Peptide was incubated at 4°C for 24 hours and centrifuged at 12,000g for 10 min. The formation of oligomers was confirmed by native polyacrylamide gel electrophoresis. After determining the concentration of the oligomeric peptide in the supernatant, the oligomers were added to the mouse primary neuronal cultures at the desired concentration on DIV13 and incubated for 2 days.
Mitochondrial dyes
To assay the electrochemical potential across the IMM of cultured cells, we used the cell-permeant dye TMRM. For colocalization studies with Mt-GFP, TMRM was used at 20 nM on DIV14 and incubated for 30 min before imaging. For the orthogonal screen of compounds, TMRM was added to primary neurons on DIV13 at 10 nM, 1 hour before pintooling selected compounds, and incubated for 24 hours. Cells were washed 2× with prewarmed fresh feeding media without B27 before measurements with a plate reader.
The cell-permeable dye MitoTracker Orange (ThermoFisher) was used to label mitochondria in live primary neurons according to the manufacturer’s protocol. Briefly, neurons at DIV14 were stained with 50 nM MitoTracker Orange dissolved in fresh feeding media and incubated for 30 min. The cells were washed 2× with fresh feeding media before imaging.
Isolation of mitochondria
For the ATP synthesis experiments of Fig. 6, forebrains from P0 pups were dissected as described above. Cells (2 × 107) were harvested for high-purity mitochondrial isolation using the Qiagen Qproteome kit. The mitochondrial pellet was resuspended in 500 μl of mitochondria storage buffer (provided by kit) and kept on ice until further use. Mitochondrial protein content was determined to be within 0.3 to 0.4 μg/μl.
Whole brains were harvested from two mice per group of 9-month-old C57BL/6J mice receiving dyclonine (25 mg/kg)–supplemented or standard water for 7 months. Mitochondria were isolated as described in (38), method “A” with modifications. Briefly, fresh brain tissue was minced and homogenized in a 40-ml Dounce homogenizer with cold isolation buffer [10 mM tris, 1 mM EDTA, and sucrose (110 g/liter) (pH 7.4)], performing all subsequent steps on ice. The supernatant from two consecutive 5-min centrifugations at 1300g were combined and centrifuged for 10 min at 21,000g. The harvested pellet was resuspended in 15% Percoll and layered above a 23% over 40% Percoll gradient. The gradient was centrifuged at 30,700g for 15 min in an SW41Ti rotor, and the mitochondrial fraction (a band at the 23%/40% Percoll interface) was aspirated. Mitochondria were washed and pelleted at 16,900g for 10 min, and the pellet was precipitated by adding bovine serum albumin (BSA) and centrifuging at 6700g for 10 min. The final mitochondrial pellet was resuspended in cold mitochondrial assay solution (MAS) without BSA [70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM Hepes, and 1 mM EGTA (pH 7.2 adjusted using KOH)]. The yield of mitochondrial protein was determined by bicinchoninic acid (BCA) assay.
Measurement of ATP production
The total amount of ATP produced by cultured primary neurons was measured by CellTiter-Glo Luminescent Cell Viability Assay (Promega). A recombinant luciferase enzyme catalyzed the mono-oxygenation of luciferin while hydrolyzing ATP produced by the cells and emitting light. We followed the manufacturer’s protocol with primary neurons on DIV14.
ATP production of isolated forebrain mitochondria of P0 pups was measured by the ATP Determination Kit (Molecular Probes). Briefly, isolated mitochondria were diluted 20× in ATP assay buffer (225 mM sucrose, 44 mM KH2PO4, 12.5 mM Mg acetate, and 6 mM EDTA) and incubated with 10 μM of compounds for 20 min. Mitochondria were further diluted 10× with the standard reaction solution (provided by the kit; includes luciferin-luciferase reagents) containing 10 μM of compounds, 100 μM pyruvate, 5 μM palmitoyl-l-carnitine, 1 mM α-ketoglutarate, and 100 μM malate in addition. The reactions were initiated by the addition of 1 mM ADP [deprived of ATP contamination by adding hexokinase (3 U/ml) to 10 mM ADP in 50 mM tris-HCl, 20 mM MgCl2, and 22 mM glucose; incubating the solution at 37°C for 1 hour; and heat-inactivating the hexokinase at 100°C for 3 min]. Luminescence was recorded every 15 to 30 s for 1 hour at 580 ± 40 nm.
Respiration of isolated mitochondria
The OCR of permeabilized neurons and isolated brain mitochondria (from 9-month-old mice) was measured by the XF96 Extracellular Flux analyzer (Seahorse Bioscience). For assays using cultured neurons, primary neurons were dissociated from the forebrains of P0 pups, seeded at 1.9 × 104 per well (PDL-coated XF96 plate), cultured to DIV13, and incubated with dyclonine or DMSO (0.1%) for 24 hours. Before the start of the OCR measurement, all but 30 μl of Neurobasal-A/B27 culture medium was removed from each well. Cells were washed twice with prewarmed MAS-BSA assay medium [fatty acid–free BSA (4 mg/ml), pH 7.2] and incubated in 180 μl at 37°C for 5 min. Following the cartridge calibration, cells were loaded into the XF96 Extracellular Flux analyzer and further equilibrated for 10 min with two cycles of 3-min mixing and 2-min resting before the first measurement of basal respiration. OCR was measured at 37°C under basal conditions followed by the sequential injection of prewarmed 10× mitochondrial substrates/ADP (20 μl), oligomycin (22 μl), FCCP (24 μl), and rotenone/antimycin A (26 μl). The final concentrations of injected compounds were as follows: 10 mM mitochondrial substrates (pyruvate/malate or succinate), 1 mM ADP, 1.5 μM oligomycin, 3 μM FCCP, and 2 μM rotenone/antimycin A. Saponin (25 μg/ml) was coinjected with substrate/ADP to permeabilize the cells and stimulate ADP-dependent respiration. Two baseline measurements were obtained before any injection, and two response measurements were collected after each injection except for after FCCP addition, which consisted of one measurement (nine total measurements in each assay). Each measurement cycle consisted of 2-min mixing, 2-min waiting, and 3-min data acquisition. This protocol allowed for sequential assessment of basal cell respiration, maximal mitochondrial respiratory capacity (State 3 with substrate/ADP), proton leak (State 4o with oligomycin), uncoupled maximal respiration (State 3U with FCCP), and nonmitochondrial respiration with the complex I/complex III inhibitors rotenone/antimycin A. Cells were washed twice with MAS buffer to remove residue BSA contained in the assay medium and lysed in 50 μl of lysis buffer [25 mM Hepes, 1 mM EGTA, 1 mM EDTA, 0.1% SDS, 1% NP-40, 1× protease, and phosphatase inhibitor (pH 7.0 adjusted with NaOH)], and total protein was determined by BCA assay. Oxygen consumption was normalized to total protein content as pmol O2 min−1 μg−1 total protein.
For OCR measurements of isolated brain mitochondria, the mitochondria were diluted to 3 μg/20 μl in cold MAS-BSA buffer + substrate (pyruvate/malate or succinate, to maintain healthy state of mitochondria) and plated onto 96-well PDL-coated plates (20 μl per well). Wells without mitochondria were used for background correction. The plate was centrifuged at 2000g for 20 min at 4°C to attach the mitochondria. After centrifugation, an additional 160 μl of MAS-BSA buffer + substrate was added to the wells. Mitochondria were checked under the microscope to ensure a homogeneous mitochondrial monolayer in each well and incubated at 37°C for 10 min. The plates were further equilibrated for 8 min by two cycles of 1-min mixing and 3-min rest before the measurement of basal respiration. Two baseline measurements were obtained before any injection, and one response measurement was obtained after each injection followed by additional 30-s mixing. The final concentrations of compounds after injections were as follows: 10 mM pyruvate/malate or succinate, 1 mM ADP, 2 μM oligomycin, 4 μM FCCP, and 2 μM rotenone/antimycin A. Each measurement cycle consisted of 30-s mixing and 3-min data acquisition except for the measurement after ADP injection, which lasted 6 min to observe the transition from State 3 to State 4 due to the depletion of ADP in the microchamber. The additional mixing step after each measurement facilitated the sensor returning to ambient O2 concentration. State 3 respiration parameters driven by mitochondrial complex I substrates (pyruvate/malate) were measured first, while complex II–driven respiration (succinate) was measured by inhibiting complex I with rotenone (2 μM). OCR was calculated by the Seahorse XF96 software package. OCR data measured from isolated brain mitochondria are displayed in the “point-to-point mode” showing a series of OCRs across each measurement period.
sEPSCs in hippocampal neurons
Primary neurons were isolated from the hippocampi of C57BL/6J mice at embryonic stage 16, plated onto PDL-coated coverslips, and cultured in complete medium [Neurobasal 1×, 2% B27, 2 mM GlutaMAX, and gentamicin (2 μg/ml)] until DIV8 to allow the development of extensive synaptic networks. The cultures were treated with selected compounds or 0.1% DMSO for 24 hours before measuring sEPSCs. Whole-cell patch-clamp recordings were performed using an Axon MultiClamp 700B amplifier, a 1440A Digidata digitizer, and data acquisition using pClamp software (Axon Instruments, Foster City, CA). The neurons were first recorded in a current clamp mode to monitor cell health. Only neurons with a resting membrane potential of less than −40 mV were used for further analysis. sEPSC recordings were made at 50 kHz and subsequently filtered at 5 kHz. The membrane potential was held at −70 mV during the recording. The extracellular bath solution for the recordings contained 135 mM NaCl, 10 mM glucose, 3 mM CaCl2, 2 mM KCl, 2 mM MgCl2, and 5 mM Hepes, adjusted pH to 7.4 with NaOH, and to 300 to 315 mOsm with sucrose. Patch pipettes were pulled from borosilicate glass with a micropipette puller (Sutter Instrument Co.) and filled with intracellular solution containing 100 mM K-gluconate, 1.7 mM KCl, 0.6 mM EGTA, 5 mM MgCl2, 10 mM Hepes, 4 mM ATP, and 0.1 mM guanosine triphosphate, adjusted pH to 7.2 with NaOH and to 300 to 315 mOsm with sucrose. Once a neuron was patched, its seal was monitored, and if the transient resistance was less than 100 mΩ, then the recording was not used for analysis. All experiments were performed at room temperature. The frequency and amplitude of sEPSCs were analyzed using the template match search (pClamp) and measured as a percentage of baseline level, calculated from the average of a 5-min baseline recording. The values of the amplitude and frequency of EPSCs for each recording were reported as an average from a 5-min recording period.
Live-dead assay
After cultured primary forebrain neurons to DIV15, media was removed from the wells and propidium iodide (PI) dissolved in HBSS was added at 100 μM. The cells were incubated for 2 min followed by the addition of equivalent volume of HBSS containing the cell-permeable fluorescein diacetate (FDA) yielding final concentrations of 12 μM for FDA and 50 μM for PI. Cells were imaged 5 min later.
Imaging
Images were captured using a GE IN Cell 6000 system [60× objective, 0.95 numerical aperture (NA), 16-bit images] equipped with environmental control (37°C and 5% CO2). Mt-GFP (green channel) was imaged by confocal imaging (three slices, Δz = 0.7 μm, aperture = 1.0 AU, λexc = 488 nm, and λem = 515 to 535 nm). Cyto-tdTomato (red channel) and MitoTracker Orange (red channel) were imaged using confocal mode (three slices, Δz = 0.7 μm, aperture = 1.0 AU, λexc = 561 nm, and λem = 569 to 641 nm, MitoTracker Orange) or with wide-field mode (Cyto-tdTomato). TMRM imaged for the colocalization experiments with Mt-GFP was imaged using the confocal mode (three slices, Δz = 0.7 μm, aperture = 1.0, λexc = 488, and λem = 569 to 641 nm). For the live-dead assay, primary neurons were imaged with a 20× (wide-field mode, NA 0.45) objective using λexc = 561 nm and λem = 569 to 641 nm for the PI-stained dead cells (red channel) and λexc = 488 nm and λem = 515 to 535 nm for the FDA-stained live cells (green channel). Ten fields were collected from each well, and four wells were imaged per condition in the live-dead assay. For all other experiments, four fields per well were imaged unless otherwise stated.
A CLARIOstar (BMG LABTECH) microplate reader was used to measure fluorescence of TMRM and luminescence produced from the ATP. Fluorescence was measured by applying 540 ± 18–nm excitation and 585 ± 20–nm emission filters in the orbital averaging well scan mode, 17 flashes per well. Luminescence was measured through an emission filter of 571 ± 80 nm, detecting light from the whole well.
Automated data analysis pipeline
Most experiments used only Mt-GFP; some used both Mt-GFP and Cyto-tdTomato. The Z-projected images were preprocessed using a custom Fiji macro. For preprocessing of Mt-GFP images, background was subtracted (rolling ball, radius = 3), images were median-filtered (radius = 1), and somatic mitochondria were removed. For removal of the somatic mitochondria, they were first segmented and then a somatic mitochondrial mask was overlaid on the image. Steps for somatic mitochondria segmentation were as follows: taking the square root of the image (equalization of the too bright and too dim somatic mitochondria), minimum filtering (kernel = 20) to erode small structures, using Gaussian filter (σ = 20) to ensure mitochondria in neurites were downscaled, Otsu thresholding, object filtering (5000 to 100,000 pixels), and binary dilation (15 cycles) of segmented somatic mitochondria. The size or shape of the mitochondria in neurites did not change during this preprocessing. For preprocessing of Cyto-tdTomato images, we applied a tubeness filter (39) and then removed the soma using the somatic mitochondrial mask from the green channel. For further details, see also (40).
Segmentation of the preprocessed images followed using GE Developer software. Images were equalized to the complete 16-bit dynamic range and segmented using object-based segmentation with a kernel size of 3. Segmented objects were classified as axonal mitochondria (green channel; length, 0.5 < Mtaxon ≤ 1.4 μm; intensity, >5000; area, >0.25 μm2; circularity, >0.6), dendritic mitochondria (green channel; Mtdend length, ≥ 2.4 μm; intensity, >5000), and neurites (red channel; intensity, >5000; circularity, <0.5). Count, length, area, and circularity were measured for axonal and dendritic mitochondria; area was measured for neurites; and the measurements were aggregated using the median for mitochondrial area, circularity, and length and a sum of the area for mitochondria (CA) and neurites.
Aggregated field values were further processed by a custom Python script. Field values containing less than 500 mitochondria were removed, along with outliers, identified by a custom-written machine learning algorithm (fig. S6, left), and field values were averaged to well values. Robust Z-scores were calculated using neutral wells (fig. S6, right) for the primary screen and DMSO-treated wells for the rescreen. Outliers were removed from the replicate plate well values, and the replicate plates were averaged. Replicate plate wells containing outliers were identified by selecting those where the SDs of the replicates’ robust Z-scores were larger than 3. In those cases, one value, which was furthest from the mean, was removed. In the rescreen, where 10 replicate plates were used instead of 4, this algorithm was run three times, allowing the removal of maximum three values from the 10 replicates. Finally, hits were selected on the basis of their robust Z-scores: for axonal mitochondrial content hits, axonal mitochondrial CA > 2.5 and dendritic mitochondrial CA between −2 and 2; for dendritic mitochondrial content hits, CA > 2.5 and axonal mitochondrial CA < 2; for elongation hits, dendritic mitochondrial length median > 2.5; and for health hits, axonal mitochondrial circularity median < −2.5.
Structural clustering of hits
Morgan fingerprints of the rescreened hits were calculated using RDKit. A distance matrix using the dice similarities of the fingerprints were created, and clustering was performed by WPGMA (Weighted Pair Group Method with Arithmetic Mean) method.
Statistical analysis
N = 4 (from one experiment) and N = 10 (from three independent experiments) replicate plates (one well per compound per plate, four fields per well, n = 1000 to 5000 mitochondria per field) were used for the primary screen and rescreen, respectively. Hits were picked on the basis of a robust Z-score cutoff of 2.5. Detailed data analysis can be found in the “Automated data analysis pipeline” section. Compounds are significantly different from control if less than or greater than 2 robust Z-scores. Data of functional assays are from N = 4 replicate plates (8 wells per compound per plate, two independent experiments). Statistical tests were performed using GraphPad Prism 7.0 (GraphPad Software Inc.)
Supplementary Material
Acknowledgments
Funding: This research was supported by NIH grants R01MH109957 and 3R01MH10997-03S1 to R.L.D. Additional support was provided by a training grant in Alzheimer’s Drug Discovery from the Lottie French Lewis Fund of the Community Foundation for Palm Beach and Martin Counties, the Coleman Hogan Fund for Memory Research, and major contributions from community philanthropists that include W. Meyer, A. Dreyfoos, and P. McGraw. We thank K. Kelly for assistance with hippocampal neuron cultures for the electrophysiological studies. Author contributions: B.H.V., M.K., K.V., and R.L.D. developed the mitochondrial dynamics assays described. K.V. performed the primary screen. B.H.V. and M.K. developed the image and data analysis pipeline and performed the rescreens, dose-response, disease model experiments, and bioinformatics. Z.L. developed and performed ATP production and TMRM and OCR assays. C.M.M. designed the vectors and generated the Mt mouse line. Y.A. and S.V.P. designed the electrophysiology experiments, and Y.A. collected the data for these. R.P. treated mice with compounds and isolated the mitochondria. B.H.V., M.K., and R.L.D. wrote the initial drafts of the manuscript. The final versions were edited and approved by all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaaw8702/DC1
Fig. S1. Optimizing the screen.
Fig. S2. Mitotoxicity and neurotoxicity with FCCP treatment.
Fig. S3. False-positive hit rate in the screen.
Fig. S4. Structural clustering of the MnMs.
Fig. S5. Dose response of Aβ(1–42) oligomers, peroxide, and glutamate treatment on neuronal mitochondrial features.
Fig. S6. Scheme of field outlier removal and using neutral wells for robust Z-score calculation.
Table S1. The effect of MnMs on neurite sprouting.
Table S2. EC50 values of selected MnMs for processes of mitostasis and neurite sprouting.
Movie S1. Time-lapse video of elongating and fusing mitochondria.
REFERENCES AND NOTES
- 1.Misgeld T., Schwarz T. L., Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitaker R. M., Corum D., Beeson C. C., Schnellmann R. G., Mitochondrial biogenesis as a pharmacological target: A new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 56, 229–249 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Nunnari J., Suomalainen A., Mitochondria: In sickness and in health. Cell 148, 1145–1159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D., Wang J., Bonamy G. M. C., Meeusen S., Brusch R. G., Turk C., Yang P., Schultz P. G., A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 51, 9302–9305 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Baek S. H., Park S. J., Jeong J. I., Kim S. H., Han J., Kyung J. W., Baik S.-H., Choi Y., Choi B. Y., Park J. S., Bahn G., Shin J. H., Jo D. S., Lee J.-Y., Jang C.-G., Arumugam T. V., Kim J., Han J.-W., Koh J.-Y., Cho D.-H., Jo D.-G., Inhibition of Drp1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer's disease model. J. Neurosci. 37, 5099–5110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Q., Tammineni P., Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J. Alzheimers Dis. 57, 1087–1103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calkins M. J., Manczak M., Mao P., Shirendeb U., Reddy P. H., Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 20, 4515–4529 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desler C., Lillenes M. S., Tønjum T., Rasmussen L. J., The role of mitochondrial dysfunction in the progression of Alzheimer's disease. Curr. Med. Chem. 25, 5578–5587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D. I., Lee K. H., Oh J. Y., Kim J. S., Han H. J., Relationship between β-amyloid and mitochondrial dynamics. Cell. Mol. Neurobiol. 37, 955–968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewerenz J., Maher P., Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 9, 469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim G. H., Kim J. E., Rhie S. J., Yoon S., The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pagani L., Eckert A., Amyloid-beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011, 925050 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iannetti E. F., Smeitink J. A. M., Beyrath J., Willems P. H. G. M., Koopman W. J. H., Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat. Protoc. 11, 1693–1710 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Iannetti E. F., Willems P. H. G. M., Pellegrini M., Beyrath J., Smeitink J. A. M., Blanchet L., Koopman W. J. H., Toward high-content screening of mitochondrial morphology and membrane potential in living cells. Int. J. Biochem. Cell Biol. 63, 66–70 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Kitami T., Logan D. J., Negri J., Hasaka T., Tolliday N. J., Carpenter A. E., Spiegelman B. M., Mootha V. K., A chemical screen probing the relationship between mitochondrial content and cell size. PLOS ONE 7, e33755 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suen D.-F., Norris K. L., Youle R. J., Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebrahimi-Fakhari D., Saffari A., Wahlster L., Di Nardo A., Turner D., Lewis T. L. Jr., Conrad C., Rothberg J. M., Lipton J. O., Kölker S., Hoffmann G. F., Han M.-J., Polleux F., Sahin M., Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 17, 1053–1070 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung C. H.-L., Cheng S. S.-Y., Cheung Y.-T., Wuwongse S., Zhang N. Q., Ho Y.-S., Lee S. M.-Y., Chang R. C.-C., A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 14, 7–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badal K. K., Akhmedov K., Lamoureux P., Liu X.-A., Reich A., Fallahi-Sichani M., Swarnkar S., Miller K. E., Puthanveettil S. V., Synapse formation activates a transcriptional program for persistent enhancement in the bi-directional transport of mitochondria. Cell Rep. 26, 507–517.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frederick R. L., Shaw J. M., Moving mitochondria: Establishing distribution of an essential organelle. Traffic 8, 1668–1675 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsutsumi R., Yoshida T., Nii Y., Okahisa N., Iwata S., Tsukayama M., Hashimoto R., Taniguchi Y., Sakaue H., Hosaka T., Shuto E., Sakai T., Sudachitin, a polymethoxylated flavone, improves glucose and lipid metabolism by increasing mitochondrial biogenesis in skeletal muscle. Nutr. Metab. (Lond). 11, 32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindahl M., Tagesson C., Flavonoids as phospholipase A2 inhibitors: Importance of their structure for selective inhibition of group II phospholipase A2. Inflammation 21, 347–356 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Novo Belchor M., Hessel Gaeta H., Fabri Bittencourt Rodrigues C., Ramos da Cruz Costa C., De Oliveira Toyama D., Domingues Passero L. F., Dalastra Laurenti M., Hikari Toyama M., Evaluation of Rhamnetin as an inhibitor of the pharmacological effect of secretory phospholipase A2. Molecules 22, E1441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farooqui A. A., Ong W.-Y., Horrocks L. A., Inhibitors of brain phospholipase A2 activity: Their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol. Rev. 58, 591–620 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Choi J. W., Jang B. K., Cho N.-c., Park J.-H., Yeon S. K., Ju E. J., Lee Y. S., Han G., Pae A. N., Kim D. J., Park K. D., Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg. Med. Chem. 23, 6486–6496 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Edmondson D. E., Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 20, 155–160 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Zheng J., Ramirez V. D., Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br. J. Pharmacol. 130, 1115–1123 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen T., Nioi P., Pickett C. B., The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmström K. M., Kostov R. V., Dinkova-Kostova A. T., The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 1, 80–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klinge C. M., Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 105, 1342–1351 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jornayvaz F. R., Shulman G. I., Regulation of mitochondrial biogenesis. Essays Biochem. 47, 69–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnold L. A., Estébanez-Perpiñá E., Togashi M., Shelat A., Ocasio C. A., McReynolds A. C., Nguyen P., Baxter J. D., Fletterick R. J., Webb P., Guy R. K., A high-throughput screening method to identify small molecule inhibitors of thyroid hormone receptor coactivator binding. Sci. STKE 2006, pl3 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Choudhary V., Kaddour-Djebbar I., Lakshmikanthan V., Ghazaly T., Thangjam G. S., Sreekumar A., Lewis R. W., Mills I. G., Bollag W. B., Kumar M. V., Novel role of androgens in mitochondrial fission and apoptosis. Mol. Cancer Res. 9, 1067–1077 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh B. K., Sinha R. A., Tripathi M., Mendoza A., Ohba K., Sy J. A. C., Xie S. Y., Zhou J., Ho J. P., Chang C.-y., Wu Y., Giguère V., Bay B.-H., Vanacker J.-M., Ghosh S., Gauthier K., Hollenberg A. N., McDonnell D. P., Yen P. M., Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 11, eaam5855 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Sureda F. X., Gabriel C., Pallàs M., Adan J., Martínez J. M., Escubedo E., Camarasa J., Camins A., In vitro and in vivo protective effect of orphenadrine on glutamate neurotoxicity. Neuropharmacology 38, 671–677 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Chang C.-R., Blackstone C., Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., Lein E. S., Zeng H., A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sims N. R., Anderson M. F., Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 3, 1228–1239 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Sato Y., Nakajima S., Shiraga N., Atsumi H., Yoshida S., Koller T., Gerig G., Kikinis R., Three-dimensional multi-scale line filter for segmentation and visualization of curvilinear structures in medical images. Med. Image Anal. 2, 143–168 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Kepiro M., Varkuti B. H., Davis R. L., High content, phenotypic assays and screens for compounds modulating cellular processes in primary neurons. Methods Enzymol. 610, 219–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaaw8702/DC1
Fig. S1. Optimizing the screen.
Fig. S2. Mitotoxicity and neurotoxicity with FCCP treatment.
Fig. S3. False-positive hit rate in the screen.
Fig. S4. Structural clustering of the MnMs.
Fig. S5. Dose response of Aβ(1–42) oligomers, peroxide, and glutamate treatment on neuronal mitochondrial features.
Fig. S6. Scheme of field outlier removal and using neutral wells for robust Z-score calculation.
Table S1. The effect of MnMs on neurite sprouting.
Table S2. EC50 values of selected MnMs for processes of mitostasis and neurite sprouting.
Movie S1. Time-lapse video of elongating and fusing mitochondria.