

The CPMC model demonstrates the regulatory role of chromatin’s physical structure on transcription and phenotypic plasticity.
Abstract
Three-dimensional supranucleosomal chromatin packing plays a profound role in modulating gene expression by regulating transcription reactions through mechanisms such as gene accessibility, binding affinities, and molecular diffusion. Here, we use a computational model that integrates disordered chromatin packing (CP) with local macromolecular crowding (MC) to study how physical factors, including chromatin density, the scaling of chromatin packing, and the size of chromatin packing domains, influence gene expression. We computationally and experimentally identify a major role of these physical factors, specifically chromatin packing scaling, in regulating phenotypic plasticity, determining responsiveness to external stressors by influencing both intercellular transcriptional malleability and heterogeneity. Applying CPMC model predictions to transcriptional data from cancer patients, we identify an inverse relationship between patient survival and phenotypic plasticity of tumor cells.
INTRODUCTION
Most perturbations a eukaryotic cell experiences occur at nonreplicative time scales. These perturbations are remarkably varied, range in intensity, and can be completely distinct from previously encountered stimuli. Examples exist throughout the human body, including within the skin, the alimentary tract, the immune system, the respiratory tract, and the reproductive system and in malignancy. Consider the epithelial lining of the digestive and respiratory systems. While both systems are constantly renewing their lining, most functional cells within these tissues persist for days to weeks after replication. During their life span, these cells are exposed to a wide range of nutrients and toxicants that necessitate the modification of gene expression to carry on basic cellular functions across these variable conditions (e.g., appropriately absorb nutrients, regulate ionic homeostasis, maintain a sufficient mucosal barrier, excrete waste products, and secrete immunoglobulins). No better example may exist than malignant tumor cells, which are remarkably adept at acclimating to a broad spectrum of cytotoxic chemotherapies and radiation exposure while evading detection from the myriad tools present within the immune system. These capabilities evoke a critical question: How do individual cells acclimate to fluctuating or completely novel conditions? Likewise, how do collections of cells, such as an organ or a tumor mass, acclimate in aggregate to a heterogeneous, rapidly evolving environment?
One widely explored mechanism to respond to these varied conditions is to have a level of predetermined functionalization: intermixing specialized cells within an organ to carry out specific roles. Beyond establishing precoordinated responses, an intriguing possibility is for cells and cell populations to have an encoded level of phenotypic plasticity to acclimate to changing environmental conditions in real time (1, 2). In the context of multicellular systems, the level of phenotypic plasticity encoded is a product of cellular malleability—the functional responsiveness of cells toward stable end states upon external stimulation—and the level of intercellular heterogeneity—the diversity of stable states that are observed within the same population at a given time. Recent advances in single-cell technologies have highlighted the remarkable levels of diversity within multicellular systems for otherwise seemingly identical cells (3–5). An extraordinary level of diversity has been demonstrated in the lungs (3), breast tissue (4), the gastrointestinal tract (5), and in malignancy (6–9). Furthermore, the cancer state is associated with considerable structural (10, 11), transcriptional (8, 12), epigenetic (9, 13), and mutational heterogeneity (6, 9)—all of which have been demonstrated to be independently linked to chemotherapeutic resistance, metastasis, survival, and resilience in multiple cancer models. Likewise, transcriptional responsiveness is concomitant with cancer cell survival in response to chemotherapy and the functional responsiveness of immune cells to microbes (14). To date, these two facets of phenotypic plasticity have largely been viewed as distinct entities, as no mechanisms have been identified that simultaneously link both diversity and responsiveness. However, at the level of gene transcription, both the malleability and intercellular heterogeneity of gene expression within cell populations could result from the physical organization of chromatin (14, 15).
To test the hypothesis that the physical organization of the genome is a regulator of both transcriptional malleability and intercellular heterogeneity, we used multiscale modeling to describe transcription as a series of chemical reactions occurring in a heterogeneous, crowded environment—a disordered chromatin-packing macromolecular crowding (CPMC) model. Pairing the CPMC model with single-cell RNA sequencing (scRNA-seq), chromatin electron microscopy tomography (ChromEM)—a DNA-specific staining technique for electron microscopy—and live-cell partial wave spectroscopic (PWS) microscopy, we demonstrate that the physical structure of chromatin packing determines both the level of transcriptional malleability and heterogeneity. In particular, the CPMC model predicts that at the supranucleosomal scale [from ~kbp (kilo–base pairs) to several Mbp (million base pairs)], the scaling behavior of chromatin packing, which is the relationship between the genomic length of a chromatin chain and its packing size in three-dimensional space, determines the level of intercellular transcriptional heterogeneity by regulating local variations in chromatin density (14, 16). Furthermore, the scaling of chromatin packing directly influences the level of transcriptional malleability by regulating both gene accessibility and the free energy of transcription reactions (17–19). Last, applying the CPMC model to interrogate the phenotypic plasticity of cancer cells, we show that increased transcriptional malleability has an impact on cancer patient mortality. Analyzing gene expression data from The Cancer Genome Atlas (TCGA) (20), we demonstrate that transcriptional divergence—a direct measure of transcriptional malleability, which is connected with chromatin packing scaling—is inversely related to patient survival in advanced (stage 3 and stage 4) colorectal, breast, and lung cancers. In summary, this work mechanistically links two distinct aspects of phenotypic plasticity, transcriptional malleability and intercellular heterogeneity, with the physical properties representing the structure of disordered chromatin packing. Using the CPMC model, we quantitatively describe the role that physical forces play on gene expression in vitro and describe a potential mechanistic relationship between structural alterations of chromatin observed in cancer and prognosis.
Model
The CPMC model considers transcription in dilute, ex vivo conditions as a series of diffusion-limited chemical reactions that use DNA, transcription factors (TFs), and RNA polymerase II (Pol-II) to produce mRNA (Fig. 1A). The total production of mRNA in these conditions will depend on the concentration of reactants ([C]tot; Fig. 1B), the rate of polymerase elongation (km; Fig. 1C), and the dissociation rates of TFs and polymerase from DNA (KD: Fig. 1D). These molecular factors are well-studied regulators of gene expression in vitro. For example, at the scale of nuclear compartments, the formation and dissipation of topologically associating domains (TADs) can alter local TF concentrations (21). In addition, posttranslational histone modifications alter nucleosomal stability, thereby influencing the rate of polymerase elongation (22). Other posttranslational modifications of RNA polymerase itself independently control polymerase activity (23). Furthermore, gene motifs determine binding affinities of polymerase and TFs, resulting in varied dissociation constants of these molecules from their respective target genes (24).
Fig. 1. The chromatin-packing macromolecular-crowding model integrates molecular and physical regulators of transcription.
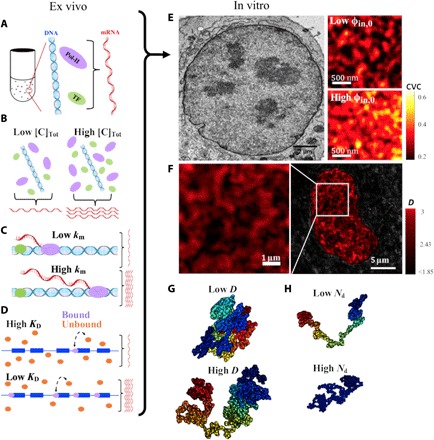
The regulators influencing transcription reactions can be generally divided into two categories: molecular regulators (km, KD, and [C]tot) (A to D) and physical regulators (D, φin,0, and Nd) (E to H). (A) The CPMC model describes transcription as a series of diffusion-limited chemical reactions. Ex vivo, expression depends on (B) concentration of transcriptional reactants [C]tot (TFs, green; Pol-II, yellow), (C) RNA polymerase elongation rate (km), and (D) the disassociation rate of Pol-II from the transcription start site (TSS; KD). (E) Left: In addition to the molecular determinants, transcriptional reactions are influenced by the highly dense and complex nuclear environment. The concentration of the main crowder with the nucleus, chromatin, can be measured by chromatin electron microscopy (ChromEM). As an example, ChromEM of a nucleus of an A549 lung adenocarcinoma cell is shown. Right: ChromEM measurements of chromatin volume concentration (CVC) demonstrate that chromatin density varies throughout the nucleus. Chromatin packing domains can be visualized as areas of higher chromatin packing density. Within each packing domain, the average volume fraction of chromatin can range from 15 to 65%. Typical domains are 100 to 200 nm in diameter and may contain, on average, ~400 kb. (F) Representative PWS image of an A549 cell demonstrating the existence of chromatin packing domains as regions of elevated chromatin packing scaling (also referred to as fractal dimension) D, which vary throughout the nucleus. (G) A polymer with a higher D (right) has a more heterogeneous density distribution and a greater accessible surface area compared to a polymer with a lower D (left). (H) Nd is the genomic size (in bps) of a chromatin packing domain and can range from less than 100 kbp to several Mbp. Packing domains are illustrated by color coding, with each color representing a separate domain.
Compared to ex vivo conditions, the eukaryotic nucleus is a highly crowded, heterogeneous environment (Fig. 1E). To model transcription reactions within such an environment requires consideration of the length scales involved. At the smallest scale (within ~20 nm of a gene, i.e., an “interaction volume”), macromolecular crowding (ϕin) influences transcription by affecting the mobility of transcriptional reactants and the dissociation rate of these molecules from DNA (19, 25, 26). In addition, the accessible surface area of chromatin determines the number of DNA binding sites available to transcriptional reactants. The probability of a gene promoter to be available for transcription depends on its local accessible surface area. At these small length scales, transcription can be modeled as a network of chemical reactions involving TFs, Pol-II, and DNA. TFs bind to their respective DNA binding sites and recruit polymerases to gene promoters, which, in turn, bind DNA. These series of reactions result in intermediary transcription complexes that stochastically transcribe genes into mRNA. Each reaction coefficient depends on local crowding effects, which can be calculated using Brownian dynamics (BD) and Monte Carlo (MC) simulations. Gene expression for particular crowding conditions is calculated by solving the steady-state network of equations that models these transcription reactions (19, 26). This modeling approach predicts a nonmonotonic dependence of transcription on crowding. This nonmonotonic behavior is influenced by the molecular factors previously discussed and is due to the opposing effects of macromolecular crowders on chemical reactions. Initially, transcription rates increase with crowding due to an enhanced binding stability of TFs and Pol-II arising from attractive depletion interactions. At higher crowding conditions, however, the crowding-induced reduction of molecular mobility dominates, lowering transcription rates. Notably, the most prevalent macromolecular crowder in the nucleus is chromatin. Thus, local chromatin density within the interaction volume of a gene should have a profound effect on transcription processes. Recent electron microscopy studies have shown that chromatin packing density is highly heterogeneous across the genome. Some genes have interaction volumes with exceedingly high densities [chromatin volume concentration (CVC) up to >60%], while others may be positioned in regions of the nucleus with CVC as low as ~10 to 20% (27). One approach to study the effect of local crowding on transcription in cells would be to experimentally measure the local density of chromatin near every gene using electron microscopy and pair these measurements with in situ mRNA levels. This, however, is beyond existing technical capabilities, and an alternate approach is needed.
Instead of experimentally mapping gene expression to locus-specific crowding conditions, the CPMC model probabilistically samples the polymeric properties of chromatin to approximate transcriptional output of an ensemble of genes under similar molecular and varying physical conditions (14, 28, 29). A combination of molecular factors influences the relative initial expression levels of these genes (19). In this work, we focus on how physical regulators further modulate transcription reactions to produce a final observed transcription rate. The model considers chromatin to be a disordered heteropolymer that is heterogeneously packed in three-dimensional (3D) space. The 3D packing of the chromatin polymer determines the volume fraction occupied by chromatin, the number of nucleotides acting together as a grouped polymeric entity (Nd), and the space-filling geometry or the scaling behavior of these polymeric entities. Nd can be considered as the number of nucleotides that are contained within a subset of the chromatin polymer that has self-similar, power law scaling properties. The power law scaling behavior exhibited by the chromatin polymer within a self-similar domain of size Nd describes the relationship between the length of a given segment (e.g., the number of nucleotides, N) and the size (R) of the physical space occupied by that segment, N ∝ RD for N ≤ Nd. The scaling factor D is frequently referred to as the fractal dimension of the polymer and is determined by the balance of the free energy of polymer-polymer and polymer-solvent interactions. D of an unconstrained free polymer may range from D = 5/3 for an excluded volume polymer to D = 2 for an ideal chain polymer in theta solvent and to D = 3 for a completely space-filling polymer. A polymer with a uniform chain structure throughout would form a single fractal domain with D determined by the properties of the chain and the solvent. Chromatin, on the other hand, is a heterogeneous polymer with variable post-translational histone modifications and DNA methylation patterns. This leads to differential interactions between the heterogeneous chromatin subunits and results in chromatin compartmentalization, potentially as a result of liquid-liquid phase separation (30). Additional topological constraints induced by chromatin-binding proteins, such as those responsible for the formation of chromatin loops or nuclear lamins, might further influence D within a given chromatin domain or compartment. Electron microscopy and super-resolution imaging studies have demonstrated the existence of spatially segregated supranucleosomal nanoscale packing domains with a variable size distribution in 3D space (27, 31). We have been able to visualize the existence of these packing domains using ChromEM (Fig. 1E) and PWS (Fig. 1F) as small (100 to 200 nm in diameter; genomic size, between 100 and 400 kb), globular regions that exhibit power law scaling behavior and tend to have higher chromatin density and D. The CPMC model considers a gene’s interaction volume to be located within these packing domains. Accordingly, the local environment of a gene’s interaction volume is determined by the encompassing packing domain, each of which may have its own average nuclear crowding density (ϕin,0) (Fig. 1E), chromatin packing scaling D (Fig. 1, F and G), and genomic size (Nd) (Fig. 1H). These local physical conditions are important determinants of gene expression. In addition, gene length (L) partially influences the size of the interaction volume of a given gene, affecting the range of crowding conditions the gene is probabilistically exposed to. The CPMC model uses these measurable physical regulators of chromatin to approximate distributions of chromatin density and accessibility to determine transcription for each gene throughout the entire nucleus, a feat that is currently experimentally infeasible (17).
The expected expression rate of a gene in vitro is the product of the steady-state mRNA expression rate of that gene (ϵ) and the probability of the gene to be on the accessible surface of the chromatin polymer (pg). Steady-state expression rate is a function of molecular features surrounding the gene of interest (; TF concentration, histone state, enhancer-promoter interactions, etc.) (Fig. 1, B to D) in the context of local physical conditions (Fig. 1, E to H) (14, 18, 19, 25). The probability of gene accessibility contributes to the likelihood of a gene to interact with transcriptional components (TFs and Pol-II) in vitro (32). It is beyond technical capabilities to measure all molecular and physical parameters of the model for specific genes at the single-cell level. Thus, we explore how a given ensemble of genes with similar molecular features (e.g., grouped by their initial expression or associated gene ontologies) would respond to changes in average measurable physical conditions. Specifically, we study how average nuclear crowding density (ϕin,0), average chromatin packing scaling (D), and genomic size of a packing domain (Nd) change the behavior of global transcription processes. It is critical to stress that the CPMC model does not assume that the chromatin polymer has the same power law scaling behavior or constant density throughout the entire nucleus, but that this is instead an approximation due to existing experimental limitations. The model can further be extended to consider that each packing domain has its own chromatin packing scaling D, as technological capabilities to co-register chromatin packing, molecular, and genomic properties advance. In addition, this model considers nuclear crowding density within each interaction volume, ϕin, is assumed to be constant relative to the time scale of transcription (in minutes), in line with recent imaging studies of chromatin mobility (33).
Given these considerations, in a population of cells, each gene will be exposed to different crowding densities ϕin. Each ϕin will be sampled from the probability distribution function f(ϕin), which is assumed to follow a normal distribution with mean ϕin,0 and variance , where rmin is the radius of the elementary unit of chromatin (e.g., a base pair) and rin is the radius of the interaction volume (Supplementary Text) (14). Because of the mass fractal nature of chromatin, for a gene of length L, where is the radius of the interaction volume for a single base pair and is approximated from previous MC simulations of crowding effects (14, 19). Thus, the expected range of crowding densities each gene is exposed to is dependent on the statistical properties of the packing domain of size Nd where the gene is located, including D and ϕin,0, and is further influenced by length L of the gene. The transcription rate ϵ itself is assumed to depend on molecular features and on local crowding density ϕin. We calculate all expression rates under the assumption that molecular features remain constant throughout the population, with physiologically relevant values used in previous MC and BD crowding simulations (table S1) (19). This gives rise to the form of , the average expression rate for an ensemble of genes that share a given as
| (1) |
Likewise, a power law model of chromatin packing scaling allows the CPMC model to calculate the probability of a unit of DNA (e.g., a gene promoter) to be on the accessible surface of chromatin, pg (28, 29)
| (2) |
Last, merging accessibility with steady-state expression rate for a group of genes, the ensemble expression rate is
| (3) |
To quantitatively analyze the effect of D on gene expression, we calculated the sensitivity of gene expression as a function of D as predicted by the CPMC model. Sensitivity (Se) is the measurement of how a dependent variable (i.e., gene expression) will change as a function of a perturbation to an independent variable (i.e., D). Se of expression rate for any group of genes to changes in chromatin packing is defined as
| (4) |
where Ei is the initial average expression rate of the group of genes sharing similar molecular features and gene length L, and Di is the initial average chromatin packing scaling within a cell population. A positive Se for a given group of genes indicates that an increase in the scaling of chromatin packing (D↑), on average, enhances their collective expression rate. The CPMC model predicts the output of transcription reactions that occur within the nucleus. Assuming that the half-life of mRNA transcripts is dictated by cytoplasmic conditions, structural changes in chromatin that alter chromatin packing scaling D are not considered to alter the degradation rate of mRNA. Thus, sensitivity should be directly related to the number of transcripts produced for any group of genes in the nucleus.
To solve Eq. 4, we used a Taylor series approximation of around ϕin,0
| (5) |
where is a nonmonotonic function of ϕin due to the competing effects of crowding on depletion interactions and molecular diffusion, and quantifies gene expression as a function of crowding within a transcriptional interaction volume. Expression rate κ = 22.6 nM/s is derived from a steady-state solution of rate equations that model transcription and whose crowding-dependent rates were determined from BD and MC simulations as described previously (14, 19). Notably, the function can be simulated by varying any or several of the components of . Although, in principle, the exact form of as a function of may depend on which component of is being varied, i.e., , in practice, κ is only weakly dependent on . In other words, depends on primarily through , with the average expression rate as the “common dominator” of multiple molecular factors. Thus, predictions of the CPMC model regarding the effects of physical regulators on ensemble gene expression should be robust to changes in molecular factors. Combining Eqs. 1 to 5, Se of expression rate becomes
| (6) |
RESULTS
Physical factors of chromatin structure regulate sensitivity of gene expression to changes in chromatin packing scaling
To first test the CPMC model predictions in vitro, we used live-cell PWS microscopy to measure D (Fig. 2, A and B) (33, 34) and ChromEM to measure ϕin,0 (Fig. 2, C and D) (27) paired with mRNA microarrays, RNA-seq, and scRNA-seq to measure gene expression of cell populations under different conditions. Specifically, average D was calculated by first averaging D values from PWS measurements within each cell nucleus and then averaging these measurements over the entire cell population for each treatment condition. Using ChromEM, average chromatin density was measured within each nucleus with ~3-nm resolution. As ϕin,0 represents the crowding contributions from both chromatin and mobile crowders within the nucleus, we added to our ChromEM measurements an additional 5% contribution from unbound macromolecules (as described in Materials and Methods). In addition, we used publicly available DNA sequencing information to obtain gene length and high-throughput chromatin conformation (Hi-C) capture data to approximate Nd from the size of TADs (35). In relation to previous work on higher-order chromatin organization, Nd could extend from tens of thousands to millions of base pairs. While Nd might not necessarily represent the organization observed in TADs, TAD size was used as an approximate measure of Nd as these domains have been shown to obey self-similar organization (36), as evidenced by power law scaling properties of contact probability within TADs (37). Combining these methods, we then tested the CPMC model’s predictions of Se against in vitro measurements for each identified physical regulator of gene expression.
Fig. 2. Comparison of the CPMC model with experimental measurements of gene expression as a function of physical regulators Di, Nd, and φin,0 and gene length L.
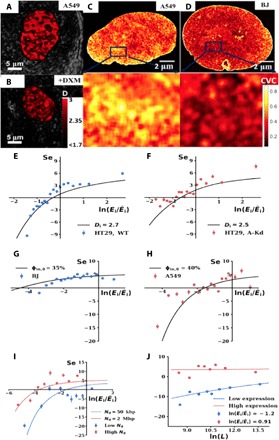
(A and B) Representative live-cell PWS microscopy images of nuclear D scaled between 2.56 and 2.66 for control (A) and 12-hour dexamethasone (DXM)–treated lung adenocarcinoma A549 cells (B). Brighter red corresponds to higher chromatin packing scaling. (C and D) Representative heat maps of CVC values from analysis of ChromEM images of cell nuclei from A549 cells (C) and human fibroblasts BJ (D). Representative magnified regions from each nucleus demonstrate an average CVC of 0.35 in A549 cells compared to 0.30 in BJ cells, which represents the chromatin contribution to the average crowding volume fraction φin,0. (E to J) Comparison between the CPMC model (solid lines) and experimentally measured (points) sensitivity of gene expression to an incremental change in chromatin packing scaling D (Se, y axis) as a function of initial gene expression (x axis). (E) Cells with chromatin with a higher initial Di = 2.7 [wild-type (WT) HT-29 cells] have a bidirectional Se curve that becomes attenuated if Di is lowered to 2.5 (short hairpin RNA knockdown Arid1a HT-29 cells) (F). Each point represents the average of 100 genes. Changes in D were induced by cell treatment with 10% FBS, 100 nM EGF, and 100 nM PMA. The CPMC model was able to explain 86% of the variance of the experimental data for WT HT-29 cells and 51% of the variance for Arid1a HT-29 cells. (G) Se in cells with a lower φin,0 [BJ cells, φin,0 = 35%; each point corresponds to 300 genes; explained variance (EV) = 59%] is attenuated in comparison to that of cells with a higher density (H) (A549 cells; φin,0 = 40%, 100 genes per point, EV = 74%). (I) Genes located within larger packing domains (Nd ~ 2 Mbp, 12 genes per point, EV = 56%) have a lower initial expression but have a larger Se in comparison to genes localized within smaller packing domains (Nd ~ 50 kbp, 12 genes per point, EV = 37%). The change in D was induced in A549 cells by treatment with 100 nM dexamethasone. Nd was approximated on the basis of corresponding TAD size: 2-Mbp TADs for the high Nd group of genes versus 50-kbp TADs for the low Nd genes. TAD size was measured using the Arrowhead function from Juicer Tools used to analyze Hi-C data. (J) Comparison between the CPMC model (solid line) with experimental results (points, 60 genes per point) in HT-29 cells showing the effect of gene length (L, x axis) on Se (y axis). In agreement with the model, shorter, initially underexpressed genes (low expression, blue curve; points, EV = 67%) are disproportionally repressed by an incremental increase in D compared to longer genes (high expression, red curve, points). Error bars represent SE from four biological replicates.
To test the role of initial Di, we performed an RNA interference knockdown of the chromatin remodeling enzyme Arid1a (A-Kd) in human colon carcinoma HT-29 cells, which resulted in a lower Di compared to wild-type (WT) cells (17). Next, we used PWS microscopy to measure changes in chromatin packing scaling D in serum-starved WT and A-Kd HT-29 cells before and 30 min after stimulation with 10% fetal bovine serum (FBS), 100 nM epidermal growth factor (EGF), and 100 nM phorbol 12-myristate 13-acetate (PMA) (14). In parallel, we measured gene expression for these conditions at 5 hours using mRNA microarrays. Genes were grouped for WT and A-Kd cells separately based on their relative initial expression during serum starvation, and the experimentally measured sensitivity Δ ln E/Δ ln D was calculated for each group of genes. As predicted by the CPMC model, experimental measurements of Se of gene expression show a bidirectional, monotonic responsiveness to D as a function of initial expression in HT-29 cells (ϕin,0 ~ 39%, approximated by dividing chromosome copy number by nuclear volume). In addition, we found that Di predominantly changes the responsiveness of initially underexpressed genes (Fig. 2, E and F). These results indicate that populations of cells with a higher D would have a higher level of transcriptional divergence (the difference between highly and low expressed genes) than lower D cells. Cancer cells across most malignancies, stem cells, and, especially, cancer stem cells are all examples of cell populations that have elevated chromatin packing scaling (2, 38). Functionally, this suggests that D can act as a means to optimize transcriptional responses as is explored in subsequent sections.
Next, we tested the effect of average nuclear crowding density, ϕin,0, on gene expression sensitivity to changes in chromatin packing scaling D. ChromEM was used to measure average chromatin density for both human lung adenocarcinoma A549 cells and differentiated BJ fibroblast cells, which had a mean CVC of 0.35 and 0.30, respectively (Fig. 2, C and D; distribution of CVC values shown in fig. S3). Approximating for an additional contribution to excluded volume interactions from mobile crowders, estimates of ϕin,0 were 40% in A549 and 35% in BJ cells. Each cell line was treated with 100 nM dexamethasone (DXM) to modulate D, which was measured by PWS. Gene expression of both cell lines with and without dexamethasone treatment was measured by RNA-seq. Sensitivity of gene expression was measured as described above for each cell line. The CPMC model predicts that cells with a lower ϕin,0 would have an attenuated bidirectional Se, an effect confirmed experimentally in the lower chromatin density BJ cells (Fig. 2G). In contrast, the higher chromatin density A549 cells (Fig. 2H) retain an asymmetric response to altered chromatin packing scaling. This suggests that cells with smaller nuclear volume, such as immune cells, or cells with increased chromosome copy number, such as malignant cancer cells, would be predisposed to produce a more pronounced bidirectional response in gene expression to stimuli that alter whole nuclear chromatin packing structure compared to cells with lower chromatin density. These results demonstrate that the net effect of increasing D and ϕin,0 is an increased transcriptional divergence between initially over- and underexpressed genes.
In addition, we tested the roles of Nd on Se. From our model, Nd determines the probability of genes being located on an exposed surface (thus allowing transcription reactions to occur), a relationship that depends nonlinearly on D (Eq. 2). Consequently, the CPMC model predicts that (i) genes in larger packing domains (e.g., Nd > 2 Mbp) would be relatively underexpressed in comparison to those within smaller Nd domains (<50 kbp) and (ii) genes within large Nd domains would be more likely to become enhanced as a function of increasing D (+Se). To test these predictions experimentally, we used the arrowhead function in Juicer tools to measure TAD sizes from Hi-C data of untreated and dexamethasone-treated A549 cells (35). As the dissociation and formation of TADs have been previously shown to alter gene expression, for our analysis, we only selected TADs that were unaltered with dexamethasone treatment. The top 20% largest (~2 Mbp) and bottom 30% smallest (~50 kbp) of these conserved TADs were chosen to produce roughly equal-sized groups of genes (~130 genes per group). Using RNA-seq to measure gene expression and PWS microscopy to measure changes in D before and after dexamethasone treatment, we analyzed the differences in sensitivity of expression between genes localized to smaller 50-kbp TADs compared to larger 2-Mbp TADs (Fig. 2I). As predicted from the CPMC model, in vitro results demonstrate that genes within larger 2-Mbp TADs have an overall higher sensitivity to changes in D (Fig. 2I) while simultaneously having lower initial expression compared to those within smaller 50-kbp TADs. Consequently, these findings suggest a regulatory role of spatially confining genes into self-similar structures, such as those found in TADs, in determining the probability of a gene to be exposed to transcriptional reactants. Given recent work indicating substantial variability in TADs from cell to cell, this would suggest yet another mechanism that cells can use to regulate their functional diversity within a population.
Last, we tested the role of gene length on sensitivity of twofold underexpressed (low) and twofold overexpressed (high) genes in the serum-starved WT HT-29 cells described above. Using the built-in Mathematica function, GenomeData, to obtain sequence length of genes, the sensitivity of gene expression to D was then calculated as a function of gene length. The model predicts that shorter genes have a smaller interaction volume, increasing the variance of crowding conditions these genes are exposed to. Consequently, an increase in D should further increase fluctuations in crowding concentrations surrounding these shorter genes, causing initially underexpressed genes to further reduce their expression in proportion to decreasing gene length L. However, genes with an initially higher expression level will be relatively unaffected by changes in gene length due to more optimal molecular characteristics (e.g., high TF and Pol-II concentrations) and initial crowding conditions these genes are exposed to. In line with the CPMC model, our experimental microarray data demonstrate that shorter, initially underexpressed genes become disproportionately underexpressed as a function of increasing D, whereas length minimally influences initially overexpressed genes (Fig. 2J).
The scaling behavior of chromatin packing regulates phenotypic plasticity through transcriptional divergence and malleability
A major implication of the CPMC model is the role physical chromatin structure plays in shaping gene expression. Thus, the model could provide a mechanistic link between two aspects of phenotypic plasticity of a population of cells: transcriptional malleability and intercellular transcriptional heterogeneity. In this case, we can consider transcriptional malleability to be the average change in expression of a gene in response to an external stimulus, while transcriptional heterogeneity can be thought of as the range in expression levels of each gene across a cell population. While there is likely to be increased complexity that results from cell-to-cell variations in average density and D, we here test how heterogeneity and malleability are influenced by the measurable features of disordered chromatin packing within a cell population. An ideal test bed for this mechanistic integration is cancer. Multiple lines of evidence have shown that chromatin structure is nearly universally transformed in malignancy (39–42). Microscale structural alterations in chromatin are currently the gold standard for histopathological diagnosis of dysplasia and malignancy (39). At the nanoscale, an increase in D has been previously reported to occur at predysplastic stages of lung, colon, esophageal, ovarian, liver, prostate, and pancreatic cancers, while the severity of the chromatin transformation has been shown to be an accurate indicator of the tumor aggressiveness (40, 42). Because (i) elevated D is a hallmark of malignancy, (ii) there is an emergent role of intercellular heterogeneity in determining chemotherapeutic responsiveness, and (iii) cancer cells rapidly alter their gene expression to overcome cytotoxic stressors (14, 16, 43), we hypothesized that cancer cells could leverage physical transformation within the nucleus to gain survival advantages. Therefore, we wanted to test whether cells could use the scaling of chromatin packing as a regulator of both transcriptional malleability and heterogeneity to achieve a rapid response to external stressors.
According to the CPMC model, the dependence of transcriptional malleability on chromatin packing scaling results from the observed asymmetric response of up-regulated and down-regulated genes to changes in D (Fig. 2), which we denote as transcriptional divergence. Here, we focus on changes in gene expression due to an external stimulus. A transcriptional response of a cell to a chemotherapeutic stress provides a case in point. Chemotherapeutic induction of apoptosis has been shown to depend on the rate of change in expression of critical genes (e.g., p53) and not their steady-state levels alone (44). Accordingly, mechanisms that increase the rate of up-regulation of these critical genes would facilitate the development of cellular resilience to stressors. Consider two populations of cells that have a baseline difference in their initial D. These two populations are then exposed to the same exogenous stressor, and a series of stress signaling pathways are activated in an attempt to overcome the perturbation. The cells’ survival now depends, in part, on the increased expression of these genes within a critical time frame. The CPMC model predicts that the population of cells with initially higher D will be more likely to up-regulate these critical genes and remain viable (Fig. 3A).
Fig. 3. The scaling of chromatin packing increases the transcriptional malleability of cancer cells.
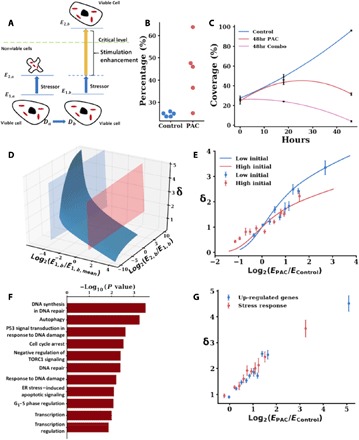
(A) In response to a stressor, such as a chemotherapeutic agent (e.g., paclitaxel), cells with a higher level of transcriptional malleability may have the ability to respond faster, which may lead to an increased survival. Cells with higher D (right, Db) have an increased a change in the rate of transcription induced by a stimulus/stressor by a factor δ (yellow arrow) relative to a change in the rate of transcription in a cell with a lower D = Da < Db. If in response to a stressor a cell may increase its probability of remaining viable by upregulating expression of pro-survival genes beyond a given threshold, a higher D cell b would increase the probability of reaching this crucial level of expression compared to cell a. (B and C) The fraction of high D cells in a cell population increases after treatment with paclitaxel (PAC) for 48 hours, suggesting that cells with higher D are more likely to survive exposure to a cytotoxic chemotherapeutic agent. (B) The percentage of cells having D in the top quartile of D values of a control cell population (y axis) increases in cells that survive treatment with paclitaxel for 48 hours. For both conditions, each dot represents the percentage of high D cells for PWS experiments on one cell population for a total number of N = 5 replicates per condition. (C) Combination treatment with celecoxib, which lowers D, and paclitaxel for 48 hours results in an increased elimination of cancer cells compared with untreated controls and cells only treated with paclitaxel. (D) CPMC model predictions of the relative transcriptional malleability coefficient δ for initially underexpressed (blue spline) and overexpressed genes (red spline) for Da = 2.3 and Db = 2.5, a difference in D relevant to experimentally observed differences in celecoxib-treated versus untreated A2780 cells. (E) scRNA-seq on A2780 cells was performed to compare transcriptional profiles of control A2780 cells (high D population) and cells treated with 75 μM of a D-lowering agent celecoxib (low D population) and their response to treatment with 5 nM paclitaxel (stressor) for 16 hours. Initially underexpressed and initially overexpressed genes are defined on the basis of control expression levels. Genes are grouped on the basis of their quantile of log2(EPAC/Econtrol), and the mean and SE of each quantile for initially underexpressed genes (blue dots, 300 genes per data point) and initially overexpressed genes (red dots, 100 genes per data point) are plotted. (F) GO analysis identified biological processes that are most significantly involved in the response to 48-hour paclitaxel treatment. Up-regulated genes were defined as those with at least twofold increase in expression. (G) Chromatin packing scaling–facilitated up-regulation (δ) of the stress response genes identified by the GO analysis (red points, 150 genes per data point) was similar to that for all up-regulated genes (blue points, 650 genes per data point).
To quantify the effect of initial D on transcriptional responsiveness, let mRNA1,a be the initial expression (the number of mRNA transcripts) for a given gene in cell a with chromatin packing state Da. At time point t = 0, a stimulus produces an increase in the gene’s rate of expression from E1,a to E2,a. Without loss of generality, we first assume that both expression rate E2,a remains stable and that the rate of mRNA degradation, v, remains constant after stimulation. The relative change in expression at time t is (mRNAa(t) − mRNA1,a)/mRNA1,a = (E2,a/E1,a − 1)(1 − e−νt), where mRNA1,a = E1,a/ν is the prestimulation steady-state expression. This relative change in expression increases with the ratio E2,a/E1,a, which is itself a function of both molecular features and the chromatin packing state surrounding the gene. This can be illustrated by comparing the response of an individual gene to an exogenous stressor in two cells a and b. Let the same gene in both cells be associated with similar molecular features but different chromatin packing states Da and Db, with Db > Da. From Eq. 4, , it follows that
| (7) |
where Sei(D) is the sensitivity of expression state Ei,a. In this situation, the effect of D on relative changes in transcription in cell b compared to cell a would be defined as
| (8) |
Within the physiological range of transcription, Se is an increasing function of E (Fig. 2), and as E2 > E1 for both cells, δ > 1. Consequently, the same stimulus will result in enhanced up-regulation of the same gene in cell b compared to cell a, driven by the differences in chromatin packing scaling between the two cells. This effect is expected to be particularly pronounced for initially underexpressed genes with Se1 < 0 that undergo a significant amplification (Se2 > 0) upon stimulation. We see that δ is directly related to transcriptional divergence and the shape of the function Se(E) (Fig. 2). A faster rise of Se as a function of E results in a higher δ. For cells a and b with similar D, δ ≈ 1 + (Se2 − Se1)(Db − Da)/Da. This implies that factors that tend to increase transcriptional divergence (e.g., high D, high crowding density, and small Nd) would be expected to result in a higher transcriptional malleability.
The functional significance of the relative transcriptional malleability coefficient δ is twofold. First, for highly amplified genes (E2/E1 ≫ 1), the relative increase in transcription at any given time after the stimulation is proportional to δ
| (9) |
Second, the time τ required to reach a given level of expression is dependent on chromatin packing scaling and inversely proportional to δ, i.e., τb/τa ≈ δ−1. This conclusion is applicable to genes that are both up-regulated and those that are down-regulated in response to a stimulus, an effect that might be especially consequential if decisions regarding cell fates must be made within a limited time period after the introduction of the stressor (44).
To experimentally explore the relationship between D and phenotypic plasticity, we performed concurrent scRNA-seq and live-cell PWS microscopy experiments on A2780 ovarian adenocarcinoma cells in response to treatment conditions that modulate chromatin packing scaling. We first tested whether chemotherapy treatment of cancer cells resulted in a preselection of high D cells. We measured changes in D using live-cell PWS in A2780 ovarian adenocarcinoma cells before and after treatment with a chemotherapeutic agent, 5 nM paclitaxel, for 48 hours. We also monitored cell coverage, which represents the survival of a cell population. Defining high D cells as those that fall within the top 25th percentile of D in the cell population before paclitaxel treatment (D = 2.47), we then measured the percentage of cells with high D at 48 hours after paclitaxel treatment. We observed that the percentage of high D cells increased in paclitaxel-treated cells compared to the control population (Fig. 3B). In combination with coverage measurements, which demonstrated significant cell death after 48 hours of paclitaxel treatment, our results indicate that high D cells have an increased survival rate when exposed to paclitaxel treatment (Fig. 3, B and C).
We then compared the transcriptional malleability of populations of cells with different initial D. As a model system, we relied on chemically induced modulation in D. To reduce D, we treated A2780 cells with 75 μM celecoxib, a nonsteroidal anti-inflammatory agent for 16 hours. Previously, we have found that celecoxib reduces D within 30 min of treatment in A2780 ovarian carcinoma cells by at least 8% compared to untreated cells (14). As a model of high-D cells, we used untreated A2780 cells. Both celecoxib-treated cells (low D) and untreated cells (high D) were then exposed to a chemotherapeutic agent, 5 nM paclitaxel, for 16 or 48 hours. scRNA-seq was conducted using Illumina NextSeq500. Raw reads were aligned, mapped, and used to calculate transcripts per million (TPM) for each condition using bowtie2 (45) and RSEM (46). Thus, as a model system, we measured transcriptional perturbation induced by a cytotoxic chemotherapy stressor in lower D (celecoxib-treated) versus higher D (untreated) A2780 cell populations.
Inputting the experimentally observed difference in D into the CPMC model, we estimated δ > 4 for initially underexpressed genes that become activated (Fig. 3D, blue manifold) and a smaller increase in δ for initially overexpressed genes that are up-regulated in response to stimulation (Fig. 3D, red manifold). We then tested whether these predicted trends are observed experimentally using scRNA-seq. The crucial window for response to chemotherapy is frequently thought to occur within 24 hours (44). Thus, we compared changes in gene expression in A2780 cells with initially higher D to initially lower D after stimulation by paclitaxel treatment for 16 hours. In agreement with the CPMC model, the stimulation of initially underexpressed genes by chemotherapy treatment in initially higher D cells (up-regulation of expression rate from control rate E1,b to 16-hour paclitaxel–treated rate E2,b) was much higher than that in lower D cells (from celecoxib-treated rate E1,a to 16-hour combo rate E2,a), resulting in δ~4 (Fig. 3E). Likewise, a similar but mitigated effect was observed in initially overexpressed genes (Fig. 3E), in strong agreement with the model predictions. Next, we tested whether these trends were independent of cell line and compound. We performed parallel experiments using propranolol as a D-lowering agent in A2780 cells and celecoxib and propranolol to decrease D in more malignant TP53 mutant A2780 (M248) cells. These additional conditions demonstrated a similar effect of D on transcriptional malleability in response to paclitaxel stimulation of high D compared to low D cells (fig. S4). Last, we tested whether the observed effect of chromatin packing scaling influences genes specifically involved in functionally relevant stress response pathways. We first identified differentially expressed genes that, on average, increased their expression at least twofold in A2780 cells treated with paclitaxel for 48 hours compared to control cells. Gene Ontology (GO) analysis of these up-regulated genes showed the activation of multiple stress response pathways after stimulation by paclitaxel treatment, including DNA repair, autophagy, cell cycle arrest, and apoptosis (P < 0.05; Fig. 3F and fig. S5). The effect of D on the activation of these established stress response genes was consistent with that observed in all up-regulated genes, with δ as high as ~4 (Fig. 3G).
The scaling behavior of chromatin packing regulates phenotypic plasticity through intercellular transcriptional heterogeneity
Another key aspect of phenotypic plasticity that can be modulated by the disordered packing of chromatin is transcriptional heterogeneity or the range of expression levels across genes exposed to similar molecular conditions. The CPMC model predicts that transcriptional heterogeneity increases as a function of D due to increased variations in both packing density () and gene accessibility (pg). To quantify this effect from the CPMC model, the variance in ϵ across any given cell population, Varϵ, is (14)
| (10) |
Consequently, intercellular transcriptional heterogeneity, i.e., the SD of steady-state expression rate E in Eq. 3 becomes
| (11) |
and the coefficient of variation (the ratio of the SD to the mean expression) is . Both H and COV increase with D, and COV also decreases as a function of expression.
To investigate the association between D and intercellular transcriptional heterogeneity, we analyzed our scRNA-seq data to quantify the spread in transcriptional states across each treatment condition. Focusing on overall transcriptional differences between cells within the same condition provides better validation to the model than analyzing the spread of all observed genes. Thus, we first used t-distributed stochastic neighbor embedding (t-SNE) combined with principal components analysis (PCA) to reduce the dimensionality of the system on all cells simultaneously (47). The dimensionality reduction mapped each cell onto a 3D projection. Distances between cells in 3D space represented overall differences in transcriptional states, as has been described by van der Maaten and Hinton (47). Intercellular transcriptional heterogeneity for each cell population was quantified by the average radius of the cluster of cells, , where ri is the position of each cell in the reduced spaced, N is the total number of cells in each treatment group, and . Intuitively, Rc can be thought of as the radius of relative genomic space. Consistent with predictions of the CPMC model, we found that transcriptional heterogeneity, as measured by the radius of genomic space, increases with D in response to paclitaxel treatment, which preselects for high D cells, as shown above. Notably, after 48 hours of paclitaxel treatment, the population of surviving cells had both higher D and increased transcriptional heterogeneity compared to control cells (Fig. 4, A to C and F). In contrast, celecoxib treatment reduces average D of a cancer cell population. Accordingly, cells treated with celecoxib for 16 hours had a lower transcriptional heterogeneity compared to control cells. In addition, when these celecoxib-primed cells with initially lower D were treated with paclitaxel for 16 hours, they had a decreased transcriptional heterogeneity compared to paclitaxel-treated control cells (Fig. 4, D to F). Although the resulting projection from t-SNE is nonunique, the trends in the radius of genomic space across conditions are robust to randomly selected choice of seed (fig. S6). Additional analyses quantifying the Euclidean distance between expression of DNA repair genes up-regulated in 48-hour paclitaxel treatment and the coefficient of variation of expression between cells in the same treatment condition demonstrate the same effect of chromatin packing scaling on transcriptional heterogeneity as the t-SNE results (fig. S7).
Fig. 4. The scaling of chromatin packing regulates intercellular transcriptional heterogeneity of cancer cells.
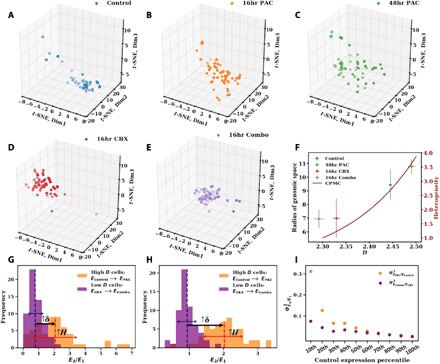
(A to E) 3D projections of scRNA-seq data (TPM values of 8275 expressed genes) onto reduced t-SNE space for five conditions: (A) control cells (n = 46), (B) cells treated with 5 nM paclitaxel for 16 hours (16hr PAC; n = 55), (C) 5 nM paclitaxel for 48 hours (48hr PAC; n = 53), (D) 75 μM celecoxib for 16 hours (16hr CBX; n = 62), and (E) combination of 75 μM celecoxib and 5 nM paclitaxel for 16 hours (16hr Combo; n = 59). The size of the cluster indicates the transcriptional heterogeneity within the population of surviving cells for each condition. (F) The radius of genomic space Rc [the radius of clusters from (A) to (E)] increases as a function of the chromatin packing scaling D. D was measured by live-cell PWS at each time point on cells before sequencing. Cells treated with paclitaxel (higher D) have greater transcriptional heterogeneity, especially when compared to cells treated with a nonsteroidal anti-inflammatory agent, celecoxib, which lowers D. Likewise, the CPMC model (red curve, right side, y axis) shows that intercellular transcriptional heterogeneity increases with D. Error bars represent the SE of D calculated from PWS measurements (x axis) and Rc (y axis) for each condition. (G) Relative expression of high D versus low D cells in response to paclitaxel treatment for genes associated with DNA repair pathways, which are up-regulated in 48-hour paclitaxel–treated cells. For each condition (Control, 16hr PAC, 2hr CBX, and 16hr Combo), TPM values of these genes (48 in total) were averaged within each cell. Next, expression of paclitaxel-stimulated cells was normalized by the average of the corresponding unstimulated population. The resulting intercellular distribution of relative expression levels is shown. Dashed lines represent the mean relative expression. Solid red and blue arrows represent the SD of distributions EPAC/EControl and ECBX/ECombo, respectively. For these stress response genes, cells with a higher initial D versus cells with a lower initial D had an increase in transcriptional malleability (↑δ) and a higher intercellular transcriptional heterogeneity (↑ H). (H) Distribution of relative expression of genes, as described in (G), in the lowest percentile (10th percentile) of control expression levels (839 in total). (I) Variance (σ2) of intercellular distribution of relative expression for each percentile of control expression levels. Initially underexpressed genes show an increased effect of chromatin packing scaling on increasing intercellular transcriptional heterogeneity in response to paclitaxel stimulation compared to that of initially overexpressed genes.
Next, we sought to investigate the effect of chromatin packing scaling on changes in transcriptional heterogeneity in response to stimulation. For higher D compared to lower D populations, the CPMC model predicts an increase in transcriptional malleability concomitant with an increase in gene expression variability in response to stimulation. As a case in point, consider the up-regulation of stress response genes due to a stressor such as chemotherapy. Both transcriptional malleability and heterogeneity may facilitate a response to the stress. An increase in the average expression (malleability) and in the SD of expression levels (heterogeneity) for these genes upon the stimulation would increase the percentage of cells that express these genes above any given level that may facilitate cell survival, regardless of the exact value of this critical level. We used scRNA-seq data on A2780 cells to analyze the distributions of transcriptional responses to paclitaxel treatment, as an example of an exogenous stressor, in cell populations with different initial D. We assessed the ratio of the up-regulated expression rate due to the stressor versus the initial expression rate (relative up-regulation, E2/E1). Focusing on transcriptional responsiveness of genes associated with DNA repair pathways that had been up-regulated in response to 48-hour paclitaxel treatment, we found that the higher D population had both an increase in the average (malleability) and the variance (heterogeneity) of relative up-regulation compared to the lower D population (Fig. 4G). Next, we examined relative expression levels of genes suppressed in the control condition, specifically those that occupied the bottom 10th percentile of gene expression, and observed a similar behavior (Fig. 4H). This D-dependent increase in intercellular transcriptional heterogeneity was itself a function of initial expression level. Specifically, genes that were underexpressed in cells before paclitaxel treatment had a more significant difference in the heterogeneity of their relative up-regulation in the high D compared to the low D populations than those that were already highly expressed before the stimulation (Fig. 4I), also in agreement with the CPMC predictions.
Transcriptional divergence is inversely associated with patient survival
As described above, D determines a cell’s responsiveness to stressors such as chemotherapeutic agents through the effect of chromatin packing scaling on phenotypic plasticity. A logical next step was to establish whether these physical regulators play a role in tumor aggressiveness in vivo. The effects of chromatin packing scaling on phenotypic plasticity may foster the ability of cancer cells to develop resilience and/or resistance to chemotherapy in vivo and may also be involved in other processes fostering increased tumor fitness and aggressiveness. Throughout carcinogenesis, tumors are frequently exposed to a wide range of stressors including attack by a host’s immune system, inadequate oxygen supply from nearby blood vessels, or an acidotic microenvironment. To test whether such a relationship between phenotypic plasticity and tumor fitness exists, we analyzed publicly available RNA-seq data collected by the TCGA Research Network (20) for lung, colorectal, and breast cancers, the three most prevalent malignancies in the United States. As the model predicts cellular responsiveness to external stressors, of which chemotherapy is an example, we focus on patients presenting with stage III and IV tumors at the time of diagnosis, as systemic therapy is the standard of care for these patients. Using the R package TCGAbiolinks (48), we quantified gene expression in units of fragments per kilobase million (FPKM) for each patient. As these data lack initial control measurements of cancer cells before initiation of systemic therapy, transcriptional malleability cannot be measured directly for each patient. In addition, we do not have information related to chromatin packing scaling and other physical regulators of transcription for these patients. However, the essence of the effect of δ is that elevated D amplifies a gene’s transcriptional response to stimuli: Overexpressed genes are enhanced, whereas underexpressed genes are suppressed (Fig. 2). Consequently, as the bidirectional behavior of Se(E) curves indicates, an elevated D widens the distribution of gene expression, resulting in increased transcriptional divergence, which, in turn, is a key determinant of transcriptional malleability (Fig. 5A). Thus, quantifying transcriptional divergence within these patient cohorts should, by proxy, measure transcriptional responsiveness, which we have shown above is linked to D. Borrowing a method from macroeconomics, transcriptional divergence can be quantified by the ratio of expression of the top 50% of genes and the bottom 50% of genes (P50/P50), for ranked expression, Ek, and total number of detected genes, N
| (12) |
Fig. 5. The relationship between transcriptional divergence (P50/P50) and patient survival in stage III and IV lung, breast, and colon cancers.
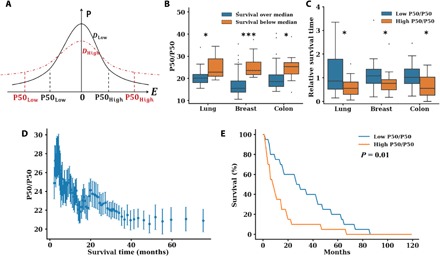
(A) From the Se curve predicted by the CPMC model, cells with high D, such as cancer cells, have a wider distribution of gene expression (transcriptional divergence). Quantitatively, this transcriptional divergence can be calculated by measuring the ratio of the expression of the top 50% of genes to that of the bottom 50% of genes (P50/P50). (B to E) Analysis of transcriptional divergence, P50/P50, in the cancer cells of patients with stage III and IV lung cancer (n = 31), breast cancer (n = 168), and colon cancer (n = 60) versus survival from the time of diagnosis based on TCGA dataset for patients <75 years old at the time of diagnosis. (B) P50/P50 was elevated in patients with a survival duration below the median for each cancer type (P = 0.021, P < 0.001, and P = 0.018 for lung, breast, and colon cancers, respectively). (C) The RST (ratio between patient survival time and that predicted by a multidimensional linear regression model based on known prognostic factors such as stage at diagnosis, race, and molecular subtypes of the tumor) is higher for patients with low P50/P50 (P50/P50 below the mean for all patients with a given cancer type). RST < 1 indicates survival shorter than expected based on demographic factors and molecular subtype (all P < 0.05). For all three malignancies, RST < 0.8 in high P50/P50 patients. RST is an independent predictor of survival duration. (D) Pooling all patients with these malignancies, we analyzed survival duration (x axis, in months) versus P50/P50 at the time of diagnosis. There was an inverse relation between P50/P50 and survival duration. Each point is an MWA of 10 patients to account for unreported variables (e.g., comorbidities). (E) The Kaplan-Meier curve measuring patient survival for the three malignancies. Patients with a high P50/P50 (P50/P50 above the mean) have a shorter survival duration (median survival = 8 months) than patients with low P50/P50 (P50/P50 below the mean, median survival = 28 months; P = 0.01).
As age is also a major predictor of cancer mortality, we restricted our analysis to patients under 75 years at the time of diagnosis. As the CPMC model predicts that higher transcriptional divergence produces more adaptable tumor cells, we would expect that patients with a shorter survival time would have tumor cells with an elevated P50/P50 ratio at the time of diagnosis. To test this hypothesis, we compared the P50/P50 ratio calculated at the time of diagnosis of patients surviving above or below the median survival time for each cancer type (Fig. 5B). We found a statistically significant inverse relationship between P50/P50 ratio and relative patient survival time for lung (Fig. 5B; P < 0.021), breast (Fig. 5B; P < 0.0001), and colon (Fig. 5B; P = 0.018) cancers.
Next, we analyzed the relative contribution of transcriptional divergence to patient survival time compared to effects of other prognostic factors (e.g., demographic factors, tumor molecular subtype, and stage) by performing a multivariate regression on each prognostic factor. We then calculated the relative survival time (RST) for each patient as the observed survival time relative to the expected survival time based on these additional prognostic factors. RST < 1 indicates that a patient’s survival is shorter than expected (e.g., RST = 0.5 indicates that their survival duration is 50% shorter than expected), whereas RST > 1 indicates a longer than expected survival time. Patients were then grouped into a high and a low P50/P50 cohort based on whether they were in the top or bottom half of P50/P50 values, respectively. Notably, high P50/P50 patients had an RST below 0.8 for all malignancies, whereas a low P50/P50 translated into a significantly higher RST > 1 (P < 0.05). Next, we analyzed the relationship between patient survival and P50/P50 directly for all malignancies. As survival depends on a multitude of factors, some of which were not available within the TCGA dataset for all patients (e.g., comorbidities), a fixed moving window average (MWA) was applied to the data (see Materials and Methods for details). We found a continuous inverse trend between P50/P50 and patient survival for all three malignancies (Fig. 5D and fig. S8). Last, Kaplan-Meier survival curves show that patients with high P50/P50 ratios have a median survival of 8 months compared to 28 months for those with a low P50/P50 (Fig. 5E; P = 0.01). In summary, these results support a strong correlation between transcriptional divergence, a facet of phenotypic plasticity that is directly affected by chromatin packing scaling, and patient survival (Fig. 5).
DISCUSSION
In this work, we combined multiscale modeling with Hi-C, scRNA-seq, chromatin electron tomography, and live-cell PWS microscopy to demonstrate the role of the disordered chromatin polymer on regulating both intercellular transcriptional heterogeneity and transcriptional malleability. On the basis of predictions from the CPMC model, which were verified experimentally, the spatial arrangement of chromatin packing affects gene expression through a number of physical regulators, including ϕin,0, Nd, and D (Figs. 1 and 2). We demonstrate, both computationally and experimentally, that a crucial role of chromatin packing is to determine the level of phenotypic plasticity within a cell population. In particular, the scaling of chromatin packing, D, modulates both the transcriptional malleability through a chromatin-mediated enhancement δ, a “tailwind effect” (Fig. 3), and the level of intercellular transcriptional heterogeneity (Fig. 4). This effect is further regulated by other physical properties of chromatin. A higher average crowding density within the nucleus suppresses the expression of initially underexpressed genes as D increases (Fig. 2, G and H). The modulatory effects of Nd are twofold. Genes localized to domains with a large Nd (Mbp range) are more suppressed than those localized to domains with small Nd (kbp range) owing to a reduced accessibility to TFs and polymerases. However, as D increases, expression of genes associated with large Nd is disproportionately enhanced (Fig. 2I). Overall, higher D, higher crowding density, and lower Nd increase both transcriptional malleability and heterogeneity, with D having a much larger effect compared to the other two chromatin packing properties.
The fact that eukaryotic cells have encoded information into the scaling behavior of chromatin packing may have important medical implications. Elevated D is a hallmark of cancer cells and could represent a mechanism by which malignancy gains nonmutational advantages over neighboring healthy cells. As observed in vitro, treating cells with a chemotherapeutic agent such as paclitaxel selects for cells with higher D (Fig. 3B), which, as demonstrated within this work, is, in part, due to increased phenotypic plasticity compared to cells with lower D (Figs. 3 and 4). This selects for tumor cell populations with a higher transcriptional adaptive potential, which, in turn, may facilitate their survival despite future exposure to new stressors. In support of this potential mechanism, our data show that transcriptional divergence, the cross-sectional measurement of transcriptional malleability, in advanced colorectal, lung, and breast cancers is associated with worse prognosis independent of demographic factors (e.g., age and gender), tumor stage, and molecular transformations (Fig. 5).
At present, experimental validation of the CPMC model relies on the measurement of average chromatin packing scaling D and crowding within the entire nucleus. While currently beyond existing experimental capabilities, subsequent studies directly comparing how local (e.g., intra-packing domain) chromatin structure affects transcriptional processes and output would be of considerable importance. Pairing gene-tracking techniques such as CRISPRainbow with imaging modalities that measure chromatin structure, such as live-cell PWS microscopy and ChromEM, and super-resolution imaging of molecular factors would help elucidate how intranuclear variations in molecular and physical regulators of transcription contribute to transcriptional heterogeneity and malleability (12, 27, 49).
Although not explored in this work, there are several implications of these results on the understanding of multicellular fitness in the context of cell biology. For example, the localization of genes into domains has been demonstrated to be a conserved, albeit heterogeneous, process (50). From the predictions of the model, cells would benefit from localizing genes into large domains that are intended to be suppressed at baseline but require rapid amplification to adapt to changing environmental conditions. Likewise, crowding density could be adjusted by cells either as a preprogrammed response by changing nuclear volume or incidentally from the retention of an extra chromosome during replication. Consequently, as has been hypothesized, this could be a mechanism linking nuclear size and density (e.g., hyperchromasia) with differential gene expression. Nuclear size, hyperchromasia, and abnormal nuclear texture are some of the most ubiquitous histological markers of neoplasia, although their etiology and functional consequences have been poorly understood (51).
In light of the CPMC model conclusions, it should be clear that disordered chromatin packing does not mean that the configurations are random or that observed patterns in gene transcription are the result of configurational noise. While it is beyond the scope of this work, the conformation of a chromatin polymer depends on the balance between chromatin-chromatin and chromatin-nucleoplasm interactions and is further shaped by active chromatin loop formation processes and other constraints such as nuclear lamins (52). The shape of the disordered chromatin polymer will ultimately depend on molecular factors such as histone modifications, transcriptional and replication-induced supercoiling, and DNA motif stiffness, as well as nucleoplasmic factors such as nuclear pH, ionic concentrations, and crowding, which collectively alter chromatin-chromatin and chromatin-nucleoplasm interactions (26, 53–55). Therefore, individual cells could use a combination of chromatin-chromatin and chromatin-nucleoplasm interactions to appropriately organize the genome while also encoding a predetermined level of phenotypic plasticity.
In addition, this work may have implications on the open question in chromatin biology regarding the importance of noncoding DNA. Some roles have since been illuminated, including the production of noncoding RNA and the distribution of transcriptional regulatory motifs such as enhancers and insulators (21, 56). In light of this work, and in relation to previously suggested hypotheses of the role of macromolecular crowding on gene expression, one of the evolutionary functions of noncoding DNA could be derived from its space-filling role. Consequently, noncoding DNA might be a critical component within the genome to determine phenotypic plasticity as it possesses the ability to modulate transcription reactions by influencing the free energy of transcription reactions and the diffusion of reactants.
Last, one could consider how D plays a role in the adaptability of cancer cells throughout carcinogenesis. Carcinogenesis depends on cells overcoming aberrations in metabolism, derangements of the microenvironment, inadequate vascular supply, immune surveillance, and acclimation to distal tissue environments during metastasis. As it could take multiple replicative generations to develop a new useful mutation within a population for each of these processes, cancer cells could additionally leverage the physical properties of chromatin packing to increase their transcriptional plasticity to acclimate to these conditions over a faster time scale. Thus, it may be worth investigating, for example, whether cancer cells with elevated D are better able to survive an immune response and acclimate to distant tissue sites during metastasis. From the therapeutic standpoint, while mutations are difficult to remove from a cell population, this work suggests that limiting cancer cell evolution might be possible pharmacologically by lowering the scaling of disordered chromatin packing.
MATERIALS AND METHODS
Gene expression analysis
mRNA microarray for HT-29 cells
HT-29 cells were serum-deprived for 5 hours before treatment with 10% (v/v) FBS, EGF (100 ng/ml), or PMA (100 ng/ml). mRNA for these treatment groups was collected by TRIzol isolation (Life Technologies, Carlsbad, CA) from 10-ml petri dishes and analyzed using Illumina human HG12-T microarray chips. The R Bioconductor package lumi was used for quality control analysis by the Northwestern Genomics Core to assess probe-level processing from the Illumina microarray data. Differentially expressed genes (2445) were identified for subsequent analysis.
RNA-seq for A549 and BJ cells
RNA-seq data for A549 and BJ cells were downloaded from ENCODE and Gene Expression Omnibus (GEO) with access codes ENCSR897XFT for A549 cells and GSE81087 for BJ cells (57, 58). Four replicates were included in both control and 12-hour dexamethasone–treated A549 cells. Gene expression was quantified using featureCounts on RNA-seq data from ENCODE for A549 cells. The length and counts for each replicate from featureCounts outputs were then changed into TPM using , where TPMi, ctsi, and Li are the TPM value, the count, and the length of gene i, respectively. The differential expression (DE) analysis for A549 cells was performed using the DESeq2 packages in R. Differentially expressed genes (2292) were found after 12 hours of 100 nM dexamethasone treatment in A549 cells using P < 0.01. Three replicates were included in the gene expression analysis for BJ cells. The processed FPKM results from three replicates each from control cells and cells treated with 100 nM dexamethasone for 32 hours were downloaded from GEO and transformed into TPM unit using , where FPKMi is the FPKM value of gene i. The same DE method was used on BJ cells, and 7601 genes were identified with P < 0.01.
RNA-seq for A2780.M248 cells
RNA extraction was performed on samples from ovarian carcinoma TP53 mutant clone A2780.M248 cells were collected from the cells treated under control, 16-hour celecoxib, 16-hour paclitaxel, 16-hour paclitaxel and celecoxib, and 48-hour paclitaxel conditions with three biological replicates per condition. Stranded mRNA sequencing was conducted in the Northwestern University NUSeq Core Facility. Briefly, total RNA quantity was determined with a Qubit fluorometer, and quality was assessed using RNA integrity numbers (RINs) generated from Agilent Bioanalyzer 2100. To proceed to sequencing library preparation, RIN must be at least 7. The Illumina TruSeq Stranded mRNA Library Preparation Kit was used to prepare sequencing libraries from 100 ng of RNA. The kit procedure was performed without modifications. This procedure includes mRNA purification and fragmentation, complementary DNA synthesis, 3′ end adenylation, Illumina adapter ligation, library polymerase chain reaction (PCR) amplification, and validation. Illumina HiSeq 4000 Sequencer was used to sequence the libraries with the production of single-end, 50–base pair reads. Single-end FASTQ reads from RNA-seq measurements were aligned and mapped to hg38 using bowtie2. TPM from mapped reads were estimated using RSEM. Significant genes that are expressed across all conditions and have fold changes larger/smaller than 2-fold/1/2-fold of control in cells treated with paclitaxel for 48 hours were selected.
scRNA-seq for A2780 cells
scRNA-seq experiments on A2780 cells were conducted using the Illumina NextSeq 500 platform with the Smart-seq protocol at the University of Illinois at Chicago Research Resources Center Cores. The paired FASTQ reads with four technical replicates of each cell were aligned to the hg38 genome using bowtie2. The gene expression levels, (TPM values) for each condition were estimated using the RSEM software package. Control cells (46 of 57), 16-hour paclitaxel cells (55 of 58), 48-hour paclitaxel cells (53 of 62), 16-hour celecoxib cells (62 of 67), and 16-hour combination (paclitaxel and celecoxib) (59 of 59) cells were selected after quality control (excluding cells with less than 4000 genes expressed). Additional quality control was performed using the expression level of housekeeping genes (59), but no additional cells were excluded. In total, 8415 genes were identified for subsequent analysis for each individual cell after removing genes expressed in less than 20% of the total cell population. To quantify the size of genomic information space at different chromatin packing conditions, 8276 genes (average fold changes relative to control are larger than 1.5 or smaller than two-thirds) were selected to do a 3D t-SNE analysis (47). The t-SNE analysis was done using “Rtsne” package in R with initial PCA step performed.
GO analysis
To perform the GO analysis, the average TPM values of each gene across all A2780 cells under each condition were normalized to the mean TPM values of control cells. The top 10% expressed genes after normalizing with 48-hour paclitaxel treatment were selected (841 genes) to conduct the GO analysis using DAVID. Twenty biological processes were shown to be significantly involved by these overexpressed genes (fig. S5). Of the 20 up-regulated biological processes, 11 of them were identified to be involved in DNA repair (Fig. 3D).
TCGA patient expression analysis
The P50/P50 ratio of each patient’s gene expression was calculated by , where FPKMk is the sorted FKPM value of the transcribed genes in each patient and N is the total number of measurably transcribed genes. Only transcribed genes (FKPM ≠ 0) were considered from RNA-seq data obtained from the TCGA database. Patients with breast, colon, and lung cancers in stages III and IV were divided into survival over/below the median survival time based on their vital status and survival time after diagnosis. The number of patients in each group can be found in table S2. Then, the P50/P50 values of patients from all three cancer types (breast, colon, and lung cancers) were pooled together to apply a fixed MWA with 15 patients per group to analyze whether an overall trend exists between P50/P50 and survival time (days). This analysis is applicable to inherently noisy data or for datasets where important covariates are not completely available (e.g., chemotherapeutic/radiation therapy status or comorbidities were not present in the dataset). A linear regression analysis using survival duration, P50/P50, and tumor stage as survival predictors was also conducted using Python, showing a significant prediction of patient survival only for P50/P50 (P < 0.05) with a negative coefficient of −33.6 days. Notably, regression analysis did not show a strong predictive power of stage at the time of diagnosis (P > 0.05) or an association between tumor stage and P50/P50 level.
Hi-C TAD analysis
The total genomic size of chromatin at the upper length scale of self-similarity Nd of genes in the 3D space was estimated using the publicly available Hi-C data on A549 cells (GEO access code: GSE92819 for control cells and GSE92811 for cells treated with dexamethasone for 12 hours) (35). Nd was approximated as the size of the TADs measured from Hi-C. The processed TADs in A549 cells from the GEO datasets were used to determine the size of TADs surrounding differentially expressed genes. Genes localized within the same TAD were assigned the same Nd. As dissolution of TADs was previously shown to alter access of TFs to DNA and we wanted to analyze the effect of Nd size, we selected only TADs that remained intact and of comparable size before and after dexamethasone treatment. Genes within these TADs of constant size were divided into two cohorts: a high Nd group and a low Nd group. Each group had ~130 genes, and the average Nd values for each group were approximately 50 kbp for the low Nd group and 2 Mbp for the high Nd group. Genes with top 5% and bottom 5% Nd were removed from each group to exclude outliers.
Live-cell PWS microscopy
HT-29 cell culture
HT-29 cells (American Type Culture Collection, Manassas, VA) were grown in Gibco formulated McCoy’s 5A medium (Life Technologies, Carlsbad, CA) supplemented with 10% (v/v) FBS (Sigma-Aldrich, St. Louis, MO) and grown at 37°C and 5% CO2. All cells in this study were maintained between passage 5 and 25. Transient HT-29 Arid1a short hairpin RNA knockdown line (A-Kd) was produced using a Lipofectamine vector. Quantitative reverse transcription PCR was used to assess for knockdown: Imaging and microarrays were performed on clones that demonstrated at least an 80% reduction in Arid1a expression compared to the control vector.
Before imaging, cells were cultured in 35-mm glass bottom petri dishes (Cellvis, Mountain View, CA) until at least 50% confluent. Cells were given at least 24 hours to readhere before 5 hours of serum deprivation. For serum deprivation, cells were grown in fresh McCoy’s 5A (Life Technologies) without serum supplementation and maintained at 37°C and 5% CO2.
A2780 cell culture
Ovarian A2780 cells were a gift from C.-P. Huang Yang and obtained from the laboratory of E. de Vries at the Albert Einstein College of Medicine. They were cultured in RPMI 1640 medium (#11875127, Thermo Fisher Scientific, Waltham, MA). All culture media were supplemented with 10% FBS (#16000044, Thermo Fisher Scientific, Waltham, MA). Cells were cultured in 35-mm six-well glass bottom plates (Cellvis, Mountain View, CA) until 60 to 85% confluent. All cells were given at least 24 hours to readhere before pharmacological treatment. Cells were treated with 75 μM celecoxib (2 and 16 hours), 5 nM paclitaxel (16 and 48 hours), or a combination of celecoxib and paclitaxel (16 hours) before trypsinization and resuspension in growth medium. Cell sorting was performed on a Fluidigm C1 Single-Cell Capture instrument. Single-cell sequencing of the sorted cells was performed by staff researchers at the University of Illinois Chicago Genomics Core.
A549 and BJ cell culture
A549 cells were cultured in Dulbecco’s modified Eagle medium (DMEM; #11965092, Thermo Fisher Scientific, Waltham, MA). BJ cells were cultured in minimum essential medium (MEM; #11095080, Thermo Fisher Scientific, Waltham, MA). All culture media were supplemented with 10% FBS (#16000044, Thermo Fisher Scientific, Waltham, MA) and penicillin-streptomycin (100 μg/ml; #15140122, Thermo Fisher Scientific, Waltham, MA). All cells were maintained and imaged at physiological conditions (5% CO2 and 37°C) for the duration of the experiment. All cell lines were tested for mycoplasma contamination with Hoechst 33342 within the past year. Experiments were performed on cells from passage 5 to 20. Before imaging, cells were cultured in 35-mm glass bottom petri dishes until approximately 70% confluent. All cells were given at least 24 hours to readhere before treatment (for treated cells) and imaging. A549 and BJ cells were treated with 100 nM dexamethasone (D6645, Sigma-Aldrich, St. Louis, MO) for 12 and 32 hours, respectively, in line with published chromatin conformation capture and RNA-seq experiments.
Live-cell PWS measurements
PWS measurements were performed on a commercial inverted microscope (Leica DM IRB) using a Hamamatsu Image EM charge-coupled device camera (C9100-13) coupled to a liquid crystal tunable filter (CRi, Woburn, MA) to acquire monochromatic spectrally resolved images that range from 500 to 700 nm at 1-nm intervals produced by a broad band illumination provided by an Xcite-120 LED lamp (Excelitas, Waltham, MA) as previously described (33, 34). Briefly, PWS measures the spectral interference resulting from internal light scattering structures within the cell, which captures the mass density distribution. To obtain the interference signal directly related to refractive index fluctuations in the cell, we normalized measurements by the reflectance of the glass medium interface, i.e., to an independent reference measurement acquired in an area without cells. PWS measures a data cube (spatial coordinates of a location within a cell and the light interference spectrum recorded from this location). The data cube then allow to measure spectral SD (Σ), which is related to the spatial variations of refractive index within a given coherence volume. The coherence volume was defined by the spatial coherence in the transverse directions (~200 nm) and the depth of field in the axial direction (~1 μm). In turn, the spatial variations of refractive index depended on the local autocorrelation function (ACF) of the chromatin refractive index. Finite-difference time-domain simulations have shown that PWS is sensitive to ACF within the 20- to 200-nm range. According to the Gladstone-Dale equation, refractive index is a linear function of local molecular crowding. Therefore, Σ depends on the ACF of the medium’s macromolecular mass density. Small molecules and other mobile crowders within the nucleus are below the limit of sensitivity of PWS, and PWS is primarily sensitive to chromatin conformation above the level of the nucleosome. To convert Σ for a given location within a nucleus to mass fractal dimension D, we modeled ACF as a power law , where is the variance of CVC (60). In general, Σ is a sigmoidal function of D. However, for fractal structures such as a chromatin packing domain where within physiological range 2 < D < 3, Σ can be approximated as a linear function of D by the relationship D ≈ D0 + aΣ, where D0 = 1.473 and is comparable to the minimal fractal dimension that an unconstrained polymer can attain and constant a ~ 7.6. The measured change in chromatin packing scaling between treatment conditions was quantified by first averaging D within each cell’s nucleus and then averaging nuclei from over 50 cells per condition.
Chromatin electron microscopy
A549 cell culture
Two cell lines were used in this work: adenocarcinomic human lung epithelial cell line (A549) and human cellosaurus cell line (BJ). The A549 cells were grown in DMEM with 10% FBS. The BJ cells were grown in MEM with 10% FBS and 1× nonessential amino acids. All cells were cultured on 35-mm MatTek dishes (MatTek Corp.) at 37°C and 5% CO2. Confluency of about 60% was reached for all experiments.
Electron microscopy sample preparation and TEM/STEM data collection
For electron microscopy experiment, all cells were prepared by the ChromEM staining protocol and embedded in Durcupan resin (EMS) (27). After curing, 40-nm-thin sections were made and deposited onto copper 200-mesh grid with carbon/formvar film (EMS). The grids were plasma-cleaned by a plasma cleaner (easiGlow, TED PELLA) before use. An HT7700 (HITACHI) transmission electron microscopy (TEM) was used to record TEM images of cell sections at 80 kV with a pixel size of 2.5 nm.
Supplementary Material
Acknowledgments
We thank Z. Ji and members of his laboratory for advice on scRNA-seq analysis. Funding: This work was supported by NSF grant EFMA-1830961 and NIH grants R01CA228272, R01CA225002, U54CA193419, and T32GM008152. Author contributions: Conceptualization and project design: W.W., L.M.A., H.K.R., I.S., and V.B.; formal analysis: R.K.A.V., L.M.A., and W.W.; experiments: L.M.A., G.M.B., Y.L., D.V., J.F., and A.E.; random medium simulations: D.Z.; writing (original draft): W.W., L.M.A., I.S., and V.B.; writing (review and editing): R.K.A.V., L.M.A., and V.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The results of patient study are based on data generated by the TCGA Research Network (www.cancer.gov/tcga). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaax6232/DC1
Section S1. CPMC model: Variance of chromatin packing density
Section S2. Random medium simulations: Calculation of D from mass density variations
Section S3. Calculation of ϕin,0 from ChromEM measurements
Fig. S1. Analysis of the relationship between D and .
Fig. S2. Random medium simulations for low and high D.
Fig. S3. CVC distributions of A549 and BJ cells as measured by ChromEM.
Fig. S4. Transcriptional malleability in A2780 and M248 cells.
Fig. S5. GO analysis of up-regulated genes.
Fig. S6. t-SNE transcriptional heterogeneity analysis is independent of seed.
Fig. S7. Transcriptional heterogeneity is increased in high D cells.
Fig. S8. Relationship between survival time and transcriptional divergence.
Table S1. Descriptions and values of CPMC model parameters.
Table S2. TCGA patient information.
REFERENCES AND NOTES
- 1.Rathert P., Roth M., Neumann T., Muerdter F., Roe J.-S., Muhar M., Deswal S., Cerny-Reiterer S., Peter B., Jude J., Hoffmann T., Boryń Ł. M., Axelsson E., Schweifer N., Tontsch-Grunt U., Dow L. E., Gianni D., Pearson M., Valent P., Stark A., Kraut N., Vakoc C. R., Zuber J., Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern S., Dror T., Stolovicki E., Brenner N., Braun E., Genome-wide transcriptional plasticity underlies cellular adaptation to novel challenge. Mol. Syst. Biol. 3, 106 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plasschaert L. W., Žilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A. M., Jaffe A. B., A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen Q. H., Pervolarakis N., Blake K., Ma D., Davis R. T., James N., Phung A. T., Willey E., Kumar R., Jabart E., Driver I., Rock J., Goga A., Khan S. A., Lawson D. A., Werb Z., Kessenbrock K., Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 9, 2028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haber A. L., Biton M., Rogel N., Herbst R. H., Shekhar K., Smillie C., Burgin G., Delorey T. M., Howitt M. R., Katz Y., Tirosh I., Beyaz S., Dionne D., Zhang M., Raychowdhury R., Garrett W. S., Rozenblatt-Rosen O., Shi H. N., Yilmaz O., Xavier R. J., Regev A., A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawson D. A., Kessenbrock K., Davis R. T., Pervolarakis N., Werb Z., Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 20, 1349–1360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C. A., Dempster J., Lyons N. J., Burns R., Nag A., Kugener G., Cimini B., Tsvetkov P., Maruvka Y. E., O’Rourke R., Garrity A., Tubelli A. A., Bandopadhayay P., Tsherniak A., Vazquez F., Wong B., Birger C., Ghandi M., Thorner A. R., Bittker J. A., Meyerson M., Getz G., Beroukhim R., Golub T. R., Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirosh I., Izar B., Prakadan S. M., Wadsworth M. H. II, Treacy D., Trombetta J. J., Rotem A., Rodman C., Lian C., Murphy G., Fallahi-Sichani M., Dutton-Regester K., Lin J.-R., Cohen O., Shah P., Lu D., Genshaft A. S., Hughes T. K., Ziegler C. G., Kazer S. W., Gaillard A., Kolb K. E., Villani A.-C., Johannessen C. M., Andreev A. Y., Van Allen E. M., Bertagnolli M., Sorger P. K., Sullivan R. J., Flaherty K. T., Frederick D. T., Jané-Valbuena J., Yoon C. H., Rozenblatt-Rosen O., Shalek A. K., Regev A., Garraway L. A., Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Easwaran H., Tsai H.-C., Baylin S. B., Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleppe A., Albregtsen F., Vlatkovic L., Pradhan M., Nielsen B., Hveem T. S., Askautrud H. A., Kristensen G. B., Nesbakken A., Trovik J., Wæhre H., Tomlinson I., Shepherd N. A., Novelli M., Kerr D. J., Danielsen H. E., Chromatin organisation and cancer prognosis: A pan-cancer study. Lancet Oncol. 19, 356–369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedin V., Adam R. L., de Sá B. C., Landman G., Metze K., Fractal dimension of chromatin is an independent prognostic factor for survival in melanoma. BMC Cancer 10, 260 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torre E., Dueck H., Shaffer S., Gospocic J., Gupte R., Bonasio R., Kim J., Murray J., Raj A., Rare cell detection by single-cell RNA sequencing as guided by single-molecule RNA FISH. Cell Syst. 6, 171–179.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aryee M. J., Liu W., Engelmann J. C., Nuhn P., Gurel M., Haffner M. C., Esopi D., Irizarry R. A., Getzenberg R. H., Nelson W. G., Luo J., Xu J., Isaacs W. B., Bova G. S., Yegnasubramanian S., DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl. Med. 5, 169ra110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almassalha L. M., Bauer G. M., Wu W., Cherkezyan L., Zhang D., Kendra A., Gladstein S., Chandler J. E., VanDerway D., Seagle B.-L. L., Ugolkov A., Billadeau D. D., O’Halloran T. V., Mazar A. P., Roy H. K., Szleifer I., Shahabi S., Backman V., Macrogenomic engineering via modulation of the scaling of chromatin packing density. Nat. Biomed. Eng. 1, 902–913 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cremer T., Cremer C., Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Almassalha L. M., Bauer G. M., Chandler J. E., Gladstein S., Szleifer I., Roy H. K., Backman V., The greater genomic landscape: The heterogeneous evolution of cancer. Cancer Res. 76, 5605–5609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almassalha L. M., Tiwari A., Ruhoff P. T., Stypula-Cyrus Y., Cherkezyan L., Matsuda H., Dela Cruz M. A., Chandler J. E., White C., Maneval C., Subramanian H., Szleifer I., Roy H. K., Backman V., The global relationship between chromatin physical topology, fractal structure, and gene expression. Sci. Rep. 7, 41061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J. S., Yethiraj A., Crowding effects on protein association: Effect of interactions between crowding agents. J. Phys. Chem. B. 115, 347–353 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Matsuda H., Putzel G. G., Backman V., Szleifer I., Macromolecular crowding as a regulator of gene transcription. Biophys. J. 106, 1801–1810 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Criscione S. W., De Cecco M., Siranosian B., Zhang Y., Kreiling J. A., Sedivy J. M., Neretti N., Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2, e1500882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatesh S., Workman J. L., Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 16, 178–189 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Chiba K., Yamamoto J., Yamaguchi Y., Handa H., Promoter-proximal pausing and its release: Molecular mechanisms and physiological functions. Exp. Cell Res. 316, 2723–2730 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Jothi R., Cuddapah S., Barski A., Cui K., Zhao K., Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 36, 5221–5231 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morelli M. J., Allen R. J., ten Wolde P. R., Effects of macromolecular crowding on genetic networks. Biophys. J. 101, 2882–2891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J. S., Szleifer I., Crowding-induced formation and structural alteration of nuclear compartments: Insights from computer simulations. Int. Rev. Cell Mol. Biol. 307, 73–108 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Ou H. D., Phan S., Deerinck T. J., Thor A., Ellisman M. H., O’Shea C. C., ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357, eaag0025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.B. B. Mandelbrot, The Fractal Geometry of Nature (Macmillan, 1983), vol. 173. [Google Scholar]
- 29.Bancaud A., Lavelle C., Huet S., Ellenberg J., A fractal model for nuclear organization: Current evidence and biological implications. Nucleic Acids Res. 40, 8783–8792 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larson A. G., Elnatan D., Keenen M. M., Trnka M. J., Johnston J. B., Burlingame A. L., Agard D. A., Redding S., Narlikar G. J., Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M. T., Wendt K. S., Okada Y., Nagai T., Maeshima K., Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Francastel C., Schübeler D., Martin D. I., Groudine M., Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 1, 137–143 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Gladstein S., Almassalha L. M., Cherkezyan L., Chandler J. E., Eshein A., Eid A., Zhang D., Wu W., Bauer G. M., Stephens A. D., Morochnik S., Subramanian H., Marko J. F., Ameer G. A., Szleifer I., Backman V., Multimodal interference-based imaging of nanoscale structure and macromolecular motion uncovers UV induced cellular paroxysm. Nat. Commun. 10, 1652 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almassalha L. M., Bauer G. M., Chandler J. E., Gladstein S., Cherkezyan L., Stypula-Cyrus Y., Weinberg S., Zhang D., Thusgaard Ruhoff P., Roy H. K., Subramanian H., Chandel N. S., Szleifer I., Backman V., Label-free imaging of the native, living cellular nanoarchitecture using partial-wave spectroscopic microscopy. Proc. Natl. Acad. Sci. U.S.A. 113, E6372–E6381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durand N. C., Shamim M. S., Machol I., Rao S. S. P., Huntley M. H., Lander E. S., Aiden E. L., Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seaman L., Chen H., Brown M., Wangsa D., Patterson G., Camps J., Omenn G. S., Ried T., Rajapakse I., Nucleome analysis reveals structure–function relationships for colon cancer. Mol. Cancer Res. 15, 821–830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanborn A. L., Rao S. S., Huang S.-C., Durand N. C., Huntley M. H., Jewett A. I., Bochkov I. D., Chinnappan D., Cutkosky A., Li J., Geeting K. P., Gnirke A., Melnikov A., McKenna D., Stamenova E. K., Lander E. S., Aiden E. L., Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 112, E6456–E6465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reya T., Morrison S. J., Clarke M. F., Weissman I. L., Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Zink D., Fischer A. H., Nickerson J. A., Nuclear structure in cancer cells. Nat. Rev. Cancer 4, 677–687 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Cherkezyan L., Stypula-Cyrus Y., Subramanian H., White C., Dela Cruz M., Wali R. K., Goldberg M. J., Bianchi L. K., Roy H. K., Backman V., Nanoscale changes in chromatin organization represent the initial steps of tumorigenesis: A transmission electron microscopy study. BMC Cancer 14, 189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu W., Radosevich A. J., Eshein A., Nguyen T.-Q., Yi J., Cherkezyan L., Roy H. K., Szleifer I., Backman V., Using electron microscopy to calculate optical properties of biological samples. Biomed. Opt. Express 7, 4749–4762 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konda V. J., Cherkezyan L., Subramanian H., Wroblewski K., Damania D., Becker V., Gonzalez M. H., Koons A., Goldberg M., Ferguson M. K., Waxman I., Roy H. K., Backman V., Nanoscale markers of esophageal field carcinogenesis: Potential implications for esophageal cancer screening. Endoscopy 45, 983–988 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaffer S. M., Dunagin M. C., Torborg S. R., Torre E. A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P. A., Xiao M., Eggan E., Anastopoulos I. N., Vargas-Garcia C. A., Singh A., Nathanson K. L., Herlyn M., Raj A., Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paek A. L., Liu J. C., Loewer A., Forrester W. C., Lahav G., Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell 165, 631–642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Maaten L., Hinton G., Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008). [Google Scholar]
- 48.Colaprico A., Silva T. C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T. S., Malta T. M., Pagnotta S. M., Castiglioni I., Ceccarelli M., Bontempi G., Noushmehr H., TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma H., Tu L.-C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T., Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 34, 528–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn E. H., Pegoraro G., Brandão H. B., Valton A.-L., Oomen M. E., Dekker J., Mirny L., Misteli T., Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176, 1502–1515.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reddy K. L., Feinberg A. P., Higher order chromatin organization in cancer. Semin. Cancer Biol. 23, 109–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Florescu A.-M., Therizols P., Rosa A., Large scale chromosome folding is stable against local changes in chromatin structure. PLOS Comput. Biol. 12, e1004987 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uhler C., Shivashankar G. V., Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 18, 717–727 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Arya G., Schlick T., Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci U.S.A. 103, 16236–16241 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerlitz G., Hock R., Ueda T., Bustin M., The dynamics of HMG protein–chromatin interactions in living cells. Biochem. Cell Biol. 87, 127–137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker S. C., Hansen L., Abaan H. O., Tullius T. D., Margulies E. H., Local DNA topography correlates with functional noncoding regions of the human genome. Science 324, 389–392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen H., Chen J., Muir L. A., Ronquist S., Meixner W., Ljungman M., Ried T., Smale S., Rajapakse I., Functional organization of the human 4D Nucleome. Proc. Natl. Acad. Sci. U.S.A. 112, 8002–8007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicot N., Hausman J.-F., Hoffmann L., Evers D., Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56, 2907–2914 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Cherkezyan L., Zhang D., Subramanian H., Capoglu I., Taflove A., Backman V., Review of interferometric spectroscopy of scattered light for the quantification of subdiffractional structure of biomaterials. J. Biomed. Opt. 22, 30901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Radosevich A. J., Yi J., Rogers J. D., Backman V., Structural length-scale sensitivities of reflectance measurements in continuous random media under the Born approximation. Opt. Lett. 37, 5220–5222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Przibilla S., Dartmann S., Vollmer A., Ketelhut S., Greve B., von Bally G., Kemper B., Sensing dynamic cytoplasm refractive index changes of adherent cells with quantitative phase microscopy using incorporated microspheres as optical probes. J. Biomed. Opt. 17, 97001-1 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Sung Y., Choi W., Lue N., Dasari R. R., Yaqoob Z., Stain-free quantification of chromosomes in live cells using regularized tomographic phase microscopy. PLOS ONE 7, e49502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaax6232/DC1
Section S1. CPMC model: Variance of chromatin packing density
Section S2. Random medium simulations: Calculation of D from mass density variations
Section S3. Calculation of ϕin,0 from ChromEM measurements
Fig. S1. Analysis of the relationship between D and .
Fig. S2. Random medium simulations for low and high D.
Fig. S3. CVC distributions of A549 and BJ cells as measured by ChromEM.
Fig. S4. Transcriptional malleability in A2780 and M248 cells.
Fig. S5. GO analysis of up-regulated genes.
Fig. S6. t-SNE transcriptional heterogeneity analysis is independent of seed.
Fig. S7. Transcriptional heterogeneity is increased in high D cells.
Fig. S8. Relationship between survival time and transcriptional divergence.
Table S1. Descriptions and values of CPMC model parameters.
Table S2. TCGA patient information.