

Abstract
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy. Although novel emerging drugs are available, the overall prognosis remains poor and new therapeutic approaches are required. PP2A phosphatase is a key regulator of cell homeostasis and is recurrently inactivated in AML. The anticancer activity of several PP2A-activating drugs (e.g., FTY720) depends on their interaction with the SET oncoprotein, an endogenous PP2A inhibitor that is overexpressed in 30% of AML cases. Elucidation of SET regulatory mechanisms may therefore provide novel targeted therapies for SET-overexpressing AMLs. Here, we show that upregulation of protein kinase p38β is a common event in AML. We provide evidence that p38β potentiates SET-mediated PP2A inactivation by two mechanisms: facilitating SET cytoplasmic translocation through CK2 phosphorylation, and directly binding to and stabilizing the SET protein. We demonstrate the importance of this new regulatory mechanism in primary AML cells from patients and in zebrafish xenograft models. Accordingly, combination of the CK2 inhibitor CX-4945, which retains SET in the nucleus, and FTY720, which disrupts the SET-PP2A binding in the cytoplasm, significantly reduces the viability and migration of AML cells. In conclusion, we show that the p38β/CK2/SET axis represents a new potential therapeutic pathway in AML patients with SET-dependent PP2A inactivation.
Subject terms: Biochemistry, Cell signalling, Acute myeloid leukaemia, Acute myeloid leukaemia
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous fatal disease that results from the enhanced proliferation and impaired differentiation of hematopoietic stem and progenitor cells1. For decades, chemotherapy consisting of cytarabine and anthracyclines has been the standard in AML care. Emerging drugs show promising results2,3; however, the outcome for AML remains poor and most patients ultimately relapse and die from disease progression despite initial sensitivity to chemotherapy. Patients older than 60 years old, who represent the main group, are refractory to cytotoxic intensive chemotherapy because of biological disease-related factors, such as increased frequency of adverse-risk cytogenetic and molecular features, and secondary AML3. Moreover, they present comorbidities that reduce their tolerance of intensive therapies, leaving few treatment options in most cases1. Even in younger patients the outcome is dismal. In patients ≤60 years old complete remission is achieved in around 70%, but a subset of patients relapse, depending on the prognostic factors, and only 5–10% survive after relapse1,4. Current efforts directed towards the genetic characterization of AML have led to the development of new targeted therapies, including FLT3, BCL2 and IDH1/2 inhibitors5–10. However, monotherapy with these drugs does not result in durable responses. Thus, further research is necessary to develop new personalized therapeutic strategies for the treatment of this aggressive disease.
Reversible phosphorylation allows the cell to maintain a proper homeostasis regulation; therefore, the balance between kinases and phosphatases is essential to control correct proliferation, apoptosis, and differentiation. Many studies have analyzed the abnormal behavior of protein kinases in AML, but the role of phosphatases remains underexplored11,12. Protein phosphatase 2A (PP2A) is a tumor suppressor that regulates several essential cell functions and counteracts most of the kinase-driven intracellular signaling pathways13. Previous results from our group and others showed that PP2A inactivation is a recurrent event in AML, and that its pharmacological activation by PP2A-activating drugs (OP499, FTY720, and its analogues) effectively antagonizes leukemogenesis14,15. Furthermore, preclinical studies show that these drugs have synergistic effects with conventional chemotherapy and tyrosine kinase inhibitors, opening new possibilities for personalized medicine in AML16,17. Interestingly, the anticancer activity of several PP2A-activating drugs depends on their ability to interact with the endogenous PP2A inhibitor SET, an oncoprotein overexpressed in ~30% of AML patients and associated with poor outcome18,19. Therefore, targeting SET allows PP2A to be reactivated indirectly, avoiding toxicity problems related to the direct activation of this complex holoenzyme. SET is a multitask oncogenic protein involved in many cellular processes20–23. However, despite the prognostic impact of SET overexpression in both hematologic and solid tumors, the mechanisms by which SET is regulated remain poorly understood. We have previously reported a novel multi-protein complex that activates SET transcription in AML24. Here, we explore the post-transcriptional regulation of SET, which may help us to develop novel targeted therapies in AML patients with PP2A inactivation and high expression of SET. Using genetic and pharmacological approaches, we found that p38β, one of the p38 family members whose function is not well known, has a dual role in the regulation of PP2A activity in AML. p38β regulates SET phosphorylation and intracellular localization through the activity of casein kinase 2 (CK2). Furthermore, p38β stabilizes the SET protein, facilitating its PP2A inhibitory role. Importantly, we validated this mechanism in vivo by demonstrating that the combination of the CK2 inhibitor CX-4945, and the PP2A-activating drug FTY720 significantly reduces the viability and migration of AML cells. This novel mechanism may constitute the basis for targeted therapy in AML patients with SET overexpression.
Materials and methods
Patient samples
The study comprised peripheral blood mononuclear cells (PB-MC) samples of 27 patients with AML at diagnosis who stated an informed consent (Supplementary Table S1). All patients were treated with standard induction chemotherapy. High-dose cytarabine, and autologous or allogeneic stem cell transplantation, when possible, were used as consolidation therapy. PB-MC samples of healthy donors were used as controls. This study is part of a project approved by the Comité Ético de Investigación Clínica, Gobierno de Navarra (2018/32). The experiments conformed to the principles set out in the WMA Declaration of Helsinki. AML patient sample cells (CD34+) were cultivated in the semisolid medium MethoCult (StemCell Technologies, Grenoble, France) supplemented with penicillin G (100 U/ml) and streptomycin (0.1 mg/ml). In the medium, different concentrations of FTY720, CX-4945 and combination were added. After 12–14 days growing at 37 °C in a 5% CO2 atmosphere, the present colonies were counted at an inverted light microscope (Leica Biosystems, Barcelona, Spain) using a grid (2700, StemCell).
In vitro kinase assay
Bacterially-expressed p38α or p38β (0.2 µg) were pre-incubated with purified MKK6 (40 ng) and then incubated with purified GST, GST-ATF2 or GST-SET (1 µg) in kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT, 0.1 mM Na3VO4, 1 mM PMSF and 10 µg/ml aprotinin and leupeptin) containing 100 μM cold ATP and 2μCi of [γ-32P]ATP (3 000 Ci/mmol) for 40 min at 30 °C. Reactions were stopped by adding sample loading buffer and boiling 5 min. Proteins were resolved by SDS-PAGE, stained with Coomassie, and analyzed by autoradiography.
Plasmids, siRNA, and transfection
siRNAs were from Ambion (Madrid, Spain): scramble siRNA (#AM4635), MAPK11/p38β (#1:s11155 and #2:11156), MAPK14/p38α (#1:s3586 and #2:s3585), and CK2 (s3638). SET siRNAs were siSET#1 (#23-2506-2/4, Eurofins, Ebersberg, Germany) and siSET#2 (#5883466, Invitrogen). Due to the high efficiency obtained with siRNAs #1 from p38α and p38β we used them for all experiments. For silencing experiments, cells were transfected using GenePulser XcellTM (Bio-Rad, Madrid, Spain) with 300 V and 1000 µF. The shRNAp38β cloned in the pINDUCER 11 (44.363 from Adgene, Teddington, UK) was shRNA1: CACGTTCAATTCCTGGTTT and shRNA2: GCGCCAGAAGGTGGCGGTGAAG.
General methodology
Details on general methodology as western blot, protein immunoprecipitation, apoptosis, and MTS assay have been previously described14,18,19,24,25. Reagents and antibodies used are displayed in Supplementary Tables S2 and S3, respectively. Nuclear and cytoplasmic proteins were extracted using the NE-PER nuclear and cytoplasmic extraction kit (Thermo-scientific, UK) according to manufactured instructions.
Cell culture and treatments
HL60, MOLM-13, and HEK293T cells were maintained in RPMI-1640 (Invitrogen, UK) supplemented with 10% FBS, penicillin G (100U/ml) and streptomycin (0.1 mg/ml). Cell lines were grown at 37 °C in a 5% CO2 atmosphere. Prior treatments, cells were plated at 100,000 cells/ml.
Immunofluorescence
100,000 cells were seeded on cover slips coated with poly-L-lysine (Sigma, Madrid, Spain), fixed with 4% paraformaldehyde (Thermo-scientific) and permeabilized with 0.1% Triton-X-100. After blocking with 5% FBS, incubation with primary and secondary antibodies were performed (Supplementary Table S3). Images were acquired using a Confocal Scanning Laser Microscopy Zeiss LSM 800 with ×63 immersion oil objective. Image quantification was performed using Fiji software26. For colocalization, the red, green and red-green colocalization volumes (um³) were quantified and referred to total cell volume. For nuclear and cytoplasmic quantification, green volume (um³) was measured in the cytoplasm and the nucleus and referred to total cell volume.
Phos-tag, immunoblot and λ-phosphatase treatment
10% acrylamide gels were prepared in the presence of 40 µM of Phos-tag (Fujifilm Wako, Neuss, Germany) and 20 µM of MnCl2. Proteins were transferred to a PVDF membrane (Immobilon-P membranes, Millipore, Madrid, Spain) using the Tank blotting system (Bio-Rad, Madrid, Spain). As a control, an aliquot of the cell lysate (15 µg) was incubated with 100 units of λ-phosphatase (Biolabs, Spain) for 1 h at 30 °C in a shaking thermoblock.
Migration assay
Migration assay was performed in a 24-transwell permeable plate with 8.0 μM pores (Corning Costar, Madrid, Spain). The lower compartment contained RPMI supplemented with 10% FBS. 500,000 treated cells were seeded in the upper insert in medium without serum and allowed to migrate for 3 h. The volume of the bottom well was collected and mixed with perfect-count microspheres (cytognos, Salamanca, Spain). The amount of viable migrated cells was determined by flow cytometry, counting 5000 microsphere-events and expressed as a percentage of the control.
Zebrafish husbandry and embryo collection
Wild-type zebrafish (Danio rerio, AB strain), from the Zebrafish International Resource Centre, were maintained in re-circulating tanks according to the standard procedures. Adult fishes were maintained at 26 °C, with a light/dark cycle of 14/10 h, and were fed twice daily, once with dry flake food (Prodac, Italy) and another with live Artemia salina (MC 450, Ive Aquaculture, USA). Zebrafish embryos were maintained in egg water at 28.5 °C, fed for 5 days with Novo Tom and with live Artemia salina at 11 days of life. All experiments were performed in compliance with the Guidelines of the European Union Council for animal experimentation (86/609/EU).
Xenograft of human leukemia cells into zebrafish embryos
Wild-type zebrafish embryos at 48hpf were anesthetized with 0.04% Tricaine (Sigma–Aldrich). Treated leukemia cells were stained with red fluorescent CM-DiI (Invitrogen) prior the injection. 50–75 labeled cells were injected into the yolk sac of dechorionated zebrafish embryos using a manual injector (Narishige). Fish with fluorescently labeled cells appearing outside the implantation area at 2hpi were excluded from analysis. All other fishes were incubated at 35 °C for 72 h and analyzed with the SteReo Lumar V12 stereomicroscope with an AxioCam MR5 camera (Carl Zeiss, Germany). Positive embryo colonization was considered when more than five human leukemia cells were present outside the yolk sac at 72hpx. Zebrafish colonization index was calculated as the proportion of embryos colonized in the treatment condition divided by the proportion of invaded embryos in the control condition. Tumor growth and proliferation were evaluated at 2 (reference) and 72hpx in a M205-FA fluorescence microscopy with a DFC365FX camera (Fujifilm Leica). Proliferation index (Fluorescence intensity medium value*fluorescence pixel number) and area were measured with a Leica Application Suite-X software.
Statistical analysis
Data represented are the mean of three independent experiments ±S.D. Statistical comparisons were carried out using the nonparametric method Kruskal–Wallis test for more than two independent samples, followed by Mann–Whitney U test to compared two groups when the distribution was not normal (Shapiro-Wilk test p < 0,05). Two-way ANOVA (Tukey’s multiple comparisons test) when the distribution was normal (zebrafish proliferation experiments). Chi-square statistical analysis was done for the invasive potential calculation in zebrafish experiments. Significance was considered when p < 0.05. For AML patient samples tested, 20% decrease in viability was chosen as the threshold as a response to the treatments.
Results
p38β overexpression regulates PP2A activity in AML through SET
To investigate the regulation of the SET oncoprotein in AML, we performed a functional drug screen using inhibitors of the main signaling pathways such as PI3K, p38, JNK, and ERK in HL60 cells. Notably, only p38 inhibition using either SB203580 or PH797804 decreased SET protein content without altering its mRNA levels (Supplementary Fig. S1a, b), suggesting that SET was regulated at post-transcriptional level. In fact, reduced phosphorylation of HSP27, a downstream target of p38, paralleled the decrease in SET protein levels (Fig. 1a). Since SET is an important inhibitor of PP2A in AML, we assessed the PP2A activity in the treated cells. As expected, both p38 inhibitors increased PP2A activity (Fig. 1b and Supplementary Fig. S1a), suggesting that p38 inhibition affects PP2A activity in AML through SET. These results were confirmed in MOLM-13, another AML cell line (Fig. 1 and Supplementary Fig. S1c).
Fig. 1. p38β is overexpressed in AML and its inhibition decreases SET protein levels, increasing PP2A activity.

a HL60 and MOLM-13 cell lines were treated with the p38 inhibitors SB203580 (2.5 µM) and PH797804 (250 nM) for 24 h. Protein expression for p-HSP27/HSP27 (p38 substrate) and SET was analyzed by western blot. b Measurement of PP2A activity after p38 inhibition by immunoprecipitation and phosphatase assay. c Silencing of p38α and p38β with specific siRNA (50 nM for 48 h), and analysis of total protein by western blot. d Measurement of PP2A activity by immunoprecipitation and phosphatase assay. e Western blot analysis of total protein in AML cell lines and AML patient samples, compared to peripheral blood mononuclear cells (PB-MC). The results are corrected by the specific loading control and are expressed as fold-change of the control, which are assigned a value of 1 and are mean values. Experiments were performed in triplicate four times. *p < 0.05.
The p38 family has four members: p38α, p38β, p38γ, and p38δ. As p38α and p38β are the main targets of SB203580 and PH797804 at the tested concentrations27,28, we focused on these two kinases. Knockdown of p38α or p38β by specific siRNAs showed that downregulation of p38β, but not p38α, significantly decreased SET protein levels and increased PP2A activity (Fig. 1c, d).
To explore the clinical relevance of this finding, we assessed p38α and p38β expression in AML. The p38β protein was highly expressed in 5 out of 7 AML cell lines (71%), and in 23 out of 27 AML patient samples (85%); whereas the p38α protein was almost equally expressed in PB and AML specimens (Fig. 1e, Supplementary Fig. S2). Correlation analysis indicated a positive co-expression between SET and p38β protein levels, which was statistically significant (R2 = 0.376 p-value 0.0014). However, no correlation was found between p38α and SET (R2 = 0.004 p-value 0.7694). Quantitative analysis confirmed that p38α was expressed at similar levels in PB and AML cell lines (20–30 ng/100 µg total protein). However, p38β was expressed at lower levels than p38α in HL60 and MOLM-13 cells (2–3 ng/100 µg total protein), but it was undetectable in PBMC (Supplementary Fig. S3). Taken together, these results suggest that p38β is overexpressed in AML and can regulate PP2A activity via SET.
p38β binds to and stabilizes SET in AML cells
We next focus on dissecting the mechanisms through which p38β regulates the SET protein. Co-immunoprecipitation experiments indicated that SET bound to p38β in both HL60 and MOLM-13 cells, and to a lesser extent, to p38α in HL60 cells (Fig. 2a). Immunofluorescence analysis confirmed high expression and cytoplasmic colocalization between p38β and SET, which disappeared after silencing p38β, whereas p38α silencing had no effect (Fig. 2b).
Fig. 2. p38β co-localizes with SET in AML cells.

a Immunoprecipitation of SET, p38α and p38β with specific antibodies in HL60 and MOLM-13 cells. Normal goat Ig was used as negative control b Knockdown of p38α and p38β with siRNA (50 nM for 48 h), using scramble siRNA as control, in HL60 and MOLM-13 cells. Immunofluorescence analysis of either p38α or p38β (red) and SET (green). Nuclei were stained with DAPI. Immunofluorescences were visualized by confocal microscopy. Quantification table of colocalization fluorescence and green fluorescence (SET) in nucleus and cytoplasm. Quantification analysis showed ~60% of SET-p38β colocalization in both cell lines, and only 12 and 29% of SET-p38α colocalization in MOLM-13 and HL60, respectively. c Immunofluorescence analysis of p38β (red) and SET (green) in peripheral blood mononuclear cells (PB-MC) and the primary AML patient samples AML-23 and AML-24. Nuclei were stained with DAPI. Quantification table of colocalization fluorescence. Immunofluorescences were visualized by confocal microscopy. The results are expressed as mean values ± SEM. Experiments were performed in triplicate four times. *p < 0.05, **p < 0.01. Scale bar represents 5 µm.
We hypothesized that p38β might phosphorylate SET. Surprisingly, in vitro kinase assays showed no direct SET phosphorylation either by p38β or p38α (Supplementary Fig. S4a). Importantly, co-immunoprecipitation in HL60 cells treated with p38 inhibitors showed that SET-p38 interaction did not require kinase activation (Supplementary Fig. S4b). For these reasons, we postulated that p38β could regulate SET stability in a kinase-independent manner. Treatment of cells with cycloheximide demonstrated that SET is stable up to 48 h (Supplementary Fig. S5a). Treatment of p38β-silenced cells with cycloheximide resulted in a significant decrease in SET (Supplementary Fig. S5b), suggesting that p38β-SET interaction is critical for SET stability. Besides, immunofluorescence analysis in samples from AML patients that overexpress SET and p38β, such as AML-23 or AML-25, demonstrated that both proteins tend to associate and colocalized in the cytoplasm. In contrast, samples from patients with no SET or p38β overexpression, such as AML-24, showed minimal colocalization (Fig. 2c, Supplementary Fig. S6a, b). These results support the biological importance of SET-p38β binding in AML, and suggest that p38β contributes to cytoplasmic SET stability. Data from our group previously reported that SET protein stability is enhanced through its binding to SETBP125. Here, we show that SETBP1 and p38β colocalized along with SET in the cytoplasm (Fig. 3a, Supplementary Fig. S7). Furthermore, we found SET-PP2Ac and PP2Ac-p38β colocalization and interaction in AML cells (Fig. 3b). Taken together, these results suggest that p38β acts as a SET stabilizing protein, together with SETBP1, allowing SET to inhibit PP2A in the cytoplasm.
Fig. 3. p38β acts as a SET stabilizing protein.
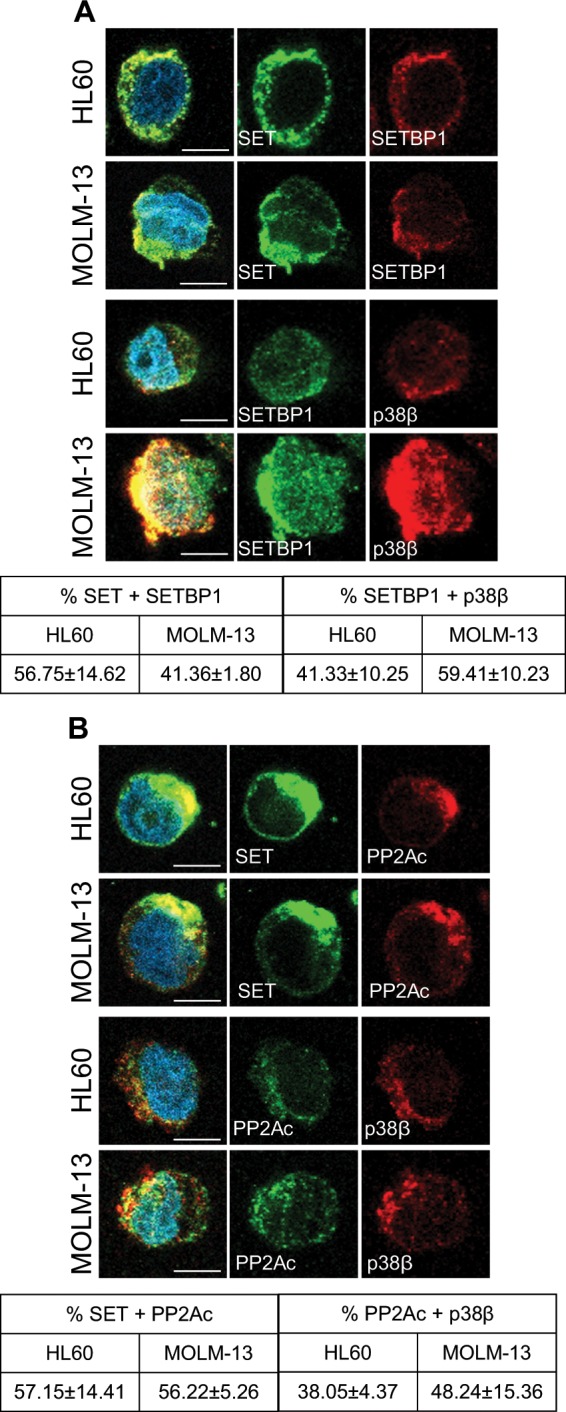
a Immunofluorescence analysis of SET (green) and SETBP1 (red), and SETBP1 (green) and p38β (red), in HL60 and MOLM-13 cells. Nuclei were stained with DAPI. Quantification of colocalization fluorescence. (b) Immunofluorescence analysis of SET (green) and PP2Ac (red) and PP2Ac (green) and p38β (red), in HL60 and MOLM-13 cells. Nuclei were stained with DAPI. Quantification of colocalization fluorescence. Immunofluorescences were visualized by confocal microscopy. The results are expressed as mean values ± SEM. Experiments were performed in triplicate four times. *p < 0.05, **p < 0.01. Scale bar represents 5 µm.
p38β regulates CK2-mediated phosphorylation of SET and facilitates its translocation to the cytoplasm
SET is mainly localized in the nucleus29, but AML cells overexpressing SET showed strong cytoplasmic half-moon-shape localization (Fig. 2b, c). It has been reported in Alzheimer’s disease models that CK2 phosphorylates Ser9 on SET, leading to its cytoplasmic translocation and inhibition of PP2A, resulting in tau phosphorylation30,31. CK2 is overexpressed in most hematological tumors, including AML32, and it is a target of p38 signaling33. This data prompted us to postulate the potential role of p38β in regulating CK2 and, consequently, the phosphorylation of SET in AML. Western blot showed that overexpression of CK2 is a recurrent event in both AML cell lines and patient samples (Fig. 4a, Supplementary Fig. S2). First, we confirmed that CK2 phosphorylation is indeed regulated by p38 in AML cells, as it is decreased after p38α or p38β knockdown (Fig. 4b). Next, we investigated whether p38β silencing or CK2 inhibition using CX-4945 affects SET phosphorylation, by using Phos-tagTM SDS-PAGE. Inhibition of CK2 and silencing of p38β, but not p38α, substantially decreased the phosphorylated forms of SET in AML cells (Fig. 4c, Supplementary Fig. S8a). Interestingly, while both p38α and p38β can potentially regulate CK2 phosphorylation, only the inhibition of p38β affected SET phosphorylation and SET interaction with CK2.
Fig. 4. p38β regulates CK2-mediated phosphorylation of SET.

a Western blot analysis of the CK2 protein in AML cells lines compared to peripheral blood mononuclear cells (PB-MC). b Silencing of p38α and p38β with specific siRNA (50 nM for 48 h) and analysis of phospho- and total CK2 by western blot in HL60 and MOLM-13 cells. c MOLM-13 cells treated with either siRNA for silencing p38α and p38β (50 nM for 48 h) or with the CK2 inhibitor CX-4945 (5 µM, 24 h) and analyzed for phosphorylated forms of SET in SDS-PAGE with Phos-TagTM. A sample treated with λ phosphatase (100 units, 1 h) was used as control. d Overexpression of p38β in HEK293T cells with 1 µg of pEFM link p38β plasmid or the empty plasmid with lipofectamine 2000 and treated with CX-4945 (3,75 µM, 24 h). Analysis of p38β, SET and phospho- and total CK2 by western blot and PP2A activity. e Silencing of SET with specific siRNA (50 nM for 72 h) and analysis of SET by western blot and PP2A activity in HL60 and MOLM-13 cells. The results are corrected by the specific loading control and are expressed as fold-change of the control, which are assigned a value of 1 and are mean values. Experiments were performed in triplicate four times. *p < 0.05 **p < 0.01.
In order to confirm that p38β regulates PP2A activity, we overexpressed p38β in HEK293T cells with the pEFM-link-p38β plasmid. The ectopic increment of p38β resulted in a significant increase in the phosphorylation of CK2, accompanied with a reduction of PP2A activity (Fig. 4d), suggesting that p38β is involved in CK2 activation and regulation of PP2A activity. To further demonstrate that CK2 is crucial in the decrease of PP2A activity produced by p38β overexpression, we inhibited CK2 by adding CX-4945 (3.75 µM) for 24 h. CK2 inhibition restored PP2A activity in cells overexpressing p38β (Fig. 4d) suggesting that CK2 is an intermediate in PP2A activity regulation by p38β. Additionally, to study whether CK2 has a direct effect on PP2A regulation, SET was silenced in both AML cell lines and then, the cells were treated with CX-4945 to inhibit CK2. Inhibition of CK2 in cells with reduced amount of SET had no effect in PP2A activity in the AML cells tested (Fig. 4e), suggesting that CK2-dependent inhibition of PP2A is through SET.
Next, we treated AML cells with either CX-4945 or CK2 siRNA, and found that CK2 inhibition or downregulation resulted in increased nuclear and decreased cytoplasmic localization of SET (Fig. 5a, b, Supplementary Fig. S8b, c), which increased PP2A activity (Fig. 5c). These results were confirmed by immunofluorescence (Fig. 5d). Furthermore, nuclear SET retention was accompanied by enhanced p38β nuclear localization (Fig. 5e). Taken together, our results show that p38β-dependent activation of CK2 leads to SET phosphorylation, enhancing its cytoplasmic localization and consequently reducing PP2A activity (Fig. 5f).
Fig. 5. CK2 inhibition retains SET in the nucleus.

a Nuclear (Nuc.) and cytoplasmic (Cyto.) protein isolated from HL60 and MOLM-13 cells treated with CX-4945 (5 µM, 24 h) and analyzed by western blot for SET localization. b HL60 and MOLM-13 cells treated with specific siRNA for CK2 (20 nM, 48 h). Nuclear (Nuc.) and cytoplasmic (Cyto.) proteins were isolated and analyzed by western blot for SET localization. c Measurement of PP2A activity by immunoprecipitation and phosphatase assay. The results are expressed as fold-change of the control, which are assigned a value of 1 and are mean values ± SEM. Experiments were performed in triplicate four times. *p < 0.05, **p < 0.01. d Immunofluorescence analysis of CK2 (red) and SET (green). Nuclei were stained with DAPI. Immunofluorescences were visualized by confocal microscopy. Scale bar represents 5 µm. Quantification table of green fluorescence (SET) in nucleus and cytoplasm. The results are expressed as mean values ± SEM. Experiments were performed in triplicate four times. *p < 0.05, **p < 0.01. e Immunofluorescence analysis of p38β (red) and SET (green). Nuclei were stained with DAPI. Immunofluorescences were visualized by confocal microscopy. Scale bar represents 5 µm. f p38β is able to activate CK2, which phosphorylates SET and, as consequence, facilities SET trafficking to the cytoplasm, contributing to PP2A inactivation in AML cells. Moreover, p38β binds to SET in the cytoplasm, contributing to its stability and leading to PP2A inactivation. Treatment with CX-4945 (CK2 inhibitor) retains SET in the nucleus, avoiding its phosphorylation. FTY720 treatment disrupts the SET-PP2A biding which remains in the cytoplasm.
Inhibition of CK2 potentiates the anticancer activity of a PP2A-activating drug on AML cells
We have shown that p38β contributes to the inactivation of PP2A in AML cells, which involves phosphorylation of SET by CK2. Therefore, we speculated that CK2 inhibition by CX-4945 could enhance the antileukemic effect of the PP2A-activating drug FTY720 that binds SET19. To test this idea, we first used immunofluorescence to analyze the time-course of SET nuclear retention after CK2 inhibition, which started at 4 h (Supplementary Fig. S9). Accordingly, AML cells were treated with CX-4945 for 4 h prior to FTY720 treatment. The combined treatment significantly decreased cell viability (Fig. 6a) and increased apoptosis (Fig. 6b) in AML cells, being more effective than either single treatment and having a synergistic effect (Supplementary Fig. 10a). These effects correlated with increased PP2A activity (Fig. 6c) and reduced cell migration ability (Fig. 6d). Immunofluorescence analysis showed that the combined treatment retained SET in the nucleus together with p38β, and disrupted the SET-PP2Ac interaction (Fig. 6e), supporting the mechanism proposed. We also evaluated the combined treatment in AML primary patient samples. According to availability, we treated patient-derived PB-MC samples (Supplementary Table S1) with FTY720, CX-4945 or both, and then performed MTS assays. When there was enough sample, we also tested the colony formation ability. We found decreased viability in 64% of the samples treated with FTY720 (15/24), and in 90% of the samples treated with CX-4945 (10/11). Importantly, all five AML patient samples that were treated with both drugs showed a significant decrease in cell viability compared to either treatment alone (a representative case shown in Supplementary Fig. S10b). Colony formation ability was also reduced in the three AML patient samples treated with both drugs that were grown in semisolid medium (Fig. 6f, Supplementary Fig. S10c).
Fig. 6. CX-4945 and FTY720 combination therapy induces apoptosis in AML cell lines and primary patient samples.

HL60 and MOLM-13 cells pretreated for 4 h with CX-4945 (5 µM) and then treated for 24 h with FTY720 (5 µM). a Cell viability was measured by MTS analysis. The results are corrected by the DMSO control and are expressed as fold-change of the control, which are assigned a value of 1 and are mean values ± SEM. b FACS analysis of apoptosis in HL60 and MOLM-13 cell lines stained with propidium iodide (PI) and Annexin V. The percentages of viable and apoptotic cells are indicated. c PP2A activity analysis performed by immunoprecipitation and activity assay. d Migration of HL60 and MOLM-13 placed in the upper well of a 8.0 μM transwell plate in RPMI without FBS. The lower chamber contained RPMI supplemented with 10% FBS. Migration assay was performed for 3 h and then assessed for cell number using flow cytometry e Immunofluorescence analysis of SET (green) and PP2A (red) and quantification table of % colocalization between red fluorescence (PP2A) and green fluorescence (SET) and % of green fluorescence (SET) in nucleus and cytoplasm. Nuclei were stained with DAPI. Immunofluorescences were visualized by confocal microscopy. Scale bar represents 5 µm. Experiments were performed in triplicate four times. *p < 0.05, **p < 0.01. f AML patient samples were cultured in semisolid medium and treated with CX-4945 (5 µM) and FTY720 (4 and 8 µM), alone or in combination. Colony formation units (CFU) were counted 12 days after seeding. Graphs of counted CFU represented as percentage of CFU related to the control, which are assigned the total CFU (100%) and are mean values ± SEM.
Finally, we validated the proposed mechanism in a zebrafish xenograft model, a robust animal system to test tumor cell behavior and drug response34–36. AML cells were evaluated for in vivo proliferation and invasion potential in zebrafish embryos upon treatment with FTY720, CX-4945 or their combination, following the scheme in Fig. 7a. Embryos were analyzed 2 h post-xenograft (hpx) to confirm proper injection, and 72hpx for proliferation and invasion. The combined treatment significantly decreased the proliferation index compared to both single treatments and the control (Fig. 7b), as well as the tumor growth area in zebrafish embryos (Fig. 7c). We also studied the colonization potential of treated AML cells by analyzing the zebrafish larvae with invasion in the tail, as illustrated in Fig. 7d. Quantifications demonstrated that treatment with CX-4945 and FTY720 significantly reduced zebrafish larvae with AML cell tail invasion (Fig. 7e). To corroborate the importance of p38β in our model, we injected zebrafish embryos with AML cells expressing two different doxycycline inducible p38β shRNAs (Fig. 7a). We found that p38β silencing decreased the proliferation and colonization index of AML cells in zebrafish embryos 72hpx (Fig. 7f, Supplementary Fig. S11), supporting the functional importance of p38β overexpression in AML cells. Taken together, our results combining FTY720 with CK2 inhibitors or using p38β shRNAs support the proposed new mechanism that regulates AML cell viability and migration.
Fig. 7. CX-4945 and FTY720 combination therapy induces AML cell death in zebrafish xenograft models.

In vivo proliferation and invasive potential of HL60 and MOLM-13 cells upon treatment with FTY720, CX-4945 or the combination of both compounds were analyzed in a xenograft zebrafish model. a Timing scheme for the xenografts of zebrafish embryos. b Measurement of proliferation index performed as fluorescence intensity medium value* RF pixel, demonstrating cell proliferation of treated cells in the xenograft model. c Representative pictures of Tumor growth of HL60 treated cells in zebrafish embryos 2 hpx (reference fluorescence) and 72hpx. Scale bars represent 0.1 mm. d Representative pictures of zebrafish embryos injected with MOLM-13 cells and treated with DMSO or combination of CX-4945 (1 µM) and FTY720 (1 µM), which show the cells that migrated to the tail after the treatment mentioned. Magnified pictures on the bottom show the invasion of cells in the tail. Scale bars from whole zebrafish picture represent 0.5 mm and from zoom 0.1 mm. e Quantification of the invasive potential of the injected cells upon drug treatment. Quantification performed as colonization index: count of zebrafish embryos with invasion of cells migrating outside the yolk sac referred to the control embryos (injection of DMSO treated cells). Hpx: hours post-xenograft. **p < 0.01, ***p < 0.001 vs. DMSO treated cells. f In vivo proliferation and invasive potential of HL60 and MOLM-13 cells upon infection with shp38β pINDUCER11 treated with and without doxycycline in a xenograft zebrafish model measured at 72hpx. **p < 0.01, ***p < 0.001 vs. control cells.
Discussion
Here, we investigated the post-transcriptional regulation of the SET oncoprotein, establishing that p38β overexpression is a common event in AML that leads to PP2A inactivation through its endogenous inhibitor SET. Furthermore, we provide evidence that p38β, but not the closely related p38α family member, controls the phosphorylation of SET by CK2, facilitating SET shuttling from the nucleus to the cytoplasm. Besides, p38β also acts as a SET stabilizing protein, facilitating PP2A inactivation. We describe a novel molecular pathway of leukemogenesis with therapeutic potential in AML patients that show SET-dependent PP2A inactivation, a subgroup with poor prognosis that represent ~30% of all AML cases. PP2A is a tumor suppressor, which regulates most of the kinase-driven intracellular signaling pathways. Thus, by targeting SET, this approach allows reactivating PP2A indirectly, avoiding toxicity issues related to the direct activation of this complex holoenzyme.
Mitogen-activated protein kinase (MAPK) cascades are important signaling pathways used by eukaryotic cells to transduce extracellular signals. Using chemical inhibitors of several MAPKs, we found that only p38 inhibitors were able to increase PP2A activity by decreasing SET protein, suggesting post-transcriptional regulation of SET. The p38 family is involved in many cellular processes, and plays a key role in the stress response37. Although p38α and p38β share 70% in amino acid sequence homology, they have different functions37,38 and differential regulatory mechanisms39. Nevertheless, the high expression of p38α in most tissues, together with the results using knockout mice deficient in p38α or p38β, suggests that p38α is the dominant form, although functional redundancy has been reported40. As a consequence, most studies have focused on p38α, and little is known about p38β37. In this regard, we have elucidated an important role of p38β in AML that has not been previously suggested. p38β is widely expressed, but usually at low levels in most tissues37, and it is not detected in monocytes, macrophages, or neutrophils41. We found that upregulation of p38β, but not p38α, is a common event in AML cases that contributes to the SET-dependent inactivation of PP2A in AML, pointing to a relevant role of p38β in this aggressive disease.
It has been reported that PP2A can regulate p38 signaling pathway, and that PP2A and p38 form complexes in the cytoplasm in basal conditions; in fact, p38 may act as scaffold protein for NMP and PP2A42–45. However, the nature of their connection varies depending on the context. Upon TNF-induced stress conditions in endothelium-derived cell lines, p38 positively regulates PP2A activity42, whereas under hypoxia and survival conditions in colorectal cancer cell lines, PP2A negatively regulates p38MAPK activity44. Here, we show for the first time that p38β controls PP2A activity through the regulation of its endogenous inhibitor SET.
Pyridinyl-imidazole inhibitors have allowed the identification of many functions regulated by p38 beyond the stress response. However, these compounds do not permit us to distinguish functions mediated by p38β from those regulated by p38α. Therefore, we used RNA interference to decipher the specific role of p38β in AML cells. Our immunoprecipitation and immunofluorescence analysis support the notion that p38β interacts with SET in AML cells, and knockdown of p38β, but not p38α, decreases SET protein levels and enhances PP2A activity. Moreover, we found that p38β binding stabilizes the SET protein in the cytoplasm, demonstrating a new role for p38β. We had previously demonstrated that SETBP1 binds to and stabilizes SET, facilitating PP2A inhibition25, and this result has been confirmed in other reports46,47. Here we further characterize this mechanism by showing that p38β co-localizes with SET, SETBP1, and PP2A, regulating PP2A activity in AML cells. Additional studies will be needed to elucidate how the interplay among these proteins regulates SET stability.
SET is mostly located in the nucleus where it regulates DNA replication, chromatin remodeling, gene transcription20, DNA repair21, migration22, and cell-cycle progression23. Here, we report a robust accumulation of SET into the cytoplasm of primary and patient-derived AML cells. Several studies in Alzheimer’s disease show that CK2 phosphorylates SET on Ser9, in the nuclear localization signal, which is key for SET cytoplasmic localization and inhibition of PP2A, leading to tau hyperphosphorylation30,31,48. Here we demonstrate in AML cells that p38β is involved in SET trafficking to the cytosol and PP2A inactivation through the activation of CK2, and that silencing of p38β but not p38α decreases CK2-dependent phosphorylation of SET. Moreover, overexpression of p38β decreased PP2A activity in a CK2-dependent manner. Consistent with these findings, pharmacological inhibition or silencing of CK2 increased the nuclear localization of SET, as well as PP2A activity, without altering total SET protein levels. Interestingly, in the absence of SET, inhibition of CK2 has no effect in PP2A activity, supporting the new mechanism described here. It should be noted that CK2 overexpression has been associated with poor prognosis in AML patients with normal karyotype32,49. Thus, our results support a model in which p38β overexpression activates CK2, which in turn phosphorylates SET, facilitating its trafficking to the cytoplasm where it inactivates PP2A. Therefore, our study identifies a novel p38β-CK2-SET signaling pathway in leukemogenesis that mediates PP2A inactivation. Interestingly, the same pathway would be probably activated in AML cases with CK2 overexpression, opening new insights into the role of CK2 in AML.
We validated the importance of this new pathway by showing that the combination of CX-4945, which inhibits CK2 allowing nuclear SET retention, and FTY720, which disrupts SET-PP2A binding, is more effective in decreasing viability and inducing apoptosis of AML cells from patients than either single treatment. In vivo studies using zebrafish xenografts as a preclinical model34 supported the importance of p38β in AML cells, and confirmed that the combination of CX-4945 and FTY720 is more effective than either treatment alone at reducing tumor growth, as well as impairing cell migration and invasion. Consistent with our results, SET phosphorylation would allow its interaction with the GTPase Rac1 and cytoplasm localization, where SET inactivates PP2A. In fact, it has been reported that Rac1 stimulated signaling required for efficient cell migration involves SET-mediated inhibition of PP2A, and that cytoplasmic targeting of SET inhibits Rac1-induced cell spreading and migration22. Taken together, our in vivo results confirm the value of targeting multiple components of the same pathway, and support the use of zebrafish xenografts to predict drug sensitivity for personalized treatments in AML cases.
In conclusion, we have identified a new role of p38β MAPK and CK2 in AML leukemogenesis. We show that p38β overexpression is a recurrent event in AML cases that contributes to PP2A inactivation by regulating the SET oncoprotein through two mechanisms: (i) p38β controls CK2-mediated phosphorylation of SET facilitating its cytoplasmic localization, and (ii) p38β binds to and stabilizes SET in the cytoplasm. Furthermore, we provide in vivo evidence of this mechanism by targeting the same pathway at different levels. We show that a combination therapy using the CK2 inhibitor CX-4945, which retains SET in the nucleus, and FTY720, which disrupts the SET-PP2A binding in the cytoplasm, re-activates PP2A, reducing the viability of AML cells. Our results therefore provide the rationale for using a combination of PP2A-activating drugs and CK2 inhibitors as a novel therapeutic option for treating a subgroup of 30% AML cases characterized by SET-dependent PP2A inactivation.
Supplementary information
Acknowledgements
This work was supported by grants from the ISCIII and Spanish Ministry of Economy and Competiveness (grants PI14/02073 and PI17/02272) (M.D.O.) and by CIBERONC (CB16/12/00489, CB16/12/00369, CB19/07/00031) (M.D.O., D.A., M.L.C.) (Co-financed with FEDER funds), Department of Health of the Government of Navarra (29/2015) (M.D.O.), Department of Industry of the Government of Navarra (0011-1365-2016-000294) (M.D.O.), Prostate Cancer Foundation (R.P.), Spanish Association Against Cancer (Fundación Científica AECC, INVES18061ODER) (C.V.), and Spanish Ministry of Economy and Competitiveness (SAF2016-81043-R) (A.R.N.).
Author contributions
E.A., C.V., R.P., E.M.B., N.M., I.P., and A.I. performed experiments, analyzed results, and made the figures. P.G.R., J.R., D.A., M.C.M., and M.L.C. analyzed results. E.A., R.P., M.L.C., A.R.N., and M.D.O. designed the research. E.A., R.P., A.R.N., and M.D.O. wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Elena Arriazu, Email: earriazu@unav.es.
María D. Odero, Email: modero@unav.es
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41408-019-0270-0).
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.Perl AE. The role of targeted therapy in the management of patients with AML. Hematol. Am. Soc. Hematol. Educ. Progr. 2017;2017:54–65. doi: 10.1182/asheducation-2017.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood. 2016;127:71–78. doi: 10.1182/blood-2015-07-604538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–495. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- 5.Pratz KW, et al. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115:1425–1432. doi: 10.1182/blood-2009-09-242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stone RM, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2:444–453. doi: 10.1182/bloodadvances.2017011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017;16:273–284. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 9.Chan SM, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei AH, Tiong IS. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood. 2017;130:2469–2474. doi: 10.1182/blood-2017-08-784066. [DOI] [PubMed] [Google Scholar]
- 11.Westermarck J. Targeted therapies don’t work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. FEBS J. 2018;285:4139–4145. doi: 10.1111/febs.14617. [DOI] [PubMed] [Google Scholar]
- 12.Bertolotti A. The split protein phosphatase system. Biochem. J. 2018;475:3707–3723. doi: 10.1042/BCJ20170726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arriazu E, Pippa R, Odero MD. Protein phosphatase 2A as a therapeutic target in acute myeloid leukemia. Front. Oncol. 2016;6:78. doi: 10.3389/fonc.2016.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cristóbal I, et al. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia. 2011;25:606–614. doi: 10.1038/leu.2010.294. [DOI] [PubMed] [Google Scholar]
- 15.Estella-Hermoso de Mendoza A, et al. Lipid nanosystems enhance the bioavailability and the therapeutic efficacy of FTY720 in acute myeloid leukemia. J. Biomed. Nanotechnol. 2015;11:691–701. doi: 10.1166/jbn.2015.1944. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal A, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014;20:2092–2103. doi: 10.1158/1078-0432.CCR-13-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith AM, et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget. 2016;7:47465–47478. doi: 10.18632/oncotarget.10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristóbal I, et al. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica. 2012;97:543–550. doi: 10.3324/haematol.2011.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pippa R, et al. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia. 2014;28:1915–1918. doi: 10.1038/leu.2014.141. [DOI] [PubMed] [Google Scholar]
- 20.Chambon J-P, et al. The PP2A inhibitor I2PP2A is essential for sister chromatid segregation in oocyte meiosis II. Curr. Biol. 2013;23:485–490. doi: 10.1016/j.cub.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Kalousi A, et al. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep. 2015;11:149–163. doi: 10.1016/j.celrep.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 22.ten Klooster JP, Leeuwen Iv, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 2007;26:336–345. doi: 10.1038/sj.emboj.7601518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chae Y-C, et al. Inhibition of FoxO1 acetylation by INHAT subunit SET/TAF-Iβ induces p21 transcription. FEBS Lett. 2014;588:2867–2873. doi: 10.1016/j.febslet.2014.06.053. [DOI] [PubMed] [Google Scholar]
- 24.Pippa R, et al. MYC-dependent recruitment of RUNX1 and GATA2 on the SET oncogene promoter enhances PP2A inactivation in acute myeloid leukemia. Oncotarget. 2017;8:53989–54003. doi: 10.18632/oncotarget.9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cristóbal I, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115:615–625. doi: 10.1182/blood-2009-06-227363. [DOI] [PubMed] [Google Scholar]
- 26.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuenda A, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-F. [DOI] [PubMed] [Google Scholar]
- 28.Xing L, et al. Structural bioinformatics-based prediction of exceptional selectivity of p38 MAP kinase inhibitor PH-797804. Biochemistry. 2009;48:6402–6411. doi: 10.1021/bi900655f. [DOI] [PubMed] [Google Scholar]
- 29.Qu D, et al. The nuclear localization of SET mediated by impalpha3/impbeta attenuates its cytosolic toxicity in neurons. J. Neurochem. 2007;103:408–422. doi: 10.1111/j.1471-4159.2007.04747.x. [DOI] [PubMed] [Google Scholar]
- 30.Yu G, et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol. Aging. 2013;34:1748–1758. doi: 10.1016/j.neurobiolaging.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q, et al. CK2 phosphorylating I2PP2A/SET mediates tau pathology and cognitive impairment. Front. Mol. Neurosci. 2018;11:146. doi: 10.3389/fnmol.2018.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buontempo F, et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia. 2018;32:1–10. doi: 10.1038/leu.2017.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sayed M, Kim SO, Salh BS, Issinger OG, Pelech SL. Stress-induced activation of protein kinase CK2 by direct interaction with p38 mitogen-activated protein kinase. J. Biol. Chem. 2000;275:16569–16573. doi: 10.1074/jbc.M000312200. [DOI] [PubMed] [Google Scholar]
- 34.Leslie M. Zebrafish larvae could help to personalize cancer treatments. Science. 2017;357:745. doi: 10.1126/science.357.6353.745. [DOI] [PubMed] [Google Scholar]
- 35.White R, Rose K, Zon L. Zebrafish cancer: the state of the art and the path forward. Nat. Rev. Cancer. 2013;13:624–636. doi: 10.1038/nrc3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Letrado P, de Miguel I, Lamberto I, Díez-Martínez R, Oyarzabal J. Zebrafish: speeding up the cancer drug discovery process. Cancer Res. 2018;78:6048–6058. doi: 10.1158/0008-5472.CAN-18-1029. [DOI] [PubMed] [Google Scholar]
- 37.Igea A, Nebreda AR. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015;75:3997–4002. doi: 10.1158/0008-5472.CAN-15-0173. [DOI] [PubMed] [Google Scholar]
- 38.Zhong W, et al. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy. 2014;10:1285–1300. doi: 10.4161/auto.28789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beenstock J, et al. The p38β mitogen-activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the α-G helix and MAPK insert. J. Biol. Chem. 2014;289:23546–23556. doi: 10.1074/jbc.M114.578237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.del Barco Barrantes I, Coya JM, Maina F, Arthur JSC, Nebreda AR. Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development. Proc. Natl Acad. Sci. USA. 2011;108:12764–12769. doi: 10.1073/pnas.1015013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J. Immunol. 1999;162:4246–4252. [PubMed] [Google Scholar]
- 42.Grethe S, Pörn-Ares MI. p38 MAPK regulates phosphorylation of Bad via PP2A-dependent suppression of the MEK1/2-ERK1/2 survival pathway in TNF-alpha induced endothelial apoptosis. Cell. Signal. 2006;18:531–540. doi: 10.1016/j.cellsig.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 43.Junttila MR, Li S-P, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 44.Lin S-P, et al. Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS ONE. 2012;7:e49605. doi: 10.1371/journal.pone.0049605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guillonneau M, et al. Oxidative stress disassembles the p38/NPM/PP2A complex, which leads to modulation of nucleophosmin-mediated signaling to DNA damage response. FASEB J. 2016;30:2899–2914. doi: 10.1096/fj.201500194R. [DOI] [PubMed] [Google Scholar]
- 46.Acuna-Hidalgo R, et al. Overlapping SETBP1 gain-of-function mutations in Schinzel-Giedion syndrome and hematologic malignancies. PLoS Genet. 2017;13:e1006683. doi: 10.1371/journal.pgen.1006683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piazza R, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013;45:18–24. doi: 10.1038/ng.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arnaud L, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett. 2011;585:2653–2659. doi: 10.1016/j.febslet.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JS, et al. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007;13:1019–1028. doi: 10.1158/1078-0432.CCR-06-1602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.