

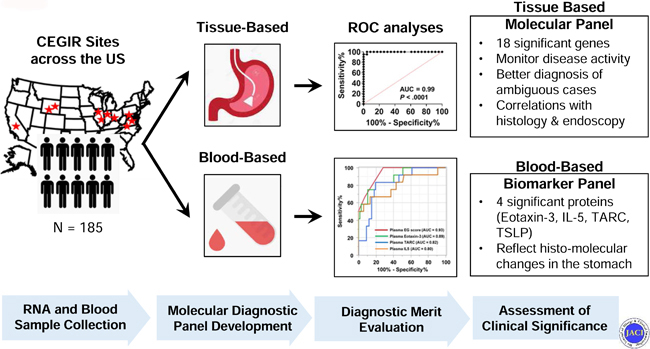
Abstract
Background:
Eosinophilic gastritis (EG) is a clinicopathologic disorder with marked gastric eosinophilia and clinical symptoms. There is an unmet need in EG for more precise diagnostic tools.
Objective:
We aimed to develop tissue- and blood-based diagnostic platforms for EG.
Methods:
Patients with EG and non-EG controls were enrolled across 9 Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)-associated sites. An EG Diagnostic Panel (EGDP; gastric transcript subset) and EG blood biomarker panel (protein multiplex array) were analyzed. The EGDP18 scores were derived from the expression of 18 highly dysregulated genes and Blood EG scores from dysregulated cytokines/chemokine levels.
Results:
Gastric biopsies and blood samples from 185 subjects (EG 74, non-EG 111) were analyzed. The EGDP a) identified patients with active EG (P < .0001, AUC ≥0.95); b) effectively monitored disease activity in longitudinal samples (P = .0078); c) highly correlated in same-patient samples (antrum vs. body, r = 0.85, P < .0001); and d) inversely correlated with gastric peak eosinophil levels (r = −0.83, P < .0001), periglandular circumferential collars (r = − 0.73, P < .0001), and endoscopic nodularity (r = −0.45, P < .0001). For blood-based platforms, eotaxin-3, TARC, IL-5, and TSLP levels were significantly increased. Blood EG scores a) distinguished patients with EG from non-EG controls (P < .0001, AUC ≥0.91); b) correlated with gastric eosinophil levels (plasma r = 0.72, P = .0002; serum r = 0.54, P = .0015); and c) inversely correlated with EGDP18 scores (plasma r = −0.64, P = .0015; serum r = −0.46, P = .0084). Plasma eotaxin-3 strongly associated with gastric CCL26 expression (r = 0.81, P < .0001).
Conclusion:
We developed tissue- and blood-based platforms for assessment of EG and uncovered robust associations between specific gastric molecular profiles and histologic and endoscopic features, providing insight and tools for this emerging rare disease.
Keywords: biomarker, diagnostic panel, eosinophil, eosinophilic gastritis, transcriptome
Graphical Abstract
Capsule summary:
We have developed molecular diagnostic platforms for EG, validated their utility for disease diagnosis and monitoring, and assessed their clinical significance by concurrent analysis of histological and endoscopic findings, providing insight into disease pathogenesis.
INTRODUCTION
Eosinophilic gastritis (EG) is one of the eosinophilic gastrointestinal disorders (EGIDs) characterized by marked eosinophil accumulation into the gastrointestinal tract1, 2 with an estimated prevalence of about 6.3 patients per 100,000 individuals, which is likely rising.3, 4 Although studies of eosinophilic esophagitis (EoE) have elucidated specific molecular, cellular, and immune mechanisms involved in its pathogenesis,5 EG is more rare than EoE and thus less well understood, with few publications even addressing diagnostic criteria, genetics, biomarkers, or disease pathogenesis. Unlike in the esophagus, eosinophils normally reside in gastric mucosa during homeostasis,6, 7 adding complexity to disease diagnosis and monitoring.
Substantial progress has been made using whole-genome transcript expression profiling (transcriptome) of tissue biopsies from patients with EGIDs, particularly EoE.8–15 We previously developed a molecular EoE diagnostic panel (EDP), a set of 96 informative transcripts that can distinguish, monitor, and endotype EoE;16, 17 however, such molecular diagnostic profiles are lacking for the other EGIDs. To date, EG studies have identified a prominent and conserved gastric transcriptome that is largely distinct from the EoE transcriptome.10, 13 In this context, we hypothesized that a tissue-based diagnostic platform based on the EG transcriptome would provide more clarity to EG diagnosis than would isolated eosinophil counts, would correlate with gastric endoscopic and pathologic parameters, and would align with specific clinical findings.
EGID diagnosis and management requires procuring tissue biopsies during endoscopy, an invasive procedure that adds costs and risk. Two unmet needs are to more precisely and objectively diagnose EG and to develop monitoring tools for disease management.18, 19 Circulating biomarkers have the potential to facilitate non-invasive disease diagnosis and monitoring and represent a pressing need in the field. Previous studies suggest that EG has potential systemic markers, for example concurrent peripheral blood eosinophilia.10, 20, 21 However, the peripheral expression of EG-related inflammatory markers and their potential to function as surrogate disease markers in EG has not been examined.
Herein, we aimed to develop tissue and blood-based diagnostic platforms for EG; validate their utility for diagnosis, monitoring, and management; assess their clinical significance by concurrent analysis of histologic and endoscopic findings; and provide insight into disease pathogenesis. To approach this aim, we examined patients with EG across multiple sites associated with the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)22 and an independent replication cohort from a single center.
METHODS
Study design and participants
This study was conducted within the context of CEGIR,22 a national collaborative network of 16 academic centers caring for adults and children with EGIDs. The CEGIR clinical study, Outcomes Measures in Eosinophilic Gastrointestinal disorders Across the ages (OMEGA), is a longitudinal cohort study aimed at understanding the natural history of EoE, EG, and eosinophilic colitis during routine clinical care. Demographic, clinical, endoscopic, and histologic data, as well as gastrointestinal and blood samples, were prospectively collected starting from 2015; all samples from any CEGIR site that contributed patients with EG were used (n = 9 sample-providing institutions). The clinical features of subjects were determined during a standard-of-care evaluation using standardized intake forms (see Supplementary Materials for details). All subjects’ clinical data were stored at the Data Management and Coordinating Center (DMCC) at the University of South Florida in Tampa, FL. Data were systematically extracted from the databases. Pediatric subjects were defined as having an age of less than 18 years. Atopy was defined on the basis of self-report of allergic rhinitis, dermatitis, asthma, or food allergy. For the validation cohort of the tissue-based platform and the plasma cohort of the blood-based platform, children and adults with EG presenting for standard care between 2007–2016 were enrolled in an independent cohort at Cincinnati Children’s Hospital Medical Center (CCHMC) using the same disease definitions. This study was approved by the institutional review boards of the participating institutions via a central institutional review board at CCHMC.
Patients were defined as having EG if they had ≥30 eosinophils/high-power field (HPF) in ≥5 HPFs.23 There were no other known causes of gastric mucosal eosinophilia as defined by standard of care, such as negative testing for other potential causes including stool culture for pathogenic bacteria or parasites, viral antibody titers and/or PCR, celiac and inflammatory bowel disease serology, and staining for H. pylori infection. Active EG was defined as gastric biopsies that showed ≥30 eosinophils/HPF in ≥5 HPFs, intermediate EG was defined as gastric biopsies that showed ≥30 eosinophils in 1–4 of 5 HPFs, and inactive EG was defined as <30 eosinophils/HPF in all HPFs in patients with a previous history of EG. Patients with EG were allowed to be included if they had gastrointestinal eosinophilia outside of the stomach.
Non-EG control subjects, from the Cincinnati Center for Eosinophilic Disorders (CCED) EGID database between 2015–2018, included children and adults who had undergone endoscopy, had no history of EG, had no pathologic evidence of EG surveyed during the index endoscopy, were not taking systemic glucocorticoid treatments in the period immediately prior to the index endoscopy, and had gastric biopsies and/or blood samples collected for research purposes during the index endoscopy. For the tissue-based platform and blood-based platform, control subjects included those with atopic comorbidities, chronic gastritis, and concurrent active EoE since these controls, with different TH2 baseline levels, would aid in identifying transcriptional changes that are specific to EG. Controls were selected with an effort of closely matching the gender and age. Gastric biopsies in the discovery and validation cohorts did not overlap.
RNA sequencing analysis
Fresh biopsy specimens collected from patients with EG and non-EG controls were stored in RNAlater until they were subjected to RNA isolation using the miRNeasy kit (Qiagen, Valencia, Calif) per the manufacturer’s instructions. Samples for RNA sequencing were selected from the total cohort on the basis of RNA quality and quantity. RNA sequencing acquired 20 million mappable, 75–base-pair reads from paired-end libraries and was performed at the DNA Sequencing and Genotyping Core Facility at CCHMC. Data were aligned to the GRCh37 build of the human genome using the Ensembl annotations as a guide for TopHat. Expression analysis was performed using DESeq2 in CLC Genomics Workbench software (CLC bio, Waltham, MA, USA). Reads per kilobase of exon per million reads mapped (RPKM) were assessed for statistical significance using a Welch t test with Benjamini–Hochberg false-discovery rate (FDR) and a threshold of P < .05 and a 2-fold–change cut-off filter. Gene ontology enrichment analysis was performed with the ToppGene suite (https://toppgene.cchmc.org/).
Tissue-based diagnostic platform
RNA from fresh gastric biopsies (from antrum and/or body) was isolated from patients with EG and non-EG controls as described above. RNA was reverse transcribed using the iScript cDNA Synthesis Kit (170–8891; Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s protocol. The transcriptomic signature of gastric biopsy samples was obtained using an EG diagnostic panel (EGDP) comprising a set of 48 gastric transcripts (including 2 housekeeping transcripts) (Table E1 in this article’s Online Repository at www.jacionline.org). In addition to the significantly dysregulated gene transcripts highly reproducible by microarrays and RNA sequencing, we considered the magnitude of dysregulation, the direction of dysregulation and cell type tissue origins to optimize the diagnostic algorithm. Moreover, we aimed to reveal the presence and function of different biological processes and cell types that are known to be involved in GI TH2 allergic disorders, so that we would be able to diagnose EG using a personalized medicine approach. TaqMan reagents for amplification of major EG signature genes were obtained from Applied Biosystems (Foster City, CA), and TaqMan real-time PCR amplification was performed on the Quant Studio 7 (Life Technologies). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an expression control for all analyzed genes. Samples with a GAPDH value of <30 CT value were considered acceptable for analysis. The expression CT value of the housekeeping gene GAPDH was subtracted from each EG gene of interest (GOI) CT value to acquire the CT. The EGDP score was calculated by summing CT values of the most highly dysregulated genes (ΣΔCT), as described previously16 and expanded upon later.
Blood-based diagnostic platform
Peripheral blood samples were collected prior to the endoscopy, separated into serum and/or plasma, and aliquots were frozen and stored at −80°C. The major difference between plasma and serum is the depletion of the coagulation components present in the blood, a process that might alter the detection of cytokine profiles in the blood. The levels of blood cytokines/chemokines — eotaxin-1 (CCL11), eotaxin-2 (CCL24), eotaxin-3 (CCL26), IL-1α, IL-4, IL-5, IL-13, IL-33, thymus- and activation-regulated chemokine/chemokine ligand 17 (TARC/CCL17), and thymic stromal lymphopoietin (TSLP) were assayed by customized immunoassays, quantified on a Sector Imager 6000 (Meso Scale Discovery, Gaithersburg, MD, USA), according to the manufacturer’s instructions. Scoring systems for plasma and serum were established separately by their respective dysregulated biomarker levels. One point was added to a score for upregulation of specific cytokines (upregulation, having a value higher than the cut-off value [pg/mL] based on the comparison between active EG and non-EG [cut-off values (plasma: eotaxin-3 > 168, IL-5 > 1.4, and TARC > 87; serum: eotaxin-3 > 32, IL-5 > 1.5, and TSLP > 6.7)]) (see Fig E8 in this article’s Online Repository at www.jacionline.org). A plasma or serum EG score is the sum of the assigned scores for each biomarker assessed, ranging from 0 to 3.
Endoscopic and histologic data
Endoscopic features were prospectively recorded in real-time using a classification and grading system specifically developed for EG by CEGIR, including erosion/ulceration, granularity, raised lesions nodularity, erythema, friability/bleeding, and fold thickness. These findings were scored (erosion/ulceration: 0–6; granularity: 0–2; raised lesions nodularity: 0–2; erythema: 0–2; friability/bleeding: 0–2; fold thickness: 0–1) for the fundus, body, and antrum. The total score for each feature was calculated as the sum of the scores for the three anatomical sites. The overall global assessment of endoscopic severity for the stomach, ranging from 0 to 10, was evaluated for each patient.
Gastric biopsies were assessed by the peak eosinophil counts and the histologic features of EG. These features included lamina propria eosinophil sheets (LPES), periglandular circumferential collars (PCC), eosinophils in surface epithelium (EoSE), eosinophil glandulitis (EoG), eosinophil gland abscess (EoGA), eosinophils in muscularis mucosa (EoM), lamina propria fibroplasia (LPF), lamina propria smooth muscle hyperplasia (LPSMH), reactive epithelial changes (REC), acute inflammatory cells (AIC), and surface erosion/ulceration (SEU) (Table E2 in this article’s Online Repository at www.jacionline.org). Each feature was scored using a 3-point scale (0 = absent, 1 = mild/moderate, 2 = marked). The maximum score for each biopsy is 22, as it is the summation of the 11 feature scores if each feature were to be scored at 2. The final score of the histologic severity, which ranges from 0–1, is the ratio of the sum of the assigned scores for each evaluated feature (0–22) divided by the maximum possible score for that biopsy (22). For example, if all 11 features have maximum scores of 2, the final score is 22/22 = 1. If a feature were not evaluated, the maximum possible score was reduced by 2 per feature not evaluated; most score reductions occurred because gastric muscularis mucosa was not present in the biopsy.
Statistical analysis
Statistical analyses were performed using the JMP v13.1 (SAS Institute, Cary, NC), CLC Genomics Workbench software (CLC bio, Waltham, MA, USA), GeneSpring GX 12.6 (Agilent Technologies, Santa Clara, CA), and GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA). Data are n (%) or median (interquartile range [IQR]) unless otherwise stated. Missing data were excluded from all formal statistical analyses. Sample size was estimated to provide 90% power to conclude that 0.8 of the area under the curve (AUC) for an individual marker was significantly greater than 0.5 while controlling the type I error rate at 1% for multiplicity. Statistical significance comparing 2 different groups was determined by the Mann-Whitney U test (nonparametric test, 2 groups), the Kruskal-Wallis test followed by a Dunn multiple-comparison test (nonparametric test, 3 groups or more), or a paired t test (for quantification of longitudinal data and different site of biopsies in the same patients). Nonparametric correlations were calculated using Spearman correlations. Receiver operating characteristic (ROC) curves were constructed, and AUC were calculated to determine the utility of the developed platforms for distinguishing patients with EG from non-EG controls. The optimal cut-off points were determined by Youden’s index. We measured sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) to assess the diagnostic accuracy of the scoring systems. A significant P value was defined as less than 0.05.
RESULTS
Patient characteristics
A total of 185 subjects (patients with EG n = 74; non-EG controls n = 111) provided 201 gastric biopsies (RNA sequencing n = 21; EGDP discovery n = 104; EGDP validation n = 76) and 155 blood samples (plasma n = 81; serum n = 74) for analyses. A flow chart of the analysis is shown in Fig 1, A. Demographic and clinical characteristics of the study cohort and subsets, including patients with EG and clinically relevant non-EG controls, are detailed in Table I.
FIG 1. Schematic illustration of the study and genome-wide screening of gastric tissue for identifying representative biomarkers.

A, Flow chart of the study design and strategy. B, The gastric transcriptome data on non-EG controls (blue) and patients with EG (red) were reduced to 3-dimensional presentation by multidimensional scaling analysis at the whole-genome level for visual presentation of the expression distance between samples. C, Heat map (red, upregulated; blue, downregulated) of 1,226 differentially dysregulated genes’ expression profiles (FDR P < .05, ≥2-fold change). Clustering analysis was performed; each column represents an individual patient or control. D, Venn diagram comparing the number of genes identified as dysregulated in EG across different platforms. E, Functional enrichment gene ontology analysis of the strongly upregulated genes (FDR P < .01, ≥10-fold change). The x-axes represent the negative log10 FDR P value. Red bars indicate cytokine/chemokine-associated terms and pathways. EG, eosinophilic gastritis; EGDP, eosinophilic gastritis diagnostic panel; FDR, false-discovery rate; GO, gene ontology.
TABLE I.
Demographic and clinical characteristics of study subjects*
| Tissue-based platform |
Blood-based platform |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All study subjects (n = 185) | All study subjects in tissue-based platform (n = 124) | Non-EG (n = 55) | Active EG (n =39) | P value | All study subjects in blood-based platform (n = 108) | Non-EG (n = 67) | Active EG (n =27) | P value | ||
| Demographics | ||||||||||
| Age at biopsy (years) | 15.2 (11.0 − 20.0) | 15.5 (11.2 − 20.5) | 14.2 (8.4 − 18.0) | 16.5 (12.3 − 21.2) | 0.06 | 15.4 (11.2 − 19.5) | 14.6 (10.5 − 19.1) | 17.7 (12.5 − 20.6) | 0.11 | |
| Pediatric subjects | 124 (67.0%) | 81 (65.3%) | 40 (72.7%) | 23 (59.0%) | 0.19 | 74 (68.5%) | 48 (71.6%) | 15 (55.6%) | 0.15 | |
| Gender | Male | 90 (48.6%) | 65 (52.4%) | 24 (43.6%) | 21 (53.8%) | 0.40 | 51 (47.2%) | 28 (41.8%) | 14 (51.9%) | 0.49 |
| Race | White | 169 (91.4%) | 111 (89.5%) | 52 (94.5%) | 34 (87.2%) | 0.27 | 100 (92.6%) | 65 (97.0%) | 24 (88.9%) | 0.14 |
| History of EGID | ||||||||||
| EG only | 22 (11.9%) | 22 (17.7%) | - | 12 (30.8%) | - | 14 (13.0%) | - | 8 (29.6%) | - | |
| EG and EoE | 47 (25.4%) | 46 (37.1%) | - | 27 (69.2%) | - | 20 (18.5%) | - | 16 (59.3%) | - | |
| EG and EC | 2 (1.1%) | 1 (0.8%) | - | - | - | 2 (1.9%) | - | 2 (7.4%) | - | |
| EG, EoE, and EC | 3 (1.6%) | 3 (2.4%) | - | - | - | 3 (2.8%) | - | 1 (3.7%) | - | |
| EoE only | 20 (10.8%) | 10 (8.1%) | 10 (18.2%) | - | 10 (9.3%) | 10 (14.9%) | - | - | ||
| Clinical symptoms at biopsy | ||||||||||
| Symptomatic (any) | 130 (70.3%) | 85 (68.6%) | 30 (54.6%) | 39 (100%) | < 0.01 | 90 (83.3%) | 53 (79.1%) | 27 (100%) | < 0.01 | |
| Abdominal pain | 91 (49.2%) | 52 (41.9%) | 19 (34.6%) | 22 (56.4%) | 0.04 | 66 (61.1%) | 41 (61.2%) | 18 (66.6%) | 0.62 | |
| Heartburn | 66 (35.7%) | 37 (29.8%) | 13 (23.6%) | 12 (30.8%) | 0.44 | 45 (44.4%) | 30 (44.8%) | 9 (33.3%) | 0.31 | |
| Nausea/vomit | 88 (47.6%) | 54 (43.6%) | 23 (41.8%) | 19 (48.7%) | 0.51 | 60 (55.6%) | 40 (59.7%) | 14 (51.9%) | 0.49 | |
| Other | 33 (17.8%) | 33 (26.6%) | 20 (36.4%) | 16 (41.0%) | 0.65 | 14 (13.0%) | 19 (28.4%) | 11 (40.7%) | 0.24 | |
| Atopic status | ||||||||||
| Atopy (any) | 129 (69.7%) | 84 (67.7%) | 36 (65.5%) | 31 (79.5%) | 0.17 | 76 (70.4%) | 51 (76.1%) | 19 (70.4%) | 0.61 | |
| Asthma | 74 (40.0%) | 49 (39.5%) | 14 (25.5%) | 19 (48.7%) | 0.03 | 40 (37.0%) | 25 (37.3%) | 11 (40.7%) | 0.82 | |
| Allergic rhinitis | 80 (43.2%) | 55 (44.4%) | 25 (45.5%) | 18 (46.2%) | 1.00 | 48 (44.4%) | 30 (44.8%) | 14 (51.9%) | 0.65 | |
| Eczema | 52 (28.1%) | 33 (26.6%) | 11 (20.0%) | 13 (33.3%) | 0.16 | 30 (27.8%) | 20 (29.9%) | 9 (33.3%) | 0.81 | |
| Food allergy | 60 (32.4%) | 58 (46.8%) | 12 (21.8%) | 28 (71.8%) | < 0.01 | 18 (16.7%) | 1 (1.5%) | 11 (40.7%) | < 0.01 | |
| Treatment at biopsy | ||||||||||
| Ongoing diet | 54 (29.2%) | 44 (35.5%) | 13 (23.6%) | 22 (56.4%) | < 0.01 | 29 (26.9%) | 10 (14.9%) | 13 (48.1%) | < 0.01 | |
| therapy | ||||||||||
| Proton pump | 93 (50.3%) | 48 (38.7%) | 19 (34.5%) | 19 (48.7%) | 0.29 | 67 (62.0%) | 48 (71.6%) | 15 (55.6%) | 0.15 | |
| inhibitor | ||||||||||
| Topical steroids | 50 (27.0%) | 49 (39.5%) | 13 (23.6%) | 19 (48.7%) | 0.03 | 19 (17.6%) | 0 (0%) | 12 (44.4%) | < 0.01 | |
| Systemic steroids | 10 (5.4%) | 8 (6.5%) | 0 (0%) | 5 (12.8%) | 0.01 | 6 (5.6%) | 0 (0%) | 2 (7.4%) | 0.08 | |
| Disease parameters in stomach | ||||||||||
| Peak eosinophil | 16 (10 − 65) | 16 (10 − 63) | 11 (6 − 15) | 124 (68 − 192) | < 0.01 | 56 (10 − 127) | 7 (5 − 9) | 123 (77 − 183) | < 0.01 | |
| counts | ||||||||||
| Average eosinophil | 11.8 (6.0 − 53.7) | 11.8 (5.9 − 51.1) | 6.6 (3.8 − 10.0) | 88.2 (61.2 − 142.6) | < 0.01 | 33.8 (5 − 77.2) | 4.3 (1.9 − 5.0) | 72.6 (62.0 − 128.6) | < 0.01 | |
| counts | ||||||||||
| Endoscopic | 0(0−3) | 0(0−3) | 0(0−0) | 4(0−7) | < 0.01 | 2(0−5) | 0(0−0) | 6(3−7) | < 0.01 | |
| severity | ||||||||||
| Histologic severity | 0.14 (0.09 − 0.35) | 0.14 (0.09 − 0.35) | 0.09 (0.05 − 0.14) | 0.41 (0.35 − 0.55) | < 0.01 | 0.35 (0.10 − 0.46) | 0.10 (0.06 − 0.10) | 0.35 (0.29 − 0.50) | < 0.01 | |
Data pertains to the time at pertains to the time at biopsy/blood procurement. Data are n (%) or median (IQR). EGID, eosinophilic gastrointestinal disorders; EG, eosinophilic gastritis; EoE, eosinophilic esophagitis; EC, eosinophilic colitis; EGDP, ophilic gastritis diagnostic panel.
Among all of the study subjects, the age ranged from 1 year to 67 years, with 124 pediatric (67%) and 61 adult (33%) subjects. There was a similar proportion of both genders, with 90 male (48.6%) and 95 female (51.4%) subjects; the majority of subjects were white (91.4%). Peak gastric eosinophil counts ranged from 0 to 352 eosinophils/HPF (active EG: 36–352; inactive EG: 2–29; non-EG controls: 0–28 eosinophils/HPF). Non-EG controls (n = 111) included subjects with atopic comorbidities (n = 47, 42.3%), chronic gastritis (n = 44, 39.6%), and active EoE without EG (n = 20, 18.0%).
There were no significantly different baseline demographic features among the cohorts for the tissue (n = 124) and blood-based (n = 108) platforms. Focusing on patients with EG (n = 74), 46 (62%) had concurrent eosinophilia in the esophagus, 2 (3%) had concurrent eosinophilia in the colon, and 3 (4%) had concurrent eosinophilia in both the esophagus and colon. In tissue- and blood-based platforms, active EG did not reveal any significant differences from non-EG controls in age, gender, race, atopic status, or proton pump inhibitor (PPI) therapy at time of biopsy, whereas active EG showed significantly higher levels in the disease parameters (peak/average gastric eosinophil counts, endoscopic severity, and histologic severity [P < .01, respectively]) and a higher rate of treatment (ongoing diet therapy, topical steroid therapy, and systemic steroid therapy).
RNA sequencing of gastric tissue for identifying representative biomarkers
In order to obtain the molecular foundation for developing the EGDP, we aimed to identify a gene set that was conserved across multiple experimental platforms. Accordingly, we generated an RNA sequencing data set of gastric tissue from active EG (n = 9) and non-EG control (n = 12) individuals. Unsupervised principal component analysis (PCA) demonstrated robust separation of the two groups (Fig 1, B) defined by 1,226 differentially expressed genes (DEG) (≥2-fold change, FDR P < .05) (Fig 1, C and see Fig E1, in this article’s Online Repository at www.jacionline.org). We then overlapped this gene signature with two recently published, microarray-based expression profiles (Fig 1, D and Supplementary Fig E1 in this article’s Online Repository at www.jacionline.org). Upregulated genes (≥10-fold change, FDR P < .05) were markedly enriched in cytokine/chemokine-associated pathways (Fig 1, E), notably including the IL-13 pathway.
Development of tissue-based platform (EGDP) and EGDP18 score based on differentially expressed genes
We manually curated the EG transcriptome with the aim of selecting 48 informative genes that could be embedded into a multiplex PCR-based panel for serially diagnosing and probing clinical samples. Accordingly, an EG Diagnostic Panel (EGDP) was generated based on the following considerations: dysregulation between EG and non-EG defined by P value and fold change, bi-directional changes of gene expression, and inclusion of genes in pathways that were likely to be involved, such as type 2 immunity. The major functional categories represented included those associated with antimicrobial defense, cell adhesion, cytokines and chemokines, epithelial-to-mesenchymal transition, hypoxia, eosinophils, epithelium, fibrosis, inflammatory response, ion transportation, mast cells, neurosensory, neutrophils, and stomach-related processes (Table E1 in this article’s Online Repository at www.jacionline.org).
Using this set of 48 informative genes, we aimed to determine the minimal number that would successfully distinguish patients with active EG (n = 21) from control individuals (n = 23) in a discovery cohort (Table E3 in this article’s Online Repository at www.jacionline.org). Using relatively stringent criteria (≥10-fold change, FDR P < .01), 18 differentially expressed genes completely separated the two groups (Fig 2, A and B). Among the 18 genes, 8 were upregulated genes related to cytokine/chemokines (CCL26, CCL18, IL13RA2, and IL5), eosinophilia (CLC), cell adhesion (CDH26), antimicrobial defense (KLK7), and epithelial-related (MUC4), and 10 were downregulated genes related to antimicrobial defense (DEFB1), fibrosis (BMP3 and COL2A1), ion transportation (SLC26A7), neurosensory activity (GABRA1, GLDN, NPY, and TAC1), and stomach-related processes (ATP4A and SST).
FIG 2. Development of tissue-based platform (EGDP) and EGDP18 score on the basis of differentially expressed genes.

A, Heat map (yellow, upregulated; blue, downregulated) based on the 18 core genes (FDR P < .01 and fold change ≥10-fold change) in the discovery cohort. B, Three-dimensional presentation by PCA between samples on the basis of the 18 core genes (blue, non-EG controls; red, patients with EG). C, Comparison of the EGDP18 score between EG and non-EG in discovery and validation cohort. D, ROC curve analysis showing the utility of the EGDP18 score for the diagnosis of EG. E, Correlation between peak gastric eosinophil counts and EGDP18 score. F, Longitudinal changes of peak gastric eosinophil counts and EGDP18 score in patients with EG at active and inactive state. G, Correlation of EGDP18 score between the gastric antrum and body mucosa from the same subjects. H, EGDP18 score as a function of different patient groups, including patients with EG with involvement of 1–5 HPFs. AUC; area under the curve; EG, eosinophilic gastritis; EGDP, eosinophilic gastritis diagnostic panel; FDR, false-discovery rate; HPF, high-power field; NPV, negative predictive value; PCA, principal component analysis; PPV, positive predictive value; ROC, receiver operating characteristic.
With the goal of developing a quantitative diagnostic cut-off, the EGDP18 score was developed to distinguish EG versus non-EG and to quantify the severity of EG. On the basis of the 18 significant and reproducible differential genes, we made CT sums of the upregulated genes and downregulated genes separately, and then combined the two sums considering their different direction of dysregulation. The EGDP18 score was significantly decreased in patients with active EG compared to non-EG patients in the discovery cohort (P < .0001) and similarly decreased in the validation cohort (P < .0001) (Fig 2, C). ROC analysis demonstrated an excellent diagnostic merit (P < .0001, AUC ≥0.95) in both cohorts (Fig 2, D). After investigation by setting the optimal cut-off points, a score of less than 0 resulted in PPV of 100% and NPV of >94% (Fig 2, D). Of note, the EGDP18 score is inversely correlated with disease severity as defined by eosinophil counts when analyzed cross sectionally (r = −0.83, P < .0001) (Fig 2, E) and longitudinally (P = .0078) (Fig 2, F). The EGDP18 score showed comparable levels and high correlation between the gastric antrum and body (n = 8, r = 0.85, P < .0001) (Fig 2, G). Among patients with active EG, the EGDP18 score showed consistency across geographically diverse sites (see Fig E2 in this article’s Online Repository at www.jacionline.org) and comparable levels across ages (pediatric vs. adult patients), atopic status (atopy vs. no atopy), co-existence with EoE (EG only vs. EG with EoE), and treatment status at biopsy (ongoing therapy including diet and steroids vs. no therapy) (see Fig E3 in this article’s Online Repository at www.jacionline.org).
Interestingly, the EGDP18 score was able to classify patients with intermediate tissue eosinophil levels (i.e., the number of HPFs with ≥30 eosinophils, n = 1–4 HPFs). When these patients were analyzed by the EGDP18 score (n = 8, all of them were clinically symptomatic), 5 patients (63%) were molecularly equivalent to active EG (Fig 2, H and see Fig E4 in this article’s Online Repository at www.jacionline.org).
Gastric transcript associations with histologic and endoscopic features
Significant correlations were noted between specific genes within the EGDP and the peak gastric eosinophil level, histologic severity, and overall global assessment of endoscopic severity (Fig 3, A and Table E4 in this article’s Online Repository at www.jacionline.org). The top 10 genes that tracked with tissue eosinophilia were CCL26, CLC, IL13RA2, BMP3, IL5, CDH26, CCL18, NPY, HPGDS, and SST; with histologic severity were CCL26, IL13RA2, CLC, SST, BMP3, IL5, CDH26, GLDN, ANXA1, and DEFB1; and with endoscopic severity were CCL26, GLDN, IL13RA2, SST, DEFB1, GABRA1, IL5, TAC1, CLC, and IL13. Notably, these gene groups included genes that overlapped between tissue eosinophilia, histologic severity, and endoscopic severity (i.e., CCL26, CLC, IL13RA2, IL5, and SST).
FIG 3. Gastric transcript associations with histologic and endoscopic features.

A, Associations between the individual genes of EGDP and diagnostic parameters. Negative log10 FDR P value of the Spearman correlation between the individual genes of EGDP and peak gastric eosinophil counts (left), histologic severity (middle), and the overall assessment of endoscopic severity (right). Red indicates a positive correlation and blue indicates a negative correlation. The dashed line indicates an FDR P value of .05. The top genes and features are labeled. B, Associations between the EGDP18 score and the individual components of histology (left) and endoscopy (right). C, Associations between the EGDP and the histologic (left) and endoscopic (right) features. Clustering tree with Spearman r–based heat diagram for the correlation at the gene level were generated. Darker red shades indicate stronger positive correlations, whereas darker blue shades indicate stronger negative correlations. The shorter the distance (tree-branch length) the more similar the expression correlation is for each feature. EG, eosinophilic gastritis; FDR, false-discovery rate; AIC, acute inflammatory cells; EoG, eosinophil glandulitis; EoGA, eosinophil gland abscess; EoM, eosinophils in muscularis mucosa; EoSE, eosinophils in surface epithelium; LPES, lamina propria eosinophil sheets; LPF, lamina propria fibroplasia; LPSMH, lamina propria smooth muscle hyperplasia; PCC, periglandular circumferential collars; REC, reactive epithelial changes; SEU, surface erosion/ulceration.
Individual components of the histologic and endoscopic features associated with the EGDP18 score. Associations were noted between the EGDP18 score and several histologic features (Spearman r range: −0.05 – −0.73; Fig 3, B left panel) with periglandular circumferential collars showing the highest magnitude of correlation with the EGDP18 score (r = −0.73, FDR P < .0001). Associations were also observed between the EGDP18 score and several endoscopic features (Spearman r range: −0.22 – −0.45; Fig 3, B right panel). The EGDP18 score inversely correlated the most with granularity (r = −0.45, FDR P < .0001) and nodularity (r = −0.45, FDR P < .0001).
Three histologic features (lamina propria eosinophil sheets, periglandular circumferential collars, and eosinophil glandulitis) showed higher correlations, based on hierarchical clustering of Spearman correlations (Fig 3, C left panel), suggesting that they might be more effective than other features at capturing biological processes underlying the EGDP. At the gene level, CCL26 showed the strongest correlation with the histologic features, most notably periglandular circumferential collars (r = 0.74, P = 7.0E-21) and eosinophil glandulitis (r = 0.68, P = 2.0E-16); in correlation strength, CCL26 was followed by IL13RA2, which correlated most notably with periglandular circumferential collars (r = 0.67, P = 7.0E-16) and lamina propria eosinophil sheets (r = 0.67, P = 5.0E-16). Interestingly, though muscularis mucosa eosinophilia showed relatively weak associations compared with epithelial and lamina propria changes, DUOX2 and DUOXA2 showed unique association with muscularis mucosa eosinophilia (r = 0.32, P = 3.8E-3) (see Fig E5 and Table E5 in this article’s Online Repository at www.jacionline.org). Some features, such as acute inflammation and erosion/ulcer, were uncommon and, possibly for that reason, did not show gene correlations.
In contrast to histology, for which gene transcripts showed association with only a limited set of histologic features, all recorded endoscopic features correlated with specific gastric transcripts (Fig 3, C right panel). CCL26 showed the strongest correlation with any endoscopic features, most notably nodularity (r = 0.55, P = 2.8E-5) and granularity (r = 0.53, P = 4.9E-5), followed by IL33, which inversely correlated most notably with granularity (r = −0.46, P = 6.1E-4) and friability and bleeding (r = −0.39, P = 3.9E-3). Interestingly, clustering separated endoscopic features into 2 general groups, one was associated with endoscopic changes, including friability/bleeding and erythema, and the other was associated with endoscopic changes, including nodularity and granularity. Endoscopic changes, including friability/bleeding and erythema, were associated with downregulation of molecular signatures (ATP4A, IL33, and SLC26A7), whereas endoscopic changes, including nodularity and granularity, were associated with upregulation of type 2 immunity and eosinophil-associated pathways (CCL26, IL13RA2, and IL5) (see Fig E6 and Table E6 in this article’s Online Repository at www.jacionline.org). A heat map showing −log10 FDR P value determined by differential expression between patients with EG with or without a specific endoscopic feature by the Mann-Whitney U test also supported this finding (see Fig E7 in this article’s Online Repository at www.jacionline.org).
Development of blood-based platforms and blood EG score based on significantly increased biomarkers
We explored the possibility that systemic levels of cytokines/chemokines might be elevated in EG. Focusing on plasma and serum samples from patient cohorts with and without EG, we designed a multiplex immunoassay containing 10 EG-relevant cytokines/chemokines, particularly those based on type 2 immunity as reflected in the functional predictions found in the EG transcripts (Fig 1, E). Notably, patients with active EG showed significantly higher levels of 3 cytokines in the plasma and 3 cytokines in the serum (plasma eotaxin-3/CCL26, IL-5, and TARC/CCL17; serum TSLP, eotaxin-3/CCL26, and IL-5) (Fig 4, A and B, and Table E7 in this article’s Online Repository at www.jacionline.org), suggesting that the activity of the disease consistently affects these cytokines systemically.
FIG 4. Development of blood-based platforms via a multiplex protein array.

A and B, Among the 10 biomarkers embedded in the platform, a statistical screening was performed between the non-EG subjects and patients with EG in the (A) plasma and (B) serum cohort, separately, resulting in 3 biomarkers (red) with adjusted P < .05 (Bonferroni correction). C and D, Levels of blood EG scores in patients with active EG (C, plasma; D, serum). E and F, Blood EG scores in patients with active EG and inactive EG (E, plasma; F, serum). G and H, ROC curves and performance of blood EG scores. The AUC was calculated for 4 conditions: (G) Plasma cohort; plasma EG score, eotaxin-3, IL-5, and TARC. (H) Serum cohort; serum EG score, eotaxin-3, IL-5, and TSLP. AUC; area under the curve; EG, eosinophilic gastritis; NPV, negative predictive value; PCA, principal component analysis; PPV, positive predictive value; ROC, receiver operating characteristic.
On the basis of the levels of these dysregulated cytokines and chemokines, we developed a circulation-based EG biomarker scoring system for plasma and serum (see Fig E8 in this article’s Online Repository at www.jacionline.org). The blood-based EG score differentiated patients with active EG from non-EG controls in both the plasma and serum cohorts (P < .0001) (Fig 4, C and D). Notably, patients with active EG showed significantly higher scores than did patients with inactive EG (plasma EG score P < .0001, serum EG score P = .0012) (Fig 4, E and F). To determine their diagnostic performances, ROC analyses were constructed to investigate the use of blood EG scores and cytokine/chemokine levels alone (Fig 4, G and H). The plasma EG score yielded an AUC of 0.93, whereas levels of eotaxin-3 alone yielded an AUC of 0.89, levels of TARC alone yielded an AUC of 0.82, and levels of IL-5 alone yielded an AUC of 0.80. The serum EG score yielded an AUC of 0.91, whereas levels of TSLP alone yielded AUC of 0.86, levels of eotaxin-3 alone yielded an AUC of 0.80, and levels of IL-5 alone yielded an AUC of 0.77.
Associations among the local and systemic molecular expressions
We were interested in exploring the association between the tissue local gene expressions and circulating systemic molecular expressions. Notably, plasma eotaxin-3 exhibited a higher magnitude of correlation with the EGDP than any other protein (P < .01) (Fig 5, A, upper panel). Using Spearman r for the correlation between the EGDP gene expressions and plasma and serum protein biomarkers, a Spearman r–based heat diagram for the correlation at the gene level was generated (Fig 5, A, lower panel). Focusing on the plasma eotaxin-3, we observed that plasma eotaxin-3 correlated with genes related to gastric cytokine/chemokines (CCL26, IL13RA2, IL1RL1, IL4, and IL5), eosinophilia (CLC and CCR3), cell adhesion (CDH26), mast cells (CPA3 and HPGDS), inflammatory response (ANXA1), other (ITLN1), neurosensory features (GLDN), fibrosis (BMP3), and antimicrobial defense (DEFB1).
FIG 5. Associations among the local and systemic features.

A, Associations between the EGDP and the blood cytokine/chemokines. Using Spearman r for the correlation between the EGDP gene expressions and (left) plasma and (right) serum cytokine/chemokines, the magnitudes of correlation with the EGDP are shown (upper). A Spearman r–based heat diagram for the correlation at the gene level are shown ( lower). Genes shown on the y axis are organized within functional groupings. Darker red shades indicate stronger positive correlations, whereas darker blue shades indicate stronger negative correlations. B, Correlation between blood EG score (left, plasma; right, serum) and peak gastric eosinophil counts, with Spearman r and P values shown. C, Correlation between blood EG scores (left, plasma; right, serum) and the EGDP18 score, with Spearman r and P values shown. EG, eosinophilic gastritis; EGDP, eosinophilic gastritis diagnostic panel; EMT, epithelial-mesenchymal transition; HPF, high-power field. *P < .01 vs. all other proteins.
The blood EG score—the circulation-based biomarker—showed significant correlations with gastric eosinophil counts (plasma r = 0.72, P = .0002; serum r = 0.54, P = .0015) (Fig 5, B) and EGDP18 score (plasma r = −0.64, P = .0015; serum r = −0.46, P = .0084) (Fig 5, C), suggesting that the systemic circulating biomarker levels reflect the local gastric inflammatory process defined by the eosinophil levels and transcript expression profiles.
DISCUSSION
In this study, we have established molecular diagnostic criteria for EG by utilizing gastric mRNA transcript and circulating protein levels. We have 1) developed a set of EG transcripts, composed of 48 genes, that robustly distinguishes EG from non-EG and whose expression is equivalent across independent and geographically diverse sites and across patient ages (including children and adults) and medical therapy; 2) determined that a diagnostic score limited to changes in a subset of 18 genes, referred to as the EGDP18 score, is sufficient to allow EG diagnosis relative to controls (sensitivity of 88–95% in discovery and validation cohort and specificity of 100%), including individuals with non-EG and patients with EGID limited to the esophagus; 3) determined that the EGDP18 score can robustly separate patients with active EG from those with inactive EG, strongly correlates with gastric eosinophil levels (r = −0.83, P < .0001), and potentially aids in diagnostic classification of patients with intermediate eosinophil levels; 4) determined that expression of specific genes tracks with tissue eosinophilia, namely expression of CCL26, CLC, IL13RA2, BMP3, IL5, CDH26, CCL18, NPY, HPGDS, and SST; 5) linked the magnitude of molecular changes to endoscopic changes, most notably associating nodularity and granularity with a subset of type 2 inflammatory genes, including CCL26 and IL13RA2, respectively; 6) linked the magnitude of molecular changes to histologic changes; most notably, the levels of CCL26 strongly associated with periglandular circumferential collars (r = 0.74, P = 7.0E-21) and eosinophil glandulitis (r = 0.68, P = 2.0E-16), whereas IL13RA2 correlated most notably with periglandular circumferential collars (r = 0.67, P = 7.0E-16) and lamina propria eosinophil sheets (r = 0.67, P = 5.0E-16); 7) identified circulating biomarkers that reflect local changes in the stomach, most notably gastric eosinophilia; and 8) demonstrated that the combined levels of plasma eotaxin-3, TARC, and IL-5 have the capacity to diagnose EG disease and monitor disease activity with high sensitivity and specificity (100% and 72%, respectively).
Herein, we analyzed more than 200 gastric tissue samples and assessed the overlap among molecular profiles. Although it is conceivable that EG and EoE share a common TH2 molecular pathogenesis, as we published earlier,10 the EG and EoE transcriptomes (as assessed by microarray analysis) only overlap by 7% despite a common IL-13 induced signature. In this study, using independent methods of RNA sequencing and qPCR arrays, we confirmed our previous observation that the overall gene expression profiles of EoE and EG are distinct at a transcription level. Previous histopathologic studies indicate that the eosinophilic infiltration in EG can be patchy and that the minimum threshold number of gastric eosinophils required for the diagnosis of EG varies, ranging from 20 eosinophils in 1 HPF to 70 eosinophils per HPF in at least 3 HPFs.21, 24 Our results were obtained by using only 1 RNA sample per patient, suggesting that molecular diagnosis is a relatively promising and sensitive method for disease diagnosis and monitoring. The EGDP18 score algorithm indicated that 63% of the histologically intermediate patients were molecularly equivalent to patients with active EG, providing evidence that 30 eosinophils/HPF in less than 5 HPFs is still associated with robust molecular inflammatory processes. These data suggest that analysis of less than 5 HPFs may be sufficient for diagnosis.
Beyond diagnostic merits, in order to understand disease pathogenesis, we also assessed correlations between molecular profiles and histologic and endoscopic features. For EG histologic features, regardless of the distribution, eosinophilic features (periglandular circumferential collars, eosinophil glandulitis, lamina propria eosinophil sheets, and eosinophils in surface epithelium) were highly associated with the EG transcriptome (especially the EGDP core 18 genes), with the strongest association occurring in periglandular circumferential collars. Not all histologic features showed strong associations with the EGDP, possibly due to the insufficient depth of biopsies resulting in many of them not including muscularis mucosa and/or to the low occurrence of some histologic features (lamina propria fibroplasia, surface erosion/ulceration, eosinophil gland abscess, and acute inflammatory cells). Moreover, certain features of endoscopic changes, such as nodularity and granularity, were notable as features uniquely related to transcript changes, particularly those enriched in inflammatory responses involving upregulation of type 2 immunity and eosinophil-related pathways (IL13RA2, CCL26, and IL5). Both CCL26 and IL13RA2 are IL-13–inducible genes; 25 controversy exists as to whether the latter is an activating or possibly inhibitory signaling molecule (including a potential inhibitor role of soluble IL-13Rα2). The prominent role of type 2 immunity–related responses provides the scientific basis for therapeutic intervention with dupilumab (anti–IL-4R α, which inhibits IL-4Rα/IL-13Rα1), anti–IL-13R α1, and/or anti–IL-13 (e.g., RPC4046 inhibiting IL-13 interactions with both IL-13Ra1 and IL-13Ra2).26 Increased expression levels of type 2 immune/eosinophil associated pathways can be seen in other atopic disorders associated with nodularity, such as chronic rhinosinusitis with nasal polyps, suggesting that they were not to be EG-specific but might function to generate these histological features in certain tissue/conditions. Interestingly, endoscopic changes, such as friability and erythema, were associated with downregulation of IL33 (epithelium-derived, pro-inflammatory alarmin), SLC26A7 (anion exchange transporter), and ATP4A (proton pump; gastric H, K-ATPase alpha subunit). Decreased expression levels of these genes may suggest injured mucosa due to tissue inflammation. Our current findings only showed minimal overlap (e.g. AREG, CXCL8, SST, and TGFBR1) compared to a prior report limited to 8 patients with EG from a single site,13 probably due to the differences in sample size, molecular platform, and definition of endoscopic features. However, in this large, cross-sectional cohort of EG, we could identify specific findings with differences in potential pathways.
Biopsy procurement is currently required to establish definitive diagnosis of EGID. The field urgently calls for developing non-invasive biomarkers. Prior findings suggested that EG is more systemic than EoE on the basis of the co-occurrence of EG with circulating eosinophilia;10 therefore, we hypothesized that circulating biomarkers may be present in patients with EG. Indeed, eotaxin-3 and IL-5 were significantly upregulated in both serum and plasma of patients with EG compared to non-EG controls, and circulating levels of eotaxin-3 were particularly correlative with tissue expression of CCL26. Of note, the plasma eotaxin-3 levels in EG were not reflective of an atopic state in general, as they were not elevated in patients with EoE. Furthermore, average circulating eotaxin-3 levels in EG appear to be substantially higher than levels reported in other atopic diseases, such as chronic rhinosinusitis with high-eosinophil mucosal infiltration (plasma 122.6 pg/mL vs. 481.2 pg/mL seen in our study),27 but in the same range for serum as seen for another rare eosinophilic disease, eosinophilic granulomatosis with polyangiitis.28 We speculate that the stomach (related disease EG) may contribute to higher circulating eotaxin-3 levels than does the esophagus (related disease EoE) due to differences in the underlying tissue architecture, with the gastric mucosa having relatively increased proximity to the vasculature, a relatively large surface area, and resident eosinophil populations during homeostasis. The reasons for the selective increase in circulating eotaxin-3 levels in EG compared with other EGIDs deserves further attention.
To our knowledge, this is the first EGID study simultaneously addressing tissue signature and circulating cytokine profiles in the same disorder with autologous samples across different collecting centers. This study was not intended to replace the histologic method but rather to provide at least two alternative platforms to more precisely and sensitively diagnose EG. It is conceivable that the circulating markers could serve as an early non-invasive test during EGID/EG screening, whereas the tissue signature profiling (EGDP) could be used for definitive diagnostic confirmation. The combination of both would provide molecular tools to diagnose, monitor, and potentially further subtype (e.g., endotype) knowledge of EG. Future studies should examine the utility of the blood-based platform to identify disease remission with treatment, which would prevent the need for repeat endoscopy.
Our study has several strengths. First, we analyzed samples from multiple sites across the USA, which increases the generalizability of the results. Second, participants were assessed with several diagnostic assessments, allowing us to examine associations between gene expressions, endoscopic, and histologic parameters. Third, we assessed not only gene expression, but also circulating blood protein. Fourth, we validated gene expression differences between EG diagnoses in an independent cohort. Our study also has limitations. First, our findings include patients with active EG with mixed treatment status or who have disease that is refractory to treatment, which might potentially influence the results. However, patients still exhibited signs of disease clinically, histologically, and molecularly. Second, most of the analyses for gene expression and biomarker were restricted to 48 genes included in the EGDP and 10 blood biomarkers. Unbiased, genome-wide transcriptome and proteome approaches would likely reveal additional genes of interest, biomarkers, and optimal combinations. Finally, the data are limited by the cross-sectional approach, highlighting the importance of additional replication, particularly in prospective and longitudinal studies.
In conclusion, we have developed and validated diagnostic panels that can diagnose EG using biopsy and blood samples. CCL26/eotaxin-3 emerged as the strongest single tissue and circulating disease biomarker. We have uncovered robust associations among the EG molecular profile, periglandular circumferential collars, and endoscopic granularity/nodularity, providing insight into the better understanding of the pathogenesis for EG. Further work is required to apply these platforms to a prospective trial in a different clinical setting, explore the feasibility and further validation, and optimize platforms for disease stratification.
Supplementary Material
Clinical Implications:
We have developed tissue- and blood-based platforms for diagnosing and monitoring eosinophilic gastritis and uncovered likely molecular pathogenesis that accounts for the distinct endoscopic and histological features of the disease.
Acknowledgments
The authors would like to thank all patients who participated in the study. The authors are also grateful to their colleagues and clinical support staff for procuring biopsies, blood samples, and clinical data.
Grant Support
This study was supported by CEGIR (U54 AI117804), which is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), and is co-funded by National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and NCATS. CEGIR is also supported by patient advocacy groups including the American Partnership for Eosinophilic Disorders (APFED), Campaign Urging Research for Eosinophilic Disease (CURED), and Eosinophilic Family Coalition (EFC).
M.E.R. is a consultant for Pulm One, Spoon Guru, Celgene, Shire, Astra Zeneca, GlaxoSmithKline, Allakos, Adare, Regeneron, and Novartis and has an equity interest in Pulm One, and ClostaBio, and Spoon Guru and royalties from reslizumab (Teva Pharmaceuticals). M.E.R. is an inventor of patents, owned by Cincinnati Children’s. G.W.F. has received research support from Celgene/Receptos, Regeneron, Takeda/Shire, and Adare and is a consultant for Adare and Takeda/Shire. M.H.C. is a consultant for Shire, Regeneron, Receptos, and Esocap and has received research funding from Shire, Regeneron, and Receptos. S.K.G. is a consultant for Abbott, Adare, Allakos, QOL, and Receptos/Celgene and receives research support from Shire. V.A.M. is a consultant for Shire and has received research funding from Shire. N.G. is a consultant for Allakos. E.S.D. is a consultant for Adare, Alivio, Allakos, AstraZeneca, Banner, Biorasi, Calypso, Enumeral, EsoCap, Gossamer Bio, GlaxoSmithKline, Receptos/Celegene, Regeneron, Salix, and Shire; has received research funding from Adare, Allakos, GlaxoSmithKline, Meritage, Miraca, Nutricia, Receptos/Celgene, Regeneron, and Shire; and has received educational grants from Allakos, Banner, and Holoclara. S.S.A. is a consultant for Regeneron, AImmune Therapeutics, and Gossamer; is an inventor of oral viscous budesonide, patented by UCSD and licensed by Shire/Takeda; and has research funding from the Ferring Research Institute. J.M.S. is a consultant for Regeneron and DBV Technology, and his research is supported by the NIH, Everbody Eats (EATS) foundation, AImmune Therapeutics, Food Allergy Research & Education (FARE), and DBV Technology. I.H. is a consultant for Regeneron, Receptos, Shire, Allakos, and Adare and has received research funding from Regeneron, Receptos, Shire, and Adare. G.T.F. is a consultant for Shire and a co-founder of EnteroTrack. J.B.W. is a consultant for Allakos.
Writing assistance
Shawna Hottinger provided editorial assistance as a medical writer funded by Cincinnati Children’s Hospital Medical Center.
Abbreviations used
- APFED
American Partnership for Eosinophilic Disorders
- AUC
area under the curve
- CCED
Cincinnati Center for Eosinophilic Disorders
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CEGIR
Consortium of Eosinophilic Gastrointestinal Disease Researchers
- CURED
Campaign Urging Research for Eosinophilic Disease
- DMCC
Data Management and Coordinating Center
- EDP
EoE diagnostic panel
- EFC
Eosinophilic Family Coalition
- EG
eosinophilic gastritis
- EGDP
EG Diagnostic Panel
- EGID
eosinophilic gastrointestinal disorders
- EMT
epithelial-mesenchymal transition
- EoE
eosinophilic esophagitis
- DEG
differentially expressed genes
- FDR
false-discovery rate
- GO
gene ontology
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GI
gastrointestinal
- GOI
gene of interest
- HPF
high-power field
- IQR
interquartile range
- NPV
negative predictive value
- OMEGA
Outcomes Measures in Eosinophilic Gastrointestinal disorders Across the ages
- ORDR
Office of Rare Diseases Research
- PCA
principal component analysis
- PPI
proton pump inhibitor
- PPV
positive predictive value
- RDCRN
Rare Diseases Clinical Research Network
- ROC
receiver operating characteristic
- RPKM
reads per kilobase of exon per million reads mapped
- TARC/CCL17
thymus and activation–regulated chemokine/chemokine ligand 17
- TSLP
thymic stromal lymphopoietin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The list of participants is provided in this article’s Supplementary Material
Conflicts of Interest
All other authors declare that they have no competing interests.
REFERENCES
- 1.Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 2004; 113:11–28. [DOI] [PubMed] [Google Scholar]
- 2.Walker MM, Potter M, Talley NJ. Eosinophilic gastroenteritis and other eosinophilic gut diseases distal to the oesophagus. The Lancet Gastroenterology & Hepatology 2018; 3:271–80. [DOI] [PubMed] [Google Scholar]
- 3.Jensen ET, Martin CF, Kappelman MD, Dellon ES. Prevalence of Eosinophilic Gastritis, Gastroenteritis, and Colitis: Estimates From a National Administrative Database. J Pediatr Gastroenterol Nutr 2016; 62:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pesek RD, Reed CC, Muir AB, Fulkerson PC, Menard-Katcher C, Falk GW, et al. Increasing Rates of Diagnosis, Substantial Co-Occurrence, and Variable Treatment Patterns of Eosinophilic Gastritis, Gastroenteritis, and Colitis Based on 10-Year Data Across a Multicenter Consortium. Am J Gastroenterol 2019; 114:984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Shea KM, Aceves SS, Dellon ES, Gupta SK, Spergel JM, Furuta GT, et al. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018; 154:333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBrosse CW, Case JW, Putnam PE, Collins MH, Rothenberg ME. Quantity and distribution of eosinophils in the gastrointestinal tract of children. Pediatr Dev Pathol 2006; 9:210–8. [DOI] [PubMed] [Google Scholar]
- 7.Matsushita T, Maruyama R, Ishikawa N, Harada Y, Araki A, Chen D, et al. The number and distribution of eosinophils in the adult human gastrointestinal tract: a study and comparison of racial and environmental factors. Am J Surg Pathol 2015; 39:521–7. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matoso A, Mukkada VA, Lu S, Monahan R, Cleveland K, Noble L, et al. Expression microarray analysis identifies novel epithelial-derived protein markers in eosinophilic esophagitis. Mod Pathol 2013; 26:665–76. [DOI] [PubMed] [Google Scholar]
- 10.Caldwell JM, Collins MH, Stucke EM, Putnam PE, Franciosi JP, Kushner JP, et al. Histologic eosinophilic gastritis is a systemic disorder associated with blood and extragastric eosinophilia, TH2 immunity, and a unique gastric transcriptome. J Allergy Clin Immunol 2014; 134:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014; 15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoda T, Matsuda A, Arai K, Shimizu H, Morita H, Orihara K, et al. Sera of patients with infantile eosinophilic gastroenteritis showed a specific increase in both thymic stromal lymphopoietin and IL-33 levels. J Allergy Clin Immunol 2016; 138:299–303. [DOI] [PubMed] [Google Scholar]
- 13.Sato M, Shoda T, Shimizu H, Orihara K, Futamura K, Matsuda A, et al. Gene Expression Patterns in Distinct Endoscopic Findings for Eosinophilic Gastritis in Children. J Allergy Clin Immunol Pract 2017; 5:1639–49 e2. [DOI] [PubMed] [Google Scholar]
- 14.Shoda T, Matsuda A, Nomura I, Okada N, Orihara K, Mikami H, et al. Eosinophilic esophagitis versus proton pump inhibitor-responsive esophageal eosinophilia: Transcriptome analysis. J Allergy Clin Immunol 2017; 139:2010–3.e4. [DOI] [PubMed] [Google Scholar]
- 15.Peterson KA, Yoshigi M, Hazel MW, Delker DA, Lin E, Krishnamurthy C, et al. RNA sequencing confirms similarities between PPI-responsive oesophageal eosinophilia and eosinophilic oesophagitis. Aliment Pharmacol Ther 2018; 48:219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, Putnam PE, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013; 145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoda T, Wen T, Aceves SS, Abonia JP, Atkins D, Bonis PA, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018; 3:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bedell A, Taft T, Craven MR, Guadagnoli L, Hirano I, Gonsalves N. Impact on Health-Related Quality of Life in Adults with Eosinophilic Gastritis and Gastroenteritis: A Qualitative Assessment. Dig Dis Sci 2018; 63:1148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hines BT, Rank MA, Wright BL, Marks LA, Hagan JB, Straumann A, et al. Minimally invasive biomarker studies in eosinophilic esophagitis: A systematic review. Ann Allergy Asthma Immunol 2018; 121:218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chehade M, Magid MS, Mofidi S, Nowak-Wegrzyn A, Sampson HA, Sicherer SH. Allergic eosinophilic gastroenteritis with protein-losing enteropathy: intestinal pathology, clinical course, and long-term follow-up. J Pediatr Gastroenterol Nutr 2006; 42:516–21. [DOI] [PubMed] [Google Scholar]
- 21.Ko HM, Morotti RA, Yershov O, Chehade M. Eosinophilic gastritis in children: clinicopathological correlation, disease course, and response to therapy. Am J Gastroenterol 2014; 109:1277–85. [DOI] [PubMed] [Google Scholar]
- 22.Gupta SK, Falk GW, Aceves SS, Chehade M, Collins MH, Dellon ES, et al. Consortium of Eosinophilic Gastrointestinal Disease Researchers: Advancing the Field of Eosinophilic GI Disorders Through Collaboration. Gastroenterology 2019; 156:838–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lwin T, Melton SD, Genta RM. Eosinophilic gastritis: histopathological characterization and quantification of the normal gastric eosinophil content. Mod Pathol 2011; 24:556–63. [DOI] [PubMed] [Google Scholar]
- 24.Pineton de Chambrun G, Gonzalez F, Canva JY, Gonzalez S, Houssin L, Desreumaux P, et al. Natural history of eosinophilic gastroenteritis. Clin Gastroenterol Hepatol 2011; 9:950–6.e1. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007; 120:1292–300. [DOI] [PubMed] [Google Scholar]
- 26.Hirano I, Collins MH, Assouline-Dayan Y, Evans L, Gupta S, Schoepfer AM, et al. RPC4046, a Monoclonal Antibody Against IL13, Reduces Histologic and Endoscopic Activity in Patients With Eosinophilic Esophagitis. Gastroenterology 2019; 156:592–603 e10. [DOI] [PubMed] [Google Scholar]
- 27.Yamada T, Miyabe Y, Ueki S, Fujieda S, Tokunaga T, Sakashita M, et al. Eotaxin-3 as a Plasma Biomarker for Mucosal Eosinophil Infiltration in Chronic Rhinosinusitis. Front Immunol 2019; 10:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polzer K, Karonitsch T, Neumann T, Eger G, Haberler C, Soleiman A, et al. Eotaxin-3 is involved in Churg-Strauss syndrome--a serum marker closely correlating with disease activity. Rheumatology (Oxford) 2008; 47:804–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.