

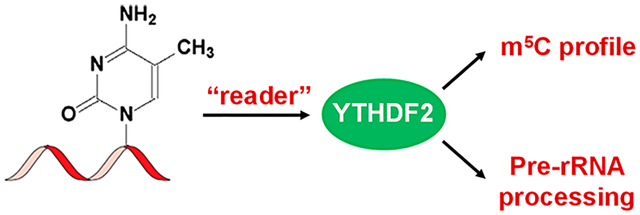
Abstract
5-Methylcytosine is found in both DNA and RNA; although its functions in DNA are well established, the exact role of 5-methylcytidine (m5C) in RNA remains poorly defined. Here we identified, by employing a quantitative proteomics method, multiple candidate recognition proteins of m5C in RNA, including several YTH domain-containing family (YTHDF) proteins. We showed that YTHDF2 could bind directly to m5C in RNA, albeit at a lower affinity than that toward N6-methyladenosine (m6A) in RNA, and this binding involves Trp432, a conserved residue located in the hydrophobic pocket of YTHDF2 that is also required for m6A recognition. RNA bisulfite sequencing results revealed that, after CRISPR-Cas9-mediated knockout of the YTHDF2 gene, the majority of m5C sites in rRNA (rRNA) exhibited substantially augmented levels of methylation. Moreover, we found that YTHDF2 is involved in pre-rRNA processing in cells. Together, our data expanded the functions of the YTHDF2 protein in post-transcriptional regulations of RNA and provided novel insights into the functions of m5C in RNA biology.
Graphical Abstract
RNA harbors more than 100 distinct types of modifications, which modulate its structure and functions.1 Recent transcriptome-wide mapping studies revealed the widespread occurrence of 5-methylcytidine (m5C),2,3 N6-methyladenosine (m6A),4,5 N6,2′-O-dimethyladenosine (m6Am),6 and pseudouridine (Ψ)7–9 in mRNA. In addition, proteins involved in the installation (writers),10–12 removal (erasers),13,14 and recognition (readers)4,15–17 of m6A have been discovered and found to play important roles in modulating the localization, stability, and translational efficiencies of mRNA. These recent exciting findings suggest that post-transcriptional modifications of RNA, similar to the methylation of cytosine in DNA and post-translational modifications of histones, may play an epigenetic role in gene expression.18
Recent studies also offered some insights into the functions of m5C in RNA.3,19,20 In this respect, m5C in tRNA and rRNA were shown to stabilize the secondary structure of tRNA and regulate translational fidelity, respectively.19,21 In addition, m5C is known to be present in mRNA, where a previous transcriptome-wide mapping study revealed the enrichment of m5C in the untranslated regions of mRNA in HeLa cells,2 though a recent study showed that m5C in mRNA may not be as widespread as initially thought.3 Hence, the functions of m5C in mRNA remain unclear. NSUN2 and TRDMT2 are two known methyltransferases for the formation of m5C in eukaryotes (writers),22,23 and m5C in RNA can be converted to 5-hydroxymethylcytidine by ten-11 translocation (Tet) enzymes (erasers).24,25 A recent study showed that m5C can interact with mRNA export adaptor ALYREF (reader).26 However, it remains unknown whether other cellular proteins are also involved in the recognition of m5C in RNA.
In this study, we discovered, by employing an unbiased quantitative proteomics method, a number of candidate protein readers of m5C, including YTH domain-containing proteins. We also showed that YTH domain-containing family protein 2 (YTHDF2) binds to m5C-carrying RNA in vitro and in cells. In addition, genetic ablation of YTHDF2 elicited substantial elevations in the levels of m5C at multiple loci in rRNA. Moreover, YTHDF2 could modulate rRNA maturation in human cells. Together, our study expanded the functions of YTHDF2 and provided a foundation for understanding better the biological functions of m5C in RNA.
EXPERIMENTAL SECTION
Cell Culture.
HeLa and HEK293T cells (ATCC) were cultured at 37 °C in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (Invitrogen) and 100 units ml−1 penicillin and 100 μg mL−1 streptomycin (Life Technologies) in an incubator containing 5% CO2.
For SILAC experiments, DMEM medium without lysine or arginine was obtained from Fisher Scientific. The complete light and heavy DMEM media were prepared by the addition of light or heavy lysine and arginine ([13C6,15N2]-l-lysine and [13C6]-l-arginine, Sigma), along with dialyzed fetal bovine serum, to the above medium. The cells were cultured in a 37 °C incubator for at least 10 days (more than 5 cell doublings) to ensure complete stable isotope incorporation.
Quantitative Discovery of m5C-Binding Proteins.
Biotin-labeled oligoribonucleotides with the sequence of 5′-biotin-ACUGGCUCCUUCCACGUCUCACXAGGCAGACAGU-3′ (X = C or m5C) were obtained from Integrated DNA Technologies (IDT). HeLa cells cultured in SILAC medium were harvested at 70–80% confluence, washed with PBS, and lysed in CelLytic M cell lysis buffer (Sigma). The lysates were centrifuged at 13 000 rpm and at 4 °C for 10 min. The supernatant was precleared at 4 °C for 1 h by incubation with streptavidin-conjugated agarose beads (Thermo Scientific). Biotinylated RNA baits (3 μg) were incubated, at 4 °C for 2 h, with precleared cell lysates in a binding buffer containing 10 mM Tris-HCl (pH 7.5), 150 mM KCl, 1.5 mM MgCl2, 0.05% (v/v) IGEPAL CA-630, 0.5 mM DTT, and 0.4 units μL−1 RNase inhibitor (New England Biolabs). Streptavidin-conjugated agarose beads were then added to the mixture, which was kept in a shaker at 4 °C for 2 h. The beads were extensively washed, and the heavy and light lysates were then combined. In forward SILAC experiments, the m5C and the control probes were incubated with the heavy and light isotope-labeled lysates, respectively. The opposite incubations were conducted in reverse SILAC experiments. The samples were separated on a 10% (w/v) SDS-PAGE gel for a short distance (1 cm) and stained with Coomassie blue. The gel was then destained. The proteins were subsequently reduced and alkylated with dithiothreitol and iodoacetamide, respectively, and then digested in gel with trypsin (Roche) at 37 °C for 16 h. The resulting tryptic peptides were subsequently extracted from the gel with 5% acetic acid, desalted, and analyzed using LC-MS/MS.
LC-MS/MS experiments were performed as previously described.27 Briefly, the peptides were separated on an EASY-nLC II and analyzed on an LTQ Orbitrap Velos mass spectrometer equipped with a nanoelectrospray ionization source (Thermo). The trapping column (150 μm × 50 mm) and separation column (75 μm × 120 mm) were both packed with ReproSil-Pur C18-AQ resin (3 μm in particle size, Dr. Maisch HPLC GmbH, Germany). The peptide samples were first loaded onto the trapping column in CH3CN/H2O (2:98, v/v) at a flow rate of 4.0 μL/min and resolved on the separation column with a 120 min linear gradient of 2–40% acetonitrile in 0.1% formic acid and at a flow rate of 300 nL/min. The LTQ-Orbitrap Velos mass spectrometer was operated in the positive-ion mode, and the spray voltage was 1.8 kV. The full-scan mass spectra (m/z 300–2000) were acquired with a resolution of 60 000 at m/z 400 after accumulation to a target value of 500 000 in the linear ion trap. MS/MS data were obtained in a data-dependent scan mode where one full MS scan was followed with 20 MS/MS scans.
Protein identification and quantification were performed using Maxquant,28 Version 1.2.2.5 against International Protein Index (IPI) database, version 3.68. The maximum number of miscleavages for trypsin was two per peptide. Cysteine carbamidomethylation and methionine oxidation were set as fixed and variable modifications, respectively. The search was performed with the tolerances in mass accuracy of 10 ppm and 0.6 Da for MS and MS/MS, respectively. The required false-positive discovery rate was set at 1% at both the peptide and protein levels, with the minimal required peptide length being set at six amino acids. For obtaining reliable results, the quantification of the protein expression ratio was based on six independent SILAC labeling experiments, which included three forward and three reverse labelings.
Vector Construction and Protein Expression.
The human YTHDF2 gene was amplified from mRNA isolated from HEK293T cells by reverse transcription-PCR to introduce a 5′ XbaI site and a 3′ BamHI site, and subcloned into pRK7–3 × FLAG vector. The pGEX-4T-1-YTHDF2 vector was a gift from Prof. Chuan He.16 The vector for the YTHDF2-W432A mutant was constructed by site-directed mutagenesis using primers containing the indicated mutations. The primers are listed in Supplementary Table S1.
Recombinant YTHDF2 and YTHDF2-W432A proteins were obtained by inducing transformed Rosetta (DE3) pLysS Escherichia coli cells with 1 mM isopropyl 1-thio-β-D-galactopyranoside when OD600 of the culture reached approximately 0.6 and culturing at room temperature overnight. Subsequently, the recombinant proteins were extracted from the lysate with glutathione agarose (Pierce) following the manufacturer’s recommended procedures. The proteins were concentrated and purified using Microcon YM-30 ultra-centrifugal filters (Millipore).
Cellular RNA Sample Preparation and LC-MS/MS Measurement.
Total RNA was extracted from HEK293T cells using TRI reagent (Sigma), and mRNA was isolated and purified by using PolyATtract mRNA Isolation System IV (Promega) and RiboMinus Transcriptome Isolation Kit (Invitrogen) according to the manufacturers’ instructions.
The in vitro pull-down experiment was performed using a previously reported method with minor changes.16 Briefly, recombinant YTHDF2 protein purified from E. coli was pretreated with RNase to remove any residual RNA from bacteria cells, washed thoroughly, and incubated with mRNA from HEK293T cells in IPP buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% IGEPAL CA-630, 0.5 mM DTT, 40 units ml−1 RNase inhibitor) at 4 °C for 2 h. GST-affinity beads (Pierce) were then added to the mixture, and the mixture was incubated at 4 °C with shaking for another 2 h. Unbound mRNA was recovered from the aqueous phase as the flow-through fraction. The beads were washed for four times and the YTHDF2-bound mRNA was extracted from the beads using TRI reagent. The procedures for in vitro RNA cross-linking and immunoprecipitation (CLIP) were similar to the in vitro RNA pull-down experiment, with the following modifications: Before immunoprecipitation with GST-affinity beads, the RNA-protein mixture was cross-linked by irradiating on ice for three times with 0.15 J/cm2 of 254 nm UV light each time; after UV cross-linking, the RNA-protein mixture was subjected to RNase T1 digestion (1 unit μL–1 RNase T1 for 8 min at 22 °C); and after immunoprecipitation and washing, the RNA was detached from GST-affinity beads by treating with proteinase K (1 mg mL−1) at 50 °C for 30 min, and the RNA was further recovered by using Zymo RNA Clean and Concentrator.
For the cellular pull-down experiment, HEK293T cells were transfected with a plasmid encoding FLAG-tagged YTHDF2 or the corresponding W432A mutant. After a 48-h incubation, the cells were washed with PBS and lysed in a lysis buffer, which contained 10 mM HEPES (pH 7.5), 150 mM KCl, 2 mM EDTA, 0.5% IGEPAL CA-630, 0.5 mM DTT, protease inhibitor (Sigma), and 40 units ml−1 RNase inhibitor. The supernatant was incubated with anti-FLAG M2 beads (Sigma) at 4 °C overnight. The beads were washed for three times with a washing buffer containing 50 mM HEPES (pH 7.5), 200 mM NaCl, 2 mM EDTA, 0.05% IGEPAL CA-630, and 0.5 mM DTT. The beads were then incubated with proteinase K (1.2 mg/mL) at 55 °C for 1 h. The sample was centrifuged at 5000 rpm for 1 min, and the RNA was recovered from the supernatant.
The LC-MS/MS measurement of RNA samples was performed using previously reported methods with some modifications.29 Briefly, 100 ng of mRNA was digested with 1 unit of nuclease P1 in a 25-μL buffer containing 25 mM NaCl and 2.5 mM ZnCl2. The mixture was then incubated at 37 °C for 2 h, and to the mixture were added 0.5 unit of alkaline phosphatase and 3 μL of 1.0 M NH4HCO3. After incubating at 37 °C for an additional 2 h, the digestion mixture was dried and reconstituted in 100 μL ddH2O. Uniformly 15N-labeled cytidine and [13C5]-m5C were employed as internal standards for the quantifications of cytidine and m5C, respectively, and 13C5-labeled adenosine and [D3]-m6A were used as internal standards for the quantifications of adenosine and m6A, respectively. The enzymes in the digestion mixture were removed by extraction using chloroform:isoamyl alcohol (24:1). The aqueous layer was dried, reconstituted in 10 μL of ddH2O, and injected for LC-MS/MS analysis on an LTQ-XL linear ion trap mass spectrometer equipped with nanoelectrospray ionization source and an EASY-nLC II (Thermo). The instrument conditions and scan events were previously described.29 For cytidine and m5C quantification, the precolumn and analytical column were packed with porous graphitic carbon (PGC) and Magic C18 AQ, respectively, where a gradient of 0–15% B in 10 min, 15–35% B in 40 min, 35–90% B in 1 min, and 90% B in 15 min was used. For adenosine and m6A quantification, both the precolumn and analytical column were packed with Magic C18 AQ, where a gradient of 0–15% B in 40 min, 55–90% B in 1 min, and 90% B in 10 min was employed.
RESULTS
A Quantitative Proteomics Method Led to the Identification of Multiple Putative m5C-Interacting Proteins.
Systematic identifications of m5C-binding proteins constitute an important step toward understanding the biological functions of m5C in RNA. Hence, we employed a quantitative proteomics method to screen for proteins in HeLa and HEK293T cells that can bind to m5C-bearing RNA, where we employed metabolic labeling with SILAC (stable isotope labeling by amino acid in cell culture) (Figure 1A).30 In this respect, we chose an RNA sequence derived from the mRNA of the human CINP gene, which was recently shown to carry an m5C at position 748 (with approximately 46% methylation in HeLa cells),2 as the probe bait and the corresponding unmethylated sequence as the control bait.
Figure 1.

Identification of m5C-interacting proteins. (A) A schematic overview of the SILAC-based quantitative proteomics method for discovering m5C reader proteins. Shown is the workflow for a forward SILAC labeling experiment. (B) A scatter plot showing the proteins identified in RNA pull-down assay in HeLa cells. Displayed are results based on three forward and three reverse SILAC labeling experiments. (C) Representative ESI-MS for the [M+2H]2+ ions of YTHDF2 peptide SINNYNPK revealing the preferential binding of YTHDF2 toward the m5C probe in both forward (up) and reverse (bottom) SILAC experiments.
Our results led to the identification of multiple proteins exhibiting preferential binding toward the m5C-bearing RNA over control (Figure 1B and Supplementary Figure S1). These proteins include mRNA cleavage stimulation factors CSTF1–3, YTH domain-containing family proteins 1–3 (YTHDF1–3), pre-mRNA splicing factors SFPQ/NONO, and others (ratio of m5C/C > 1.4, Supplementary Tables S2 and S3). Figure 1C depicts the representative electrospray ionization-mass spectrometry (ESI-MS) results for a tryptic peptide derived from YTHDF2 (MS/MS for the peptide are shown in Supplementary Figure S2), which supports the preferential binding of YTHDF2 toward the m5C probe. Consistent with the findings made from the SILAC-based quantitative proteomic analysis, Western blot results showed the presence of higher levels of YTHDF1–3 and CSTF1–3 proteins in the pull-down mixture using the m5C bait than control (Supplementary Figure S3).
YTHDF2 Is a Reader for m5C-Containing RNA.
Because YTHDF2 was previously found to be a reader protein for m6A and N1-methyladenosine,4,31 we decided to choose this protein for further investigation. In this context, we examined whether YTHDF2 can bind directly to m5C in RNA by performing electrophoretic mobility shift assay (EMSA) with recombinant YTHDF2 protein purified from E. coli. Our results showed that YTHDF2 binds more strongly to an m5C-carrying RNA substrate than its unmethylated counterpart, though the binding affinity is much weaker than that toward m6A-containing RNA (Supplementary Figure S4).
The X-ray crystal structure of YTHDF2 revealed three aromatic amino acid residues in the hydrophobic pocket of YTHDF2 that are crucial for its recognition of m6A.32,33 To explore whether this hydrophobic pocket also assumes an important role in binding toward m5C, we conducted an EMSA experiment with a mutant form of YTHDF2 protein where the conserved Trp432 was mutated to an alanine (W432A). The result indeed showed that the mutation led to a reduction in binding affinity toward the m5C-containing probe (Supplementary Figure S4), suggesting that m5C may bind to the same hydrophobic pocket in YTHDF2 that is required for m6A recognition.
To further substantiate the above findings, we performed an in vitro pull-down assay to determine if recombinant YTHDF2 can allow for the enrichment of m5C-carrying mRNA. LC-MS/MS analysis of the mononucleoside mixture arising from the enzymatic digestion of the poly(A)-tailed mRNA samples revealed that the level of m5C was significantly higher in the YTHDF2-bound fraction than the input or flow-through fraction (Figure 2A). When the YTHDF2 protein was cross-linked with its associated RNA using UV light and the cross-liked RNA was partially digested using RNase T1, the enrichment of m5C in the YTHDF2-bound fraction was increased compared to that without RNase T1 digestion (Figure 2A). Similar observations were made for m6A (Figure 2B), which is in keeping with the previous finding.16 We further expressed FLAG-tagged wild-type YTHDF2 and the W432A mutant in HEK293T cells, immunoprecipitated the proteins using anti-FLAG beads, and quantified the levels of m5C in the total RNA samples isolated from the immunoprecipitated proteins (Supplementary Figure S5). Our results showed that the levels of m5C were significantly higher in the pull-down samples of wild-type YTHDF2 than those of the W432A mutant (Figure 2C). Together, the above results support that YTHDF2 can bind directly to m5C, and this binding entails the intact hydrophobic pocket of YTHDF2 that is also involved with m6A binding.
Figure 2.

YTHDF2 is an m5C-binding protein. (A) LC-MS/MS quantification results showed that recombinant YTHDF2 protein can enrich m5C-containing RNA from poly(A)-tailed mRNAs of HEK293T cells. When YTHDF2 was cross-linked with its associated RNA by UV light and partially digested using RNase T1, the enrichment of m5C in the YTHDF2-bound fraction was increased compared to that without RNase T1 treatment. (B) LC-MS/MS results showed that m6A was enriched in YTHDF2-bound mRNA than in the input or flow-through samples. (C) Relative enrichment of m5C in total RNA products immunoprecipitated with wild-type YTHDF2 over W432A mutant protein from HEK293T cells. The data in (A–C) represent the mean and SEM of results from three (A, B) or six (C) technical replicates, i.e., three parallel pull-down experiments each for data in (A) and (B), and six independent transfection and pull-down experiments for data in (C). “*”, P < 0.05; “**”, P < 0.01. The P values were calculated using unpaired (A, C) or paired (B) two-tailed Student’s t-test. (D, E) Closed-up view of the docking models of the YTH domain of YTHDF2 with the 3-mer RNA housing an m6A (D) or m5C (E) in similar orientation. The comparison shows a similar mode of recognition of m6A and m5C. RNAs and the residues involved in the recognition are depicted in stick representation. Hydrogen bonds are shown in dashed lines. The nitrogen and oxygen atoms are colored in blue and red, respectively. YTH domain is colored in green, m6A- and m5C-containing RNA, i.e., G(m6A)C and G(m5C)C, are displayed in yellow and cyan, respectively.
We realized that the exact mechanism of recognition of m5C by YTHDF2 necessitates structural studies. We have attempted but failed to obtain the crystal structure of YTHDF2 in complex with m5C-bearing RNA. Therefore, we performed structural modeling for YTHDF2-m5C-RNA using the crystal structures of YTH domain in complex with m6A-carrying RNA as a reference model.33,34 The results showed that m6A is docked in the aromatic cage of the YTH domain composed of W432, W486, and W491 (Figure 2D). Aside from the hydrophobic interaction, the side chain carbonyl oxygen of D422 forms a hydrogen bond with the N1 of m6A at an average distance of 3.0 Å. The mode of binding is nearly identical to that observed in the crystal structure,33 which validates our docking method. The results from the corresponding docking of m5C-carrying RNA showed that m5C is sandwiched between W432 and W491 at the hydrophobic cage (Figure 2E). In addition, the side-chain carbonyl oxygen of D422 forms a hydrogen bond with the exocyclic N4 nitrogen of m5C at an average distance of 3.0 Å. Hence, the comparison of the docking structures showed a similar mode of recognition of m5C and m6A by the YTH domain of YTHDF2.
YTHDF2 Regulates the m5C Profile in RNA of Human Cells.
Others revealed that YTHDF2 interacts physically with components of m6A writers35 and it protects the m6A in the 5′ UTR of stress-induced transcripts by limiting the FTO-mediated demethylation of m6A.36 Thus, we next investigated how the absence of YTHDF2 in HEK293T cells alters the distribution of m5C in RNA by employing a recently reported method of bisulfite conversion and next-generation sequencing analysis,2,3 where we knocked out the YTHDF2 gene by using the CRISPR-Cas9 system and conducted the experiments in three replicates.
To process the bisulfite sequencing data, we employed a recently reported statistical method to calculate the adjusted p values,3 which was used to define the final list of the “authentic” m5C sites, and the mean m5C rate, which was used for comparing the levels of m5C between HEK293T cells and the isogenic YTHDF2 knockout cells. To examine whether there is a global change in m5C level, we performed a linear regression analysis using the m5C rates between HEK293T and YTHDF2 knockout cells in three groups of loci derived from rRNA, mitochondrial RNA (mtRNA), and others (primarily mRNAs). The results revealed no appreciable change in m5C rates in mtRNAs upon deletion of YTHDF2 gene [f(x) = 1.0046x + 0.0009; R2 = 0.9928, where “x” and “f(x)” represent m5C rates in HEK293T and YTHDF2 knockout cells, respectively] (Supplementary Figure S6A). This result underscores that the sample processing (bisulfite treatment, cloning, and sequencing) and data analysis workflow does not elicit a global bias favoring the YTHDF2 knockout cells or the parental HEK293T cells.
The m5C levels in the rRNAs, however, increased globally in the YTHDF2 knockout cells over the parental cells, where there is an approximately 1.6-fold increase in m5C levels upon genetic ablation of YTHDF2 [f(x) = 1.6327x + 0.0028 and R2 = 0.9307. Figure 3B]. Interestingly, the sites with high m5C levels are clustered in five regions of mature rRNAs, including one on the 18S rRNA and four on the 28S rRNA (Figure 3A). In this regard, LC-MS/MS analysis of 18S rRNA also revealed that the level of m5C, but not m6A, in 18S rRNA was significantly higher in the YTHDF2 knockout cells (Figure 3C). In contrast, we observed a mild (~20%) decrease in the m5C levels of mRNAs in the YTHDF2-depeleted cells over HEK293T cells [f(x) = 0.7810x + 0.0014; R2 = 0.9514] (Supplementary Figure S6B). Hence, the opposite trends in the alterations in the levels of m5C in rRNAs and mRNAs again suggest that the increase in m5C levels in rRNA upon knockout of YTHDF2 is not attributable to a systematic bias arising from the bisulfite treatment and/or PCR/sequencing errors.
Figure 3.

YTHDF2 regulates the m5C profile in rRNA. (A) The locations and levels of m5C in mature rRNA of HEK293T and the isogenic YTHDF2 knockout cells. The sites with high m5C levels are clustered in 5 regions, including one in the 18S rRNA and four in the 28S rRNA. (B) Linear regression analysis in the rRNAs showing that an approximately 1.6- fold increase in m5C levels upon genetic ablation of YTHDF2. “x” (X axis) and “f(x)” (Y axis) represent m5C rates in HEK293T and YTHDF2 knockout cells, respectively. (C) LC-MS/MS quantification results showing the total levels of m5C (left) and m6A (right) in 18S rRNA of HEK293T and the isogenic YTHDF2 knockout cells. Data represent the mean ± SEM (n = 3). “*”, p < 0.05; The p values were calculated using unpaired two-tailed Student’s t-test.
Our results led to the identification of a total of 1350 m5C sites, among which 208 are significantly regulated by YTHDF2 (n = 3, p < 0.05) (Supplementary Table S4). Among them, 78 and 130 sites displayed significantly increased and decreased levels of m5C, respectively, after YTHDF2 knockout (Supplementary Table S4). Of the candidate m5C sites identified in HEK293T cells, 118 were previously identified as NSUN2-methylated sites in the same cells with the miCLIP method, which relies on formation of covalent intermediate formed between methylated cytosine and the C271A mutant of NSUN2 (Supplementary Table S5).37 This is in reasonably good agreement viewing that the expression levels of NSUN2 are different in the two assay systems (miCLIP involves ectopic overexpression of NSUN2) and that not all m5C sites in coding and noncoding RNA are induced by NSUN2.
YTHDF2 Is Involved in Pre-rRNA Processing.
Ribosomal genes are first transcribed by RNA polymerase I to yield a large 47S precursor transcript that is converted to the 18S, 5.8S, and 28S mature rRNAs after elimination of two external and two internal spacers (i.e., 5′ETS, 3′ETS, ITS1, and ITS2) by a series of endonucleolytic and exonucleolytic cleavages (Figure 4A).38,39 Proteins engaged in pre-rRNA processing mainly include endo- and exoribonucleases and ribosomal proteins, including RPL10, RPL18, RPL26, and so on.40 Additionally, rRNA modifications are also required for rRNA maturation, though the underlying molecular mechanisms remain unclear.40
Figure 4.

YTHDF2 is involved in pre-rRNA processing. (A) Pre-rRNA processing pathways in HeLa cells. The 45S primary transcript is converted to the 18S, 5.8S, and 28S mature rRNAs by two alternative pathways. Two external and two internal spacers were shown as 5′ETS, 3′ETS, ITS1, and ITS2, respectively. Arrows indicate the cleavage sites. The scheme and nomenclature of the pre-RNAs were adapted from those of Hadjiolova et al.38 and Rouquette et al.39 (B, C) Northern blot analysis of total RNA extracted from HEK293T and the isogenic YTHDF2 knockout cells. Northern blots probed with oligonucleotides complementary to the 5′ of the ITS1 (B) and the ITS2 (C). PTP, primary transcript plus (47S, 46S, 45S). Right panels show the quantification data. Error bars represent the SEM (n = 3). *, p < 0.05; **, p < 0.05. The p values were calculated using unpaired two-tailed Student’s t test.
On the grounds that loss of YTHDF2 elicits increases in m5C levels at multiple sites in rRNA, we next asked whether YTHDF2 affects the maturation of rRNA. Hence, we analyzed the processing of pre-rRNA in HEK293T and the isogenic YTHDF2 knockout cells by Northern blot. The RNAs were first analyzed using a probe complementary to the 5′ of the ITS1 sequence. As expected, this probe revealed several 18S rRNA precursors, including the 45S, 41S, 30S, 21S, and 18S-E species (Figure 4B). Interestingly, genetic ablation of YTHDF2 results in a reduction in the amount of 30S pre-rRNA when compared with parental HEK293T cells (Figure 4B and Supplementary Figure S7). Using a probe complementary to the ITS2, we also observed a decrease in 12S pre-rRNA, the precursor to the 5.8S rRNA, in YTHDF2 knockout cells (Figure 4C and Supplementary Figure S7). Together, the above results support that YTHDF2 assumes an important role in rRNA processing in human cells, which may involve its binding to m5C in RNA and its regulation of m5C level in rRNA.
DISCUSSION
m5C is one of the most prevalent post-transcriptional modifications of RNA in human cells.3,19,20 In this study, we employed a state-of-the-art bioanalytical method, which involves SILAC-based quantitative proteomics, to screen for interacting proteins of m5C, and this experiment led to the identification of multiple putative readers of m5C, including three YTH domain-containing proteins (YTHDF1–3, Figure 1). We further showed that the purified recombinant YTHDF proteins can bind directly to m5C-containing RNA in vitro (Figure S4). Moreover, robust quantification by using LC-MS/MS with the stable isotope-dilution method revealed that affinity pull-down using recombinant YTHDF2 and RNA-protein photo-cross-linking followed by pull-down of ectopically expressed YTHDF2 both lead to enrichment of m5C (Figure 2). Similar LC-MS/MS measurement showed that genetic ablation of YTHDF2 led to augmented levels of m5C in 18S rRNA (Figure 3).
Different members of the YTH domain-carrying proteins are known to exert distinct functions, where bindings of m6A-bearing mRNA with YTHDF2 and YTHDF1 result in mRNA decay16 and promotion of translation,17 respectively. The characterizations of reader proteins of m5C is an important step toward understanding the biological functions of m5C in RNA.
Our findings, along with recent structural and functional studies of YTH-domain family proteins, support that YTHDF2 is a versatile reader that can recognize m6A, m1A and m5C,4,31 though the binding affinity toward m5C is much weaker than that toward m6A. In this vein, several published structural studies of YTH domain-containing proteins uncovered a hydrophobic pocket comprised of two or three aromatic residues that are instrumental for the recognition of methyl group in m6A.32–34,41,42 It is conceivable that the same hydrophobic pocket may be able to accommodate the 5-methyl group of m5C. This is indeed supported by our findings that mutation of one of the conserved aromatic residues, i.e., W432, at the hydrophobic pocket to an alanine reduces markedly the binding affinity of the protein toward m5C-carrying RNA. In addition, our docking studies revealed a very similar mode of recognition of m5C and m6A by the YTH domain of YTHDF2. Furthermore, all YTH domain-housing proteins, except YTHDC1 which preferentially binds m6A in GG(m6A)C sequence, bind m6A regardless of sequence context,41 despite the fact that m6A is known to exist in RRAC (“R” is a G or A) and RGAC consensus sequence motifs in mammalian and yeast species, respectively.4,5 The lack of recognition of the consensus flanking nucleobase(s) of m6A is consistent with the notion that YTH domain-containing proteins can recognize, in addition to m6A, other post-transcriptionally modified ribonucleosides (i.e., m1A and m5C).
We also found that YTHDF2 modulates the distribution of m5C in both coding and noncoding RNA, where its depletion led to pronounced increases in the levels of m5C at multiple sites in rRNA and decreases in m5C levels at many sites in mRNA. These results suggest that YTHDF2 may play versatile roles in regulating m5C levels. In this vein, previous reports showed that binding of YTHDF2 with m6A-bearing mRNA decreases m6A levels by P-body-mediated mRNA degradation16 but increases the m6A levels of some stress-induced transcripts by protecting them from demethylation.43 Additionally, genetic ablation of YTHDF2 resulted in substantial perturbations in rRNA maturation by diminishing the formation of 30S and 12S intermediates from the 47S rRNA precursor. Moreover, m5C in rRNA is known to modulate translational fidelity;21 hence, the findings made from the present study suggest that the function of m5C in this process may involve, at least partly, its recognition by YTHDF2. In this context, a limitation of our study resides in that we have not revealed the detailed molecular mechanisms through which genetic depletion of YTHDF2 gives rise to elevated levels of m5C in rRNA and perturbs rRNA maturation; future studies are needed for addressing these mechanistic issues.
In summary, we identified, by using an unbiased quantitative proteomic method, YTHDF2 protein as a reader of m5C in RNA, which expands the repertoire of RNA modifications that can be regulated by this protein. In this vein, polymorphism in the YTHDF2 gene was previously found to be associated with longevity in humans;44 thus, the results from the present study suggest that the function of YTHDF2 in this process could be attributed, in part, to its recognition of m5C in RNA. Furthermore, the data from our SILAC-based quantitative proteomic screening showed that m5C may also be recognized by other cellular proteins. Further characterizations of these proteins in m5C recognition and RNA metabolism will promote our understanding about the biological functions of m5C in RNA.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Prof. Chuan He for providing pGEX-4T-1-YTHDF2 plasmid. This work was supported by the National Institutes of Health (R01 ES025121). X.D. was supported by an NRSA institutional training grant (T32 ES018827).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b04505.
Detailed experimental procedures for Western blot, bisulfite sequencing, Northern blot, structure-based docking, and electrophoretic mobility shift assay; LC-MS/MS quantification data; and Western blot images (PDF)
A list of proteins with relative binding ratios toward m5C-over C-containing RNA identified from SILAC-based affinity screening experiments with the use of lysate of HeLa cells (XLSX)
A list of proteins with relative binding ratios toward m5C-over C-containing RNA identified from SILAC-based affinity screening experiments with the use of lysate of HEK293T cells (XLSX)
A list of m5C sites in HEK293T cells and the isogenic YTHDF2 knockout cells, as obtained from bisulfite sequencing analysis (XLSX)
A list of m5C sites that are commonly identified from the current bisulfite sequencing experiment and from previously published miCLIP analysis (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Cantara WA; Crain PF; Rozenski J; McCloskey JA; Harris KA; Zhang X; Vendeix FA; Fabris D; Agris PF Nucleic Acids Res. 2011, 39, D195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Squires JE; Patel HR; Nousch M; Sibbritt T; Humphreys DT; Parker BJ; Suter CM; Preiss T Nucleic Acids Res. 2012, 40, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Legrand C; Tuorto F; Hartmann M; Liebers R; Jacob D; Helm M; Lyko F Genome Res. 2017, 27, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M; Sorek R; Rechavi G Nature 2012, 485, 201–206. [DOI] [PubMed] [Google Scholar]
- (5).Meyer KD; Patil DP; Zhou J; Zinoviev A; Skabkin MA; Elemento O; Pestova TV; Qian SB; Jaffrey SR Cell 2015, 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mauer J; Luo X; Blanjoie A; Jiao X; Grozhik AV; Patil DP; Linder B; Pickering BF; Vasseur JJ; Chen Q; Gross SS; Elemento O; Debart F; Kiledjian M; Jaffrey SR Nature 2017, 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schwartz S; Bernstein DA; Mumbach MR; Jovanovic M; Herbst RH; Leon-Ricardo BX; Engreitz JM; Guttman M; Satija R; Lander ES; Fink G; Regev A Cell 2014, 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Carlile TM; Rojas-Duran MF; Zinshteyn B; Shin H; Bartoli KM; Gilbert WV Nature 2014, 515, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Khoddami V; Yerra A; Mosbruger TL; Fleming AM; Burrows CJ; Cairns BR Proc. Natl. Acad. Sci. U. S. A 2019, 116, 6784–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bokar JA; Shambaugh ME; Polayes D; Matera AG; Rottman FM RNA 1997, 3, 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- (11).Liu J; Yue Y; Han D; Wang X; Fu Y; Zhang L; Jia G; Yu M; Lu Z; Deng X; Dai Q; Chen W; He C Nat. Chem. Biol 2014, 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ping XL; Sun BF; Wang L; Xiao W; Yang X; Wang WJ; Adhikari S; Shi Y; Lv Y; Chen YS; Zhao X; Li A; Yang Y; Dahal U; Lou XM; Liu X; Huang J; Yuan WP; Zhu XF; Cheng T; et al. Cell Res. 2014, 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jia G; Fu Y; Zhao X; Dai Q; Zheng G; Yang Y; Yi C; Lindahl T; Pan T; Yang YG; He C Nat. Chem. Biol 2011, 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zheng G; Dahl JA; Niu Y; Fedorcsak P; Huang CM; Li CJ; Vagbo CB; Shi Y; Wang WL; Song SH; Lu Z; Bosmans RP; Dai Q; Hao YJ; Yang X; Zhao WM; Tong WM; Wang XJ; Bogdan F; Furu K; et al. Mol. Cell 2013, 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schwartz S; Agarwala SD; Mumbach MR; Jovanovic M; Mertins P; Shishkin A; Tabach Y; Mikkelsen TS; Satija R; Ruvkun G; Carr SA; Lander ES; Fink GR; Regev A Cell 2013, 155, 1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang X; Lu Z; Gomez A; Hon GC; Yue Y; Han D; Fu Y; Parisien M; Dai Q; Jia G; Ren B; Pan T; He C Nature 2014, 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang X; Zhao BS; Roundtree IA; Lu Z; Han D; Ma H; Weng X; Chen K; Shi H; He C Cell 2015, 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).He C Nat. Chem. Biol 2010, 6, 863–865. [DOI] [PubMed] [Google Scholar]
- (19).Motorin Y; Lyko F; Helm M Nucleic Acids Res. 2010, 38, 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hussain S; Aleksic J; Blanco S; Dietmann S; Frye M Genome Biol. 2013, 14, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chow CS; Lamichhane TN; Mahto SK ACS Chem. Biol 2007, 2, 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Brzezicha B; Schmidt M; Makalowska I; Jarmolowski A; Pienkowska J; Szweykowska-Kulinska Z Nucleic Acids Res. 2006, 34, 6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goll MG; Kirpekar F; Maggert KA; Yoder JA; Hsieh CL; Zhang X; Golic KG; Jacobsen SE; Bestor TH Science 2006, 311, 395–398. [DOI] [PubMed] [Google Scholar]
- (24).Fu L; Guerrero CR; Zhong N; Amato NJ; Liu Y; Liu S; Cai Q; Ji D; Jin SG; Niedernhofer LJ; Pfeifer GP; Xu GL; Wang YJ Am. Chem. Soc 2014, 136, 11582–11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huber SM; van Delft P; Mendil L; Bachman M; Smollett K; Werner F; Miska EA; Balasubramanian S ChemBioChem 2015, 16, 752–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yang X; Yang Y; Sun BF; Chen YS; Xu JW; Lai WY; Li A; Wang X; Bhattarai DP; Xiao W; Sun HY; Zhu Q; Ma HL; Adhikari S; Sun M; Hao YJ; Zhang B; Huang CM; Huang N; Jiang GB; et al. Cell Res. 2017, 27, 606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dai X; Otake K; You C; Cai Q; Wang Z; Masumoto H; Wang YJ Proteome Res. 2013, 12, 4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cox J; Mann M Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (29).Fu L; Amato NJ; Wang P; McGowan SJ; Niedernhofer LJ; Wang Y Anal. Chem 2015, 87, 7653–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ong SE; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (31).Dai X; Wang T; Gonzalez G; Wang Y Anal. Chem 2018, 90, 6380–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhu T; Roundtree IA; Wang P; Wang X; Wang L; Sun C; Tian Y; Li J; He C; Xu Y Cell Res. 2014, 24, 1493–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li F; Zhao D; Wu J; Shi Y Cell Res. 2014, 24, 1490–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xu C; Wang X; Liu K; Roundtree IA; Tempel W; Li Y; Lu Z; He C; Min J Nat. Chem. Biol 2014, 10, 927–929. [DOI] [PubMed] [Google Scholar]
- (35).Schwartz S; Mumbach MR; Jovanovic M; Wang T; Maciag K; Bushkin GG; Mertins P; Ter-Ovanesyan D; Habib N; Cacchiarelli D; Sanjana NE; Freinkman E; Pacold ME; Satija R; Mikkelsen TS; Hacohen N; Zhang F; Carr SA; Lander ES; Regev A Cell Rep. 2014, 8, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhou J; Wan J; Gao X; Zhang X; Jaffrey SR; Qian SB Nature 2015, 526, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hussain S; Sajini AA; Blanco S; Dietmann S; Lombard P; Sugimoto Y; Paramor M; Gleeson JG; Odom DT; Ule J; Frye M Cell Rep. 2013, 4, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hadjiolova KV; Nicoloso M; Mazan S; Hadjiolov AA; Bachellerie JP Eur. J. Biochem 1993, 212, 211–215. [DOI] [PubMed] [Google Scholar]
- (39).Rouquette J; Choesmel V; Gleizes PE EMBO J. 2005, 24, 2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Aubert M; O’Donohue MF; Lebaron S; Gleizes PE Biomolecules 2018, 8, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xu C; Liu K; Ahmed H; Loppnau P; Schapira M; Min JJ Biol. Chem 2015, 290, 24902–24913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Luo S; Tong L Proc. Natl. Acad. Sci. U. S. A 2014, 111, 13834–13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhou J; Wan J; Gao X; Zhang X; Jaffrey SR; Qian SB Nature 2015, 526, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cardelli M; Marchegiani F; Cavallone L; Olivieri F; Giovagnetti S; Mugianesi E; Moresi R; Lisa R; Franceschi CJ Gerontol., Ser. A 2006, 61, 547–556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.