

Graphical Abstract
INTRODUCTION TO GAS-PHASE ION/ION REACTIONS
While the study of reactions between oppositely-charged ions has a long history, particularly within the context of plasmas and interstellar chemistry, the exploitation of such reactions for analytical applications was made possible with the advent of electrospray ionization (ESI).1,2 ESI provides a means for generating abundant multiply-charged ions from a wide variety of molecular classes. It was then possible to use ESI to generate multiply-charged analyte or reagent ions for reactions with ions of opposite polarities without resulting in complete neutralization. The first description of ion/ion reaction studies using ESI was given by Loo et al. using a Y-tube reactor leading to the inlet of an atmosphere/vacuum interface coupled with a quadrupole mass filter.3,4 This was an early example of ion/ion chemistry being effected prior to sampling ions into a mass spectrometer. The first implementation of ion/ion reactions within the context of a MSn experiment was demonstrated at Oak Ridge National Lab.5,6 This was accomplished through the use of electrodynamic ion traps, which allow ions of opposite polarity to be stored in overlapping regions of space. Since those early reports, the use of ion/ion reactions, both in vacuo and ex vacuo, within the context of molecular analysis has expanded significantly.
A number of characteristics make ion/ion reactions attractive for analytical mass spectrometry. For example, given the long-range Coulomb attraction associated with oppositely-charged ions, the cross-sections for ion/ion collisions are very large such that reactions can be driven on the millisecond to sub-millisecond time scales, depending upon ion densities and extents of ion overlap. Another important characteristic is that mutual neutralization is highly exothermic for virtually all combinations of oppositely-charged gas-phase ion/ion reactions. Hence, ion/ion reactions are highly efficient and always result in some kind of reaction. Given the diversity of ion types that can be generated by the suite of ionization methods now available, the range of ion/ion reactions that can be effected is extremely large, even larger than that of ion/molecule reactions, which are limited by volatility constraints. In the case of ion/ion reactions that take place within a tandem mass spectrometer, such as an ion trap or hybrid instrument, the fact that ions are readily manipulated on a time-dependent basis (i.e., they can be selected or ejected either selectively or non-selectively) enables a high degree of control over the identities of the reactants and the times over which they are exposed to one another.
The attractive characteristics of ion/ion reactions for multiple analytical applications have been illustrated in reports going back to the original works described above. Many of these have been reviewed previously.7-9 In this review, we emphasize developments that have taken place largely within the past decade with particular emphasis on the last five years. These have included developments in tools used to implement ion/ion reactions for one or more types of mass spectrometry experiments, an expansion in the range of ion/ion reaction types, and the growth in analytical applications of ion/ion chemistry. The review is organized with descriptions of instrument development, proton transfer chemistry, electron transfer chemistry, and reactions that can proceed only through long-lived complexes.
DEVELOPMENTS IN ION/ION REACTION INSTRUMENTATION
Scientific progress is directly related to and dependent on the development of instrumentation. In this respect, the increasing adoption of ion/ion reactions as an analytical tool within the past half-decade stems from the development of novel instrumentation equipped to perform such experiments. The commercialization of electron transfer dissociation (ETD), and more recently proton transfer charge reduction (PTCR), has emerged as, perhaps, the most significant advancement in ion/ion reaction instrumentation, bringing ion/ion reaction capabilities to laboratories around the world. Previously, research in the area of ion/ion reaction chemistry was exclusively limited to home-built instruments or heavily modified commercial instruments. The evolution of such instrumentation has been reviewed.7-10 In this section, rather than providing a complete history of ion/ion reaction instrumentation, we focus on the recent developments since the last comprehensive review of instrumentation in 2008.10 These developments are described in the context of three categories: ion/ion reactions performed outside the mass spectrometer, ion/ion reactions performed in vacuo, and the analysis of high m/z ions.
Front-end Ion/Ion Reaction Instrumentation
Several criteria must be met for a gas-phase ion/ion reaction to occur, chief among them is the temporal and spatial overlap of oppositely charged ions. In the case of reactions proceeding at or near atmospheric pressure, this is accomplished prior to the introduction of the ions into the vacuum system. As such, the modifications required to perform gas-phase ion/ion chemistry at atmospheric pressure are localized to the ionization region, specifically prior to the entrance to the atmosphere/vacuum interface. Recently, there have been several reports of modified or repurposed commercial ionization sources used for front-end ion/ion reactions.11-13
Two reports of instrumentation highly analogous to that described by L. M. Smith and co-workers14,15 involved the modification of commercial ionization sources. In one report, the ESI source of a LC/MS platform was outfitted with a polonium-210 (210Po) α-particle source.11 The emitted α-particles initiate ion/molecule reactions involving ambient gas and solvent molecules generating ions of both polarities. Subsequent reactions between the α-particle progeny ions and the analyte ions generated via ESI result in charge reduced analyte ions, which are then sampled into the mass spectrometer for mass analysis. In another report, both an ESI and a nESI source of a traveling wave ion mobility mass spectrometr (TWIM MS) were modified to contain a corona discharge probe.12 The discharge generates anions from a nitrogen gas supply that is flowed over the platinum wire/discharge plate. Ensuing proton transfer ion/ion reactions result in the charged reduced protein species. The extent of charge reduction can be modulated by varying the flow rate of nitrogen or the applied voltage to the discharge needle. Robb et al. have demonstrated similar charge reduction reactions by simply repurposing a commercial atmospheric pressure ECD source.13
In addition to modifying commercial ionization sources as described above, commercial sources can be replaced altogether with modular front-end ionization sources designed to perform ion/ion reactions. For example, the Brodbelt group constructed a free-standing dual ionization source reactor that can be mounted to the front-end of any mass spectrometer.16 The reactor consists of two electrosonic spray ionization sources fixed to a U-shaped rail. Permitting enhanced control, the angle of the two sources relative to one another and relative to the inlet of the mass spectrometer can be adjusted (Figure 1). The first source is integrated with the instrument such that the voltage and polarity are controlled with the instrument software. The voltage of the second source is supplied by an external dual polarity high voltage power supply.
Figure 1.

Dual source reactor used to perform ion/ion reactions (a) mounted to the front end of a mass spectrometer and (b) free standing to be adapted to any mass spectrometer platform. Reprinted from the supporting information of Cotham, V. C.; Shaw, J. B.; Brodbelt, J. S. Anal. Chem. 2015, 87, 9396-9402 (ref 16). Copyright 2015 American Chemical Society.
With this setup, ion/ion reactions are conducted in dual spray mode where the two sources are operated simultaneously. One source generates a plume of multiply charged peptide cations while the other generates a mixture of singly and doubly charged reagent anions. The two sources are oriented at an angle such that collisions between oppositely charged droplets, pseudodroplets, and gas-phase ions are maximized. Experimental evidence suggests that the reactions proceed exclusively through an ion/ion mediated pathway; the abundance of the peptide/reagent complex is directly related to the anion source voltage. Additionally, when the anion source voltage is zero, reminiscent of an ion/molecule reaction, the peptide/reagent complex is not observed. Stutzman and co-workers have performed similar experiments to demonstrate the charge reduction of synthetic polymers with a bipolar dual spray setup.17 Despite the setup being highly analogous to the dual spray reactor described above, we note a lack of conclusive evidence suggesting exclusively ion/ion chemistry. Nonetheless, it represents an intriguing avenue for charge state manipulation performed ex vacuo.
In Vacuo Ion/Ion Reaction Instrumentation
While instrumentation equipped to perform front-end ion/ion reactions offer some advantages (i.e. compatibility with any mass analyzer, simplicity of modifications, etc.), the vast majority of ion/ion reactions are performed in vacuo. There are several distinct advantages to performing experiments within the confines of an electrodynamic trap: (1) independent control/optimization of reactant species, (2) well defined reaction conditions, and (3) MSn capabilities in conjunction with ion/ion reactions. Ion/ion reactions performed within the mass spectrometer have historically been conducted in either a 3-D quadrupole ion trap or a 2-D linear ion trap. Compared to 3-D traps, linear ion traps have a larger ion capacity and are more efficiently coupled to ionization sources and other devices. Consequently, apart from an IT-IM-TOF instrument which couples a 3-D ion trap with an ion-mobility cell,18 reactions performed in linear ion traps constitute the bulk of recent literature.
Much of the work surrounding ion/ion reactions performed in both quadrupolar and hexapolar linear ion traps originates from the development and optimization of the now commercialized ETD instruments. ETD equipped instrumentation has been discussed in detail by Riley and Coon.19 Therefore, it is outside the scope of this review to thoroughly discuss the instrumentation and methodological developments of ETD. The ion traps in which ETD reactions are performed, however, can serve as reaction regions for any ion/ion reaction. In this context, some of the same instrumentation is mentioned as it pertains to ion/ion reactions other than ETD.
As mentioned above, any electrodynamic ion trap with mutual storage capabilities can be used to perform ion/ion experiments, provided that ions of opposite polarity can be generated and transported to the same trap. This can be accomplished by the generation of oppositely-charged ions from two different sources and subsequently introducing ions through two separate orifices or through a common orifice. Instruments of both types have been described in recent years. He et al. described a dual polarity ion trap mass spectrometer of the former type.20 The instrument consists of two ionization sources and two sets of ion optical systems separated by a segmented linear ion trap in the middle. Ions are generated using the two sources and can be transported to the linear ion trap where the reaction proceeds. Application of dipolar DC to one rod set enables the simultaneous detection of positive and negative ions of the same reaction experiment.
Lin et al. also described an instrument which ions are introduced through separate orifices.21 Their home-built mass spectrometer consists of two discontinuous atmospheric pressure interfaces (DAPI) 180° from one another, one rectilinear ion trap, and one detector. Analyte ions are generated by DAPI I, introduced into the trap through its respective orifice, and, if desired, isolated using custom waveforms. Reagent anions generated by DAPI II are then introduced into the trap. Next, the two populations are allowed to react. However, opening the pinch valve of the DAPI results in an increased pressure of the trap which, in turn, could have deleterious effects on the ion/ion reactions. In fact, the proton transfer product ion spectrum shows two low abundance fragment ions which likely originate from the energy deposition by gas flow resulting from the sudden change in pressure from opening of DAPI II. A static higher base pressure is more desirable as it serves to enhance ion/ion reaction efficiency. However, Campbell and Hager have demonstrated ion/ion reactions in a low pressure reaction cell.22 They created an evanescent ion/ion reaction region within the low pressure region of Q3 of a hybrid triple quadrupole/linear ion trap mass spectrometer by adding DC and RF voltages to the optical components surrounding Q3 and by adding a pulsed valve for the addition of a cooling gas. The results of the ion/ion reactions performed in this low pressure region were similar to that of the higher pressure region.
Early versions of ETD equipped instrumentation contained a chemical ionization source at the rear of the instrument.23,24 Once generated, the reagent radical anions are delivered through the C-trap and into the segmented linear ion trap where the ETD reaction would take place. It wasn’t long before this platform was used for proton transfer reactions.25 In 2013, Hunt and co-workers described a front-end ETD source.26 Here, cations and anions can be generated and introduced through a common orifice at the front of the instrument. Since its inception, the front-end chemical ionization source has been implemented on a number of research grade instruments including the 21 Tesla (T) Fourier transform-ion cyclotron resonance (FTICR) mass spectrometer at the National High Magnetic Field Laboratory.27 Additionally, this source serves as the basis for the newest Orbitrap hybrid systems, including those with PTCR capabilities.
A common characteristic of all the instruments described above is the use of a linear ion trap as the reaction vessel. Ion/ion reactions are not restricted to linear ion traps though. The use of a traveling wave (T-wave) cell as a reaction vessel has been reported.28-31 Cations and anions are generated at the front of the instrument. The optical polarities are alternately switched, and the quadrupole ion guide is set to transmit either exclusively precursor cations or reagent anions into the T-wave trap cell. The reaction proceeds in this cell as the ion populations are propelled through one another by the T-wave. Similarly, a traveling wave structure for lossless ion manipulations (TW SLIM) device capable of performing ion/ion reactions has been described.32 Here, one SLIM surface contains four alternating phase RF electrodes interweaved with three traveling wave electrodes (Figure 2a). A second SLIM surface contains an identical configuration carrying the opposite phase on every RF electrode as shown in Figure 2b. Simulations suggest ion/ion reactions in this device are feasible but have yet to be demonstrated experimentally.
Figure 2.

(a) Schematic of the SLIM boards used for dual polarity ion confinement looking at the x-y plane (left) and z-y plane (right). (b) 3-D view of the SLIM device showing the ion conduits. The green line represents the equidistant line between the SLIM surfaces. Reprinted by permission from Springer Nature: Journal of the American Society for Mass Spectrometry, Garimella, S. V. B.; Webb, I. K.; Prabhakaran, A.; Attah, I. K.; Ibrahim, Y. M.; Smith, R. D. J. Am. Soc. Mass Spectrom. 2017, 28, 1442-1449 (ref 32). Copyright 2017.
Analysis of High m/z Product Ions from Ion/Ion Reactions
Ion/ion reaction product ions can be one or more orders of magnitude higher in m/z than the reactant ions. This is especially true for proton transfer ion/ion reactions involving proteins of relatively high mass (e.g. > 30 kDa). There is, however, a number of challenges associated with generating and analyzing high mass-to-charge ions, both directly from electrospray ionization and from ion/ion proton transfer reactions. In the case of an ion/ion reaction experiment in trapping instruments, the upper m/z limit is dependent on (1) the detector response, (2) the range over which ions of disparate m/z can be mutually stored, and (3) the efficiency in which high m/z ions can be mass selectively ejected towards the detector. Our group has demonstrated a waveform switching technique that directly addresses the third challenge.33 A schematic of the instrument is provided in Figure 3. A high frequency sine wave of 1.008 MHz is applied to the ring electrode during ion injection and mutual storage. After the ion/ion reaction period, a low frequency square wave is applied to the end cap electrodes, the sine wave is turned off, and product ions are mass selectively ejected via a frequency scan of the square wave. Switching from a high frequency sine wave operation of the trap to a low frequency square wave scan during mass analysis eliminates an upper m/z limit associated with voltage constraints. Additionally, the reduction in frequency places the product ions at higher q values, thereby, generating deeper well depths and smaller ion packets despite using lower amplitudes. Using this method, the upper m/z limit of the ion/ion reaction was improved by a factor of 2-3.
Figure 3.

Schematic of the instrumental setup for the waveform switching experiments. The sine wave is applied to the ring electrode and the square wave is applied to the end cap electrodes. Reprinted by permission from Springer Nature: Journal of the American Society for Mass Spectrometry, Lee, K. W.; Eakins, G. S.; McLuckey, S. A. J. Am. Soc. Mass Spectrom. 2019, 30, 1126-1132 (ref 33). Copyright 2019.
In addition to the development in high m/z analysis described above, recent modifications to the Orbitrap mass spectrometer platform have been aimed at increasing the transmission, isolation, fragmentation, and resolution of high m/z ions.34-38 Modifications include orthogonal ion injection into the instrument to reduce instrument contamination, in-source trapping to facilitate ion desolvation and to minimize the ions’ axial momentum, reduction of the RF frequencies applied to the bent flatapole, quadrupole, transfer multipole, C-trap, and HCD-cell, lengthening ion injection times from the C-trap to the Orbitrap mass analyzer, and changes to the image current preamplifier to detect a broader range of ion frequencies. Implementation of these modifications have enabled the detection of high m/z ions up to m/z 70,000.39 To date, no ion/ion reactions have been performed on this platform. However, it stands to reason these developments can lend themselves to eventually couple ion/ion reactions to high mass-to-charge protein or protein complex ions.
PROTON TRANSFER ION/ION REACTIONS
The first proton transfer ion/ion reactions were conducted in the early 1990’s using the front-end Y-tube reactor.3,4 Since then, altering ion charge states via gas-phase proton transfer has developed into a robust and mature technique that has found utility in biological mass spectrometry, primarily in the charge state manipulation of proteins. Much of the early work devoted to proton transfer for protein analysis was performed in 3-D ion traps.6,40-43 As the resolution of ion traps is generally limited, identification of ion charges greater than two can be a nontrivial task when adjacent charge states from an analyte of interest are not clearly identifiable. Proton transfer ion/ion reactions, therefore, have historically been used to simplify spectra containing highly charged ions by decreasing ion charges to mostly 1+ or 2+ and by dispersing ions across a wide mass-to-charge range. Increased resolving power can enable charge state determination via measurement of the isotope spacings within a single charge state. Yet, it has been shown that even high-resolution instruments, such as the Orbitrap and FT-ICR mass spectrometers, can benefit from using proton transfer reactions when complex mixtures of ions are present.44 In recent years, as the complexity of protein mixtures subjected to ESI has increased, there has been an extension of proton transfer workflows to high-resolution instruments. Applications of proton transfer ion/ion reactions include MSn product ion analysis, precursor ion charge state manipulation, and precursor ion signal concentration. Additionally, proton transfer reactions are used in structural proteomic studies. The following sections feature recent work describing proton transfer reactions in all these application areas.
Product Ion Analysis
Several activation methods, including electron transfer dissociation and ultraviolet photodissociation (UVPD), have emerged with a common objective to enhance structural characterization by maximizing the number of sequence informative product ions. However, fragment ions produced using these techniques typically result in a product ion spectrum in which fragments are confined to a relatively narrow m/z range centered about the precursor. When combined with proteins of relatively large size and the multiplicity of fragment ion charge states, the resulting product ion spectrum can be quite complex. In 2005, the Hunt group used sequential ion/ion reactions (i.e. electron transfer dissociation followed by proton transfer reactions) for protein identification.25 ETD of the [M+13H]13+ charge state of ubiquitin performed on a Finnigan LTQ mass spectrometer generated a spectrum that was uninterpretable. However, using proton transfer on the ETD products, approximately 90% of the residues could be sequenced. The utility of this ETD-proton transfer workflow is further demonstrated by the identification of forty-six E. coli 70S ribosomal proteins.45
Expanding upon the work performed in ion trap mass spectrometers,25,46-48 Hunt and co-workers have recently coupled the ETD-proton transfer workflow with Orbitrap mass analysis.49 In this experiment, a multiply charged protein ion was isolated and fragmented by electron transfer dissociation, resulting in a complex product ion spectrum, as shown in Figure 4a. Following ETD, product ions were charge reduced via proton transfer to sulfur hexafluoride radical anions. Figures 4b - 4e show the resulting charge reduced spectra under identical ETD parameters but increasing proton transfer reaction durations. Interestingly, the most informative reaction duration was observed to be the 20 ms ion/ion proton transfer reaction. ETD followed by proton transfer on the Orbitrap platform has been used in only a few other studies,50-52 likely due to the lack of a commercial instrument optimized to perform both types of ion/ion reactions. An increase in the number of applications utilizing ETD followed by proton transfer can be anticipated with the commercialization of PTCR on ETD equipped instruments.
Figure 4.

ETD product ion spectrum of the [M + 26H]26+ charge state of apomyoglobin followed by sequential ion/ion proton transfer (IIPT) reaction for (a) 0 ms, (b) 20 ms, (c) 40 ms, (d) 80 ms, and (e) 160 ms. Reprinted from Int. J. Mass Spectrom., Vol. 377, Anderson, L. C.; English, A. M.; Wang, W.; Bai, D. L.; Shabanowitz, J.; Hunt, D. F. Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry, pp. 617–624 (ref 49). Copyright 2015, with permission from Elsevier.
Precursor Charge State Manipulation
In addition to reducing spectral complexity during product ion analysis, proton transfer can be used to produce simpler spectra during MS1 experiments. Laszlo and Bush demonstrate that proton transfer can increase accuracy in assigning charge states of native proteins and protein complexes generated via ESI.53 With ESI under denaturing conditions, as the size of the analyte increases, the propensity for generating multiply charged ions increases potentially creating overlap between charge state distributions of different species even for relatively simple mixtures. For complex mixtures, the overlap can be quite extensive, complicating data interpretation. This problem is exacerbated with polydisperse polymers, where the distributions are broad spanning, in some cases, tens of thousands of Da. Recently, proton transfer ion/ion reactions have been used to alleviate the spectral congestion of large synthetic polymers.12,13 In those studies, extensive charge reduction via proton transfer was shown to enable facile data interpretation for polymers up to 40 kDa. Other ion/ion charge reduction strategies have been reported for polymer characterization, though it is unclear if the charge reduction occurs via proton transfer.11,17
Most examples described above (i.e. product ion analysis and polymer analysis) utilize proton transfer reactions to charge reduce analyte ions to mainly the 1+ charge state. However, in some scenarios, it is desirable to perform less extensive charge reduction. For instance, our group has used proton transfer reactions to manipulate precursor ion charge in order to study the charge state dependent collisional activation of proteins.54-59 This technique has been adopted to study the charge state dependent fragmentation of proteins utilizing ETD and UVPD activation methods.60-63
In 2016, the Brodbelt group reported on the photodissociation patterns of native proteins following gas-phase proton transfer.62 While only minor changes in fragmentation were observed, this report demonstrates another attractive application for proton transfer reactions. ESI of bovine superoxide dismutase (SOD)/CuZn complexes under native conditions generates the mass spectrum shown in the top panel of Figure 5a. Close examination of the peak at approximately m/z 3140 shows an overlap of 5+ monomers with 10+ dimers (Figure 5b, top). This overlap inhibits the independent MS/MS analysis of the monomer or dimer distribution. Proton transfer, on the other hand, can generate purified distributions of monomer and dimer. For example, a clean 5+ monomer distribution and a clean 10+ dimer distribution is generated from proton transfer of the 12+/6+ distribution (Figure 5, middle row) and from proton transfer from the 11+ dimer (Figure 5, bottom row), respectively. Here, proton transfer is used to purify charge state distributions in the gas-phase. Gas-phase charge state purification via proton transfer reactions involving protein mixtures under denaturing conditions has also been demonstrated,41,42 and the Coon group extended this approach to proteomic quantitation with isobaric tagging.64,65
Figure 5.

Mass spectra of SOD/CuZn complexes (a) as observed under native MS conditions (top), isolation and 25 ms proton transfer reaction of the 12+/6+ species (middle), and isolation and 25 ms proton transfer reaction of the 11+ species (bottom) with (b) a 60 m/z wide zoomed view of the 10+/5+ region. Adapted from Holden, D. D.; Brodbelt, J. S. Anal. Chem. 2016, 88, 12354-12362 (ref 62). Copyright 2016 American Chemical Society.
The kinetics of ion/ion reactions can play a major role in determining product ion distributions, particularly when mixtures of ions of widely different charges are subjected simultaneously to reaction. Again, using the SOD/CuZn data as an illustrative case, when both the 12+ dimer and 6+ monomer are subjected to proton transfer ion/ion reactions, the resulting spectrum shows only a 5+ monomer distribution with no evidence of the isobaric 10+ dimer species. This follows from the charge squared dependence associated with ion/ion reaction rates.40,66 That is to say, the sequential charge reduction reactions of the 12+ dimer react at a faster rate than the 6+ monomer species. Consequently, the entire 10+ population is depleted, having reacted to lower charge states, leaving a relatively enriched purified monomer distribution.
A degree of control over ion/ion reaction kinetics can be effected in a process termed “ion parking.”67 This is accomplished by applying a low amplitude auxiliary RF signal at the secular frequency of the desired product ion. The applications of ion parking are numerous. Many of the studies in the past decade involving ion/ion reactions utilized ion parking to enhance sensitivity, including some examples discussed above.51,61,62 Recently, we have used ion parking to concentrate precursor ion signal prior to moving the ions to a m/z region likely to maximize cleavage at aspartic acid and proline residues.68 The Hunt group implemented “parallel ion parking”69,70 during the on-line HPLC, data-dependent MS/MS analysis of the E. coli ribosome (Figure 6).71 Here, a broadband parallel ion parking waveform is applied to concentrate ion signal without a priori knowledge of the protein (compare signal intensities of Figures 6a and 6b). Campbell and Le Blanc employed ion parking to impart an additional degree of selectivity during protein quantitation,72 and in 2008, L. M. Smith and co-workers combined fixed-charge derivatization and ion parking to generate an abundant precursor at a charge state not generated under conventional electrospray conditions.73 Ion parking provides a high degree of flexibility in mixture analysis applications due both to the concentration of multiple charge states into one or a few charge states, which is attractive for sensitivity, and the simplification of the precursor ion spectrum, which can improve specificity.
Figure 6.

MS/MS analysis scheme of 50S L22 E. coli ribosomal protein. (a) Positive electrospray mass spectrum. (b) 100 ms proton transfer and parallel ion parking of the 600 – 1000 m/z isolation window. (c) HCD product ion spectrum of the data-dependently selected 10+ precursor. Reproduced from Ugrin, S. A.; English, A. M.; Syka, J. E. P.; Bai, D. L.; Anderson, L. C.; Shabanowitz, J.; Hunt, D. F. J. Am. Soc. Mass Spectrom. 2019 DOI: 10.1007/s13361-019-02290-8 (ref 71).
Structural Proteomic Studies
Native mass spectrometry is increasingly being adopted as a tool for structural biology, allowing researchers to extract more information from a mass spectrometry experiment than simply molecular weight.74-77 Coupling ion-mobility with native MS facilitates native protein and protein complex collisional cross section measurements. Those measurements, however, are obtained from the gas-phase structures of the analyte, and unfortunately, there is still a great deal of ambiquity regarding the relation of gas-phase structure with those observed in solution. Expanding upon the early work of Badman and co-workers,18,78 the Bush group is combining native mass spectrometry, gas-phase proton transfer, and ion-mobility to study structure/charge relationships and protein folding.79-83 Lermyte et al. perform similar experiments, but, in their case, an electron transfer reagent anion, 1,4-dicyanobenzene, is used for charge reduction.84 Using 1,4-dicyanobenzene leads to a combination of electron transfer and proton transfer.
ELECTRON TRANSFER ION/ION REACTIONS
Because of the implementation of ETD across multiple commercially available instrument platforms, electron transfer ion/ion reactions can be considered the most commonly employed type of ion/ion reaction. The ETD process begins with an ion/ion reaction involving the transfer of an electron from a reagent radical anion to a multiply charged analyte cation, viz.:
| (1) |
Fragmentation of the charge-reduced product via radical directed mechanisms results, in the case of multiply-charged polypeptide cations, in c- and z•-type fragment ions and extensive primary sequence information.23 Since its introduction in commercially available instruments, ETD has become a standard approach for the structural characterization of bio-ions. The majority of electron transfer ion/ion reactions are discussed in the context of ETD and negative electron transfer dissociation (NETD). Consequently, ETD and NETD have been extensively reviewed,19,85-88 with the latest review being published in 2018.19 Therefore, ETD is not discussed here. Instead, we highlight other electron transfer ion/ion reactions and their analytical utilities. Specifically, charge reduction via electron transfer and charge transfer dissociation are discussed.
Electron Transfer Charge Reduction
Electron transfer from a reagent radical anion to a multiply charged cation does not always lead to spontaneous dissociation to generate product ions. Non-dissociative electron transfer, often referred to as “electron transfer no dissociation” (ETnoD), gives rise to an intact charge reduced product ion. The extent of ETnoD has been shown to be dependent on several factors including protein ion conformation, precursor ion charge state, degree of supplemental activation, etc.84,89 This non-dissociative channel is undesirable when ETD is the objective, yet several studies exploit the formation of ETnoD products. The Kaltashov group, for example, employ native electrospray ionization in combination with electron transfer90-93 or electron capture94,95 charge reduction to reduce the spectral complexity of heterogeneous samples. Figure 7 shows the native ESI mass spectrum of an antithrombin-III/heparin complex (AT/heparin) in gray. Mass selection of the precursor ion and subsequent electron transfer charge reduction generates the product ion spectra shown in pink and cyan (Figure 7). The value of this strategy stems from the ability to correctly assign charges from the resulting mass spectrum that is reduced in spectral complexity. By selecting a narrower population of precursor ions, interpretable mass information can be extracted (compare magenta and cyan traces).91 Lermyte et al. performed similar electron transfer ion/ion reactions, though, in their case, precursor ions were reduced to the 1+ charge state.96
Figure 7.

The electrospray mass spectrum of a AT/heparin complex is shown in gray. Ion charge state assignment is facilitated by limited electron transfer charge reduction using wide (pink) and narrow (cyan) precursor ion selection windows. The species labeled A, B, and C represent resolved charge-reduced species present in the precursor isolation windows. Reproduced from Zhao, Y.; Abzalimov, R. R.; Kaltashov, I. A. Anal. Chem. 2016, 88, 1711-1718 (ref 91). Copyright 2016 American Chemical Society.
Beyond charge assignment and mass determination, electron transfer charge reduction has proven to be useful in both protein conformational studies and in the generation of metal species in unusual oxidation states. In the former case, the effect of charge reduction on protein conformation has been studied by coupling ETnoD with ion-mobility, providing insights into the importance of net charge in the gas-phase on the extent of protein compaction.84,97,98 In the latter case, the transfer of a single electron from a radical anion to a metal-ligand complex, M(II)Lx, serves to reduce the metal forming the M(I)Lx complex ion. Gronert first used this technique to study the reactivity of metals in the 1+ oxidation state towards allyl iodide.99 Later, Oomens and co-workers used gas-phase reduction via electron transfer to study the coordination environment of several reduced metals and their ligands.100,101
Charge Transfer Dissociation
Charge transfer dissociation (CTD), pioneered by the Jackson lab, is another electron transfer based fragmentation technique. Unlike most ion/ion and ion/electron fragmentation techniques in which reactions proceed through cation/anion interactions, the electron transfer of CTD occurs between two cationic species.102 Reactions of ions of like charge are characterized by a large Coulomb barrier that must be overcome by a large relative translation. Specifically, analyte cations are irradiated with a beam of 6 keV helium cations, leading to the abstraction of an electron by the helium ion:
| (2) |
Ultimately, this electron hole initiates radical directed fragmentation. Using cations at 6 keV overcomes the electrostatic repulsive barrier associated with cation/cation collisions. Additionally, helium is intentionally chosen as the cation of choice for CTD due to its adiabatic recombination energy of 24.6 eV, the largest of any 1+ ion, which drives the electron abstraction process from other singly charged cations. In addition to the one-electron oxidation pathway discussed above, experimental evidence exists to suggest a one-step two-electron oxidation pathway.103 Compared to ETD, CTD is a unique high energy dissociation method with the advantage of inducing fragmentation of singly charged precursor ions. To date, CTD has been applied to the analysis of peptides,102-104 phospholipids,105 and oligosaccharides.106,107
REACTIONS THAT PROCEED THROUGH COMPLEX FORMATION
Gas-phase ion/ion reactions between oppositely charged ions proceed through the formation of a Coulombically-bound orbit.108 Initially, the distance between the ions in the orbit can exceed that necessary for chemistry to occur. As translational energy is removed via collisions and/or tidal effects,109 the size of the orbit decreases until either a small charged particle (i.e. proton or electron) is transferred at a crossing point on the potential energy surface or a physical collision occurs resulting in formation of a collision complex. While all types of ion/ion reactions can take place through complex formation, many reaction types can only take place through complex formation. More so, complex lifetime is largely determined by the strength of the electrostatic interactions, the number of degrees of freedom of the complex, and both the emissive and collisional cooling rates. Examples of ion/ion reactions that can proceed only through a complex include, but are not limited to, metal ion transfer, charge inversion, and covalent chemistries. Recent work from all areas are highlighted below.
Metal Ion Transfer
Ion/ion reactions involving metal-ligand complexes can be used to insert metal cations into analyte ions. These reactions have been demonstrated in both polarities:
| (3) |
| (4) |
where M represents the analyte, Met represents the metal cation, L represents a singly charged anionic ligand, and Phen represents the neutral 1,10-phenanthroline ligand.110-115 In the case where the analyte is a singly deprotonated anion, ion/ion reaction with a doubly charged cationic metal phenanthroline complex results in the charge inversion of the analyte:
| (5) |
This case will be discussed in further detail in the charge inversion subsection below.
The reaction presented in Equation 3, for example, has been used to incorporate gold cations into disulfide containing polypeptides using AuCl2−.116-118 In all cases, gold cationization showed a preference for cleavage of disulfide bonds upon collisional activation, in contrast with the collisional activation of the corresponding protonated species. Recently, gold (I) cations have been incorporated into polypeptides lacking disulfide bonds.119,120 It was found that gold (I) cationization facilitates peptide oxidation at a neutral lysine residue via the loss of gold hydride and a molecule of ammonia. The resulting structure contains a fixed charge cyclic imine, which weakens the adjacent amide bond. Upon subsequent activation, facile fragmentation N-terminal to the oxidized residue is oberseved.119 This so called “weak-spot” at lysine was exploited for cyclic peptide analysis providing a site-specific ring opening pathway. Collisional activation of the oxidized cyclic peptide was shown to lead to unambiguous sequence information.120
Whereas metal transfer from a reagent to the analyte is highlighted above, gas-phase ion/ion reactions for selective alkali metal removal have also been demonstrated.121,122 Luongo et al. investigated a series of weakly coordinating anions and demonstrated that, of the reagents examined, carborane anions (e.g., CHB11Cl11−) were the most selective for the removal of alkali metals.122 Later, Betancourt et al. simplified the electrospray mass spectra of Polysorbate 80 via ion/ion reaction with carborane anions.123 ESI of Polysorbate 80 shows overlapping distributions from multiple ion types and multiple charge states below m/z 1400. The ion/ion reaction between Polysorbate 80 cations and carborane anions pushed the charge states to largely singly charged species. That is to say, doubly charged species adducted one carborane anion and triply charged species adducted two carborane anions. Broadband collisional activation of the product ions results in the removal of the metal cations with carborane. The product ion spectrum is less congested than the electrospray mass spectrum and shows clear distributions of singly charged, metal cationized species.123
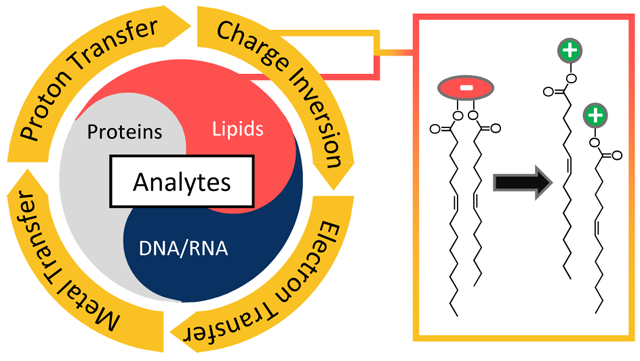
Charge Inversion
Multiple charges (i.e. two or more) can be transferred during a single intimate collision between two ions. For example, ion/ion reaction between a singly protonated phosphatidylethanolamine (PE) and doubly deprotonated 1,4-phenylenedipropinoic acid (PDPA) generates the singly deprotonated PE via the transfer of two protons from the PE cation to the PDPA anion.124 In this case, the charge of the PE analyte ion is inverted from positive to negative polarity. Charge inversion reactions of both ion polarities have been reported.125,126 Our group has been investigating the utility of charge inversion ion/ion reactions and have demonstrated their applications in sulfo- and phosphopeptide characterization,127,128 concentration of multiple cation types to a single anion type,129 and reduction of chemical noise.130 Sequential charge inversion reactions have been used to increase ion charge.131,132
Charge inversion has proven particularly useful in altering the ion type of fatty acids and glycerophospholipids to yield structurally informative ion types. For example, Stutzman et al. used PDPA dianions to convert phosphatidylcholine (PC) monocations to the structurally informative [PC − CH3]− anion species. Ion/ion reaction between PC cations and PDPA dianions formed the [PC − H + PDPA]− complex anion, and subsequent CID of the charge inverted anion resulted in concomitant proton transfer and methyl cation transfer.133 Here, the demethylated PC anions afforded acyl chain information, whereas the PC monocations fragment predominantly to give a choline head group product ion. Reactions with PDPA have also been applied to isomeric mixtures of PC and PE lipids. Here, an isomeric mixture of PC and PE cations are reacted in the gas-phase with PDPA dianions, ultimately resulting in the chemical separation of PC and PE upon charge inversion due to the proton/methyl cation transfer unique to PC cations and the double proton transfer pathway undertaken by PE cations.124 While a [PE − H + PDPA]− complex was not directly observed, the [PE − H]− product anion is not likely to be formed from two consecutive single-proton transfer reactions as neutralization would occur after the first reaction and ionization of the neutral from a second encounter is improbable. Rather, two proton transfer reactions within a single collision complex with subsequent dissociation of the complex is far more likely. Combining charge inversion with other techniques, like trimethylation enhancement using 13C-diazomethane (13C-TrEnDi) or the Paternò-Büchi reaction, can result in enhanced structural characterization of PC and PE phospholipids.134,135
In the above cases, charge inversion was used to convert phospholipid cations to structurally informative anions. Recently, gas-phase charge inversion ion/ion reactions have also been applied to convert fatty acid anions to metalated cations.136,137 These reactions have successfully been applied in a shotgun (i.e., direct infusion ESI-MS) approach for fatty acid (FA) profiling, permitting unambiguous FA identification, isomeric distinction, and relative quantitation of isomeric FA. In the special case of magnesium phenanthroline complexes, charge inversion analogous to that of Equation 5 can be achieved where only two phenanthroline ligands are lost:
| (6) |
Singly deprotonated FA anions, either derived from non-esterified (i.e., free) FA or complex lipid decomposition via solution-based hydrolysis or gas-phase broadband collisional activation, undergo charge inversion when subjected to reactions with tris-phenanthroline magnesium dications to generate [FA − H + MgPhen]+ cations. Subsequent isolation and ion-trap CID of the charge-inverted FA complex cation yields a reproducible, predictable product ion spectrum, that upon spectral matching to a developed FA mass spectral library composed of [FA − H + MgPhen]+ product ion spectra, facilitates localization of carbon-carbon double bond positions and confident FA identification. Furthermore, using multiple linear regression analysis paired with the library data and in conjunction with data derived from FA mixtures, relative abundances of isomeric FA can be sensitively determined (Figure 8).137 Collectively, this approach provides a relatively rapid and sensitive approach to lipid analysis based entirely on gas-phase chemistries.
Figure 8.

Product ion spectra of [18:1 − H + MgPhen]+ for the isomeric mixture of 18:1 n-9/n-7 at the molar ratios of (a) 5/95 and (b) 95/5. Product ions used in the multiple linear regression for relative quantitation are shown in red. Calculated molar ratios (mean ± standard deviation, n = 3) are shown in green. The lightning bolt corresponds to the species subjected to CID. Adapted from Randolph, C. E.; Foreman D. J.; Blanksby, S. J.; McLuckey, S. A. Anal. Chem. 2019, 91, 9032-9040 (ref 137). Copyright 2019 American Chemical Society.
Oxidation Reactions
Reduction/oxidation reactions in terms of electron transfer have been discussed above. However, oxidation can also be discussed in terms of oxygen transfer (i.e. oxidation is the gain of oxygen) and hydrogen transfer (i.e. oxidation is the loss of hydrogen). The gas-phase oxidation of multiply protonated peptide ions via oxygen transfer has recently been described. The periodate anion, IO4−, was used to selectively oxidize methionine residues, and to a lesser extent, tryptophan residues,138 alkylated cysteine residues,139 and disulfide bonds.140 Under favorable conditions, the periodate anion can be used to oxidize neutral basic residues in the gas-phase.141
In the case of oxygen transfer to disulfide bonds, subsequent activation of the [M + H + O]+ species results in the cleavage of the oxidized disulfide bonds at the S(O)─S bond or the C─S(O) bond, generating a fragmentation pattern indicative of the presence of a disulfide linkage.140 Tryptic digestion of disulfide intact lysozyme yields a total of three peptides; two peptides contain one intermolecular disulfide bond and one peptide contains one intermolecular and one intramolecular disulfide bond. The product ion spectra from the activation of the three oxidized species are shown in Figure 9. In all cases, cleavage of the intermolecular disulfide bond is evidenced by the presence of the A and B chain peptides.
Figure 9.

Collisional activation of the ion/ion reaction complex formed between the reaction of the periodate anion with (a) doubly protonated doubly GYSLGNWVCAAK/CK, (b) triply protonated CELAAAMK/GCR, and (c) triply protonated WWCNDGR/NLCNIPCSALLSSDITASVNCAK. The degree signs correspond to water losses, and the lightning bolts correspond to the species subjected to CID. Reproduced from Pilo, A. L.; McLuckey, S. A. Anal. Chem. 2016, 88, 8972-8979 (ref 140). Copyright 2016 American Chemical Society.
Similar to the periodate anion, a suite of reagents derived from persulfate can be used to oxidize peptides in the gas-phase; HSO5− generates [M + H + O]+, HS2O8− generates [M − H]+ and [M + H + O]+, and SO4−• generates [M]+•.142 Notably, the sulfate radical anion, denoted SO4−•, is used to generate molecular radical cations. For polypeptides/proteins, CID of radical cations can give rise to the loss of radical side chains from several amino acids, forming dehydroalanine.87 Fragmentation of peptide ions containing dehydroalanine has been noted to produce abundant c- and/or z- ions N-terminal to dehydroalanine.143 In 2017, Peng et al. introduced dehydroalanine into multiply protonated ubiquitin ions via ion/ion reaction with SO4−•.144 The so called “dehydroalanine effect” upon fragmentation led to site specific c-/z- fragment ions. In addition to the sulfate radical anion, other reagent ions have been described to generate radical containing analyte ions via ion/ion reaction.114,115,145 Whatever the reagent, though, introduction of selective cleavage at dehydroalanine may prove useful in several situations. For example, a database search incorporating dehydroalanine fragmentation could provide enhanced specificity in top-down workflows.
Covalent Chemistry: Bond Formation in the Gas-Phase
A relatively new area of ion/ion reaction research involves the formation of covalent bonds in the gas-phase. While some of the examples above (e.g., oxidation via oxygen transfer) involved the formation of new covalent bonds, not all oxidation examples included the formation of new bonds and were therefore not discussed in the context of covalent chemistry. The first selective covalent bond forming ion/ion reaction was reported in 2009 using 4-formyl-1,3- benzenedisulfonic acid (FBDSA) to generate a Schiff base.146 Much of the early work surrounding Schiff base formation via ion/ion reactions with FBDSA and the monosulfonic acid derivative, 2-formylbenzenemonosulfonic acid (FBMSA) has been previously reviewed.9 However, since that review, there have been several reports surrounding Schiff base formation via ion/ion reaction. In one example, Wang et al. identified a rearrangement of Schiff base modified peptide ions that is 19 Da lower in mass than the protonated or deprotonated peptide, which could be mistaken as a water loss.147 The Brodbelt group has demonstrated how ion/ion Schiff base formation can be used to increase UVPD efficiency compared to the unmodified peptide ions by binding a UV chromophore to the polypeptide.16 Lastly, the Brodbelt group has also shown how the electrostatic interactions of the sulfonate group of FBDSA with basic residues can be used to retain the phosphate group upon CID of phosphopeptide cations.148
In the gas-phase, N-hydroxysuccinimide (NHS) ester-based reagents have been utilized to selectively derivatize nucleophilic sites, such as unprotonated arginine residues and unprotonated primary amines, in the gas-phase. Like Schiff base formation, ion/ion reactions involving NHS ester reagents have been reviewed.9 Thereafter, Bu and co-workers explored the potential energy surfaces associated with NHS ester reagents and primary amines or guanidine groups.149 In that work, they explored two scenarios: (1) a case where the transition state barrier to covalent modification is relatively high and (2) a case where the transition state barrier is low and covalent reaction efficiencies are high. Interestingly, the efficiencies of covalent reactions in the former case could be significantly increased by implementing a long, slow-heating activation step prior to complex dissociation.149 In later work, Bu and co-workers examined new reagents that exhibited enhanced reactivity towards amines and guanidines, attributed to the lower transition state barrier using triazole ester based reagent ions.150 In addition to reactivity towards primary amines and guanidines, the gas-phase reactivity of both NHS esters and triazole esters towards carboxylates has been described in recent years.151,152 The latter chemistry has proven useful for the identification carboxylate groups present in salt bridges and/or zwitterions.152
A variety of other ion/ion reactions have been described that result in covalent bond formation including alkyl cation transfer,153 1,3-dipolar cycloaddition (i.e. click chemistry),154 and reactions with Woodward’s reagent K (wrk).155 The equivalent solution-phase chemistry of wrk was harnessed to prepare a reagent anion for the C-terminal peptide extension via gas-phase ion/ion reactions.156 Our group also demonstrated the gas-phase N-terminal peptide extension using NHS reagents.157 In all cases of gas-phase covalent bond formation, reactions were fast and efficient. Additionally, gas-phase ion/ion reactions offer a degree of selectivity that is not obtained with solution-phase reactions. Specifically, reactants can be isolated using a quadrupole mass filter prior to reaction, and the extent of reaction can be controlled.
REAGENT ION SELECTION
Mutual storage of oppositely charged ions can result in competitive reaction processes (e.g., proton transfer versus electron transfer) leading to several different product ions, where the major reaction pathways are dependent on both the analyte ion and reagent ion. For example, ETD sequence coverage is influenced by precursor ion charge60,158-160, size,161 and structure.162,163 In another example, sodium cationized arginine residues are reactive towards NHS reagent anions while protonated arginine residues are not reactive.164 In cases where the analyte ion-type is not readily varied, the partitioning of reaction products is determined by the characteristics of the reagent ion. This latter scenario is of particular interest, as the analyte ion-type is determined by the ionization method and, in most cases, the ionization method is selected based on the approach that leads to the greatest analyte ion yield. Thus, it is important to understand the characteristics of reagent ions that influence favored reaction pathways in order to efficiently transform the analyte ion to a product ion-type of interest.165 Here, several reagent characteristics as they pertain to different reaction types are discussed.
Electron transfer dissociation has been used extensively due to its ability to generate a high degree of structural information from bio-ions. The ideal electron transfer reagent would exclusively transfer an electron to generate fragment ions with minimal formation of non-dissociative product ions. However, depending on the nature of the electron transfer reagent ion, proton transfer can compete with electron transfer.89,166,167 In fact, a variety of ETD reagents have been explored and, to the best of our knowledge, every ETD anion has shown some degree of proton transfer.23,25,166-168In recognition of the importance of the reagent in ETD, much attention has been devoted to studying ETD reagents.48,89,166-169 The likelihood of observing electron transfer is governed largely by the Franck-Condon overlap between the reagent anion and corresponding neutral and the electron affinity of the corresponding neutral.167 If these parameters are not optimized, that is to say, if the Franck-Condon overlap is minimal and the electron affinity is relatively large, proton transfer will tend to dominate. 167
As highlighted in the above proton transfer section, there are scenarios where proton transfer is highly desirable. In such experiments there should be little contribution from fragmentation. Generally, fragmentation is not observed following proton transfer from a multiply protonated bio-ion to a singly charged reagent anion. Conversely, protonation of a multiply charged anion via ion/ion reaction has been shown to result in fragmentation and the extent of fragmentation was found to be related to the reaction exothermicity.170 Fragmentation could be minimized by reducing the proton affinity of the cationic reagent.170 Additionally, for proton transfer, it is often desirable that upon neutralization of the reagent ion, no long-lived complexes survive. Using the experiment performed by Bush and co-workers as an example, proton transfer was used to increase the accuracy of charge state assignment.53 Here, if the reagent was “sticky” and a long-lived complex was formed, the mass of the reagent would be added to the analyte with each reaction. Of course, if every reaction resulted in adduct formation, the mass of the analyte could be readily deduced, yet not all cases result in this ideal scenario. Adduct formation in proton transfer experiments can be especially problematic if the reaction results in a mixture of product ion types (i.e. proton transfer and adduct formation). This situation has been observed with the iodide anion, I−.171 When examining a mixture of product ion types, and without a priori knowledge of the analyte, deducing mass information may be challenging.
Several types of gas-phase ion/ion reactions rely on long-lived complex formation. One example is covalent modification in the gas-phase. Han et al. demonstrated the importance of forming a long-lived complex during Schiff base modification reactions.146 The sulfonate group of singly deprotonated 4-formyl-1,3-benzenedisulfonic acid (FBDSA) interacts strongly with the doubly protonated peptide ion. This interaction allows enough time for the imine bond formation to occur. When reagents with carboxylate groups were used, which interact less strongly with cations, proton transfer was the sole process observed likely due to a lower barrier from proton transfer relative to that for Schiff base formation.146 Sulfonate groups and fixed-charge ammonium ions are widely used to promote long-lived complexes. In addition to a sticky group, reagents for covalent modification require a reactive site that undergo chemical reactions with the analyte ion.
The above examples discussed characteristics crucial to generating the product ion-type of interest. For instance, an electron transfer experiment cannot be successful if the reagent ion results only in proton transfer. Sometimes, there are cases in which multiple reagents lead to comparable results and selecting a reagent often comes down to subtle, yet important, differences. Sulfo-benzoyl-1-hydroxy-7-azabenzotriazole ester (HOAt), sulfo-benzoyl-1-hydroxybenzotriazole ester (HOBt), and sulfo-benzoyl-N-hydroxysulfosuccinimide ester (NHS) all undergo acyl substitution reactions at primary amines and guanidine in the gas-phase.150 Figure 10 demonstrates that HOAt and HOBt are more reactive towards amines than their NHS counterpart, yet all reagents result in a covalently modified product ion shaded in yellow. The enhanced reactivity results in less selectivity. Choosing triazole ester reagents versus NHS ester reagents will depend on the experiment. In another example, calcium, strontium, barium, and magnesium phenanthroline complexes can be used to charge invert monounsaturated FAs and identify the site of unsaturation.136 However, the magnesium phenanthroline complex was the preferred reagent as it showed the lowest degree of water adduction in the collision cell from adventitious water, and therefore resulted in more straightforward product ion spectra.136 Prentice et al. demonstrated the use of reagent cluster ions for multiple modifications performed in one ion/ion collision.172 Similar results could be obtained via sequential ion/ion reactions. The difference between the two, however, is the charge “cost.” Using the reagent cluster ion results in the reduction of the analyte charge by only one charge whereas consecutive reactions require a charge for each encounter, thus limiting the extent to which multiple modifications can be performed. It is clear that for a given analyte ion, the identity of the reagent ion is crucial. Therefore, choosing the correct reagent is key for any experiment.
Figure 10.

Product ion spectrum from CID of the complex formed between doubly protonated KGAGGKGAGGKL and (a) HOAt, (b) HOBt, and (c) NHS. The m/z region corresponding to proton transfer is shaded in red, the m/z region corresponding to covalent modification is shaded in yellow, and the m/z region corresponding to the complex ion is shaded in green. Reprinted by permission from Springer Nature: Journal of the American Society for Mass Spectrometry, Bu, J.; Peng, Z.; Zhao, F.; McLuckey, S.A. J. Am. Soc. Mass Spectrom. 2017, 28, 1254-1261 (ref 150). Copyright 2017.
FUTURE OUTLOOK
Reactions that are fast, efficient, and provide useful information have a long history in analytical chemistry. Over the past twenty-five years, gas-phase ion/ion reactions have proven to be fast, efficient, and remarkably powerful as means for converting analyte ions into forms that facilitate analysis and/or structural characterization. Single proton transfer applications to simplify mixture analysis, to concentrate charge into a one or a few species, to generate charge states that are not formed directly, etc. were the first to receive extensive attention. Following the discovery of electron transfer reagents by the Hunt group, attention was extended to structural characterization applications analogous to those employing electron capture. The commercial introduction of products that support ETD experiments greatly expanded the availability of ion/ion reactions to the analytical mass spectrometry community. The commercial availability of instruments that are also optimized for proton transfer ion/ion reactions will likely lead to growing use of charge manipulation applications, particularly in top-down proteomics applications.
Single proton transfer and electron transfer ion/ion reactions are relatively mature in terms of analytical applications. Reactions that require the formation of a long-lived complex, such as charge inversion, metal ion transfer, and selective covalent reaction, on the other hand, are far less explored. However, they illustrate the diversity of reaction types that can be accessed via ion/ion reactions. No instrument vendor currently supports these reaction types as the ion sources generally used to generate proton transfer and electron transfer reagents are not suitable for the generation of reagent ions that lead to these reaction types. Two ESI sources have generally been used to generate the reactants for charge inversion, metal transfer, and covalent reaction. However, commercial platforms that include a separate ion source for mass calibration purposes can, in principle, be adapted for dual-ESI ion/ion reaction experiments, as recently described by Webb et al.31 As more laboratories gain access to such capabilities, the range of reactions and applications thereof are likely to grow significantly. As analytical mass spectrometry continues to expand to larger analytes and more complex mixtures, reactions that enhance specificity, simplify mixture analysis, and facilitate structural characterization will find use. Novel gas-phase ion/ion reactions for analytical applications in the future are likely to process through long-lived complex formation.
ACKNOWLEDGMENTS
The authors would like to acknowledge all group members, specifically Caitlin Randolph, for their helpful discussions and contributions towards this review. We would also like to acknowledge support for ion/ion reaction research in our laboratory from NIH Grants GM R37-45372 and GM R01-118484 for protein and lipid chemistries, respectively, and from Sciex for instrument development efforts.
Biography
Scott A. McLuckey received a B.S. degree in chemistry from Westminster College in New Wilmington, PA in 1978 and a Ph.D. degree in chemistry from Purdue University in 1982, with Professor Graham Cooks serving as thesis advisor. Following a one-year post-doctoral appointment at the F.O.M. Institute for Atomic and Molecular Physics, he joined the Analytical Chemistry Division at Oak Ridge National Laboratory as a Wigner Fellow, which later transitioned into a staff position. He spent sixteen years at Oak Ridge having served as a group leader and section head. In 2000, he joined the faculty at Purdue University as a full professor and was named John A. Leighty Distinguished Professor in 2008. His mass spectrometry related research has been recognized with the 2012 Field and Franklin Award in mass spectrometry, the 2016 ASMS Distinguished Contribution Award, and the 2016 Thompson Medal from the International Mass Spectrometry Foundation.
David J. Foreman obtained his B.S. in chemistry and a minor in mathematics from Merrimack College in North Andover, MA in 2011. He worked in the pharmaceutical industry for four years before beginning his graduate studies at Purdue University in 2015. He is currently a Ph.D. candidate in the research laboratory of Scott A. McLuckey. His research is focused on the introduction of selective fragmentation pathways into bio-ions via gas-phase ion chemistry.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Yamashita M, Fenn JB: Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem 88, 4451–4459 (1984) [Google Scholar]
- 2.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM: Electrospray ionization for mass spectrometry of large biomolecules. Science. 246, 64 (1989) [DOI] [PubMed] [Google Scholar]
- 3.Loo RRO, Udseth HR, Smith RD: Evidence of charge inversion in the reaction of singly charged anions with multiply charged macroions. J. Phys. Chem 95, 6412–6415 (1991) [Google Scholar]
- 4.Ogorzalek Loo RR, Udseth HR, Smith RD: A new approach for the study of gas-phase ion-ion reactions using electrospray ionization. J. Am. Soc. Mass. Spectrom 3, 695–705 (1992) [DOI] [PubMed] [Google Scholar]
- 5.Herron WJ, Goeringer DE, McLuckey SA: Product Ion Charge State Determination via Ion/Ion Proton Transfer Reactions. Anal. Chem 68, 257–262 (1996) [DOI] [PubMed] [Google Scholar]
- 6.Stephenson JL, McLuckey SA: Simplification of Product Ion Spectra Derived from Multiply Charged Parent Ions via Ion/Ion Chemistry. Anal. Chem 70, 3533–3544 (1998) [DOI] [PubMed] [Google Scholar]
- 7.McLuckey SA, Stephenson JL: Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom. Rev 17, 369–407 (1998) [DOI] [PubMed] [Google Scholar]
- 8.Pitteri SJ, McLuckey SA: Recent developments in the ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom. Rev 24, 931–958 (2005) [DOI] [PubMed] [Google Scholar]
- 9.Prentice BM, McLuckey SA: Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem. Commun 49, 947–965 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia Y, McLuckey SA: Evolution of Instrumentation for the Study of Gas-Phase Ion/Ion Chemistry via Mass Spectrometry. J. Am. Soc. Mass. Spectrom 19, 173–189 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stutzman JR, Crowe MC, Alexander JN, Bell BM, Dunkle MN: Coupling Charge Reduction Mass Spectrometry to Liquid Chromatography for Complex Mixture Analysis. Anal. Chem 88, 4130–4139 (2016) [DOI] [PubMed] [Google Scholar]
- 12.Campuzano IDG, Schnier PD: Coupling electrospray corona discharge, charge reduction and ion mobility mass spectrometry: From peptides to large macromolecular protein complexes. Int. J. Ion Mobil. Spec 16, 51–60 (2013) [Google Scholar]
- 13.Robb DB, Brown JM, Morris M, Blades MW: Method of Atmospheric Pressure Charge Stripping for Electrospray Ionization Mass Spectrometry and Its Application for the Analysis of Large Poly(Ethylene Glycol)s. Anal. Chem 86, 9644–9652 (2014) [DOI] [PubMed] [Google Scholar]
- 14.Scalf M, Westphall MS, Krause J, Kaufman SL, Smith LM: Controlling Charge States of Large Ions. Science. 283, 194 (1999) [DOI] [PubMed] [Google Scholar]
- 15.Scalf M, Westphall MS, Smith LM: Charge Reduction Electrospray Mass Spectrometry. Anal. Chem 72, 52–60 (2000) [DOI] [PubMed] [Google Scholar]
- 16.Cotham VC, Shaw JB, Brodbelt JS: High-Throughput Bioconjugation for Enhanced 193 nm Photodissociation via Droplet Phase Initiated Ion/Ion Chemistry Using a Front-End Dual Spray Reactor. Anal. Chem 87, 9396–9402 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stutzman JR, Bain RM, Hagenhoff S, Woodward WH, O’Brien JP, Lesniak M: Microdroplet Fusion Chemistry for Charge State Reduction of Synthetic Polymers via Bipolar Dual Spray with Anionic Reagents. J. Am. Soc. Mass. Spectrom 30, 1742–1749 (2019) [DOI] [PubMed] [Google Scholar]
- 18.Zhao Q, Soyk MW, Schieffer GM, Fuhrer K, Gonin MM, Houk RS, Badman ER: An ion trap-ion mobility-time of flight mass spectrometer with three ion sources for ion/ion reactions. J. Am. Soc. Mass. Spectrom 20, 1549–1561 (2009) [DOI] [PubMed] [Google Scholar]
- 19.Riley NM, Coon JJ: The Role of Electron Transfer Dissociation in Modern Proteomics. Anal. Chem 90, 40–64 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He M, Jiang Y, Guo D, Xiong X, Fang X, Xu W: Dual-Polarity Ion Trap Mass Spectrometry: Dynamic Monitoring and Controlling Gas-phase Ion–Ion Reactions. J. Am. Soc. Mass. Spectrom 28, 1262–1270 (2017) [DOI] [PubMed] [Google Scholar]
- 21.Lin Z, Tan L, Garimella S, Li L, Chen T-C, Xu W, Xia Y, Ouyang Z: Characterization of a DAPI-RIT-DAPI System for Gas-Phase Ion/Molecule and Ion/Ion Reactions. J. Am. Soc. Mass. Spectrom 25, 48–56 (2014) [DOI] [PubMed] [Google Scholar]
- 22.Campbell JL, Hager JW: Creating an evanescent ion/ion reaction region within a low-pressure linear ion trap. Int. J. Mass Spectrom 323-324, 14–20 (2012) [Google Scholar]
- 23.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 101, 9528–9533 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAlister GC, Berggren WT, Griep-Raming J, Horning S, Makarov A, Phanstiel D, Stafford G, Swaney DL, Syka JEP, Zabrouskov V, Coon JJ: A Proteomics Grade Electron Transfer Dissociation-Enabled Hybrid Linear Ion Trap- Orbitrap Mass Spectrometer. J. Proteome Res 7, 3127–3136 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF: Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 102, 9463 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Earley L, Anderson LC, Bai DL, Mullen C, Syka JEP, English AM, Dunyach J-J, Stafford GC, Shabanowitz J, Hunt DF, Compton PD: Front-End Electron Transfer Dissociation: A New Ionization Source. Anal. Chem 85, 8385–8390 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weisbrod CR, Kaiser NK, Syka JEP, Early L, Mullen C, Dunyach J-J, English AM, Anderson LC, Blakney GT, Shabanowitz J, Hendrickson CL, Marshall AG, Hunt DF: Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis. J. Am. Soc. Mass. Spectrom 28, 1787–1795 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams JP, Brown JM, Campuzano I, Sadler PJ: Identifying drug metallation sites on peptides using electron transfer dissociation (ETD), collision induced dissociation (CID) and ion mobility-mass spectrometry (IM-MS). Chem. Commun 46, 5458–5460 (2010) [DOI] [PubMed] [Google Scholar]
- 29.Rand KD, Pringle SD, Morris M, Engen JR, Brown JM: ETD in a Traveling Wave Ion Guide at Tuned Z-Spray Ion Source Conditions Allows for Site-Specific Hydrogen/Deuterium Exchange Measurements. J. Am. Soc. Mass. Spectrom 22, 1784 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lermyte F, Verschueren T, Brown JM, Williams JP, Valkenborg D, Sobott F: Characterization of top-down ETD in a travelling-wave ion guide. Methods. 89, 22–29 (2015) [DOI] [PubMed] [Google Scholar]
- 31.Webb IK, Morrison LJ, Brown J: Dueling electrospray implemented on a traveling-wave ion mobility/time-of-flight mass spectrometer: Towards a gas-phase workbench for structural biology. Int. J. Mass Spectrom 444, 116177 (2019) [Google Scholar]
- 32.Garimella SVB, Webb IK, Prabhakaran A, Attah IK, Ibrahim YM, Smith RD: Design of a TW-SLIM Module for Dual Polarity Confinement, Transport, and Reactions. J. Am. Soc. Mass. Spectrom 28, 1442–1449 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee KW, Eakins GS, Carlsen MS, McLuckey SA: Increasing the Upper Mass/Charge Limit of a Quadrupole Ion Trap for Ion/Ion Reaction Product Analysis via Waveform Switching. J. Am. Soc. Mass. Spectrom 30, 1126–1132 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosati S, Rose RJ, Thompson NJ, van Duijn E, Damoc E, Denisov E, Makarov A, Heck AJR: Exploring an Orbitrap Analyzer for the Characterization of Intact Antibodies by Native Mass Spectrometry. Angew. Chem. Int. Ed 51, 12992–12996 (2012) [DOI] [PubMed] [Google Scholar]
- 35.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL: From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem 85, 11163–11173 (2013) [DOI] [PubMed] [Google Scholar]
- 36.Snijder J, van de Waterbeemd M, Damoc E, Denisov E, Grinfeld D, Bennett A, Agbandje-McKenna M, Makarov A, Heck AJR: Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J. Am. Chem. Soc 136, 7295–7299 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Nissan G, Belov ME, Morgenstern D, Levin Y, Dym O, Arkind G, Lipson C, Makarov AA, Sharon M: Triple- Stage Mass Spectrometry Unravels the Heterogeneity of an Endogenous Protein Complex. Anal. Chem 89, 4708–4715 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Waterbeemd M, Fort KL, Boll D, Reinhardt-Szyba M, Routh A, Makarov A, Heck AJR: High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nature Methods. 14, 283 (2017) [DOI] [PubMed] [Google Scholar]
- 39.Fort KL, van de Waterbeemd M, Boll D, Reinhardt-Szyba M, Belov ME, Sasaki E, Zschoche R, Hilvert D, Makarov AA, Heck AJR: Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst. 143, 100–105 (2018) [DOI] [PubMed] [Google Scholar]
- 40.Stephenson JL, McLuckey SA: Ion/Ion Proton Transfer Reactions for Protein Mixture Analysis. Anal. Chem 68, 4026–4032 (1996) [DOI] [PubMed] [Google Scholar]
- 41.Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA: Gas-Phase Concentration, Purification, and Identification of Whole Proteins from Complex Mixtures. J. Am. Chem. Soc 124, 7353–7362 (2002) [DOI] [PubMed] [Google Scholar]
- 42.He M, Reid GE, Shang H, Lee GU, McLuckey SA: Dissociation of Multiple Protein Ion Charge States Following a Single Gas-Phase Purification and Concentration Procedure. Anal. Chem 74, 4653–4661 (2002) [DOI] [PubMed] [Google Scholar]
- 43.Amunugama R, Hogan JM, Newton KA, McLuckey SA: Whole Protein Dissociation in a Quadrupole Ion Trap: Identification of an a Priori Unknown Modified Protein. Anal. Chem 76, 720–727 (2004) [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Chrisman PA, Erickson DE, McLuckey SA: Relative Information Content and Top-Down Proteomics by Mass Spectrometry: Utility of Ion/Ion Proton-Transfer Reactions in Electrospray-Based Approaches. Anal. Chem 79, 1073–1081 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chi A, Bai DL, Geer LY, Shabanowitz J, Hunt DF: Analysis of intact proteins on a chromatographic time scale by electron transfer dissociation tandem mass spectrometry. Int. J. Mass Spectrom 259, 197–203 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drabik A, Bodzon-Kulakowska A, Suder P: Application of the ETD/PTR reactions in top-down proteomics as a faster alternative to bottom-up nanoLC-MS/MS protein identification. J. Mass Spectrom 47, 1347–1352 (2012) [DOI] [PubMed] [Google Scholar]
- 47.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ: Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci 105, 4093 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartmer R, Kaplan DA, Gebhardt CR, Ledertheil T, Brekenfeld A: Multiple ion/ion reactions in the 3D ion trap: Selective reagent anion production for ETD and PTR from a single compound. Int. J. Mass Spectrom 276, 82–90 (2008) [Google Scholar]
- 49.Anderson LC, English AM, Wang W-H, Bai DL, Shabanowitz J, Hunt DF: Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry. Int. J. Mass Spectrom 377, 617–624 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, English AM, Bai DL, Ugrin SA, Shabanowitz J, Ross MM, Hunt DF, Wang W-H: Analysis of Monoclonal Antibody Sequence and Post-translational Modifications by Time-controlled Proteolysis and Tandem Mass Spectrometry. Mol. Cell. Proteomics 15, 1479 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson LC, Karch KR, Ugrin SA, Coradin M, English AM, Sidoli S, Shabanowitz J, Garcia BA, Hunt DF: Analyses of Histone Proteoforms Using Front-end Electron Transfer Dissociation-enabled Orbitrap Instruments. Mol. Cell. Proteomics 15, 975 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhattacharjee S, Liu W, Wang W-H, Weitzhandler I, Li X, Qi Y, Liu J, Pang Y, Hunt DF, Chilkoti A: Site-Specific Zwitterionic Polymer Conjugates of a Protein Have Long Plasma Circulation. ChemBioChem. 16, 2451–2455 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laszlo KJ, Bush MF: Analysis of Native-Like Proteins and Protein Complexes Using Cation to Anion Proton Transfer Reactions (CAPTR). J. Am. Soc. Mass. Spectrom 26, 2152–2161 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wells JM, Stephenson JL, McLuckey SA: Charge dependence of protonated insulin decompositions. Int. J. Mass Spectrom 203, A1–A9 (2000) [Google Scholar]
- 55.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA: Charge-State-Dependent Sequence Analysis of Protonated Ubiquitin Ions via Ion Trap Tandem Mass Spectrometry. Anal. Chem 73, 3274–3281 (2001) [DOI] [PubMed] [Google Scholar]
- 56.Wells JM, Reid GE, Engel BJ, Pan P, McLuckey SA: Dissociation reactions of gaseous ferro-, ferri-, and apo-cytochrome c ions. J. Am. Soc. Mass. Spectrom 12, 873–876 (2001) [DOI] [PubMed] [Google Scholar]
- 57.Newton KA, Chrisman PA, Reid GE, Wells JM, McLuckey SA: Gaseous apomyoglobin ion dissociation in a quadrupole ion trap: [M + 2H]2+-[M + 21H]21+. Int. J. Mass Spectrom 212, 359–376 (2001) [Google Scholar]
- 58.Engel BJ, Pan P, Reid GE, Wells JM, McLuckey SA: Charge state dependent fragmentation of gaseous protein ions in a quadrupole ion trap: bovine ferri-, ferro-, and apo-cytochrome c. Int. J. Mass Spectrom 219, 171–187 (2002) [Google Scholar]
- 59.Hogan JM, McLuckey SA: Charge state dependent collision-induced dissociation of native and reduced porcine elastase. J. Mass Spectrom 38, 245–256 (2003) [DOI] [PubMed] [Google Scholar]
- 60.Rožman M, Gaskell SJ: Charge state dependent top-down characterisation using electron transfer dissociation. Rapid Commun. Mass Spectrom 26, 282–286 (2012) [DOI] [PubMed] [Google Scholar]
- 61.Holden DD, McGee WM, Brodbelt JS: Integration of Ultraviolet Photodissociation with Proton Transfer Reactions and Ion Parking for Analysis of Intact Proteins. Anal. Chem 88, 1008–1016 (2016) [DOI] [PubMed] [Google Scholar]
- 62.Holden DD, Brodbelt JS: Ultraviolet Photodissociation of Native Proteins Following Proton Transfer Reactions in the Gas Phase. Anal. Chem 88, 12354–12362 (2016) [DOI] [PubMed] [Google Scholar]
- 63.Bashyal A, Sanders JD, Holden DD, Brodbelt JS: Top-Down Analysis of Proteins in Low Charge States. J. Am. Soc. Mass. Spectrom 30, 704–717 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wenger CD, Lee MV, Hebert AS, McAlister GC, Phanstiel DH, Westphall MS, Coon JJ: Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nature Methods. 8, 933 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vincent CE, Rensvold JW, Westphall MS, Pagliarini DJ, Coon JJ: Automated Gas-Phase Purification for Accurate, Multiplexed Quantification on a Stand-Alone Ion-Trap Mass Spectrometer. Anal. Chem 85, 2079–2086 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLuckey SA, Stephenson JL, Asano KG: Ion/Ion Proton-Transfer Kinetics: Implications for Analysis of Ions Derived from Electrospray of Protein Mixtures. Anal. Chem 70, 1198–1202 (1998) [DOI] [PubMed] [Google Scholar]
- 67.McLuckey SA, Reid GE, Wells JM: Ion Parking during Ion/Ion Reactions in Electrodynamic Ion Traps. Anal. Chem 74, 336–346 (2002) [DOI] [PubMed] [Google Scholar]
- 68.Foreman DJ, Dziekonski ET, McLuckey SA: Maximizing Selective Cleavages at Aspartic Acid and Proline Residues for the Identification of Intact Proteins. J. Am. Soc. Mass. Spectrom 30, 34–44 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chrisman PA, Pitteri SJ, McLuckey SA: Parallel Ion Parking: Improving Conversion of Parents to First-Generation Products in Electron Transfer Dissociation. Anal. Chem 77, 3411–3414 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chrisman PA, Pitteri SJ, McLuckey SA: Parallel Ion Parking of Protein Mixtures. Anal. Chem 78, 310–316 (2006) [DOI] [PubMed] [Google Scholar]
- 71.Ugrin SA, English AM, Syka JEP, Bai DL, Anderson LC, Shabanowitz J, Hunt DF: Ion-Ion Proton Transfer and Parallel Ion Parking for the Analysis of Mixtures of Intact Proteins on a Modified Orbitrap Mass Analyzer. J. Am. Soc. Mass. Spectrom, (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Campbell JL, Le Blanc JCY: Targeted ion parking for the quantitation of biotherapeutic proteins: Concepts and preliminary data. J. Am. Soc. Mass. Spectrom 21, 2011–2022 (2010) [DOI] [PubMed] [Google Scholar]
- 73.Frey BL, Krusemark CJ, Ledvina AR, Coon JJ, Belshaw PJ, Smith LM: Ion–ion reactions with fixed-charge modified proteins to produce ions in a single, very high charge state. Int. J. Mass Spectrom 276, 136–143 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heuvel R.H.H.v.d., Heck AJR: Native protein mass spectrometry: from intact oligomers to functional machineries. Curr. Opin. Chem. Biol 8, 519–526 (2004) [DOI] [PubMed] [Google Scholar]
- 75.Sharon M, Robinson CV: The Role of Mass Spectrometry in Structure Elucidation of Dynamic Protein Complexes. Annu. Rev. Biochem 76, 167–193 (2007) [DOI] [PubMed] [Google Scholar]
- 76.Sharon M: How Far Can We Go with Structural Mass Spectrometry of Protein Complexes? J. Am. Soc. Mass. Spectrom 21, 487–500 (2010) [DOI] [PubMed] [Google Scholar]
- 77.Leney AC, Heck AJR: Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass. Spectrom 28, 5–13 (2017) [DOI] [PubMed] [Google Scholar]
- 78.Zhao Q, Schieffer GM, Soyk MW, Anderson TJ, Houk RS, Badman ER: Effects of ion/ion proton transfer reactions on conformation of gas-phase cytochrome c ions. J. Am. Soc. Mass. Spectrom 21, 1208–1217 (2010) [DOI] [PubMed] [Google Scholar]
- 79.Laszlo KJ, Munger EB, Bush MF: Folding of Protein Ions in the Gas Phase after Cation-to-Anion Proton-Transfer Reactions. J. Am. Chem. Soc 138, 9581–9588 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laszlo KJ, Buckner JH, Munger EB, Bush MF: Native-Like and Denatured Cytochrome c Ions Yield Cation-to-Anion Proton Transfer Reaction Products with Similar Collision Cross-Sections. J. Am. Soc. Mass. Spectrom 28, 1382–1391 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Laszlo KJ, Bush MF: Interpreting the Collision Cross Sections of Native-like Protein Ions: Insights from Cation-to- Anion Proton-Transfer Reactions. Anal. Chem 89, 7607–7614 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laszlo KJ, Munger EB, Bush MF: Effects of Solution Structure on the Folding of Lysozyme Ions in the Gas Phase. J. Phys. Chem. B 121, 2759–2766 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gadzuk-Shea MM, Bush MF: Effects of Charge State on the Structures of Serum Albumin Ions in the Gas Phase: Insights from Cation-to-Anion Proton-Transfer Reactions, Ion Mobility, and Mass Spectrometry. J. Phys. Chem. B 122, 9947–9955 (2018) [DOI] [PubMed] [Google Scholar]
- 84.Lermyte F, Łącki MK, Valkenborg D, Gambin A, Sobott F: Conformational Space and Stability of ETD Charge Reduction Products of Ubiquitin. J. Am. Soc. Mass. Spectrom 28, 69–76 (2017) [DOI] [PubMed] [Google Scholar]
- 85.Kim M-S, Pandey A: Electron transfer dissociation mass spectrometry in proteomics. Proteomics. 12, 530–542 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhurov KO, Fornelli L, Wodrich MD, Laskay UA, Tsybin YO: Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev 42, 5014–5030 (2013) [DOI] [PubMed] [Google Scholar]
- 87.Tureček F, Julian RR: Peptide Radicals and Cation Radicals in the Gas Phase. Chem. Rev 113, 6691–6733 (2013) [DOI] [PubMed] [Google Scholar]
- 88.Qi Y, Volmer DA: Electron-based fragmentation methods in mass spectrometry: An overview. Mass Spectrom. Rev 36, 4–15 (2017) [DOI] [PubMed] [Google Scholar]
- 89.Lermyte F, Łącki MK, Valkenborg D, Baggerman G, Gambin A, Sobott F: Understanding reaction pathways in top-down ETD by dissecting isotope distributions: A mammoth task. Int. J. Mass Spectrom 390, 146–154 (2015) [Google Scholar]
- 90.Abzalimov RR, Kaltashov IA: Electrospray Ionization Mass Spectrometry of Highly Heterogeneous Protein Systems: Protein Ion Charge State Assignment via Incomplete Charge Reduction. Anal. Chem 82, 7523–7526 (2010) [DOI] [PubMed] [Google Scholar]
- 91.Zhao Y, Abzalimov RR, Kaltashov IA: Interactions of Intact Unfractionated Heparin with Its Client Proteins Can Be Probed Directly Using Native Electrospray Ionization Mass Spectrometry. Anal. Chem 88, 1711–1718 (2016) [DOI] [PubMed] [Google Scholar]
- 92.Muneeruddin K, Bobst CE, Frenkel R, Houde D, Turyan I, Sosic Z, Kaltashov IA: Characterization of a PEGylated protein therapeutic by ion exchange chromatography with on-line detection by native ESI MS and MS/MS. Analyst. 142, 336–344 (2017) [DOI] [PubMed] [Google Scholar]
- 93.Minsky BB, Abzalimov RR, Niu C, Zhao Y, Kirsch Z, Dubin PL, Savinov SN, Kaltashov IA: Mass Spectrometry Reveals a Multifaceted Role of Glycosaminoglycan Chains in Factor Xa Inactivation by Antithrombin. Biochemistry. 57, 4880–4890 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang G, Johnson AJ, Kaltashov IA: Evaluation of Electrospray Ionization Mass Spectrometry as a Tool for Characterization of Small Soluble Protein Aggregates. Anal. Chem 84, 1718–1724 (2012) [DOI] [PubMed] [Google Scholar]
- 95.Abzalimov RR, Bobst CE, Salinas PA, Savickas P, Thomas JJ, Kaltashov IA: Studies of pH-Dependent Self- Association of a Recombinant Form of Arylsulfatase A with Electrospray Ionization Mass Spectrometry and Size-Exclusion Chromatography. Anal. Chem 85, 1591–1596 (2013) [DOI] [PubMed] [Google Scholar]
- 96.Lermyte F, Williams JP, Brown JM, Martin EM, Sobott F: Extensive Charge Reduction and Dissociation of Intact Protein Complexes Following Electron Transfer on a Quadrupole-Ion Mobility-Time-of-Flight MS. J. Am. Soc. Mass. Spectrom 26, 1068–1076 (2015) [DOI] [PubMed] [Google Scholar]
- 97.Jhingree JR, Beveridge R, Dickinson ER, Williams JP, Brown JM, Bellina B, Barran PE: Electron transfer with no dissociation ion mobility–mass spectrometry (ETnoD IM-MS). The effect of charge reduction on protein conformation. Int. J. Mass Spectrom 413, 43–51 (2017) [Google Scholar]
- 98.Jhingree JR, Bellina B, Pacholarz KJ, Barran PE: Charge Mediated Compaction and Rearrangement of Gas-Phase Proteins: A Case Study Considering Two Proteins at Opposing Ends of the Structure-Disorder Continuum. J. Am. Soc. Mass. Spectrom 28, 1450–1461 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parker ML, Gronert S: Investigating reduced metal species via sequential ion/ion and ion/molecule reactions: The reactions of transition metal phenanthrolines with allyl iodide. Int. J. Mass Spectrom 418, 73–78 (2017) [Google Scholar]
- 100.Munshi MU, Craig SM, Berden G, Martens J, DeBlase AF, Foreman DJ, McLuckey SA, Oomens J, Johnson MA: Preparation of Labile Ni+(cyclam) Cations in the Gas Phase Using Electron-Transfer Reduction through Ion–Ion Recombination in an Ion Trap and Structural Characterization with Vibrational Spectroscopy. J. Phys. Chem. Lett 8, 5047–5052 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Munshi MU, Martens J, Berden G, Oomens J: Gas-Phase Infrared Ion Spectroscopy Characterization of Cu(II/I)Cyclam and Cu(II/I)2,2′-Bipyridine Redox Pairs. J. Phys. Chem. A 123, 4149–4157 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoffmann WD, Jackson GP: Charge Transfer Dissociation (CTD) Mass Spectrometry of Peptide Cations Using Kiloelectronvolt Helium Cations. J. Am. Soc. Mass. Spectrom 25, 1939–1943 (2014) [DOI] [PubMed] [Google Scholar]
- 103.Li P, Kreft I, Jackson GP: Top-Down Charge Transfer Dissociation (CTD) of Gas-Phase Insulin: Evidence of a One- Step, Two-Electron Oxidation Mechanism. J. Am. Soc. Mass. Spectrom 29, 284–296 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li P, Jackson GP: Charge Transfer Dissociation (CTD) Mass Spectrometry of Peptide Cations: Study of Charge State Effects and Side-Chain Losses. J. Am. Soc. Mass. Spectrom 28, 1271–1281 (2017) [DOI] [PubMed] [Google Scholar]
- 105.Li P, Jackson GP: Charge transfer dissociation of phosphocholines: gas-phase ion/ion reactions between helium cations and phospholipid cations. J. Mass Spectrom 52, 271–282 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ropartz D, Li P, Fanuel M, Giuliani A, Rogniaux H, Jackson GP: Charge Transfer Dissociation of Complex Oligosaccharides: Comparison with Collision-Induced Dissociation and Extreme Ultraviolet Dissociative Photoionization. J. Am. Soc. Mass. Spectrom 27, 1614–1619 (2016) [DOI] [PubMed] [Google Scholar]
- 107.Ropartz D, Li P, Jackson GP, Rogniaux H: Negative Polarity Helium Charge Transfer Dissociation Tandem Mass Spectrometry: Radical-Initiated Fragmentation of Complex Polysulfated Anions. Anal. Chem 89, 3824–3828 (2017) [DOI] [PubMed] [Google Scholar]
- 108.Wells JM, Chrisman PA, McLuckey SA: Formation and Characterization of Protein–Protein Complexes in Vacuo. J. Am. Chem. Soc 125, 7238–7249 (2003) [DOI] [PubMed] [Google Scholar]
- 109.Bates DR, Morgan WL: New recombination mechanism: Tidal termolecular ionic recombination. Phys. Rev. Lett 64, 2258–2260 (1990) [DOI] [PubMed] [Google Scholar]
- 110.Newton KA, McLuckey SA: Gas-Phase Peptide/Protein Cationizing Agent Switching via Ion/Ion Reactions. J. Am. Chem. Soc 125, 12404–12405 (2003) [DOI] [PubMed] [Google Scholar]
- 111.Newton KA, McLuckey SA: Generation and manipulation of sodium cationized peptides in the gas phase. J. Am. Soc. Mass. Spectrom 15, 607–615 (2004) [DOI] [PubMed] [Google Scholar]
- 112.Newton KA, Amunugama R, McLuckey SA: Gas-Phase Ion/Ion Reactions of Multiply Protonated Polypeptides with Metal Containing Anions. J. Phys. Chem. A 109, 3608–3616 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hodges BDM, Liang X, McLuckey SA: Generation of di-lithiated peptide ions from multiply protonated peptides via ion/ion reactions. Int. J. Mass Spectrom 267, 183–189 (2007) [Google Scholar]
- 114.Barlow CK, Hodges BDM, Xia Y, O’Hair RAJ, McLuckey SA: Gas-phase ion/ion reactions of transition metal complex cations with multiply charged oligodeoxynucleotide anions. J. Am. Soc. Mass. Spectrom 19, 281–293 (2008) [DOI] [PubMed] [Google Scholar]
- 115.Crizer DM, Xia Y, McLuckey SA: Transition metal complex cations as reagents for gas-phase transformation of multiply deprotonated polypeptides. J. Am. Soc. Mass. Spectrom 20, 1718–1722 (2009) [DOI] [PubMed] [Google Scholar]
- 116.Gunawardena HP, O’Hair RAJ, McLuckey SA: Selective Disulfide Bond Cleavage in Gold(I) Cationized Polypeptide Ions Formed via Gas-Phase Ion/Ion Cation Switching. J. Proteome Res 5, 2087–2092 (2006) [DOI] [PubMed] [Google Scholar]
- 117.Mentinova M, Han H, McLuckey SA: Dissociation of disulfide-intact somatostatin ions: the roles of ion type and dissociation method. Rapid Commun. Mass Spectrom 23, 2647–2655 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mentinova M, McLuckey SA: Cleavage of multiple disulfide bonds in insulin via gold cationization and collision-induced dissociation. Int. J. Mass Spectrom 308, 133–136 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Foreman DJ, Betancourt SK, Pilo AL, McLuckey SA: Novel peptide ion chemistry associated with gold (I) cationization: Preferential cleavage at lysine residues. Int. J. Mass Spectrom 427, 114–122 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Foreman DJ, Lawler JT, Niedrauer ML, Hostetler MA, McLuckey SA: Gold(I) Cationization Promotes Ring Opening in Lysine-Containing Cyclic Peptides. J. Am. Soc. Mass. Spectrom, (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Newton KA, He M, Amunugama R, McLuckey SA: Selective cation removal from gaseous polypeptide ions: proton vs. sodium ion abstraction via ion/ion reactions. Phys. Chem. Chem. Phys 6, 2710–2717 (2004) [Google Scholar]
- 122.Luongo CA, Bu J, Burke NL, Gilbert JD, Prentice BM, Cummings S, Reed CA, McLuckey SA: Selective Removal of Alkali Metal Cations from Multiply-Charged Ions via Gas-Phase Ion/Ion Reactions Using Weakly Coordinating Anions. J. Am. Soc. Mass. Spectrom 26, 404–414 (2015) [DOI] [PubMed] [Google Scholar]
- 123.Betancourt SK, Pilo AL, Bu J, McLuckey SA: Simplification of electrospray mass spectra of Polysorbate 80 via cation transfer to carborane anions. J. Mass Spectrom 51, 453–458 (2016) [DOI] [PubMed] [Google Scholar]
- 124.Rojas-Betancourt S, Stutzman JR, Londry FA, Blanksby SJ, McLuckey SA: Gas-Phase Chemical Separation of Phosphatidylcholine and Phosphatidylethanolamine Cations via Charge Inversion Ion/Ion Chemistry. Anal. Chem 87, 11255–11262 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.He M, Emory JF, McLuckey SA: Reagent Anions for Charge Inversion of Polypeptide/Protein Cations in the Gas Phase. Anal. Chem 77, 3173–3182 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Emory JF, McLuckey SA: Charge inversion of polypeptide anions using protein and dendrimer cations as charge inversion reagents. Int. J. Mass Spectrom 276, 102–109 (2008) [Google Scholar]
- 127.Gunawardena HP, Emory JF, McLuckey SA: Phosphopeptide Anion Characterization via Sequential Charge Inversion and Electron-Transfer Dissociation. Anal. Chem 78, 3788–3793 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shih M, McLuckey SA: Ion/ion charge inversion/attachment in conjunction with dipolar DC collisional activation as a selective screen for sulfo- and phosphopeptides. Int. J. Mass Spectrom 444, 116181 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hassell KM, LeBlanc Y, McLuckey SA: Conversion of multiple analyte cation types to a single analyte anion type via ion/ion charge inversion. Analyst. 134, 2262–2266 (2009) [DOI] [PubMed] [Google Scholar]
- 130.Hassell KM, LeBlanc YC, McLuckey SA: Chemical Noise Reduction via Mass Spectrometry and Ion/Ion Charge Inversion: Amino Acids. Anal. Chem 83, 3252–3255 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.He M, McLuckey SA: Two Ion/Ion Charge Inversion Steps To Form a Doubly Protonated Peptide from a Singly Protonated Peptide in the Gas Phase. J. Am. Chem. Soc 125, 7756–7757 (2003) [DOI] [PubMed] [Google Scholar]
- 132.He M, McLuckey SA: Increasing the Negative Charge of a Macroanion in the Gas Phase via Sequential Charge Inversion Reactions. Anal. Chem 76, 4189–4192 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stutzman JR, Blanksby SJ, McLuckey SA: Gas-Phase Transformation of Phosphatidylcholine Cations to Structurally Informative Anions via Ion/Ion Chemistry. Anal. Chem 85, 3752–3757 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Betancourt SK, Canez CR, Shields SWJ, Manthorpe JM, Smith JC, McLuckey SA: Trimethylation Enhancement Using 13C-Diazomethane: Gas-Phase Charge Inversion of Modified Phospholipid Cations for Enhanced Structural Characterization. Anal. Chem 89, 9452–9458 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Franklin ET, Betancourt SK, Randolph CE, McLuckey SA, Xia Y: In-depth structural characterization of phospholipids by pairing solution photochemical reaction with charge inversion ion/ion chemistry. Anal. Bioanal. Chem 411, 4739–4749 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Randolph CE, Foreman DJ, Betancourt SK, Blanksby SJ, McLuckey SA: Gas-Phase Ion/Ion Reactions Involving Tris-Phenanthroline Alkaline Earth Metal Complexes as Charge Inversion Reagents for the Identification of Fatty Acids. Anal. Chem 90, 12861–12869 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Randolph CE, Foreman DJ, Blanksby SJ, McLuckey SA: Generating Fatty Acid Profiles in the Gas Phase: Fatty Acid Identification and Relative Quantitation Using Ion/Ion Charge Inversion Chemistry. Anal. Chem 91, 9032–9040 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pilo AL, McLuckey SA: Oxidation of Methionine Residues in Polypeptide Ions Via Gas-Phase Ion/Ion Chemistry. J. Am. Soc. Mass. Spectrom 25, 1049–1057 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pilo AL, Zhao F, McLuckey SA: Selective Gas-Phase Oxidation and Localization of Alkylated Cysteine Residues in Polypeptide Ions via Ion/Ion Chemistry. J. Proteome Res 15, 3139–3146 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pilo AL, McLuckey SA: Selective Gas-Phase Ion/Ion Reactions: Enabling Disulfide Mapping via Oxidation and Cleavage of Disulfide Bonds in Intermolecularly-Linked Polypeptide Ions. Anal. Chem 88, 8972–8979 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pilo AL, Bu J, McLuckey SA: Gas-Phase Oxidation of Neutral Basic Residues in Polypeptide Cations by Periodate. J. Am. Soc. Mass. Spectrom 27, 1979–1988 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pilo AL, Bu J, McLuckey SA: Transformation of [M+2H]2+ Peptide Cations to [M − H]+, [M+H+O]+, and M+• Cations via Ion/Ion Reactions: Reagent Anions Derived from Persulfate. J. Am. Soc. Mass. Spectrom 26, 1103–1114 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pilo AL, Peng Z, McLuckey SA: The dehydroalanine effect in the fragmentation of ions derived from polypeptides. J. Mass Spectrom 51, 857–866 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peng Z, Bu J, McLuckey SA: The Generation of Dehydroalanine Residues in Protonated Polypeptides: Ion/Ion Reactions for Introducing Selective Cleavages. J. Am. Soc. Mass. Spectrom 28, 1765–1774 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gilbert JD, Fisher CM, Bu J, Prentice BM, Redwine JG, McLuckey SA: Strategies for generating peptide radical cations via ion/ion reactions. J. Mass Spectrom 50, 418–426 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Han H, McLuckey SA: Selective Covalent Bond Formation in Polypeptide Ions via Gas-Phase Ion/Ion Reaction Chemistry. J. Am. Chem. Soc 131, 12884–12885 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang N, Pilo AL, Zhao F, Bu J, McLuckey SA: Gas-phase rearrangement reaction of Schiff-base-modified peptide ions. Rapid Commun. Mass Spectrom 32, 2166–2173 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cotham VC, McGee WM, Brodbelt JS: Modulation of Phosphopeptide Fragmentation via Dual Spray Ion/Ion Reactions Using a Sulfonate-Incorporating Reagent. Anal. Chem 88, 8158–8165 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bu J, Fisher CM, Gilbert JD, Prentice BM, McLuckey SA: Selective Covalent Chemistry via Gas-Phase Ion/ion Reactions: An Exploration of the Energy Surfaces Associated with N-Hydroxysuccinimide Ester Reagents and Primary Amines and Guanidine Groups. J. Am. Soc. Mass. Spectrom 27, 1089–1098 (2016) [DOI] [PubMed] [Google Scholar]
- 150.Bu J, Peng Z, Zhao F, McLuckey SA: Enhanced Reactivity in Nucleophilic Acyl Substitution Ion/Ion Reactions Using Triazole-Ester Reagents. J. Am. Soc. Mass. Spectrom 28, 1254–1261 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Peng Z, McGee WM, Bu J, Barefoot NZ, McLuckey SA: Gas Phase Reactivity of Carboxylates with N-Hydroxysuccinimide Esters. J. Am. Soc. Mass. Spectrom 26, 174–180 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pitts-McCoy AM, Harrilal CP, McLuckey SA: Gas-Phase Ion/Ion Chemistry as a Probe for the Presence of Carboxylate Groups in Polypeptide Cations. J. Am. Soc. Mass. Spectrom 30, 329–338 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gilbert JD, Prentice BM, McLuckey SA: Ion/Ion Reactions with “Onium” Reagents: An Approach for the Gas-phase Transfer of Organic Cations to Multiply-Charged Anions. J. Am. Soc. Mass. Spectrom 26, 818–825 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bu J, Pilo AL, McLuckey SA: Gas phase click chemistry via ion/ion reactions. Int. J. Mass Spectrom 390, 118–123 (2015) [Google Scholar]
- 155.Peng Z, Pilo AL, Luongo CA, McLuckey SA: Gas-Phase Amidation of Carboxylic Acids with Woodward’s Reagent K Ions. J. Am. Soc. Mass. Spectrom 26, 1686–1694 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Peng Z, McLuckey SA: C-terminal peptide extension via gas-phase ion/ion reactions. Int. J. Mass Spectrom 391, 17–23 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.McGee WM, McLuckey SA: Efficient and directed peptide bond formation in the gas phase via ion/ion reactions. Proc. Natl. Acad. Sci, 111, 1288–1292 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA: Electron Transfer Ion/Ion Reactions in a Three-Dimensional Quadrupole Ion Trap: Reactions of Doubly and Triply Protonated Peptides with SO2•. Anal. Chem 77, 1831–1839 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu J, Gunawardena HP, Huang T-Y, McLuckey SA: Charge-dependent dissociation of insulin cations via ion/ion electron transfer. Int. J. Mass Spectrom 276, 160–170 (2008) [Google Scholar]
- 160.Liu J, McLuckey SA: Electron transfer dissociation: Effects of cation charge state on product partitioning in ion/ion electron transfer to multiply protonated polypeptides. Int. J. Mass Spectrom 330-332, 174–181 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Good DM, Wirtala M, McAlister GC, Coon JJ: Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Mol. Cell. Proteomics. 6, 1942 (2007) [DOI] [PubMed] [Google Scholar]
- 162.Xia Y, Gunawardena HP, Erickson DE, McLuckey SA: Effects of Cation Charge-Site Identity and Position on Electron-Transfer Dissociation of Polypeptide Cations. J. Am. Chem. Soc 129, 12232–12243 (2007) [DOI] [PubMed] [Google Scholar]
- 163.Tureček F, Chung TW, Moss CL, Wyer JA, Ehlerding A, Holm AIS, Zettergren H, Nielsen SB, Hvelplund P, Chamot-Rooke J, Bythell B, Paizs B: The Histidine Effect. Electron Transfer and Capture Cause Different Dissociations and Rearrangements of Histidine Peptide Cation-Radicals. J. Am. Chem. Soc 132, 10728–10740 (2010) [DOI] [PubMed] [Google Scholar]
- 164.Prentice BM, McGee WM, Stutzman JR, McLuckey SA: Strategies for the gas phase modification of cationized arginine via ion/ion reactions. Int. J. Mass Spectrom 354-355, 211–218 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McLuckey SA: The Emerging Role of Ion/Ion Reactions in Biological Mass Spectrometry: Considerations for Reagent Ion Selection. Eur. J. Mass Spectrom 16, 429–436 (2010) [DOI] [PubMed] [Google Scholar]
- 166.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF: Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. Int. J. Mass Spectrom 236, 33–42 (2004) [Google Scholar]
- 167.Gunawardena HP, He M, Chrisman PA, Pitteri SJ, Hogan JM, Hodges BDM, McLuckey SA: Electron Transfer versus Proton Transfer in Gas-Phase Ion/Ion Reactions of Polyprotonated Peptides. J. Am. Chem. Soc 127, 12627–12639 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Compton PD, Strukl JV, Bai DL, Shabanowitz J, Hunt DF: Optimization of Electron Transfer Dissociation via Informed Selection of Reagents and Operating Parameters. Anal. Chem 84, 1781–1785 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Martens J, Berden G, Oomens J: Structures of Fluoranthene Reagent Anions Used in Electron Transfer Dissociation and Proton Transfer Reaction Tandem Mass Spectrometry. Anal. Chem 88, 6126–6129 (2016) [DOI] [PubMed] [Google Scholar]
- 170.Bowers JJ, Hodges BDM, Saad OM, Leary JA, McLuckey SA: Proton hydrates as soft ion/ion proton transfer reagents for multiply deprotonated biomolecules. Int. J. Mass Spectrom 276, 153–159 (2008) [Google Scholar]
- 171.Stephenson JL, McLuckey SA: Gaseous Protein Cations Are Amphoteric. J. Am. Chem. Soc 119, 1688–1696 (1997) [Google Scholar]
- 172.Prentice BM, Stutzman JR, McLuckey SA: Reagent Cluster Anions for Multiple Gas-Phase Covalent Modifications of Peptide and Protein Cations. J. Am. Soc. Mass. Spectrom 24, 1045–1052 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]