

Abstract
Background:
Atopic dermatitis (AD) is more common among African American children. Whether there are racial/ethnic difference among adults with AD, and causes for disparities are unclear.
Objective:
To examine the relationship between self-reported race/ethnicity and AD, and determine whether African genetic ancestry is predictive of these outcomes among African Americans.
Methods:
We analyzed data from two independent multi-ethnic longitudinal studies: 86,893 individuals age 18–100 from the Kaiser Permanente GERA cohort, and 5,467 individuals age 2–26 from the national PEER cohort. The primary outcomes were physician-diagnosed AD in GERA, and repeated measures of self-reported disease control among patient with physician-diagnosed AD at 6-month intervals in PEER. We examined whether self-identified African American race/ethnicity was predictive of these outcomes, and then tested whether a continuous measure of African genetic ancestry was associated with the outcomes within the African American group.
Results:
Atopic dermatitis was more common among self-identified African Americans than non-Hispanic whites in GERA (4.4% vs 2.1%, OR 2.06, 95% CI 1.70–2.48), and less well-controlled in PEER (odds of one level worse control: 1.91, 95% CI 1.64–2.22). African genetic ancestry, however, was not associated with AD risk or control among self-identified African Americans in either cohort. Nor did an AD polygenic risk score or genetic skin pigment score explain the AD disparities in AD.
Conclusion:
Ancestry-related genetic effects do not explain increased AD prevalence or poorer disease control among African Americans.
Keywords: atopic dermatitis, eczema, genetic ancestry, disease control, racial/ethnic disparities
Graphical Abstract
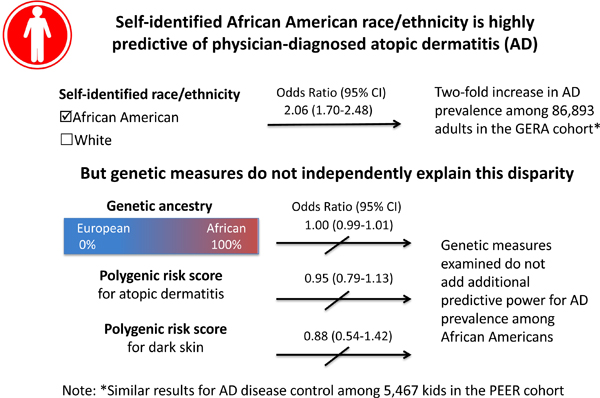
Capsule summary:
Genetic ancestry does not explain the increased prevalence and poorer disease control of atopic dermatitis among African Americans in two large US cohorts.
INTRODUCTION
Data from cross-sectional studies suggest that atopic dermatitis (AD, synonymous with atopic eczema)(1) is more common and more severe among African and African American children than non-Hispanic whites, but there are conflicting reports about whether AD remains more common among African Americans as they age, and little data about why racial/ethnic disparities may exist in AD.(2–7) Data on physician-diagnosed disease are sparse, and existing estimates may be biased by self-report and are limited to a single time point.(8) Using a longitudinal registry of children and young adults with physician-diagnosed mild-to-moderate AD, we previously found that African American race/ethnicity was one of the strongest predictors of a pattern of persistently active (vs resolving) AD during adolescence and early adulthood.(9) Because AD often waxes and wanes in severity, with extended periods of apparent disease remission in some patients, additional studies are needed to understand the nature of racial/ethnic disparities in both children and adults.
Investigations into the nature and causes of disparities in AD may elucidate mechanisms of disease susceptibility and persistence, and can inform targeted intervention strategies. AD is known to have strong genetic, environmental, and sociocultural influences,(10) but there are limited data on the extent to which these factors might impact racial/ethnic disparities. To date, most genetic research has been performed in European and Asian populations; however one recent GWAS found a novel locus for AD in African Americans.(11) Additionally, some studies suggest that filaggrin null mutations (the strongest known genetic risk factor for AD) are less common in populations of African descent, though other studies suggest these findings may be due to technical genotyping limitations.(12–16) Therefore, additional research is needed to understand whether the increased burden of AD in African Americans is due to other genetic or non-genetic factors.
Race and ethnicity are complex constructs that incorporate a range of social, cultural and genetic factors. For ancestrally admixed populations, measures of individual genetic ancestry can be calculated that represent differentially distributed genetic factors that vary according to historical geographic separations. African Americans typically have a mix of genetic ancestry from European and African populations, and a score ranging from 0–100% that represents the ancestry proportions from those two populations can be calculated for each individual.(17) An individual’s proportion of a particular genetic ancestry, which is continuous, may offer a more nuanced method for understanding disparities in outcomes beyond simple dichotomous self-identified racial and ethnic categories, and genetic ancestry analyses are increasingly being used to understand the genetic contribution to disease traits in admixed populations.(18–20) For example, African genetic ancestry has been shown to predict lung function and asthma exacerbations among African Americans better than self-reported race/ethnicity alone.(19, 21) To our knowledge, no studies have yet examined the role of genetic ancestry in AD.
The objectives of this study were to 1) determine whether there are differences in AD prevalence and control between self-identified US African Americans and whites, and 2) determine whether African genetic ancestry and/or polygenic risk scores underlying AD and skin pigment are predictive of these outcomes among African Americans in two large US cohorts. The rationale for a two-stage approach is to understand whether genetic factors provide additional explanatory evidence regarding AD disparities beyond self-identified race/ethnicity alone.
METHODS
Cohorts
The Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort, described in detail elsewhere, is health-system based and includes adults age 18 and older.(22) It was created to enable studies of genetic and environmental influences on health. Briefly, adult members from Kaiser Permanente Northern California, an integrated health delivery system with a membership of about 4.1 million representative of the general population in northern California, were asked to complete a written survey, completed a broad written consent and provided a saliva sample for extraction of DNA to be linked to their electronic health records (EHRs). The mean length of EHR follow-up is 23.5 years. Broad written consent was obtained, and the study was approved by the Institutional Review Board of the Kaiser Foundation Research Institute.
The Pediatric Eczema Elective Registry (PEER) is an ongoing prospective observational registry of patients age 2–26 with physician-diagnosed mild-to-moderate AD from across the US. The study was designed to investigate whether pimecrolimus, a topical calcineurin inhibitor, is associated with cancer, and an inclusion criterion was pimecrolimus use for at least 6 weeks prior to enrollment.(23) The diagnosis of AD was confirmed with the UK Working party criteria.(24) Patients are followed with mailed surveys every 6 months for up to 10 years, and follow-up is independent of physician visits and treatment decisions. After enrollment into the cohort, participants were asked to provide additional consent and saliva samples via mail for DNA extraction. The study was approved by the University of Pennsylvania Institutional Review Board. We included data from participants with at least one follow-up survey in the registry between 2004 and 2016; the mean duration of follow-up was 6.2 years.
In both cohorts, participants were asked in a baseline questionnaire to select one or more race/ethnicity/nationality categories that best described themselves. For this study, we only included individuals who self-identified as African American and/or non-Hispanic white. In the GERA cohort, 93.8% of participants endorsed a single race/ethnicity/nationality group and 6.2% endorsed two or more, and in the PEER cohort 93.1% of participants endorsed a single race/ethnicity group and 6.9% endorsed two or more. Individuals who endorsed more than one race/ethnicity group were included in the African American group for the primary analyses, provided that African American/Black was at least one of the categories endorsed. In a sensitivity analysis, we repeated the main analyses excluding these individuals.
DNA samples were available by design for all of the GERA participants, and for 755 (13%) individuals enrolled in the PEER cohort. For the latter, those who provided samples were more likely to self-identify as white, have a history of atopy, have a younger age at onset, higher family income at baseline, and longer duration of follow-up in the cohort (Supplemental Table 1).
Genetic ancestry estimation
In GERA, a principal components analysis was performed using the smartpca program, which is part of the EIGENSOFT 4.2 software package, on data genotyped with Affymetrix Axiom arrays.(25) Individual ancestral admixture proportions were then estimated using the full maximum-likelihood software package frappe to calculate the probability of a set of genome-wide genotypes in an individual as a weighted average of allele frequencies of putative ancestors, where the weights represent the admixture proportions.(17, 26, 27) In PEER, a panel of 186 previously validated ancestry informative markers (AIMS)(28, 29) genotyped with Illumina’s GoldenGate technology was used to estimate genetic ancestry. Ancestry estimates were calculated assuming 3 ancestral populations using a model-based clustering approach with the program STRUCTURE.(30)
AD Polygenic Risk Score
A genome-wide genetic ancestry estimate might not accurately reflect the specific genetic variants that underlie risk for AD. Therefore, using the most recently published genome-wide association study of AD,(31) we constructed a polygenic risk score (PRS) for AD risk, using 27 SNPs and weights based on the regression coefficients in that study.
Skin Pigment Polygenic Risk Score
We also tested the hypothesis that differences in skin pigment between European and African Americans could underlie the difference in risk of AD between the two racial/ ethnic groups. To test this hypothesis, we created a dark versus light skin pigment polygenic risk score (PRS) based on 39 SNPs from a recent study of skin color.(32)
Primary outcomes
The primary outcome in GERA was a physician diagnosis of AD, defined on the basis of two or more ICD-9 codes for AD (691.8) on separate dates. In sensitivity analyses, we tested the impact of varying the number of codes required to meet the definition from one to three.
In PEER, all of the participants had physician-diagnosed AD, and the primary outcome was a repeating measure of disease control. Patients were asked “during the last 6 months would you say your (or your child’s) skin has shown complete disease control, good disease control, limited disease control, or uncontrolled disease?” and whether they had used any prescription treatments for their AD. Inactive disease was defined as self-reported complete control without treatment. The disease control question was chosen because it is easily interpreted and comprehensive, and it has been shown to have good overlap with validated measures of AD severity recommended for use in clinical trials.(33–35)
Multivariable adjusted models included socio-demographic factors and atopic comorbidities previously shown to be related to AD: sex, history of atopy (yes/no), age at assessment (continuous), age at enrollment into the cohort (continuous), self-reported income at enrollment (3-level categorical), education (3-level categorical, GERA only), duration of follow-up (continuous, PEER only), and age at disease onset (continuous, PEER only). In GERA, atopic history was based on the presence of two or more ICD-9 codes for allergic rhinitis and/or asthma. Asthma was defined using ICD9 codes 493, 493.01, 493.1, 493.11, 493.82, 493.9, 493.91, and 493.92. Allergic rhinitis was defined using ICD9 codes 477, 477.1, 477.2, 477.8, and 477.9. In PEER, atopic history was based on a self-reported history of physician-diagnosed asthma or seasonal allergies.
Statistical analyses
Associations with self-reported race/ethnicity were examined using logistic regression models to estimate the odds of AD (yes/no) in African Americans as compared to non-Hispanic whites in GERA, and ordinal multilevel mixed effects models were used to estimate the subject-specific odds of a 6-month period of worse disease control in PEER. These models were then repeated among only self-identified African Americans to test for an association between a continuous measure of African genetic ancestry and atopic dermatitis prevalence (GERA) or control (PEER). The rationale for this approach was to test the additional predictive value of genetic ancestry beyond self-identified race/ethnicity, and has been used in similar analyses for asthma and lung function.(21, 36)
In post-hoc analyses, we replicated our models including information on genetic ancestry for all participants (including self-identified whites). In addition, we repeated the models with two new polygenic risk scores (described above) to further test whether genetic factors are related to disparities in AD risk. First, we tested the AD PRS for association with AD within the Europeans and African Americans by regression analysis, and also whether its inclusion in the regression model attenuated the association of self-reported race/ethnicity with AD in the GERA cohort. Second, we included a skin phenotype PRS as a covariate in a regression analysis including both the whites and African Americans in GERA, to see if the effect of self-reported race/ethnicity was attenuated, and also in an analysis of the African Americans alone to see if the pigment PRS was predictive of AD risk within that population. Additional methodological details are included in the supplemental material. Statistical analyses were performed using Stata 15 and R,(37, 38) and all tests were two-sided.
RESULTS
Participant characteristics
This study included 86,893 subjects who self-identified as non-Hispanic white and/or African American in GERA and 5,467 individuals who self-identified as non-Hispanic white and/or African American in PEER. Participants from GERA were older, had higher incomes, and lower overall rates of atopy than participants in PEER (Table 1). The distribution of demographic characteristics varied by self-reported race/ethnicity: in the GERA cohort, compared to whites, African Americans had lower incomes, lower levels of education, were younger at enrollment, and had higher rates of allergies and asthma. In the PEER cohort, compared to whites, African Americans were more likely to be female, have lower family incomes, less atopic history, and enroll at an older age (though they had similar reported age at AD onset).
Table 1.
Participant characteristics
| GERA Cohort | Total N=86,893 |
African Am* N=3,380 |
White N=83,513 |
p-value** |
|---|---|---|---|---|
| Female sex, N (%) | 50,225 (57.8%) | 1,950 (57.7%) | 48,275 (57.8%) | 0.896 |
| Annual income in US dollars, N (%) | ||||
| <$20,000 | 3890 (4.5%) | 231 (6.8%) | 3,659 (4.4%) | <0.001 |
| $20,000–60,000 | 24,113 (27.8%) | 1,116 (33.0%) | 22,997 (27.5%) | |
| > $60,000 | 51,719 (59.5%) | 1,781 (52.7%) | 49,938 (59.8%) | |
| Missing | 7,171 (8.3%) | 252 (7.5%) | 6,819 (8.3%) | |
| Education, N (%) | ||||
| Less than high school | 1545 (1.8%) | 110 (3.3%) | 1,435 (1.7%) | <0.001 |
| High School or equivalent | 31,957 (36.8%) | 1,419 (42.0%) | 30,538 (36.6%) | |
| College or graduate school | 48,363 (55.7%) | 1,579 (46.7%) | 46,784 (56.0%) | |
| Missing | 5,028 (5.8%) | 272 (8.0%) | 4,756 (5.7%) | |
| History of atopy, N (%) | 29,254 (33.7%) | 1,346 (39.8%) | 27,908 (33.4%) | <0.001 |
| Age at cohort enrollment, mean years (SD) | 63.6 (14.3) | 60.5 (14.3) | 63.8 (14.3) | <0.001 |
| PEER Cohort |
Total N= 5,467 |
African Am* N= 3,251 |
White N = 2,216 |
p-value** |
| Female sex, N (%) | 2,914 (53.3%) | 1,844 (56.7%) | 1,070 (48.3%) | <0.001 |
| Annual income in US dollars, N (%) | ||||
| <$25,000 | 2,222 (40.6%) | 1,911 (58.8%) | 311 (14.0%) | <0.001 |
| $25,000–50,000 | 810 (14.8%) | 445 (13.7%) | 365 (16.5%) | |
| > $50,000 | 1,009 (18.5%) | 218 (6.7%) | 791 (35.7%) | |
| Missing | 1,426 (26.1%) | 677 (20.8%) | 749 (33.8%) | |
| History of atopy, N (%) | 4,033 (73.8%) | 2,291 (70.5%) | 1,742 (78.6%) | <0.001 |
| Age at cohort enrollment, mean years (SD) | 7.21 (4.12) | 7.31 (4.15) | 7.06 (4.07) | 0.027 |
Notes:
Includes 516 individuals in GERA and 184 individuals in PEER who self-reported both African American and another race.
p-value from chi2 test for categorical variables and t-test for continuous variables.
When we compared individuals with and without AD in GERA, we found that cases were more likely to be female and have a history of other atopic comorbidities, and these effects were consistent in each race/ethnicity group. Among African Americans, cases were also more likely to enroll in the cohort at a younger age, and among whites, cases were more likely to have lower incomes. There were no significant differences in educational attainment (Table 2).
Table 2.
Multivariate model results
| GERA Cohort | ||
|---|---|---|
| Model 1: Odds of AD among full cohort (N=76,060) | Model 2: Odds of AD among African Americans (N=3,161) | |
| Odds Ratio (95% CI) | ||
| Percent African ancestry (by 1-unit increase) | N/A | 1.00 (0.99, 1.01) |
| Self-identified race (African American vs white) | 2.06 (1.70, 2.48) | N/A |
| Sex (female vs male) | 1.25 (1.13, 1.39) | 1.70 (1.11, 2.59) |
| Income | ||
| <$20,000 | ref | ref |
| $20,000–60,000 | 0.86 (0.69, 1.08) | 0.80 (0.36, 1.77) |
| >$60,000 | 0.77 (0.62, 0.96) | 1.09 (0.50, 2.36) |
| Education | ||
| Less than high school | ref | ref |
| High school or equivalent | 0.95 (0.66, 1.39) | 3.02 (0.40, 22.7) |
| College or graduate school | 1.06 (0.73, 1.54) | 2.69 (0.36, 20.4) |
| History of atopy | 2.19 (1.99, 2.42) | 1.92 (1.31, 2.81) |
| Age (each additional year) | 1.00 (1.00, 1.00) | 1.00 (0.98, 1.01) |
| PEER Cohort | ||
| Model 1: Odds of 1 level worse disease control among full cohort (N=2,041) | Model 2: Odds 1 level worse disease control among African Americans (N= 257) | |
| Odds Ratio (95% CI) | ||
| Percent African Ancestry (by 1-unit increase) | N/A | 1.00 (0.99, 1.02) |
| Self-identified race (African American vs white) | 2.02 (1.71, 2.37) | N/A |
| Sex (female vs male) | 1.20 (1.06, 1.37) | 1.83 (1.09, 3.07) |
| Income | ||
| $<25K | ref | ref |
| $25–50K | 0.94 (0.79, 1.12) | 0.93 (0.48, 1.81) |
| $>50K | 0.73 (0.61, 0.88) | 0.66 (0.32, 1.39) |
| History of atopy | 1.20 (1.04, 1.39) | 1.46 (0.78, 2.70) |
| Age (each additional year) | ||
| Age at cohort enrollment | 1.23 (1.21, 1.25) | 1.16 (1.07, 1.25) |
| Age at survey | 0.82 (0.81, 0.83) | 0.88 (0.84, 0.92) |
Self-reported race/ethnicity and AD
AD was more common and under worse control among African Americans than whites. The primary outcomes of physician diagnosis in GERA and disease control in PEER are shown by race/ethnicity in Supplemental Table 2 and Figure 1. In the GERA cohort, 4.4% of African Americans vs 2.1% of whites (p<0.001) met the definition of AD. In the PEER cohort, compared to whites, African Americans reported worse disease control at all ages, and the mean proportion of surveys in which respondents reported active disease during the prior 6 months was higher among African Americans (94.5% vs 84.4%, p<0.001).
Figure 1. Association between self-reported race/ethnicity and atopic dermatitis control in the PEER cohort.
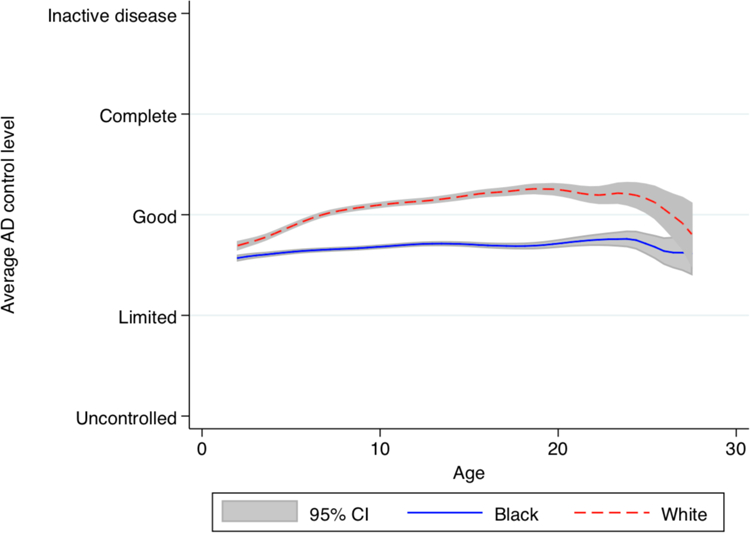
The graph is a local polynomial smoothed plot with grey shading for the 95% confidence intervals generated from cross-sectional calculations of the average atopic dermatitis control level at each age.
In multivariable models, we found African Americans had an approximately two-fold increase in both the odds of AD (OR 2.06, 95% CI 1.70–2.48) in GERA, and the odds of one level worse control (2.02, 95% CI 1.71–2.37) in PEER (Table 2).
African genetic ancestry and AD
The distribution of African genetic ancestry among self-identified African Americans (including 516 individuals in GERA and 184 individuals in PEER who self-reported African American plus at least one other racial/ethnic group) ranged from 0–100% and was similar in both cohorts (mean 74%, SD 15% in GERA; and mean 76%, SD 17% in PEER; Supplemental Figure 1 and Supplemental Table 3).
African genetic ancestry showed no association with the AD outcomes among African Americans in either cohort (Table 2). Genetic ancestry was modeled as a continuous variable ranging from 0–100% and the primary results reflect the odds of a 1% increase in genetic ancestry. To make the magnitude of association clearer, we also calculated the odds on a larger scale. In GERA, the odds of AD for each quartile increase in African ancestry were 1.07, 95% CI 0.83–1.36, and in PEER, the odds of poorer disease control for each quartile increase in ancestry were 1.05, 95%CI 0.72–1.53.
As described in the methods section, for the principal analysis, we modeled the effect of African genetic ancestry only among African Americans. When we included all participants (self-identified African Americans and whites) in the African genetic ancestry models, we again found no significant association between genetic ancestry and AD (Supplemental Table 4).
Similarly, we found that predicted skin color was not independently associated with AD beyond self-identified race/ethnicity alone in an analysis that included both whites and African Americans, nor was it associated with AD within only the African Americans group (Supplemental Table 5). Finally, we found that although a polygenic risk score was highly predictive of AD overall, it was not correlated with genetic ancestry, it did not attenuate the association of self-reported race/ethnicity in the regression model of AD risk in the entire sample, nor was it predictive of AD among self-identified African Americans in contrast to its strong association with AD risk in whites (Supplemental Table 6).
Sensitivity analyses
We repeated our analyses in the GERA cohort, varying the number of ICD9 code diagnoses required to meet the definition of AD, and found that the strength of association between self-reported race/ethnicity and AD increased with more codes. However, there continued to be no association between African genetic ancestry and AD outcomes among self-identified African Americans (Supplemental Table 7). We also repeated the analyses excluding 25 individuals from PEER who had self-identified as both African American and white, and again found similar results (Supplemental Table 8).
DISCUSSION
We found that AD was more common among adults and under worse control among children who self-identified as African American as compared to non-Hispanic whites in two large US cohorts. However, these racial/ethnic differences were not associated with a continuous measure of genetic ancestry, nor were they associated with specific genetic factors underlying AD risk or skin pigment. Our results suggest that genetic factors that vary according to historical geographic separations are unlikely to explain the observed differences, and that social and environmental factors that vary by race/ethnicity warrant additional research.
It is important to note that self-identified race/ethnicity and genetic ancestry are two distinct but correlated constructs. As shown in Supplemental Figure 1, there was a great deal of variation in genetic ancestry among African Americans, and the rationale for examining this more nuanced measure was to test whether genetic data offered additional predictive power beyond self-identified race/ethnicity alone.
To our knowledge, this is the first study to examine the relationship between genetic ancestry and AD. Although we did not find evidence for an association between global genetic ancestry and AD in African Americans, we cannot rule out the possibility that there may be genetic differences by race/ethnicity not fully captured by the ancestry estimates. We found that an AD polygenic risk score was highly predictive of AD in whites but not African Americans, similar to a recent GWAS of AD in African Americans that was not able to replicate known AD and allergy genetic loci in an African American population.(11) These results suggest that the genetic architecture of risk in African Americans may be quite distinct from that in whites, and underscores the need for additional genetic studies of AD in African Americans, in whom the prevalence is greatest. Of note, the lack of any strong specific gene association findings in that study(11) also suggests that location specific-as opposed to genome-wide genetic ancestry is unlikely to explain the higher rate of AD in African Americans. Additionally, there may be racial/ethnic differences in gene transcription. A recent study that sought to characterize the global molecular profile of AD patients found significant variation in cellular infiltrates as well as expression of immune and barrier genes between African American and white subjects.(39) The extent to which such changes are due to environmental and behavioral factors should be explored in future work.
The fact that the skin pigment polygenic risk score did not correlate with AD risk in GERA provides evidence that AD is likely independent of skin pigment, per se, and that the pigment difference between African Americans and whites appears to have no role in the AD prevalence difference. New research showing that darker skin pigmentation results in better barrier function and higher inflammatory thresholds may help to explain this finding.(40)
Our results suggest that social and environmental factors are likely to play an important role in atopic dermatitis disparities and warrant additional research. In both cohorts in our study, lower income was associated with AD outcomes, independent of self-reported race/ethnicity. Our findings contrast with those from a systematic review and meta-analysis that found AD was associated with higher socioeconomic position.(41) Of note, the majority of studies included in the review came from European populations with less racial/ethnic diversity, and there was a great deal of heterogeneity between estimates. US studies on income and AD are mixed, likely due to differences in the way AD is defined (by self-report vs physician-diagnosis) and whether studies adequately controlled for differential access to health care. Two US studies found income had no impact on childhood AD after controlling for race/ethnicity and insurance status,(2, 4) one found higher household income was associated with adult AD,(6) and another found the opposite in a large population-based sample of individuals ages 0–100.(5) Additional work needs to be done to understand the impact of socio-environmental factors including whether differential exposure to microbes, pollutants, stress, differences in health care access, and treatment explain disparities.
We chose to use the GERA and PEER cohorts because they have complementary strengths and limitations. Patients enrolled into PEER met the widely accepted and validated UK Working Party diagnostic criteria,(24, 42) and had detailed measures of disease control over time. AD is an episodic condition, meaning that the symptoms wax and wane, and patients may experience periods without any clinical signs or symptoms. Therefore, repeated measures of disease activity and control are important to understanding the course and burden of the disease. By contrast, diagnoses were limited to ICD codes in GERA, which may have induced misclassification bias. When we performed stratified analyses by number of diagnostic codes, we found that effect sizes increased with the number of diagnostic codes (which may reflect both the validity and severity of the diagnosis) in our primary analyses of self-reported race/ethnicity, but even with increasing numbers of diagnostic codes, there remained no association between genetic ancestry and AD among African Americans. Ancestry was estimated using genome-wide data in the GERA cohort, however the PEER cohort had less robust measures based on a limited number of ancestry informative markers. Nonetheless, the distribution of genetic ancestry was similar in both cohorts. Genetic data was only available for a subset of the PEER cohort. While we cannot rule out the possibility of selection bias, the probability that the contribution of a biospecimen was differentially correlated with degree of African genetic ancestry is small. Finally, although participants in the PEER cohort may have had differential access to health care, participants in GERA were all members of the Kaiser Permanente Health provider system. An overall strength of our study is use of two large longitudinal cohorts with physician-diagnosed AD, and the consistency of results across both populations is reassuring.
Our results are important in the context of the treatment revolution currently underway in AD.(43) African Americans are historically underrepresented in clinical research, and as new agents enter clinical testing, it is important to consider the extent to which trial populations are representative of the actual population of affected individuals. Our results imply that sub-analyses to detect heterogeneous treatment responses should be based on social, environmental, and demographic factors, in addition to biomarkers or genetic polymorphisms.
Supplementary Material
Clinical implication:
Self-report of race/ethnicity can help to identify patients at higher risk of AD and poor AD control.
Acknowledgments
Funding sources: KA is supported by grants from the Robert Wood Johnson Foundation and NIAMS (K23 AR073915). EJ is supported by grants from the National Eye Institute (R01 EY027004) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK116738). We are grateful to the Kaiser Permanente Northern California members who have generously agreed to participate in the Kaiser Permanente Research Program on Genes, Environment, and Health. Support for participant enrollment, survey completion, and biospecimen collection for the RPGEH was provided by the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, and Kaiser Permanente National and Regional Community Benefit programs. Genotyping of the GERA cohort was funded by a grant from the National Institute on Aging, National Institute of Mental Health, and the National Institute of Health Common Fund (RC2 AG036607 to Cathy Schaefer and NR). The funding organizations were not involved in the design and conduct of the study, collection, management, analysis, and interpretation of the data, or preparation, review or approval of the manuscript.
Abbreviations:
- AD
Atopic dermatitis
- AIM
Ancestry informative marker
- GERA
Genetic Epidemiology Research on Adult Health and Aging
- GWAS
Genome-wide association study
- PEER
Pediatric Eczema Elective Registry
- PRS
Polygenic risk score
- SNP
Single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: KA receives research funding for atopic dermatitis from the NIH and the Robert Wood Johnson Foundation, and is a consultant to TARGETPharma, a company developing a prospective atopic dermatitis registry. DM receives funding for research on atopic dermatitis from the NIH and Valeant, and he is a consultant for Sanofi and GlaxoSmithKline. The rest of the authors have nothing to declare.
REFERENCES
- 1.Silverberg JI, Thyssen JP, Paller AS, Drucker AM, Wollenberg A, Lee KH, et al. What’s in a name? Atopic dermatitis or atopic eczema, but not eczema alone. Allergy 2017;72(12):2026–30. [DOI] [PubMed] [Google Scholar]
- 2.Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol 2011;131(1):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI, Group IPTS. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol 2009;124(6):1251–8 e23. [DOI] [PubMed] [Google Scholar]
- 4.Silverberg JI, Simpson EL. Associations of childhood eczema severity: a US population-based study. Dermatitis 2014;25(3):107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanifin JM, Reed ML, Eczema P, Impact Working G. A population-based survey of eczema prevalence in the United States. Dermatitis 2007;18(2):82–91. [DOI] [PubMed] [Google Scholar]
- 6.Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol 2013;132(5):1132–8. [DOI] [PubMed] [Google Scholar]
- 7.Mahdavinia M, Fox SR, Smith BM, James C, Palmisano EL, Mohammed A, et al. Racial Differences in Food Allergy Phenotype and Health Care Utilization among US Children. J Allergy Clin Immunol Pract 2017;5(2):352–7 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kramer U, Schafer T, Behrendt H, Ring J. The influence of cultural and educational factors on the validity of symptom and diagnosis questions for atopic eczema. Br J Dermatol 1998;139(6):1040–6. [DOI] [PubMed] [Google Scholar]
- 9.Abuabara K, Hoffstad O, Troxel AB, Gelfand JM, McCulloch CE, Margolis DJ. Patterns and predictors of atopic dermatitis disease control past childhood: An observational cohort study. J Allergy Clin Immunol 2017. [DOI] [PMC free article] [PubMed]
- 10.Weidinger S, Novak N. Atopic dermatitis. Lancet 2016;387(10023):1109–22. [DOI] [PubMed] [Google Scholar]
- 11.Almoguera B, Vazquez L, Mentch F, March ME, Connolly JJ, Peissig PL, et al. Novel locus for atopic dermatitis in African Americans and replication in European Americans. J Allergy Clin Immunol 2019;143(3):1229–31. [DOI] [PubMed] [Google Scholar]
- 12.Winge MC, Bilcha KD, Lieden A, Shibeshi D, Sandilands A, Wahlgren CF, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br J Dermatol 2011;165(5):1074–80. [DOI] [PubMed] [Google Scholar]
- 13.Polcari I, Becker L, Stein SL, Smith MS, Paller AS. Filaggrin gene mutations in African Americans with both ichthyosis vulgaris and atopic dermatitis. Pediatr Dermatol 2014;31(4):489–92. [DOI] [PubMed] [Google Scholar]
- 14.Margolis DJ, Gupta J, Apter AJ, Hoffstad O, Papadopoulos M, Rebbeck TR, et al. Exome sequencing of filaggrin and related genes in African-American children with atopic dermatitis. The Journal of investigative dermatology 2014;134(8):2272–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eaaswarkhanth M, Xu D, Flanagan C, Rzhetskaya M, Hayes MG, Blekhman R, et al. Atopic Dermatitis Susceptibility Variants in Filaggrin Hitchhike Hornerin Selective Sweep. Genome biology and evolution 2016;8(10):3240–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margolis DJ, Mitra N, Gochnauer H, Wubbenhorst B, D’Andrea K, Kraya A, et al. Uncommon Filaggrin Variants Are Associated with Persistent Atopic Dermatitis in African Americans. J Invest Dermatol 2018;138(7):1501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 2015;200(4):1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta J, Johansson E, Bernstein JA, Chakraborty R, Khurana Hershey GK, Rothenberg ME, et al. Resolving the etiology of atopic disorders by using genetic analysis of racial ancestry. J Allergy Clin Immunol 2016;138(3):676–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Pacheco N, Flores C, Oh SS, Burchard EG, Pino-Yanes M. What Ancestry Can Tell Us About the Genetic Origins of Inter-Ethnic Differences in Asthma Expression. Curr Allergy Asthma Rep 2016;16(8):53. [DOI] [PubMed] [Google Scholar]
- 20.Mountain JL, Risch N. Assessing genetic contributions to phenotypic differences among ‘racial’ and ‘ethnic’ groups. Nat Genet 2004;36(11 Suppl):S48–53. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R, Seibold MA, Aldrich MC, Williams LK, Reiner AP, Colangelo L, et al. Genetic ancestry in lung-function predictions. N Engl J Med 2010;363(4):321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, Connell S, et al. Genotyping Informatics and Quality Control for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 2015;200(4):1051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapoor R, Hoffstad O, Bilker W, Margolis DJ. The frequency and intensity of topical pimecrolimus treatment in children with physician-confirmed mild to moderate atopic dermatitis. Pediatr Dermatol 2009;26(6):682–7. [DOI] [PubMed] [Google Scholar]
- 24.Williams HC, Burney PG, Hay RJ, Archer CB, Shipley MJ, Hunter JJ, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. I. Derivation of a minimum set of discriminators for atopic dermatitis. Br J Dermatol 1994;131(3):383–96. [DOI] [PubMed] [Google Scholar]
- 25.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS genetics 2006;2(12):e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genetic epidemiology 2005;28(4):289–301. [DOI] [PubMed] [Google Scholar]
- 27.Choquet H, Paylakhi S, Kneeland SC, Thai KK, Hoffmann TJ, Yin J, et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nature communications 2018;9(1):2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiner AP, Carlson CS, Ziv E, Iribarren C, Jaquish CE, Nickerson DA. Genetic ancestry, population sub-structure, and cardiovascular disease-related traits among African-American participants in the CARDIA Study. Human genetics 2007;121(5):565–75. [DOI] [PubMed] [Google Scholar]
- 29.Stefflova K, Dulik MC, Barnholtz-Sloan JS, Pai AA, Walker AH, Rebbeck TR. Dissecting the within-Africa ancestry of populations of African descent in the Americas. PLoS One 2011;6(1):e14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. American journal of human genetics 2000;67(1):170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015;47(12):1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaitanya L, Breslin K, Zuniga S, Wirken L, Pospiech E, Kukla-Bartoszek M, et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic science international Genetics 2018;35:123–35. [DOI] [PubMed] [Google Scholar]
- 33.Chang J, Bilker WB, Hoffstad O, Margolis DJ. Cross-Sectional Comparisons of Patient-Reported Disease Control, Disease Severity, and Symptom Frequency in Children with Atopic Dermatitis. Br J Dermatol 2017. [DOI] [PubMed]
- 34.Eichenfield LF, Lucky AW, Boguniewicz M, Langley RG, Cherill R, Marshall K, et al. Safety and efficacy of pimecrolimus (ASM 981) cream 1% in the treatment of mild and moderate atopic dermatitis in children and adolescents. J Am Acad Dermatol 2002;46(4):495–504. [DOI] [PubMed] [Google Scholar]
- 35.Kapp A, Papp K, Bingham A, Folster-Holst R, Ortonne JP, Potter PC, et al. Long-term management of atopic dermatitis in infants with topical pimecrolimus, a nonsteroid anti-inflammatory drug. J Allergy Clin Immunol 2002;110(2):277–84. [DOI] [PubMed] [Google Scholar]
- 36.Pino-Yanes M, Thakur N, Gignoux CR, Galanter JM, Roth LA, Eng C, et al. Genetic ancestry influences asthma susceptibility and lung function among Latinos. J Allergy Clin Immunol 2015;135(1):228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.StataCorp. Stata Statistical Software: Release 15 College Station, TX: StataCorp LLC; 2017. [ [Google Scholar]
- 38.Team RC. R: A language and environment for statistical computing Vienna, Austria: R Foundation for Statistical Computing; 2013. [Available from: http://www.R-project.org/. [Google Scholar]
- 39.Sanyal RD, Pavel AB, Glickman J, Chan TC, Zheng X, Zhang N, et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann Allergy Asthma Immunol 2019;122(1):99–110 e6. [DOI] [PubMed] [Google Scholar]
- 40.Lin TK, Man MQ Abuabara K, Wakefield J, Sheu H, Tsai J, Li C, Elias PM B By protecting against cutaneous inflammation, deep epidermal pigmentation provided additional advantages for ancestral humans. J Invest Dermatol May 2019;139(5):S137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uphoff E, Cabieses B, Pinart M, Valdes M, Anto JM, Wright J. A systematic review of socioeconomic position in relation to asthma and allergic diseases. Eur Respir J 2015;46(2):364–74. [DOI] [PubMed] [Google Scholar]
- 42.Vakharia PP, Chopra R, Silverberg JI. Systematic Review of Diagnostic Criteria Used in Atopic Dermatitis Randomized Controlled Trials. Am J Clin Dermatol 2018;19(1):15–22. [DOI] [PubMed] [Google Scholar]
- 43.Paller AS, Kabashima K, Bieber T. Therapeutic pipeline for atopic dermatitis: End of the drought? J Allergy Clin Immunol 2017;140(3):633–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.