

Abstract
Even though the majority of population studies in environmental health focus on a single factor, environmental exposure in the real world is a mixture of many chemicals. The concept of “exposome” leads to an intellectual framework of measuring many exposures in humans, and the emerging metabolomics technology offers a means to read out both the biological activity and environmental impact in the same dataset. How to integrate exposome and metabolome in data analysis is still challenging. Here, we employ a hierarchical community network to investigate the global associations between the metabolome and mixed exposures including DDTs, PFASs and PCBs, in a women cohort with sera collected in California in the 1960s. Strikingly, this analysis revealed that the metabolite communities associated with the exposures were non-specific and shared among exposures. This suggests that a small number of metabolic phenotypes may account for the response to a large class of environmental chemicals.
Introduction
The health impact from environmental exposures is rarely isolated. Most human beings live under the exposure of a complex mixture of many chemicals, and their physiological conditions contribute to the biological responses. The concept of exposome is to obtain comprehensive measurements of environmental exposures [1–3]. This is accompanied by the advancing of other high-dimensional molecular data in systems biology, which can be used to understand the biological responses to exposome. Metabolomics is an emerging data type that is of great interest to environmental research [4, 5]. LC-MS (liquid chromatograph – mass spectrometry), the dominant metabolomics technology, is now capable of routine measurement of over 10,000 features in biological matrices. Besides biological metabolites, the data also capture dietary intake, use of pharmaceuticals and other substances, microbial products and increasingly environmental chemicals. Because these measurements of small molecules serve as a readout of both gene activities and environmental impacts, metabolomics is positioned as an important layer between the interaction of gene and environment (G x E). We therefore postulate that the more actionable model of “G x E” is “G x M x E”, where M stands for metabolome. Metabolome-wide association studies (MWAS) are now part of the toolbox for epidemiology [6–9]. The integration of exposome and metabolome is still new and challenging.
The Child Health and Development Studies (CHDS) followed a birth cohort in California. During 1959-1967, over 15,000 pregnant women in the Kaiser Permanente Health Plan joined the CHDS. Over time, exposure data and health records were collected in multiple generations. Several studies have reported link between early-life DDT (dichlorodiphenyltrichloroethane) exposure and breast cancer risk based on the CHDS pregnancy cohort [10–13]. High level of o,p’-DDT in maternal serum corresponded to nearly four fold increase of breast cancer risk in the daughters [12]. In this special issue of Reproductive Toxicology, Hu et al have reported the MWAS results on DDTs in this cohort [14], that amino acid pathways were associated with levels of p,p’-DDT and o,p’-DDT, while mitochondrial pathways of carnitine and lipid metabolism were associated with p,p’-DDE. The MWAS analysis of PFAS (poly and perfluoroalkyl substances) compounds was also reported in this issue, that carnitines and urea cycle metabolites were positively associated with PFOS (perfluorooctane sulfonic acid) levels [15]. Overall in this cohort, we have targeted measurement of 39 environmental chemicals. Besides DDTs and PFASs, oxychlordane, hexachlorobenzenes, and a number of polychlorinated biphenyl compounds are among the available measurements (Suppl Table 1). This data collection thus provides an opportunity to systematically assess the mutual influence between exposome and metabolome. Specifically, what metabolic pathways are associated with what chemical classes, and how quantitative and specific are the associations?
To answer this question, several methodological challenges need to be addressed. In -omics data, it’s common that many measured variables are not independent from each other, harboring a large degree of inter-correlation and redundancy. When exposome intersects with other high-dimensional molecular data, the combinatorial space explodes and the complexity becomes overwhelming. The biological interpretation is confounded by the built-in properties of living organisms: they can tolerate and adapt to exposures, and toxicity is not necessarily manifested under most conditions [16]. A more specific challenge in the field of metabolomics is that a large number of metabolite features are not identified in the data, thus the interpretation can be severely restricted [17, 18]. Therefore, we apply to this CHDS dataset unbiased multivariate analyses, and assess the association between metabolome and mixed exposures in a hierarchical community network model. The metabolic pathways connected with the exposures are inferred using the mummichog software [19], and the hierarchical community network is used to investigate the breast cancer risk in the daughters’ generation.
Result
Variance analysis of the metabolome reveals both redundancy and specificity in exposure effects
The initial CHDS study followed pregnant women during 1960’s in California, when exposure to DDT was at a historically high level. Besides the DDT compounds, we measured 39 environmental chemical exposures in 467 CHDS participants using archived peripheral blood samples (Suppl Table 1). The metabolomics analysis was performed on the peripheral blood samples of 397 participants, using high-resolution mass spectrometry coupled with liquid chromatography. This is a subset of the 467 participants for which we described the exposure data above, as not all samples were available for the metabolomics analysis. The samples used in this study were from women of an average age of 25, mostly nonobese and non-African-American, and the detailed demographics was given in Hu et al in this issue [14, 15].
Principal component analysis (PCA) of the serum metabolome of 397 participants shows that principal component 1 captures 58.5% of the variance, with small contributions from other components (Figure 1, left). The levels of each environmental chemicals were regressed against each principal component of the metabolome, to assess their influence on the metabolome. As seen in the heatmap in Figure 1, several chemicals, such as PFOA, PFOSA and HCB, do not show significant association with any of the top ten principal components of the metabolome. DDE is associated with principal components 1 through 4, and DDT with component 9. Typically, each exposure explains less under 5% of each principal component. The three lipid measurements, total lipid (TL), triglycerol (TG) and total cholesterol (TC), were included as a control, and they show the highest association with principal component 4 of the metabolome (explaining ~14% variance therein). Of note, when common linear regression models are used to test the association between a metabolite and an exposure, they operate mostly on principal component 1. The result here indicates that the influence of exposures is not limited to principal component 1, but specific to a principal component by different exposures. The effect size is small in general, and could also be easily masked by other variances. The variables associated with the same principal component (e.g. many PCB chemicals on principal component 4) are likely to be driven by inter-correlation. This redundancy in the exposure data is further examined in the next section.
Figure 1. Variance analysis of the metabolome and environmental exposures.

Principal component analysis was performed on the serum metabolomics data from 397 CHDS participants, and the contribution of top 10 principal components is shown on the left. Corresponding to each of the components, the variance explained by each exposure (R2) is colored coded in the heatmap on the right. Data are only shown for features with p < 0.05 in Pearson correlation analysis. The lipid measures, TG, TC and TL, are included as control.
These data here provide the justification for the PLS (project to latent structures, or partial least squares) methods that are now popular in multi-omics integration. Because different data types may not associate on the same principal component, and PLS gives more flexible projection than PCA. Still, pairwise feature association disregards the inter-correlations and creates a large combination space (3,121 × 39 here). Therefore, we resort to a hierarchical community network approach [20, 21], to combine PLS regression with identifying the granular community structures within each data types. Instead of feature-level associations, the individual features are organized as members of communities, and the associations are tested at community level across exposome and metabolome.
Community structures detected in the exposome
To assess the correlation between all 39 chemicals, we computed pairwise Spearman rank correlations, and unsupervised hierarchical clustering was performed on the correlation coefficients (Figure 2A). The community structure is visually clear in the heatmap, as most PCB congeners form the largest cluster, and DDT compounds and PFAS compounds form their own clusters. In addition, we included TL, TG and TC measurements of in the analysis, because they add an important reference and the level of many environmental chemicals is influenced by these lipid levels. Not surprisingly, TL, TG and TC form their own cluster in the upper-left corner of the map (Figure 2A).
Figure 2. Exposure communities detected by quantitative correlations.
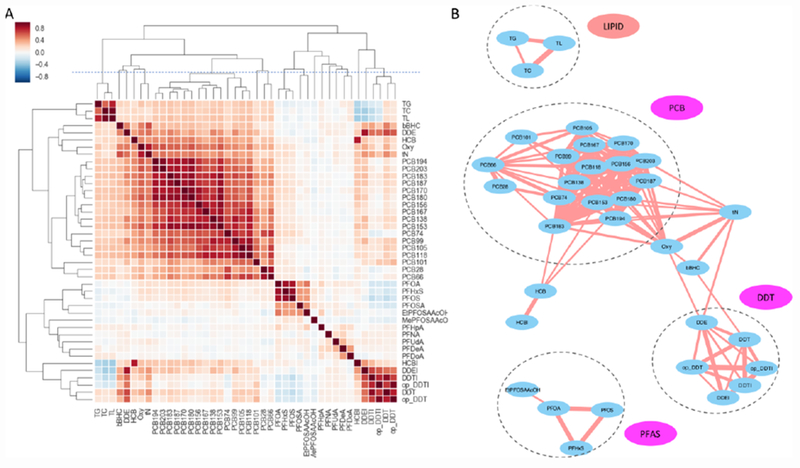
Each variable was measured in the CHDS cohort by conventional assays, either enzymatic or targeted mass spectrometry. (A) Unsupervised hierarchical clustering of exposure measurements, aside with several clinical variables, using Spearman correlation. (B) Communities detected among the variables from the correlation network, which was defined by all pairs of variables with Spearman correlation coefficient above 0.5. The result is similar to the clusters identified by the dendrogram (above the dashed line) in A.
To investigate the community structure beyond pairwise relationships, we converted the data into a correlation network. That is, an edge was formed between two nodes (variables) when their correlation coefficient was greater than a threshold of 0.5, and all edges defined the network in Figure 2B. A community was then defined as a set of nodes that have more connections within themselves than with the other members in the network. Many algorithms of community detection were developed in recent years. The application of a common Girvan-Newman algorithm [22] resulted in the detection of four communities, marked by dashed circles in Figure 2B. These correspond to communities of lipids, PCB, DDT and PFAS compounds. While these 39 measurements are far from a complete exposome, they provide a proxy to test the methodologies in investigating how complex mixtures impact the human physiology.
A hierarchical community network model for Metabolome x Exposures
We next sought to identify the communities within the metabolomics data. This approach helps to organize both the biological and analytical redundancy. Since these are untargeted metabolomics data, a metabolite can be represented in the data as multiple features (adduct ions and isotopes). The community grouping is an appealing approach to compartmentalize the redundancy in metabolomics data. We previously reported a method [20] to incorporate retention time in the distance function in hierarchical clustering, because chemicals of similar chromatographic retention time are more likely to belong to the same class or pathway. This clustering method was used to define 34 metabolite communities in this dataset (Suppl Figure 1, Suppl Table 2).
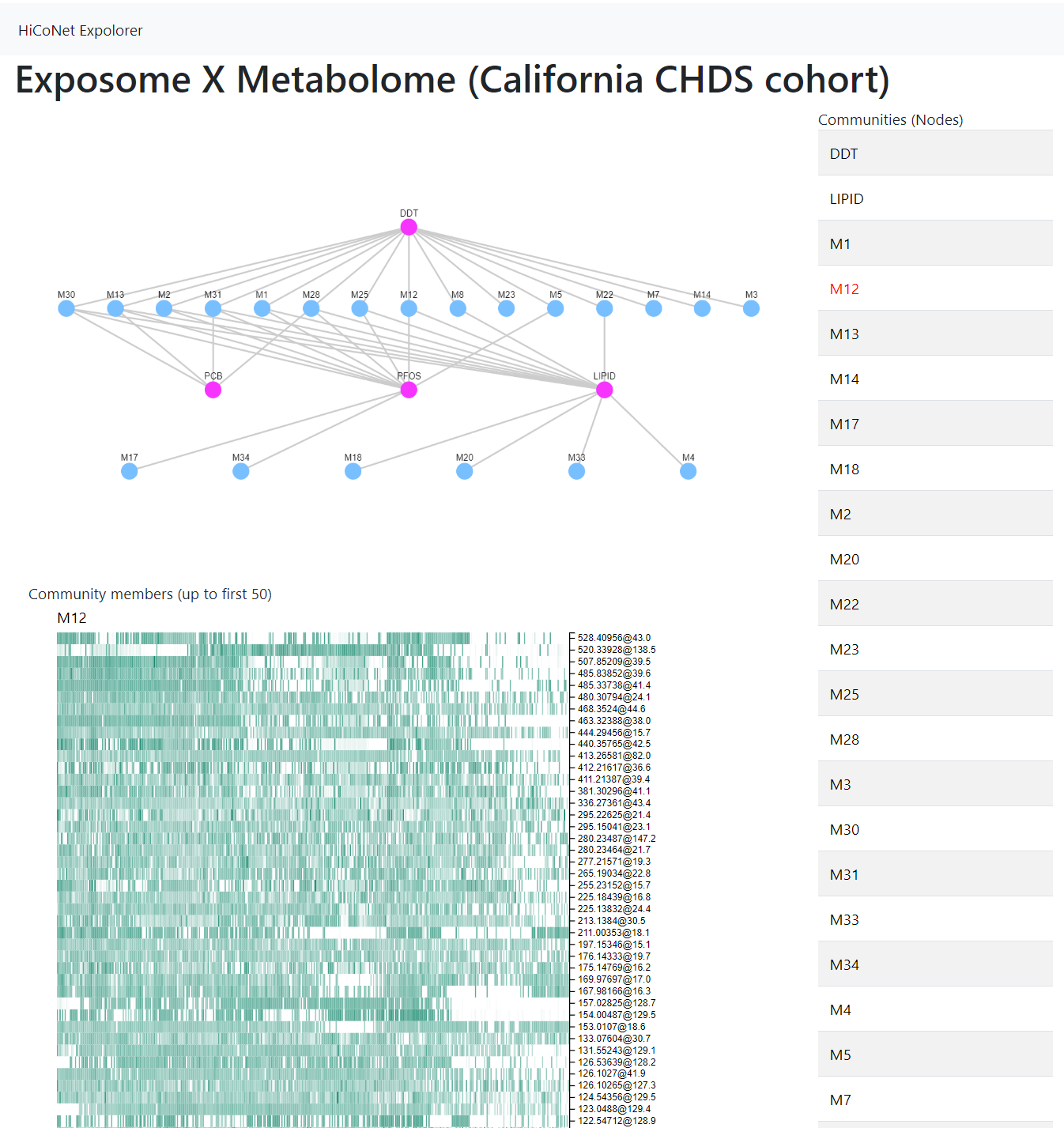
The association between each exposure community and each metabolite community was assessed by PLS regression, as described previously [20, 21]. Briefly, the goodness of using one data matrix (e.g. a metabolite community) to predict another data matrix (e.g. an exposure community) was recorded as PLS scores. These scores were compared against those obtained on randomly resampled data and statistical significance was assigned as a p-values. Taking all associations with p < 0.01, a network was formed between the exposure communities and metabolite communities (Figure 3). This network is hierarchical, because each node is a community on its own, and contains another level of members as depicted in Figure 2B. The connections in Figure 3 are visualized according to their significance. A number of metabolite communities showed strong associations with DDT and PFAS communities (and lipids), but the connection between the PCB community and the metabolome was less significant. Of note, the association by PLS can be either positive or negative, while each community contains members that are only positively correlated with each other. To accommodate the rich information from this type of analyses, we have also created a supplemental web tool to support interactive exploration of this hierarchical network (Suppl Figure 2, https://hiconet.appspot.com/exposome).
Figure 3. Hierarchical community network between exposures and metabolomics in the CHDS cohort.
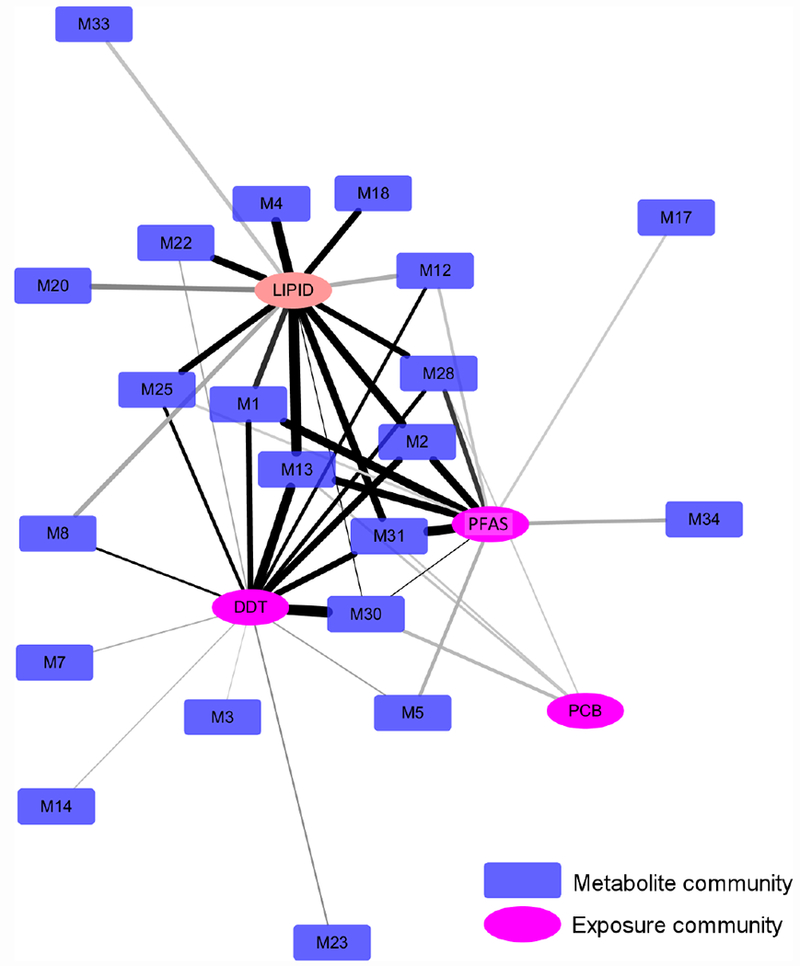
Each node represents one exposure community or metabolite community. The lipid community is included as a reference. The width of edges is proportional to PLS score linking between two communities, and color of edges proportional to −log10(p-value) with darker shades indicating more significance.
To exemplify the connections in this hierarchical community network, a metabolite community (M4) specifically associated with the lipid community is shown in Figure 4. Using the mummichog software [19], the metabolites in M4 were shown to be enriched for several fatty acid pathways, as expected (Figure 4B). Here, the lipid community consists of three members (TL, TG and TC), and they are shown in Figure 4C, ordered by TL. The metabolite community M4 consists of 103 LC-MS features, shown in the bottom heatmap in Figure 4C, with participants matched to the lipid data. Although multivariate project methods are not always visually intuitive, this shows a general trend that the member metabolites have higher intensity in the participants of higher lipid level (Figure 4C).
Figure 4. A metabolite community (M4) strongly associated with the lipid community.
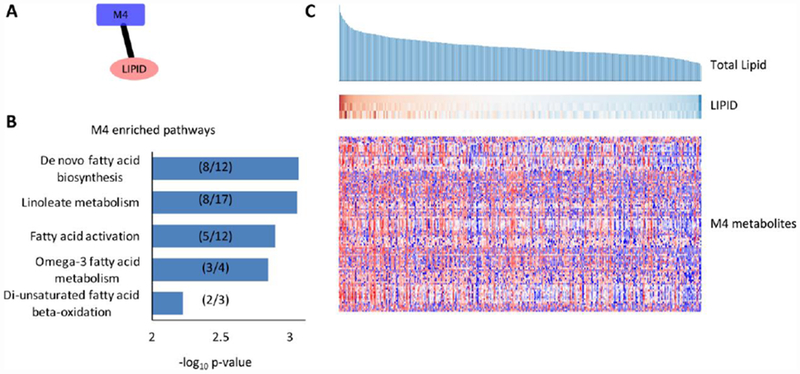
(A) A representative association link taken from Figure 3. (B) The metabolite features in the M4 community were tested by the mummichog software for their pathway enrichment. Significantly enriched metabolic pathways are plotted by their significance (−log10(p-value)). The numbers in parentheses indicate the number of significant metabolites/the number of detected metabolites for each pathway. (C) Each column represents a study participant, sorted by their blood level of total lipid in descending order (top). The levels of the three members of the LIPID community are shown in the middle and the intensities of metabolites of the M4 community are shown in the bottom heatmap, whereas each row represents a feature. In all panels, each column is matched to the same participant
Metabolome x Exposures converges on common metabolic phenotypes
A striking property of the metabolome – exposure network is that most connections are commonly shared and few are specific to one exposure community (Figure 3). The most significant connections are with the DDT and PFAS communities, and they are shared with the lipid community (shown in isolation in Figure 5A). Although the lipophilic properties of the exposure chemicals may account for some shared associations with the lipid community, this does not explain the majority of the connections, because PFASs are not lipophilic [23], while many of the PCBs are lipophilic but share few connections in Figure 3. This raises the question if a small number of metabolic phenotypes dictates the interaction with and response to the exposures.
Figure 5. Common metabolite communities associated with PCB, PFAS and LIPID communities.
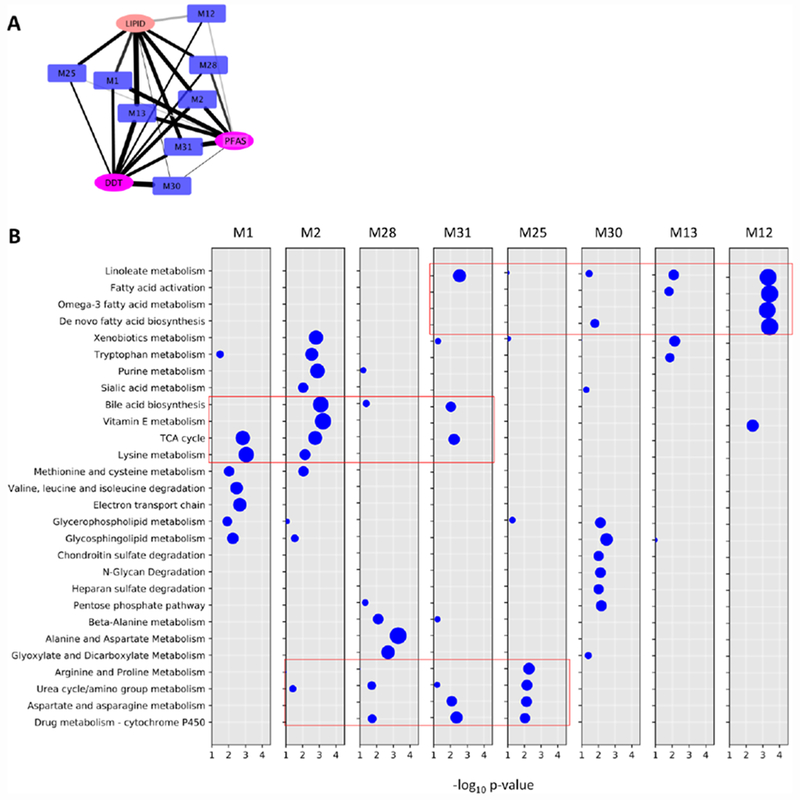
(A) The association links from Figure 2. (B) Metabolic pathways enriched in the top metabolite communities in A, tested by the mummichog software showing commonly shared clusters of pathways among communities (red box).
We used mummichog to identify the enriched pathways for each metabolite communities in Figure 5A, and they are shown collectively in Figure 5B. Among the metabolite communities, Ml is more enriched for mitochondria related pathways, M28 more for alanine and aspartate pathways, M12 more for fatty acid pathways. Xenobiotics metabolism is most significant in M2 and M13. A few common clusters are marked in red boxes in Figure 4B. The fatty acid cluster agrees with a previous MWAS study on p,p’-DDE using a different platform in a Swedish cohort [24]. Although there are specific preferences, e.g. M30 is more significantly associated with the DDT community, and the M4 community in Figure 4 is only significantly associated with the lipid community, the shared connections dominate the M x E network. Intriguingly, these metabolite communities show a rather sparse pattern of pathways in Figure 5B.
Association of exposure and metabolite communities with breast cancer occurrence in offspring generation
The goal of integrating M x E is to better understand the impact on human health. Within the 397 participants in Figure 3, the daughters of 50 were diagnosed for breast cancer in follow-up studies. Our dataset provides an opportunity to examine the relationship of the detailed molecular phenotypes embedded in metabolomics, and how they relate to environmental exposures. As described previously [21], we used the GSA algorithm [25] to query the hierarchical community network. Each community was treated like a geneset, and the GSA score represented how the community members collectively predicted an outcome, in this case, breast cancer (BC) in daughters. Permutation was performed on community members and participant labels to estimate null distribution. As shown in Figure 6A, the PFAS, lipid and PCB communities showed significant associations with BC occurrence, while the DDT community didn’t reach statistical significance (Suppl Table 3). Using the same approach, seven metabolite communities showed significant association with BC with p-value under 0.01 (Figure 6B, Suppl Table 3). The M16 community, the most significant, had 24 member features with yet no clear annotation.
Figure 6. Exposure and metabolite communities associated with breast cancer occurrence in offspring generation.
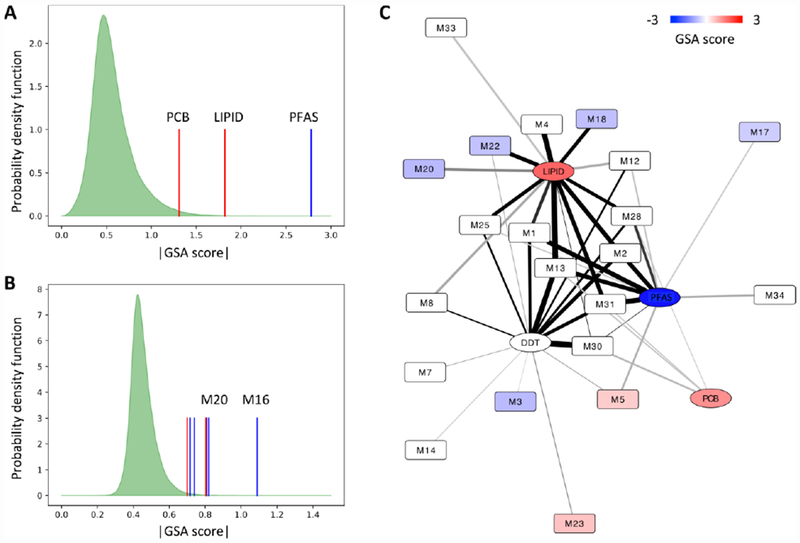
(A) The significant association between exposure communities and breast cancer occurrence in offspring, based on GSA (gene set analysis) test of each community. The green area shows the distribution of permutation data, and vertical bars of exposure communities (red higher in breast cancer cases, blue lower in breast cancer cases, DDT community not significant). (B) The significant association between metabolite communities and breast cancer occurrence in offspring, based on GSA test of each community. The green area shows the distribution of permutation data, and vertical bars of metabolite communities (red higher in BC cases, blue lower in BC cases, only communities with p < 0.01 shown). (C) The significant communities (p<0.05) mapped on the hierarchical community network as in Figure 2, recolored based on the association with breast cancer occurrence in offspring. Red higher in breast cancer cases; blue lower in breast cancer cases.
These communities associated with BC were then mapped back to the M x E network from Figure 3. This revealed that the metabolite communities M20, M22 and M18 were connected with the lipid community, suggesting a concerted role in either contributing or reacting to BC risk related physiology. Of note, the connections in Figures 3 and 6C are not directional: PLS regression captures both positive and negative associations. While the PFAS community was negatively associated with BC occurrence, its network neighbor M5 was positively associated with BC occurrence. While the DDT community itself showed no significant association with BC occurrence, four DDT-linked metabolite communities were significantly associated. These results demonstrate that both the exposures and the endogenous metabolism are statistically predictive of BC occurrence. The colored metabolite communities in Figure 6C may shed lights on the metabolic phenotypes that are prone or resistant to breast cancer.
Discussion
The CHDS cohort has been featured in many studies and conferences. With a large list of exposure measurements and new metabolomics data, we sought to use this cohort to explore how to effectively integrate exposome and metabolome. Our network model revealed that most metabolite communities were not specific to the association with a particular exposure, and many are shared between exposures. They could reflect the modes of toxic actions other than serving as biomarkers. These data are concordant with observations in other exposure MWAS studies [26, 27], and the concept of a systems response to exposures [16]. This poses an interesting question on how investigators should approach these MWAS analyses. It is possible that a small number of metabolic phenotypes are associated with a large number of exposures. As we proposed in the “G x M x E” model, the metabolome carries information from both ends. The metabolite communities we identified in this study could reflect both the endogenous metabolic capability and the result of varying exposure levels. The exposure data in this study were still limited, but these are chemicals of concern that have been reported to impact human health [28]. It will be interesting to see how MWAS data on more chemicals turn out.
As the measurement of exposures becomes high-dimensional, new approaches of analyzing and interpreting data are needed. In general, the integration of multi-omics data holds the promise to obtain more comprehensive understanding of the biological problem, to better identify the biological mechanisms at the intersection of data types, and to cross validate the findings from multiple data types [29–31]. The practice, however, has to be tailored to specific study design. The biological connection between enzymatic activity and metabolite concentration is important to integrate gene/protein data and metabolite measures in the same model. But this is rarely relevant in populations studies due to sampling. For example, it is common that gene expression is measured from blood cells and metabolomics from serum. Such mechanistic coupling cannot be assumed because serum metabolites may be regulated by enzymes in the liver not in monocytes. In the absence of a reasonable biological model, data-driven integration becomes necessary. Due to the different variance structure from different data types, many methods of data-driven integration employ some kind of decomposition or projection. Sparse methods are also popular to reduce the data dimension to a manageable and interpretable scale. The examples include the mixOmics package [32], group sparse CCA [33], multiple factor analysis (MFA, [34]), Co-Inertia analysis (CIA, [35]), sparse partial least squares [36], a joint latent variable model for integrative clustering [37] and a structured sparse CCA method [38]. Instead of removing statistically redundant features, an alternative is to group them in hierarchical networks. We have applied this hierarchical community network approach successfully in vaccine and infection studies [20, 21], and extended it here to the integration of exposome and metabolome. While PLS based methods are popular in the field of multi-omics integration, our innovation is to combine PLS with community structures within each data type, thus improving granularity of knowledge extraction and interpretation. This strikes a balance between specificity and redundancy. Not all redundancy is bad in - omics data, because multiple biological pathways can converge in statistical space. For example, we observed strong correlation between TLR signaling and glycerophospholipid metabolism in our previous vaccine study [21]. By keeping both in the data interpretation, it led to a new appreciation of these metabolic pathways in the context of immune response. This approach of hierarchical community networks is expected to be useful in gaining a global view of exposome – metabolome interactions, and to complement feature selection and biomarker studies. This is analogous to measuring the locales and variety of a forest instead of focusing on individual trees. A web supplement is provided for this dataset, and the software development is continued (https://github.com/shuzhao-li/hiconet).
We did not address the covariates such as age, gender and life styles. The age distribution in this birth cohort is very narrow, and gender cannot be accounted for in an all-female cohort. But it’s not the goal of this paper to assess their impact on the metabolome, and the metabolome is rather viewed as a phenotype than an outcome here. A compendium of 39 chemicals is a start to think of the exposome problem but nowhere close to the real complexity. Indeed, as shown in Figure 1, these exposures only account for a few percentages of variance in the metabolome. However, this is a static picture and does exclude the possibility of disease causality to be manifested at a different time or by a different data type. While the complete information may never be available, a useful step is to model the measured complexity properly. Despite of the disparities in measured exposures, we found that metabolite communities shared associations with the chemical exposures after the inter-correlation of the latter was removed by community detection. Because the exposures can modify the metabolome and vice versa, it is useful to view them as an interaction model, which can be applied to investigate outcomes such as breast cancer risk. The communities in our model are a useful tool of dimension reduction, which opens doors to further mediation and interaction models using the communities as computing units.
This study has major limitation in sampling. The samples were archived for over 50 years under suboptimal storage conditions, which were reflected in the smaller number of quality metabolite measurements compared to those from fresh samples on the same platform. The data were also from pregnant women who delivered daughters. Therefore, we could not study associations in men or compare differences between men and women. Our study investigated the metabolome in about 50% of the available cases and thus may have limited power for detecting associations of BC with the metabolome. Nonetheless, the metabolite association with the DDT community was concordant with the previous published studies [10–12, 39, 40]. The methodology is expected to be useful for broader studies of exposome.
Materials and Methods
Study Population
This study is made possible by a 54-year follow-up of 20 754 pregnancies, resulting in 9300 live-born female offspring in the CHDS pregnancy cohort. The CHDS was designed to examine the association between prenatal exposures and health and development over the life course for parents and children. The CHDS recruited women residing in the area of Oakland, California, who were members of the Kaiser Foundation Health Plan and received obstetric care for pregnancies between 1959 and 1967 [41]. More than 98% of all eligible women enrolled. CHDS founding mothers (F0) voluntarily participated in an in-person interview and gave permission to researchers for medical record access for themselves and their children. Their blood specimens were collected at several times through pregnancy and 1–3 days after delivery. As part of the Three Generations Study (3Gs) study 118 breast cancer cases in the daughters (F1) and 3 controls per case (matched on birth year and trimester of maternal blood draw, n=354) were chosen to for environmental chemicals analysis. For our current analysis, we chose to use all controls and 50 cases were randomly selected. We did not use all cancer cases to ensure that he that the MWAS didn’t get skewed from normal population. We also wanted to keep an oversampling of cases to ensure enough power to compare exposure and metabolite communities associated with cancer cases.
Cancer Cases
Breast cancer cases were identified by linkage to the California Cancer Registry and the California Vital Status Records as previously described [10, 42] and by self-report during a survey of CHDS daughters conducted from 2010 to 2013. All names for each CHDS subject were submitted for cancer linkages using fixed (ie, birth date, sex, race, and name) and changeable (ie, address and patient record number) identifiers. Cases were defined as CHDS daughters with incident invasive or noninvasive breast cancer diagnosed by age 52 years, identified through surveillance and through self-report through March of 2013. There were 137 cases who met this case definition, diagnosed as of 2012.
Serum Environmental Exposure Assays
Sample order was randomly assigned within and across batches and case-control strata were analyzed in the same batches to minimize differences due to laboratory drift. The laboratory was blind as to case or control status of the samples. Dichlorodiphenyltrichloroethane (DDTs), polychlorinated biphenyls (PCBs), Per- and Polyfluoroalkyl Substances (PFASs) and serum lipids were measured in nonfasting maternal perinatal serum samples that had been collected from 1959 through 1967. We preferred to use the early postpartum samples (collected within 1–3 d after delivery) when available to conserve serum for future studies when timing within pregnancy is more critical. Prior work has established that organochlorine levels are consistent across all trimesters of pregnancy and soon after delivery within women [43].
Serum assays methods have previously been described in detail [10–12, 44]. Briefly, the laboratory of the California Department of Toxic Substances Control (CDTSC) assayed our prenatal samples. DDT compounds (o,p’-DDT, p,p’-DDT, and p,p’-DDE) were analyzed on a DB-5ms column (30 m × 0.25 mm inner diameter, 0.25 μm film thickness; Agilent Technologies) installed in an Agilent gas chromatograph-tandem mass spectrometer (7890/7000B series). We used CDTSC’s online SPE-HPLC-MS/MS method to analyze samples for PFASs [45].
Total cholesterol and triglycerides were measured enzymatically on a Roche P Modular system using reagents and calibrators from Roche Diagnostics at the Clinical and Epidemiologic Research Laboratory (CERLab) at Boston Children’s Hospital, which is certified by the Centers for Disease Control and Prevention/National Heart, Lung, and Blood Institute Lipid Standardization Program.
LC-MS metabolomics analysis
Samples were thawed and analyzed by high-resolution liquid chromatography mass spectrometry (LC-MS) at Emory University as previously described [6, 46]. Briefly, sample aliquots (65 μL) were treated separately with acetonitrile containing a mixture of 14 stable isotopes. Samples were kept on ice for 30 minutes prior to centrifugation for 10 minutes at 13,400 × rpm at 4°C. The supernatant was removed and placed into autosampler vials. Mass spectral data for the samples from the CHDS cohort were collected with a 10-minute gradient on a Dionex U3000 coupled with a Thermo QExactive (Thermo Fisher, San Diego, CA) with an m/z range of 85 to 1275 and a resolution of 70,000. Three technical replicates were run for each sample, using C18 column in positive electrospray ionization (ESI) mode.
Following LC-MS, the data were collected and pre-processed using apLCMS [47] and xMSanalyzer [48] for feature detection and extraction. A metabolite feature was defined as a specific mass-to-charge ratio (m/z) along with its retention time and associated ion intensity. Data were log2 transformed and subjected to standard quality assessment including exclusion of data for technical replicates with overall Pearson correlation (r) < 0.70. The metabolite features were averaged for replicates. After filtering for missing values (by > 50 percent presence), 3,121 metabolite features were retained for subsequent data analysis.
The reporting of metabolite annotation adheres to the five confirmation levels in metabolomics literature [49]. Level 1 annotation applies to the metabolites confirmed by matching both m/z (mass accuracy under 10ppm) and retention time to that of authenticated chemical standards, previously characterized in our laboratory. Additional putative annotation was performed by m/z matching to KEEG database (mass accuracy under 10ppm – annotation level 3). The metabolomics data are accessible at the Metabolomics Workbench (http://www.metabolomicsworkbench.org).
Statistical and bioinformatics analyses
The data analysis was performed using Python (2.7 or 3.7), with libraries scipy and statsmodels (statistical implementation), pandas (data I/O), seaborn (heatmaps and bubble panels), and scikit-learn (PCA and PLS). Networks visualization was carried out using Cytoscape 3.4.0 (http://cytoscape.org).
The community detection in the exposure data was based on Girvan-Newman algorithm [22]. The community detection in metabolomics data was performed as previously described [20]. The PLS regression was performed similarly as previously described [21]. The significance of associations from PLS regression was assessed by permutation on both community member and sample labels. The mummichog software (version 1.0.9) was used for metabolic pathway enrichment analysis (mass accuracy under 10 ppm, which uses a probabilistic algorithm different from feature level statistics and produces pathway p-values based on permutation.
Supplementary Material
{kind=link}
{kind=link}
Highlights.
hierarchical community network to integrate exposome and metabolome
Fatty acid and mitochondria related pathways are among associations with DDTs and PFASs
Breast cancer risk in offspring positively associated with PCB compounds, negatively with PFASs
small number of metabolic phenotypes may account for large number of exposures
Acknowledgement:
This project has been funded in part by the California Breast Cancer Research Program 21UB-8002 (Cohn), the National Institutes of Health S10OD18006 (Jones), UH2AI132345 (Li) and U01OD026489 (Li).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Miller GW, Jones DP, The nature of nurture: refining the definition of the exposome, Toxicological Sciences 137(1) (2013) 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A, The blood exposome and its role in discovering causes of disease, Environmental health perspectives 122(8) (2014) 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wild CP, The exposome: from concept to utility, International journal of epidemiology 41(1) (2012) 24–32. [DOI] [PubMed] [Google Scholar]
- [4].Johnson CH, Ivanisevic J, Siuzdak G, Metabolomics: beyond biomarkers and towards mechanisms, Nature reviews Molecular cell biology 17(7) (2016) 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lankadurai BP, Nagato EG, Simpson MJ, Environmental metabolomics: an emerging approach to study organism responses to environmental stressors, Environmental Reviews 21(3) (2013) 180–205. [Google Scholar]
- [6].Gardinassi LG, Cordy RJ, Lacerda MV, Salinas JL, Monteiro WM, Melo GC, Siqueira AM, Val FF, Tran V, Jones DP, Metabolome-wide association study of peripheral parasitemia in Plasmodium vivax malaria, International Journal of Medical Microbiology 307(8) (2017) 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Flolmes E, Wilson ID, Nicholson JK, Metabolic phenotyping in health and disease, Cell 134(5) (2008) 714–717. [DOI] [PubMed] [Google Scholar]
- [8].Rhee EP, Ho JE, Chen M-H, Shen D, Cheng S, Larson MG, Ghorbani A, Shi X, Helenius IT, O’Donnell CJ, A genome-wide association study of the human metabolome in a community-based cohort, Cell metabolism 18(1) (2013) 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yap IK, Brown IJ, Chan Q, Wijeyesekera A, Garcia-Perez I, Bictash M, Loo RL, Chadeau-Hyam M, Ebbels T, Iorio MD, Metabolome-wide association study identifies multiple biomarkers that discriminate north and south Chinese populations at differing risks of cardiovascular disease: INTERMAP study, Journal of proteome research 9(12) (2010) 6647–6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cohn BA, Wolff MS, Cirillo PM, Sholtz RI, DDT and breast cancer in young women: new data on the significance of age at exposure, Environ Health Perspect 115(10) (2007) 1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cohn BA, Terry MB, Plumb M, Cirillo PM, Exposure to polychlorinated biphenyl (PCB) congeners measured shortly after giving birth and subsequent risk of maternal breast cancer before age 50, Breast cancer research and treatment 136(1) (2012) 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cohn BA, La Merrill M, Krigbaum NY, Yeh G, Park J-S, Zimmermann L, Cirillo PM, DDT Exposure in Utero and Breast Cancer, The Journal of Clinical Endocrinology & Metabolism 100(8) (2015) 2865–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cohn BA, Cirillo PM, Terry MB, DDT and Breast Cancer: Prospective Study of Induction Time and Susceptibility Windows, JNCI: Journal of the National Cancer Institute (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hu X, Li S, Cirrilo P, Krigbaum N, Tran V, Ishikawa T, La Merrill MA, Jones DP, Cohn B, Metabolome Wide Association Study of serum DDT and DDE in Pregnancy and Early Postpartum, Reproductive Toxicology (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu X, Li S, Cirillo PM, Krigbaum NY, Tran V, Jones DP, Cohn BA, Metabolome Wide Association Study of Serum Poly and Perfluoroalkyl Substances (PFASs) in Pregnancy and Early Postpartum, Reproductive Toxicology 87 (2019)70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Go YM, Fernandes J, Hu X, Uppal K, Jones DP, Mitochondrial network responses in oxidative physiology and disease, Free radical biology & medicine 116 (2018) 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barnes S, Benton HP, Casazza K, Cooper SJ, Cui X, Du X, Engler J, Kabarowski JH, Li S, Pathmasiri W, Training in metabolomics research. I. Designing the experiment, collecting and extracting samples and generating metabolomics data, Journal of Mass Spectrometry 51(7) (2016) 461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barnes S, Benton HP, Casazza K, Cooper SJ, Cui X, Du X, Engler J, Kabarowski JH, Li S, Pathmasiri W, Training in metabolomics research. II. Processing and statistical analysis of metabolomics data, metabolite identification, pathway analysis, applications of metabolomics and its future, Journal of mass spectrometry 51(8) (2016) 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, Pulendran B, Predicting Network Activity from High Throughput Metabolomics, PLOS Computational Biology 9(7) (2013)el003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gardinassi LG, Arevalo-Herrera M, Herrera S, Cordy RJ, Tran V, Smith MR, Johnson MS, Chacko B, Liu KH, Darley-Usmar VM, Integrative metabolomics and transcriptomics signatures of clinical tolerance to Plasmodium vivax reveal activation of innate cell immunity and T cell signaling, Redox biology 17 (2018) 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li S, Sullivan NL, Rouphael N, Yu T, Banton S, Maddur MS, McCausland M, Chiu C, Canniff J, Dubey S, Metabolic phenotypes of response to vaccination in humans, Cell 169(5) (2017) 862–877. el7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Girvan M, Newman ME, Community structure in social and biological networks, Proceedings of the national academy of sciences 99(12) (2002) 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, De Voogt P, Jensen AA, Kannan K, Mabury SA, van Leeuwen SP, Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins, Integrated environmental assessment and management 7(4) (2011) 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Salihovic S, Ganna A, Fall T, Broeckling CD, Prenni JE, van Bavel B, Lind PM, Ingelsson E, Lind L, The metabolic fingerprint of p,p’-DDE and HCB exposure in humans, Environment International 88 (2016)60–66. [DOI] [PubMed] [Google Scholar]
- [25].Efron B, Tibshirani R, On testing the significance of sets of genes, The annals of applied statistics 1(1)(2007)107–129. [Google Scholar]
- [26].Fernandes J, Hu X, Smith MR, Go Y-M, Jones DP, Selenium at the redox interface of the genome, metabolome and exposome, Free Radical Biology and Medicine 127 (2018) 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu X, Chandler JD, Fernandes J, Orr ML, Hao L, Uppal K, Neujahr DC, Jones DP, Go Y-M, Selenium supplementation prevents metabolic and transcriptomic responses to cadmium in mouse lung, Biochimica et Biophysica Acta (BBA) - General Subjects (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gore AC, Chappell V, Fenton S, Flaws JA, Nadal A, Prins G, Toppari J, Zoeller R, EDC-2: the Endocrine Society’s second scientific statement on endocrine-disrupting chemicals, Endocr Rev 36(6) (2015)E1–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dumas M-E, Metabolome 2.0: quantitative genetics and network biology of metabolic phenotypes, Molecular BioSystems 8(10) (2012) 2494–2502. [DOI] [PubMed] [Google Scholar]
- [30].Karczewski KJ, Snyder MP, Integrative omics for health and disease, Nature Reviews Genetics 19 (2018) 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D, Methods of integrating data to uncover genotype–phenotype interactions, Nature Reviews Genetics 16(2) (2015) 85. [DOI] [PubMed] [Google Scholar]
- [32].Rohart F, Gautier B, Singh A, Le Cao K-A, mixOmics: An R package for ‘omics feature selection and multiple data integration, PLoS computational biology 13(11) (2017) el005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lin D, Zhang J, Li J, Calhoun VD, Deng H-W, Wang Y-P, Group sparse canonical correlation analysis for genomic data integration, BMC bioinformatics 14(1) (2013) 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].De Tayrac M, Le S, Aubry M, Mosser J, Husson F, Simultaneous analysis of distinct Omics data sets with integration of biological knowledge: Multiple Factor Analysis approach, BMC genomics 10(1) (2009) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Culhane AC, Perriere G, Higgins DG, Cross-platform comparison and visualisation of gene expression data using co-inertia analysis, BMC bioinformatics 4(1) (2003) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lê Cao K-A, Martin PG, Robert-Granie C, Besse P, Sparse canonical methods for biological data integration: application to a cross-platform study, BMC bioinformatics 10(1) (2009) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shen R, Olshen AB, Ladanyi M, Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis, Bioinformatics 25(22) (2009)2906–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Safo SE, Li S, Long Q, Integrative analysis of transcriptomic and metabolomic data via sparse canonical correlation analysis with incorporation of biological information, Biometrics 74(1) (2018) 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cano-Sancho G, Salmon AG, La Merrill MA, Association between Exposure to p,p’-DDT and Its Metabolite p,p’-DDE with Obesity: Integrated Systematic Review and Meta-Analysis, Environ Health Perspect 125(9) (2017) 096002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].La Merrill M, Karey E, Moshier E, Lindtner C, La Frano MR, Newman JW, Buettner C, Perinatal Exposure of Mice to the Pesticide DDT Impairs Energy Expenditure and Metabolism in Adult Female Offspring, PLOS ONE 9(7) (2014) e103337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van den Berg BJ, Christianson RE, Oechsli FW, The California child health and development studies of the School of Public Health, University of California at Berkeley, Paediatric and perinatal epidemiology 2(3) (1988) 265–282. [DOI] [PubMed] [Google Scholar]
- [42].Cohn BA, Cirillo PM, Christianson RE, van den Berg BJ, Siiteri PK, Placental characteristics and reduced risk of maternal breast cancer, Journal of the National Cancer Institute 93(15) (2001) 1133–1140. [DOI] [PubMed] [Google Scholar]
- [43].Longnecker MP, Klebanoff MA, Gladen BC, Berendes HW, Serial levels of serum organochlorines during pregnancy and postpartum, Archives of environmental health 54(2) (1999) 110–4. [DOI] [PubMed] [Google Scholar]
- [44].Barbara Cohn MLM, Nickilou Krigbaum, Miaomiao Wang, June-Soo Park, Myrto Petreas, Greg Yeh, Russell Hovey, Eileen Johnson, Lauren, In Utero Exposure to Poly and Perfluoroalkyl Substances (PFASs) and Subsequent Breast Cancer, under review, 2019. [Google Scholar]
- [45].Wang M, Park J-S, Petreas M, Temporal changes in the levels of perfluorinated compounds in California women’s serum over the past 50 years, Environmental science & technology 45(17) (2011) 7510–7516. [DOI] [PubMed] [Google Scholar]
- [46].Hoffman JM, Soltow QA, Li S, Sidik A, Jones DP, Promislow DE, Effects of age, sex, and genotype on high-sensitivity metabolomic profiles in the fruit fly, D rosophila melanogaster, Aging Cell 13(4) (2014) 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yu T, Park Y, Johnson JM, Jones DP, apLCMS—adaptive processing of high-resolution LC/MS data, Bioinformatics 25(15) (2009) 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T, Jones DP, xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data, BMC Bioinformatics 14(1) (2013) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schymanski EL, Jeon J, Guide R, Fenner K, Ruff M, Singer HP, Hollender J, Identifying small molecules via high resolution mass spectrometry: communicating confidence, ACS Publications, 2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.