

Abstract
Background: Sorafenib is one of the most commonly used systemic therapies for hepatocellular carcinoma (HCC), but the acquired resistance towards sorafenib found in HCC patients usually led to failure of treatment and poor prognosis. Therefore, there is an urgent need to study the molecular mechanism caused by the acquired resistance. Previous studies demonstrated that P62 plays an important role in tumor cell resistance towards systemic therapies including chemotherapy and targeted therapy. However, the role of P62 in acquired resistance to sorafenib in HCC has not been clearly investigated. Materials and methods: In this study we screened the most sensitive HCC cell lines towards sorafenib using CCK8. Then on this cell line, we analyzed the relationship between P62 expression level and the sensitivity towards sorafenib by western blot and CCK8. After knockdown and overexpression of P62 in HCC cells, cells were then treated with sorafenib. After that, we detect changes of sensitivity towards sorafenib. HCC samples were used to investigate the expression of P62 and their survival time. Results: Among four HCC cell lines in our lab, HepG2 cell line with the highest sensitivity to sorafenib was screened and selected. After treatment with sorafenib, the expression of P62 was significantly increased. In HCC cells, we found that significant up-regulation of P62 was correlated with the reduction of sorafenib sensitivity. In HCC samples, we found that the expression of P62 was associated with sorafenib resistance and a shorter survival time. Conclusion: The up-regulation of P62 could reduce the sensitivity of HCC towards sorafenib. Thus, P62 could be therapeutic target to overcome sorafenib acquired resistance in the future.
Keywords: Hepatocellular carcinoma, resistance, sorafenib, P62
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related deaths worldwide, leading to over 600,000 deaths annually [1]. Only a minority of patients with HCC are amenable to liver resection because most patients are diagnosed with advanced stage and no longer suitable for surgery. Treatment for advanced HCC patients includes chemotherapy, transcatheter arterial chemoembolization (TACE), and radiofrequency ablation. Sorafenib has been used as the only standard systemic treatment for advanced HCC at the current stage as it can target multiple kinases required for tumor growth, angiogenesis, and metastasis [2]. Although sorafenib demonstrated survival benefit, the prognosis of HCC was still not satisfactory [3]. The poor prognosis was partly caused by drug resistance, which resulted in a curative effect reduction and therefore, led to chemotherapy treatment failure [4]. The intricate mechanism of anticancer drug resistance has been broadly explored in recent years and has yet to be fully elucidated.
Some studies showed that sustained sorafenib therapy leads to increased intratumor hypoxia, which has been associated with reduced sorafenib sensitivity through HIF stabilization in HCC [5]. Thus, targeting HIF can improve sorafenib efficacy. Some other reports showed that the activation of downstream signaling pathways contributed to the resistance to sorafenib. A study by Chen et al. demonstrated that sorafenib-resistant HCC cells had an increased expression of pAkt and p85 when compared with the parental sensitive cells and the resistance to sorafenib could be reversed by gene knockdown of Akt and Akt inhibitor MK-2206 [6]. Another study by Chen et al. showed that increased phosphorylation of Jak2 and Stat3 was detected in LX2 co-cultured Huh7 cells. The Jak inhibitor tofacitinib reversed sorafenib resistance by blocking Jak2 and Stat3 activation [7]. However, the underlying mechanism of sorafenib resistance has not been fully investigated.
P62 (also known as SQSTM1) is a multifunctional stress-induced scaffold protein involved in a variety of cellular processes. Its functions are strictly regulated by a wide range of post-translational modifications. Previous studies have found that P62 may play an important role in drug resistance in melanoma, non-small cell lung cancer, liver cancer, and other type of cancers [8-10]. In lung cancers, glioma, breast cancer, prostate cancer, and other cancers, P62 has found to play the role as onco-genes [11-13]. However, its role in HCC has been rarely reported and the underlying role in drug resistance towards sorafenib remains unclear.
Here, we propose that the over-expression of P62 would alleviate the sensitivity of hepatocellular carcinoma cells to sorafenib in HCC cells.
Materials and methods
Patients
From the year 2011 to 2013, 30 cases of advanced HCC who were diagnosed by pathological biopsy and immunohistochemistry in Nanjing Drum Tower Hospital Affiliated to the Medical College of Nanjing University were included. All patients had oral sorafenib as the first-line treatment. The formalin fixed tissue and fresh tissue solidified in liquid nitrogen were studied in this study. Survival information was obtained through active follow-up on the basis of identifying the patient’s life state. Overall survival rate (OS) is defined as the time between the start of treatment and death or the last follow-up to March 1, 2018. This study was conducted in accordance with the ethical standards approved by the ethics committee of our hospital and informed consent of all participants was obtained.
Cell culture and cell treatment
All the cell lines were obtained from the Hepatobiliary Research Institute of Nanjing Drum Tower Hospital affiliated to the Medical College of Nanjing University (Jiangsu, China). The cell lines HepG2, Huh7, Hep3B, and 7402 were banked upon receipt and passaged for fewer than 6 months before use without further authentication. All the cells were cultured in Dulbecco’s modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C with a humidified atmosphere of 5% CO2.
Screening sensitive cell lines
HCC cell lines HepG2, Huh7, Hep3B, and 7402 were implanted into 96-well plates and treated with sorafenib (concentrations of 5, 10, 15, 20, 25, 50, 100 and 200 ug/ml, respectively) for 24 h, 48 h and 72 h. Cell viability was assessed using the Cell Counting Kit 8 (CCK-8). In brief, the cells were seeded into the 96-well plates with an initial density of 5 × 103 cells/well for 1-3 days. Afterwards, 90 ul of fresh serum-free medium and 10 ul of CCK-8 reagent (Tongren Chemistry, Japan) were added to each well after decanting the previous medium, and the culture was continued at 37°C for 1 h. The optical density was determined using a microplate reader (Promega, Madison, WI, USA) at 450 nm. IC50 values were calculated.
Cell growth measurements and viability assays
A sensitive HCC cell line was planted on a 24-well plate, treated with sorafenib (appropriate concentration according to the above mentioned condition) for 24 h, 48 h and 72 h, and the proliferation rate of cancer cells was detected by EDU kit (Invitrogen, USA). Cell viability was expressed as a percentage of the absorbance measured in the control cells. All experiments were performed in duplicate and repeated three times.
Immunohistochemistry
Immunohistochemistry was performed according to the standard protocols. Briefly, paraffin sections were deparaffinised in xylene and transferred through two changes of 100% ethanol. Heat-induced antigen retrieval was performed in 10 mM citrate buffer at 100°C for 2 min and cooled for 30 min at room temperature. Endogenous peroxidase activity was blocked with peroxidase-blocking reagent containing 3% hydrogen peroxide and serum. After incubation, the slides were washed in PBS 3 times. Then, slides were incubated with P62 polyclonal antibody (1:100; Proteintech Inc., USA) overnight at 4°C. After being washed, the sections were incubated with biotin-goat anti-rabbit antibody at room temperature. The peroxidase reaction was developed with 3,3’-diaminobenzidine (DAB) chromogen solution in DAB buffer substrate. The sections were counterstained with hematoxylin, mounted in neutral gum, and analyzed using a bright field microscope (Olympus IX71, Tokyo, Japan). The immunostaining was microscopically evaluated by two independent pathologists. A semiquantitative scoring system was based on the staining intensity and proportion of positive cells.
Western blotting assay
Cell proteins were prepared using cell lysis buffer. Equal amounts of protein (50 mg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (Merck Millipore) by electro-blotting. The membranes were blocked with 5% nonfat dry milk in TBST for 1 h, and then incubated with primary antibodies overnight at 4°C before subsequent incubation with second antibodies (Cell Signaling Technology) for 1 h at 37°C. Protein bands were visualized with ECL plus chemiluminescence kit (Millipore, Bedford, MA, USA).
Quantitative real-time PCR
Total RNA from cells was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). After total RNA was extracted, cDNA was synthesized using the Primer-Script™ One Step RT-PCR Kit (TaKaRa, Dalian, China). The cDNA template was amplified by real time-qPCR using the SYBR® Premix Dimmer Eraser kit (TaKaRa, Dalian, China) carried out on ABI 7500 system (Applied Biosystems, CA, USA) using a SYBR Premix Ex Taq II kit (TaKaRa) according to the manufacturer’s instructions. GAPDH was measured as an internal control, and mRNA values were normalized to GAPDH. The relative expression fold change of mRNAs was calculated by the 2 -ΔΔCt method.
Statistical analysis
All statistical analyses were carried out using the SPSS 16.0 statistical software package (Chicago, IL, USA). Comparisons between groups for statistical significance were performed with a Student’s t-test. Survival curve was plotted using the Kaplan-Meier method and compared by the log-rank test. P < 0.05 was considered statistically significant in all cases.
Results
Relationship between the expression of P62 and the treatment sensitivity of sorafenib and its clinical significance
First, we compared the expression of p62 in HCC samples with adjacent normal tissues. As the Figure 1A showed that, compared with normal tissue, the expression of P62 was significantly higher in HCC samples. We also did the IHC to show the protein expression of P62 on HCC samples and normal tissues. Consistent with the mRNA expression, P62+ was found in more HCC samples, compared with adjacent normal tissues. Then we analyzed the relationship between expression of P62 and overall survival time. As Figure 1C showed that overexpression of P62 was correlated with shorter survival and the difference was statistically significant.
Figure 1.

Increased expression of P62 in HCC is associated with patients’ survival. A. The normalized relative expression of P62 in the tumor and adjacent normal tissues (30 cases) of HCC by qRT-PCR analysis. B. The normalized relative expression of P62 in the tumor and adjacent normal tissues analyzed by Immunohistochemistry. C. High expression of P62 in HCC is associated with poor patients’ survival.
Sensitivity of different HCC cell lines to sorafenib
Then, we performed the in vitro screening of four cell lines, HepG2, 7402, huh7 and hep3B sensitivity to sorafenib. CCK-8 test was performed. As Figure 2A. showed that sorafenib (from 0-200 ug/ml) was used to intervene for 24 h, 48 h and 72 h. The cell viability was the lowest when the concentration of sorafenib reached to 100 ug/ml. The HepG2 cell viability reduced to approximately 0 when the concentration of sorafenib was 100 ug/ml, while the other 3 cell lines kept 50% of cell viability. These results suggested that for four cell lines in our lab, HepG2 was the most sensitive to sorafenib. The immunofluorescence results of cells (Figure 2B) showed that sorafenib can also significantly inhibit the proliferation of HepG2 cells.
Figure 2.
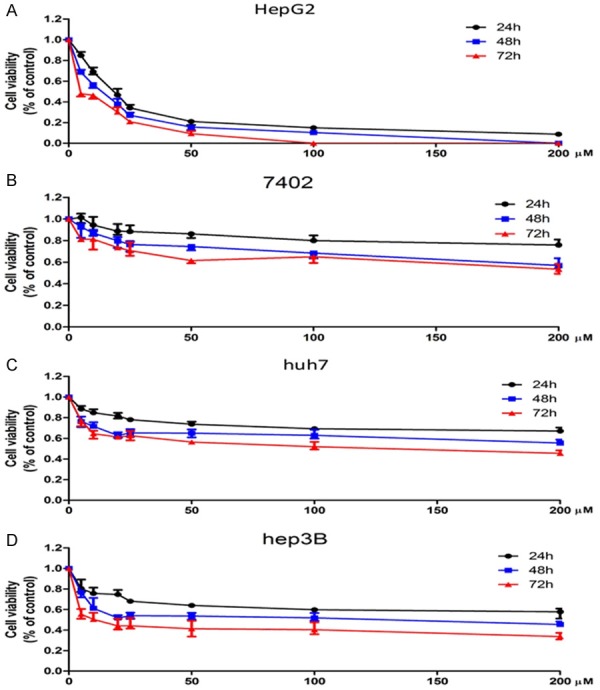
Sensitivity of different HCC cell lines towards sorafenib. A. Cell viability declined to approximately 0 after 72 hours cultured with sorafenib at a concentration of 100 um in HepG2 cell line. B. Cell viability was approximately 70% of previous after 72 hours cultured with sorafenib at a concentration of 100 um in 7402 cell line. C. Cell viability decreased to 50% of previous after 72 hours cultured with sorafenib at a concentration of 100 um in huh7 cell line. D. Cell viability was down to 40% after 72 hours cultured with sorafenib at a concentration of 100 um in hep3B cell line.
P62 was significantly up-regulated after treated with sorafenib, Real-time PCR and Western Blot were used to detect the expression levels of P62 genes and proteins in HCC cells after treated with sorafenib. As Figure 3A showed that after treated with sorafenib for 24 hours, P62 was significantly up-regulated compared with treating with control in four cell lines, but in HepG2, P62 was more significantly increased. Consistent with the mRNA expression, the protein level was also increased significantly in HepG2 as Figure 3 showed.
Figure 3.
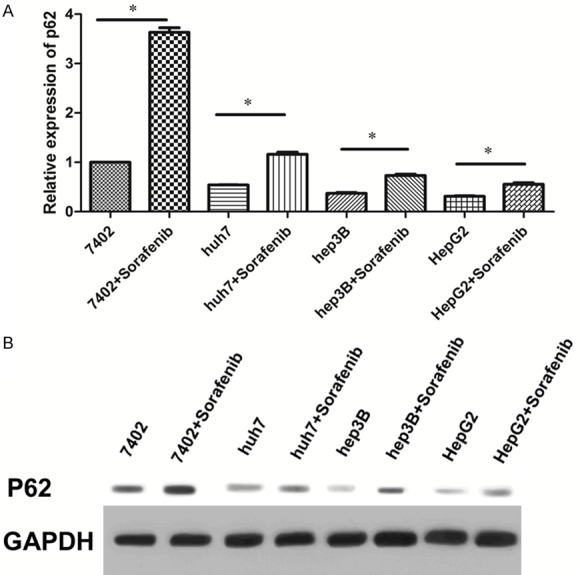
P62 was significantly up-regulated after treated with sorafenib. A. The expression of P62 all significantly increased when treated with sorafenib after 72 h, but in HepG2 cell line, P62 was not as significantly increased compared with the other three cell lines analyzed by qRT-PCR. B. Consistent with the mRNA expression, the protein expression of P62 was also significantly increased when cultured with sorafenib, and P62 was the less significantly increased compared with the other three cell lines.
Changes in the sensitivity of HCC cell lines towards sorafenib treatment after P62 intervention
Based on the above mentioned results, HepG2 was the most sensitive cell line to sorafenib and P62 was the most significantly increased when treated with sorafenib. We then used HepG2 as the cell line for further experiments. Next, we over-expressed or down-regulated the expression of P62. The CCK-8 test showed that in HepG2 cell lines with sorafenib treatment for 24 h, the knockdown of P62 significantly reduced the sensitivity of HepG2 cells, while the overexpression of P62 significantly increased the sensitivity of HepG2 cells (Figure 4A). The EDU test showed that the knockdown of P62 significantly reduced the proliferation ability of HepG2 cells and the overexpression of P62 significantly increased the proliferation ability of HepG2 cells (Figure 4B). Flow cytometry (Figure 4C) also showed that the knockdown of P62 significantly increased the apoptosis level of HepG2 cells, and the overexpression of P62 significantly reduced the apoptosis level of cancer cells.
Figure 4.
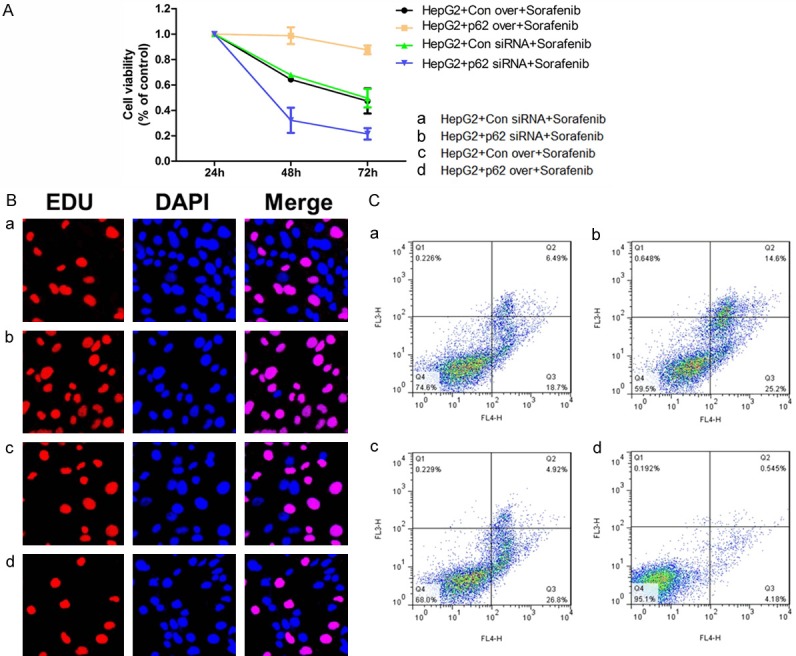
Changes in the sensitivity of HCC cell lines towards sorafenib treatment after P62 intervention. A. The cell viability after knockdown or up-regulation of P62 in HepG2 cell line. B. The cell growth after knockdown or up-regulation of P62 in HepG2 cell line. C. Cell apoptosis of HCC cells after knockdown or up-regulation of P62 in HepG2 cell line.
Discussion
Sorafenib is the only standard molecular targeted therapy for patients with advanced HCC at this current stage. Sorafenib is anti-angiogenic and anti-proliferative by inhibiting kinases such as Raf kinase, vascular endothelial growth factor receptor (VEGFR), and platelet derived growth factor receptor (PDGFR)-β tyrosine kinase [14]. Although sorafenib has improved the survival of patients with HCC, no significant difference in overall survival has been reported. Resistance to sorafenib has become one of the major obstacles to improving its efficacy.
The primary resistance of HCC to sorafenib, which because of drug resistance factors existing in tumor tissues or HCC cells of patients before drug treatment, is mainly due to genetic heterogeneity. The acquired/secondary resistance is caused by resistance factors developed during sorafenib treatment. The mechanisms of sorafenib resistance may include the following aspects. Autophagy is a large number of cellular degradation processes that can guide cells to digest damaged proteins and/or dysfunctional organelles. In HCC, experimental and clinical studies have found the effect of sorafenib on autophagy induction [15]. Sorafenib therapy increases the expression of autophagosome formation and autophagy related genes (ATGs). In one study, ATG7 knockout or combination with the pharmacological autophagy inhibitor chloroquine significantly enhanced sorafenib-induced apoptosis [16]. These studies suggest that sorafenib-induced autophagy is a cellular protective mechanism. However, Beclin1 silencing weakened the inhibitory effect of sorafenib derivatives on cell viability and the expression of autophagy markers such as LC3, ATG5, VPS34, and Beclin1 in sorafenib resistant cells was decreased, so the opposite conclusion was drawn. Also, the promotion of c-jun in sorafenib resistance was reported in two molecular studies. C-jun expression was activated in HCC cells treated with sorafenib and the inhibition of c-jun enhanced the apoptosis of HCC cells induced by sorafenib [17]. Furthermore, a member of the receptor tyrosine kinase (RTKs) family, epidermal growth factor receptor (EGFR) has been proven to be a potential predictor of sorafenib resistance in HCC cells. Down-regulation of EGFR expression or inhibition of EGFR kinase activity can help increase the sensitivity of EGFR-positive cells to sorafenib [18]. Other studies indicated that the hypoxic microenvironment may play important roles in inducing sorafenib resistance, the surviving tumor cells become more deteriorated and resistant to chemotherapy [19]. Apart from these factors, Epithelial-mesenchymal transformation (EMT) is a critical cellular process that contributes not only to cell migration, but also to drug resistance [20].
P62/SQSTM1, as the center of many signaling pathways regulating cell life activities, is closely related to the occurrence and development of tumors in abnormal expression and regulation. P62 structures containing important interacting domains demonstrated the ability of this protein to regulate the activation of these signaling pathways during tumorigenesis and transmission. P62 also acts as a molecular adapter between the autophagy mechanism and its substrate [21]. According to most studies, the expression of P62 in lung cancer, prostate cancer, gastric cancer, colorectal cancer, pancreatic cancer, diffuse large b-cell lymphoma, skin melanoma, and some other tumors is significantly higher than that in corresponding normal tissues. two authors reported that P62 protein levels in colorectal cancer and gastric cancer were lower than those in corresponding normal tissues. In addition to direct survival analysis, P62 expression was associated with lymph node status, distant metastasis, disease recurrence, and drug resistance [22]. High levels of P62 also allowed cancer cells to escape apoptosis and exacerbate cancer by activating the NF-κB/NRF2 signaling pathway [23].
After the treatment of the HepG2 cell line with sorafenib, we found that the expression of P62 significantly increased. Previous literatures have reported that P62 plays the role of oncogene in tumors and is also highly expressed in liver cancer. In addition, the knockdown of P62 led to apoptosis of HepG2 cells treated with sorafenib, suggesting that P62 may reduce the sensitivity of HepG2 cells to sorafenib. The mechanism by which P62 may participate in drug resistance was proposed by some studies. Shimizu et al. found that sorafenib treatment led to the accumulation of autophagosome and the activation of autophagy flux, which could be proved by increasing LC3 lipid and Huh7 and significantly decreasing the autophagy substrate P62 in HLF and PLC/PRF/5 cells, thereby promoting the survival of HCC cells and limiting the efficiency of sorafenib. In squamous cell carcinoma cells and melanoma tumor cells, chloroquine induced the activation of NF-κB and the expression of its target genes HIF-1α, IL-8, BCL-2, and BCL-xl through the accumulation of autophagosomes. The activation of NF-κB further increased the expression of P62 gene. Knockdown of P62 gene or inhibition of NF-κB made tumor cells sensitive to CQ, leading to increased apoptotic cell death after treatment [16].
To overcome the low efficacy of sorafenib in the treatment of advanced HCC, the applicability of different types of small molecules as a combination therapy with sorafenib was tested. For example, Chen et al. reported that 0.5 mg/kg bortezomib synergistically suppressed xenografted PLC/PRF/5 tumors when treated in combination with 5 mg/kg sorafenib by approximately 50% compared with 5 mg/kg sorafenib treatment alone [24]. Huynh et al. observed the sensitizing efficacy of rapamycin (1 mg/kg) in combination with 50 mg/kg sorafenib on the suppression of xenografted patient-derived HCC [25]. In addition, the selective MEK inhibitor AZD6244 (25 mg/kg) showed about 30-40% higher anti-cancer efficacy with sorafenib (25 mg/kg) compared with the sorafenib-treated group in patient-derived xenograft models [26].
The results of our study provide a strong theoretical basis for finding a new therapeutic target to overcome sorafenib resistance in HCC.
Acknowledgements
This study was funded by the National Key Research and Development Program of China (Grant no. 2016YFC1302603) and National Natural Science Foundation of China (Grant no. 81670566 and 81500478).
Disclosure of conflict of interest
None.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394–9. doi: 10.3322/caac.21161. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Decaens T, Raoul JL, Boucher E, Kudo M, Chang C, Kang YK, Assenat E, Lim HY, Boige V, Mathurin P, Fartoux L, Lin DY, Bruix J, Poon RT, Sherman M, Blanc JF, Finn RS, Tak WY, Chao Y, Ezzeddine R, Liu D, Walters I, Park JW. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 2013;31:3509–16. doi: 10.1200/JCO.2012.47.3009. [DOI] [PubMed] [Google Scholar]
- 5.Mendez-Blanco C, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50:134. doi: 10.1038/s12276-018-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH, Chen PJ, Cheng AL. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2011;337:155–61. doi: 10.1124/jpet.110.175786. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Wu J, Shi H, Wang Z, Zhang G, Cao Y, Jiang C, Ding Y. Hepatic stellate cell coculture enables sorafenib resistance in Huh7 cells through HGF/c-Met/Akt and Jak2/Stat3 pathways. Biomed Res Int. 2014;2014:764981. doi: 10.1155/2014/764981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Son YO, Pratheeshkumar P, Roy RV, Hitron JA, Wang L, Zhang Z, Shi X. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J Biol Chem. 2014;289:28660–75. doi: 10.1074/jbc.M114.595496. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Mao J, Ma L, Shen Y, Zhu K, Zhang R, Xi W, Ruan Z, Luo C, Chen Z, Xi X, Chen S. Arsenic circumvents the gefitinib resistance by binding to P62 and mediating autophagic degradation of EGFR in non-small cell lung cancer. Cell Death Dis. 2018;9:963. doi: 10.1038/s41419-018-0998-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Tan P, Ye Y, He L, Xie J, Jing J, Ma G, Pan H, Han L, Han W, Zhou Y. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018;16:e3000051. doi: 10.1371/journal.pbio.3000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang MA, Patel V, Gwede M, Morgado M, Tomasevich K, Fong EL, Farach-Carson MC, Delk NA. IL-1beta induces p62/SQSTM1 and represses androgen receptor expression in prostate cancer cells. J Cell Biochem. 2014;115:2188–97. doi: 10.1002/jcb.24897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 15.Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485. doi: 10.1038/cddis.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, Hiramatsu N, Fujita N, Yoshimori T, Hayashi N. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131:548–57. doi: 10.1002/ijc.26374. [DOI] [PubMed] [Google Scholar]
- 17.Haga Y, Kanda T, Nakamura M, Nakamoto S, Sasaki R, Takahashi K, Wu S, Yokosuka O. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS One. 2017;12:e0174153. doi: 10.1371/journal.pone.0174153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ezzoukhry Z, Louandre C, Trecherel E, Godin C, Chauffert B, Dupont S, Diouf M, Barbare JC, Maziere JC, Galmiche A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131:2961–9. doi: 10.1002/ijc.27604. [DOI] [PubMed] [Google Scholar]
- 19.Kim JY, Lee JY. Targeting tumor adaption to chronic hypoxia: implications for drug resistance and how it can be overcome. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18091854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du B, Shim JS. Targeting epithelial-mesenchymal transition (emt) to overcome drug resistance in cancer. Molecules. 2016;21 doi: 10.3390/molecules21070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. 2012;2:397–413. [PMC free article] [PubMed] [Google Scholar]
- 22.Ju LL, Zhao CY, Ye KF, Yang H, Zhang J. Expression and clinical implication of Beclin1, HMGB1, p62, survivin, BRCA1 and ERCC1 in epithelial ovarian tumor tissues. Eur Rev Med Pharmacol Sci. 2016;20:1993–2003. [PubMed] [Google Scholar]
- 23.Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun. 2015;6:5779. doi: 10.1038/ncomms6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Thng CH, Chow P, Ong HS, Chung A, Soo KC. Sorafenib and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J Cell Mol Med. 2009;13:2673–83. doi: 10.1111/j.1582-4934.2009.00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Toh HC, Thng CH, Chow P, Ong HS, Chung A, Goh BC, Smith PD, Soo KC. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC) J Hepatol. 2010;52:79–87. doi: 10.1016/j.jhep.2009.10.008. [DOI] [PubMed] [Google Scholar]