

Abstract
Long non-coding RNAs (lncRNAs) play important roles in the pathogenesis of various diseases, including diabetic nephropathy (DN). However, the detailed mechanism is still largely unknown. High-glucose treated SV40-MES13 cells was used to mimic diabetic nephropathy in vitro. qRT-PCR was introduced to measure Hottip, collagen type I (Col. I), collagen type IV (Col. IV), fibronectin (FN), PAI-1, miR-455-3p and Wnt2B, IL-6, TNF-α mRNA level. Ellisa was used to examine the expression level of IL-6, TNF-α in the cell culture medium. Western blotting was employed to detect the protein level of Col. I, Col. IV, FN, PAI-1, Wnt2B, β-catenin and cyclin D1. Cell viability was examined by MTT assay, luciferase reporter assay were used to determine the relationship between Hottip, miR-455-3p and Wnt2B. In the results, Hottip and Wnt2B was upregulated in db/db DN mice and high-glucose treated mouse mesangial cells (MMCs) while miR-455-3p was downregulated. High glucose treatment could enhance cell proliferation, and inflammation, increase fibrosis-related protein expression and active Wnt2B/β-catenin/cyclin D1 pathway, while Hottip silencing reversed all the effects caused by high-glucose treatment. miR-455-3p was a sponge target of Hottip while Wnt2B was a downstream target of miR-445-3p. miR-445-3p inhibitor could suppress the effect of Hottip knockdown in cell proliferation, inflammation and fibrosis-related protein expression. Our data supported lncRNA Hottip/miR-455-3p/Wnt2B axis plays an important role in cell proliferation, inflammation, and extracellular matrix (ECM) accumulation in diabetic nephropathy.
Keywords: Hottip, diabetic nephropathy, miR-455-3p, renal fibrosis, Wnt2B
Introduction
Diabetic nephropathy (DN), a chronic kidney disease, is the main cause of end-stage renal disease in diabetic patients worldwide [1-5]. About 30%-40% diabetes patients will suffer diabetic nephropathy [6]. The main pathologic characteristics of DN are increased extracellular matrix formation, podocyte loss, increased thickness of basement membrane, glomerular and tubular cell injury which lead to albuminuria, glomerular matrix accumulation, glomerular hypertrophy and kidney fibrosis or failure [7-10]. DN is a complicated pathologic process which involves many different molecules, and cells [11]. For example, immune inflammatory response [12-15], epithelial-mesenchymal transition [16], apoptosis [17,18]. Emerging evidence demonstrated that epigenetics is also involved in DN. High glucose stimulation may lead to epigenetic modifications such as DNA methylation [19], histone modification [20,21], chromatin remodeling [22], and non-coding RNA regulation [23-25].
Non-coding RNAs are transcripts with limited protein coding ability which constitute about 98% of all the transcripts [26]. Long non-coding RNAs, a type of non-coding RNAs, are a group of RNAs with over 200 nucleotides in length. They may modulate the expression of protein-coding transcripts at transcriptional and posttranscriptional levels [27]. It has been shown that lncRNAs are widely involved in biologic processes such as epigenetic regulation, cell cycle, cell differentiation, and apoptosis [28]. Aberrant expression of lncRNAs is related with various diseases, including DN [29]. However, the mechanism of lncRNA in DN is still largely unknown.
In present work, we validated that Hottip was upregulated in db/db DN mice and high-glucose treated mouse mesangial cells (MMCs). Then we hypothesized Hottip was involved in the pathogenesis of diabetic nephropathy and further study revealed that hottip/miR-455-3p/Wnt2B played an important role in cell proliferation, apoptosis, and fibrosis-protein expression in DN which provided a novel view of DN pathogenesis.
Materials and methods
Animal model
Diabetic mouse model C57BL/KsJ-db/db and matched normal db/m mice were obtained from the Laboratory Animal Services Center, Yancheng City No. 1 People’s Hospital. All mice were kept in the animal facility of Yancheng City No. 1 People’s Hospital following the animal protocol. The animal experiment was approved by the Ethics Committee of the Animal Research Institute of Yancheng City No. 1 People’s Hospital and was performed in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The kidney cortex was extracted from the kidney tissues of the db/db and db/m mice for analysis.
Cell culture
Mouse mesangial cell lines (MMCs, SV40-MES13) were purchased from American Type Culture Collection (Manassas, VA). SV40-MES13 was cultured in Dulbecco’s modified eagle medium (DMEM) medium containing with 2 mM glutamine, 50 mM β-mercaptoethanol, 20% fetal bovine serum (FBS), 0.1% penicillin/streptomycin antibiotics. SV40-MES13 cells were treated with low glucose (LG) or high glucose (HG), in low-glucose group, SV40-MES13 were stimulated with 5.5 mM glucose together with 19.5 mM mannitol; and in high-glucose group, MCs were stimulated with 25 mM glucose. High glucose treatment was performed to mimic the DN cells. HEK293T cells were cultured with DMEM medium containing 10% FBS and 0.1% penicillin/streptomycin antibiotics. SV40-MES13 and HEK293T cells were cultured in humidified 95% air with 5% carbon dioxide at 37°C and the medium was changed every 2 days.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from mouse model and MMCs using Trizol reagent. For miRNA, cDNA was reversely transcribed by miScript II RT Kit (Qiagen). For mRNA and lncRNAs, cDNA was reversely transcribed by Reverse Transcription Reagents (Applied Biosystems). Hottip, miR-455-3p, WNT2B, Col. I, Col. IV, FN, and PAI-1 expression levels were measured by SYBR Green (Promega, Madison, WI, USA) according to the manufacturer’s protocol. QuantStudio™ 3 Real-Time PCR Systems (ThermoFisher, USA) was used to detect the fluorescence and the relative expression was calculated by 2-ΔΔCt method. U6 and 18s rRNA were used to normalize the expression. Primer sequences are: mouse Hottip, forward 5’-GCACCATTCACTCACACTCCTG-3’, reverse 5’-CAAAACGGAATGCAACAGTGGA-3’; mouse WNT2B, forward 5’-CCGACGTGTCCCCATCTTC-3’, reverse 5’-GCCCCTATGTACCACCAGGA-3’; mmu-miR-455-3p, forward 5’-ACACTCCAGCTGGGGCAGTCCACGGGCATATACAC-3’; mouse Col. I (COL1A1), forward 5’-GCTCCTCTTAGGGGCCACT-3’, reverse 5’-CCACGTCTCACCATTGGGG-3’; mouse Col. IV (COL4A1), forward 5’-CTGGCACAAAAGGGACGAG-3’, reverse 5’-ACGTGGCCGAGAATTTCACC-3’; mouse Fibronectin (FN, FN1), forward 5’-GATGTCCGAACAGCTATTTACCA-3’, reverse 5’-CCTTGCGACTTCAGCCACT-3’; mouse PAI-1 (Serpine1), forward 5’-TTCAGCCCTTGCTTGCCTC-3’, reverse 5’-ACACTTTTACTCCGAAGTCGGT-3’; mouse IL-6, TGATGGATGCTTCCA AACTG, GAGCATTGGAAGTTGGGGTA; mouse TNF-α, ACTGAACTTCGGGGTGATTG, GCTTGGTGGTTTGCTACGAC; mouse 18S rRNA, forward 5’-GGCCCTGTAATTGGAATGAGTC-3’, reverse 5’-CCAA-GATCCAACTACGAGCTT-3’; mouse U6 forward 5’-GTTGACATCCGTAAAGACC-3’, reverse 5’-GGAGCCAGGGCAGTAA-3’.
Transient transfection
Si-Hottip, miR-455-3p mimic, pcDNA3.1-Hottip, miR-455-3p inhibitor and the negative controls were purchased from GenePharma Co., Ltd. (Shanghai, China). All of the oligos and plasmids were transfected into MMCs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Western blot analysis
Total protein was extracted from MMCs using RIPA lysis buffer. Equal protein samples were loaded and separated with 8%-10% sodium dodecyl sulfate polyacrylamide-gel electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). After blocking with 5% dried non-fat milk in TBS for 1 hr at room temperature, the membranes were incubated with different primary antibodies including and anti-Col. I (ab34710, 1:1000), anti-Col. IV (ab6586, 1:1000), anti-fibronectin (anti-FN, ab2413, 1:1000), anti-PAI-1 (ab66705, 1:1000), anti-Wnt2B (ab50575, 1:1000), anti-β catenin (ab32572, 1:1000), anti-cyclin D1 (ab16663, 1:1000) and anti-GAPDH (CST#5174, 1:1000) at 4°C overnight. On second day, the membranes were washed with TBS-T 3 times for 10 min each time and incubated with HRP-conjugated IgG secondary antibody at room temperature for 1 hr. After washing with TBST three times, the protein signals were detected using PierceTM ECL western blotting substrate (ThermoFisher Scientific).
Luciferase reporter assay
Hottip-WT, Hottip-MUT, WNT2B-WT and WNT2B-MUT were constructed into pGL3 vector and cotransfected with miR-455-3p into HEK293T cells by Lipofectamine 2000 (Invitrogen). After 48 hrs, the luciferase activity was measured using dual-luciferase reporter system (Promega, Madison, WI, USA).
Cell viability
MTT assay kit (Sigma-Aldrich) was used to measure the cell viability. The transfected cells were seeded into 96-well plates (Corning Costar, Corning, NY, USA) in a number of 2 × 103. On different time points, 100 μL fresh media and 1 μL MTT solution containing 12 mM MTT was added into each well and incubated for 2 h at 37°C. After that, 100 μL MTT solvent (4 mM HCl, 0.1% NP40 in isopropanol) was added into each well and incubated for 2 h at 37°C. Then we measured the absorbance at 490 nanometers using the Microplate Reader (MG LABTECH, Durham, NC, USA).
Enzyme-linked immunosorbent assay (ELISA)
The levels of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) were detected by ELISA in culture medium. ELISA kits for TNF-α and IL-6 (BD Biosciences, San Jose, CA) were used according to the manufacturers’ instructions. Results were read at an optical density of 450 nm using a Spectra Max Plus plate reader (Molecular Devices, Sunnyvale, CA).
Statistical analysis
Student t-test was used for statistical analysis. Statistical significance means P value < 0.05 or as indicated. All data were displayed as mean ± SD (standard deviation) which was analyzed by GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA).
Results
Hottip was upregulated in DN mice and high-glucose treated MMCs
To check the expression of Hottip in diabetic nephropathy, we firstly extracted the kidney cortex from db/db DN mice and db/m non-DN mice. qRT-PCR results demonstrated that Hottip was increased in db/db DN mice about 2 fold compared with db/m non-DN mice (Figure 1A). To mimic DN in vitro, we used low glucose (LG, 5 mM) and high glucose (HG, 25 mM) to treat SV40-MES13 which is one mouse mesangial cell line (MMCs) for 0 h, 6 h, 12 h, 24 h and 48 h. Hottip expression increased in a time-dependent manner significantly (Figure 1B).
Figure 1.
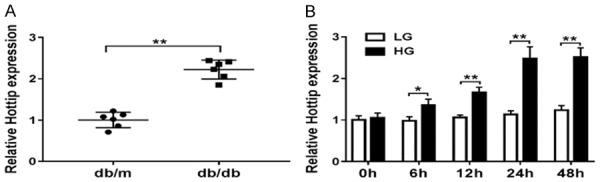
Hottip expression in DN mice and glucose treated MMCs. A. The expression of lncRNA Hottip in kidney cortex of db/db and db/m mouse. B. Mouse mesangial cells (MMCs, SV40-MES13) were treated with high-glucose (25 mM) and low-glucose (5 mM) for 0, 6, 12, 24 and 48 hrs and the expression of Hottip was measured by qRT-PCR. *P < 0.05. **P < 0.01.
Hottip knockdown inhibited cell proliferation and inflammation in high-glucose treated MMCs
To explore the function of Hottip, we used small interfering RNA (siRNA) to knock down Hottip in SV40-MES13 cells and treated with high glucose (HG, 25 mM) (Figure 2A). The proliferation was inhibited significantly by Hottip knockdown compared with control (Figure 2B). Then, RT-PCR results showed that HG could induce inflammation in SV40-MES13, which was inhibited by Hottip knockdown group compared with si-NC control group (Figure 2C and 2D). Next, we examined IL-6, and TNF-α expression in cell culture medium by ELISA and found that high glucose treatment increased IL-6, TNF-α expression which was decreased by Hottip knockdown (Figure 2E and 2F). The results suggested that Hottip knockdown could inhibit cell proliferation and inflammation in a DN model in vitro.
Figure 2.
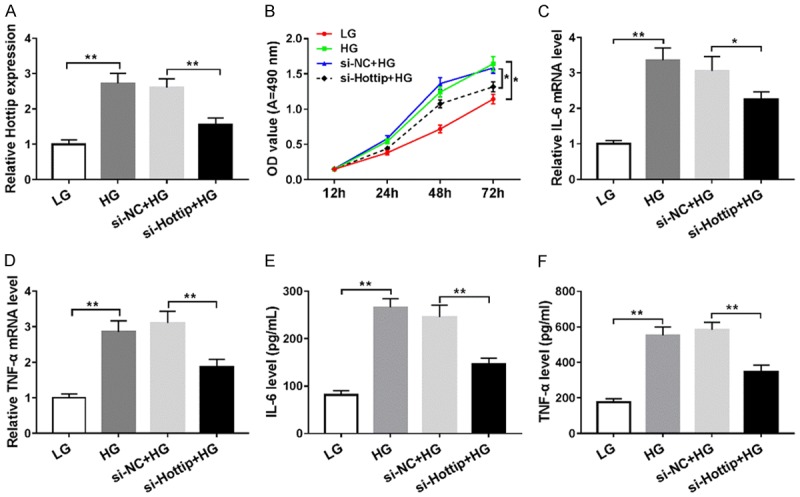
Hottip knockdown inhibited cell proliferation and inflammation in glucose treated SV40-MES13 cells. SV40-MES13 cells were transfected with si-Hottip and treated with high glucose (HG) for 24 hr. (A) The expression of Hottip in SV40-MES13 cells after si-Hottip transfection and high-glucose treatment. (B) Proliferation of SV40-MES13 cells after si-Hottip transfection and high-glucose treatment. RT-PCR (C and D) and ELISA (E and F) assay to detect the IL-6, and TNF-α levels in SV40-MES13. *P < 0.05. **P < 0.01.
Hottip knockdown suppressed the fibrosis-related protein expression and extracellular matrix accumulation in high-glucose treated MMCs
Fibrosis is one pathologic basis of extracellular matrix accumulation and mesangial cell proliferation. So we checked the expression of fibrosis-related proteins including collagen type I (Col. I), collagen type IV (Col. IV), fibronectin (FN) and PAI-1. The mRNA level of these four proteins were upregulated in db/db DN mice significantly (Figure 3A). Meanwhile, in vitro the mRNA (Figure 3B) and protein level (Figure 3C) of Col. I, Col. IV, FN and PAI-1 were increased in high glucose treated SV40-MES13 cells. But, Hottip knockdown could partially inhibit their expression (Figure 3C and 3D). These results demonstrated that Hottip silencing suppressed the fibrosis-related protein expression in vitro.
Figure 3.
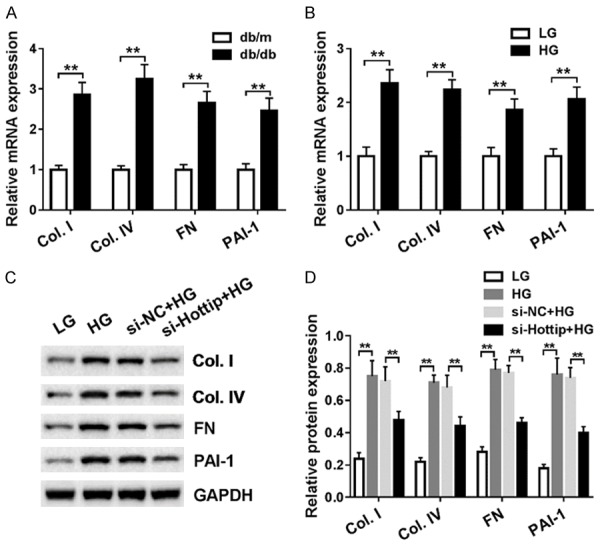
Hottip knockdown suppressed the fibrosis-related protein generation and extracellular matrix (ECM) accumulation in SV40-MES13 cells. A. The mRNA level of fibrosis-related protein including Col. I, Col. IV, FN and PAI-1 in kidney cortex of db/db and db/m mouse. B. The mRNA level of fibrosis-related protein including Col. I, Col. IV, FN and PAI-1 in mouse mesangial cells (MMCs, SV40-MES13) treated with high-glucose (25 mM) and low-glucose (5 mM) for 24 hr. C. Fibrosis-related protein expression in SV40-MES13 after Hottip knockdown and HG treatment. D. Grey scale analysis of fibrosis-related proteins. *P < 0.05. **P < 0.01.
miR-455-3p acted as a target of Hottip
To further understand the function of Hottip, we found that miR-455-3p was a sponge target miRNA of Hottip by LncBase (Figure 4A). Then we constructed luciferase reporter vector containing wild-type Hottip (Hottip-WT) and mutant Hottip (Hottip-MUT). The luciferase activity was decreased when the Hottip-WT and miR-455-3p mimics were cotransfected into HEK293T cells but not Hottip-MUT (Figure 4B). Then, qRT-PCR results showed that miR-455-3p was downregulated in db/db DN mice in vivo (Figure 4C) and high glucose (HG) treated SV40-MES13 cells in vitro (Figure 4D). In addition, knockdown of Hottip induced the expression of miR-455-3p (Figure 4E) while overexpression of Hottip inhibited miR-455-3p expression (Figure 4F). These results indicated that miR-455-3p was a target of Hottip and regulated by Hottip.
Figure 4.

mmu-miR-455-3p is a target of Hottip. A. The relationship between Hottip and miR-455-3p was predicted by LncBase. B. Luciferase assay confirmed the relationship between Hottip and miR-455-3p. C. miR-455-3p expression in kidney cortex of db/db and db/m mouse. D. miR-455-3p expression in mouse mesangial cells (MMCs, SV40-MES13) treated with high-glucose (25 mM) and low-glucose (5 mM) for 24 hr. E. miR-455-3p expression after Hottip silencing. F. miR-455-3p expression after Hottip overexpression. *P < 0.05. **P < 0.01.
Hottip/miR-455-3p regulated cell proliferation, inflammation and ECM accumulation in MMCs
Next, we introduced a rescue experiment using miR-455-3p inhibitor. We treated SV40-MES13 cells in 4 different ways: low glucose (LG), high glucose (HG), high glucose (HG) + si-Hottip + miR-NC inhibitor, high glucose (HG) + si-Hottip + miR-455-3p inhibitor. miR-455-3p inhibitor indeed inhibited miR-455-3p expression compared with control (Figure 5A). Cell proliferation of SV40-MES13 cotransfected with si-Hottip and miR-455-3p inhibitor slightly and significantly increased compared with that only transfected with si-Hottip on day 4 after high glucose treatment (Figure 5B). The IL-6, TNF-α mRNA levels were increased after miR-455-3p inhibitor transfection compared with the si-Hottip + miR-NC groups (Figure 5C and 5D). Meanwhile, the expression of IL-6, TNF-α was reversed partly in high glucose (HG) + si-Hottip + miR-455-3p inhibitor group compared with high glucose (HG) + si-Hottip + miR-NC inhibitor group (Figure 5E and 5F). Moreover, fibrosis-related proteins including collagen type I (Col. I), collagen type IV (Col. IV), fibronectin (FN) and PAI-1 were all upregulated after miR-455-3p inhibitor transfection (Figure 5G). Therefore, these data indicated that Hottip regulated cell proliferation, inflammation and fibrosis-protein expression by modulating miR-455-3p in MMCs.
Figure 5.

Hottip/miR-455-3p regulates cell proliferation, inflammation, and ECM accumulation in SV40-MES13 cells. SV40-MES13 cells were divided into 4 groups and treated with 1) low glucose (5 mM), 2) high glucose (25 mM); 3) si-Hottip + miR-NC inhibitor + high glucose; 4) si-Hottip + miR-455-3p inhibitor + high glucose. A. miR-455-3p expression in SV40-MES13 with different treatments as above. B. Proliferation curve of treated SV40-MES13. C-F. RT-PCR and ELISA assay to detect the IL-6, and TNF-α levels in SV40-MES13. G. Fibrosis-related protein expression in SV40-MES13 in these four group. *P < 0.05. **P < 0.01.
Wnt2B is a target of miR-455-3p
MicroRNAs usually play functions by binding 3’UTR of specific mRNA to regulate the transcription or mediate the degradation. So we used TargetScan to predict the target of miR-455-3p and found that WNT2B contains complementary sequence (Figure 6A). The luciferase activity was decreased when WNT2B-WT and miR-455-3p mimics were cotransfected into HEK293T (Figure 6B). Then qRT-PCR demonstrated that WNT2B was upregulated in db/db DN mice in vivo (Figure 6C) and high glucose (HG) treated SV40-MES13 cells in vitro (Figure 6D). Meanwhile, we found that WNT2B protein level was downregulated by overexpression miR-445-3p (Figure 6E) and upregulated by transfection with miR-445-3p inhibitor (Figure 6F).
Figure 6.
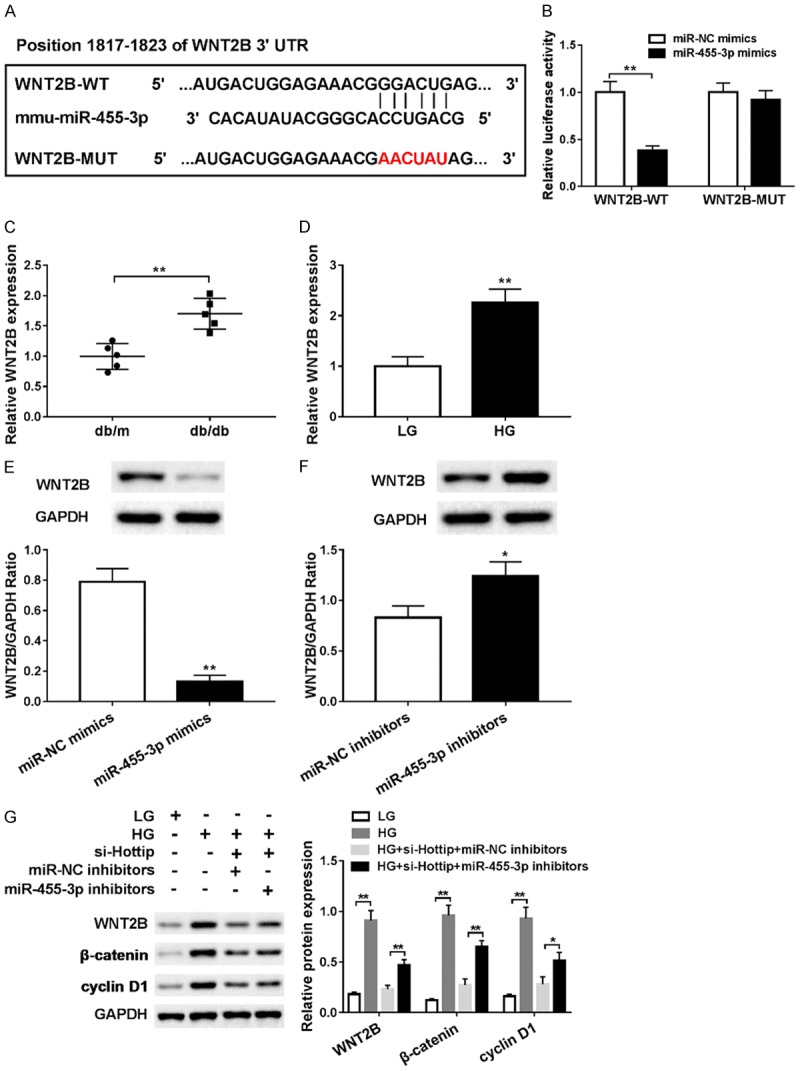
Wnt2B was a target of miR-455-3p. A. The relationship between miR-455-3p and Wnt2B predicted by TargetScan. B. Luciferase assay was used to confirm the relationship between miR-455-3p and Wnt2B. C. Wnt2B mRNA expression in kidney cortex of db/db and db/m mouse. D. The mRNA of Wnt2B in mouse mesangial cells (MMCs, SV40-MES13) treated with high-glucose (25 mM) and low-glucose (5 mM) for 24 hr. E. Wnt2B protein expression in SV40-MES13 after transfection of miR-455-3p mimic. F. Wnt2B protein expression in SV40-MES13 after transfection of miR-455-3p inhibitor. G. Wnt2B, β-catenin and cyclin D1 expression in low-glucose, high-glucose, high-glucose + si-Hottip + miR-NC inhibitor and high-glucose + si-Hottip + miR-455-3p inhibitor treated SV40-MES13 cells. *P < 0.05. **P < 0.01.
Wnt2B is a key regulator in Wnt signaling pathway of which β-catenin and cyclin D1 are downstream target. So next, we checked the expression of Wnt2B, β-catenin and cyclin D1. After high glucose treatment, all of Wnt2B, β-catenin and cyclin D1 expressions were upregulated significantly, but Hottip silencing could inhibit their expression which could be reversed by miR-455-3p inhibitor (Figure 6G). These data indicated that Wnt2B was a target of miR-455-3p and Wnt pathway was involved in DN.
Discussion
Diabetic nephropathy (DN) is a microvascular complication in diabetic patients and is a leading cause of chronic renal failure [30]. Mutifactors are involved in the pathogenesis of DN. In this study, we used the db/db DN mice [31] model and high-glucose treated mouse mesangial cell line (MMCs, SV40-MES13) [32] to study the underlying molecular mechanism of DN.
Long non-coding RNAs draw a lot attention in recent years. Although lncRNAs are not capable of protein coding, they may regulate the expression of genes at transcriptional and posttranscriptional level in DN. For example, long non-coding RNA metastasis associated lung adenocarcinoma transcript 1 (MALAT1) is increased in kidney cortex in streptozocin-induced diabetic nephropathy in mice and in cultured mouse podocytes stimulated with high glucose, knockdown of MALAT1 partially restored the function of podocytes [33]. CYP4B1-PS1-001 is significantly decreased in DN and overexpression inhibits the proliferation and fibrosis of mesangial cells [34]. ENSMUST00000147869 overexpression could reverse the proliferation and fibrosis-related protein expression in mesangial cells [35]. Long non-coding RNA taurine-upregulated gene 1 (Tug1) overexpression is associated with improvements in mitochondrial bioenergetics in podocytes [36] and alleviates extracellular matrix accumulation by regulating miR-377 in mesangial cells [37]. In our study, we found long non-coding RNA Hottip was upregulated in kidney cortex in db/db DN mice and high glucose treated MMCs SV40-MES13 cells. We further found knockdown of Hottip could inhibit cell proliferation and inflammation as well as decrease fibrosis-related protein in high-glucose treated MMCs by regulating miR-455-3p and Wnt2b.
Extracellular matrix includes collagen, laminin, fibronectin and proteoglycans. Mesangial matrix changes include disrupted expression of collagen I, collagen IV, fibronectin, PAI-1 and others [38,39]. Recent studies indicated that some signaling pathways or molecules are involved in the regulation of ECM in DN. For example, Notch1 and Jagged1 are significantly increased in tubular epithelial cells in DN patients [40]. HG treatment can activate the PI3K/Akt/mTOR pathway and subsequently promotes the expression of ECM laminin-β1. TGF-β, Col. IV and NF-κB expression were markedly decreased in db/db mice treated withTLR4 inhibitor [41]. In addition, various investigations suggested that Wnt/β-catenin pathway is relevant to the evolution of renal fibrosis because its inhibition reduces ECM expression [42-44].
Wnt signaling pathways include a group of highly conserved signal transduction pathways which may regulate various biologic processes in different tissues including kidney. Wnt pathways regulate cell-to-cell interactions in physiologic and pathologic conditions, such as inflammation, angiogenesis, and fibrosis [45]. Wnt signaling pathways have been characterized into two major transduction cascades: the Wnt/β-catenin-dependent or canonical pathway and the β-catenin-independent or non-canonical pathway [46]. Canonical Wnt pathway involves the accumulation of β-catenin but this is not true of the non-canonical pathway. Disrupted Wnt pathways are related to various diseases such as cancer [47], neurodegeneration [48], and cardiometabolic disorders [49]. Previous studies demonstrated that Wnt family members including Wnt2b, 4, 5b, 6, 7b, 9b and 11 have been expressed in kidney ontogeny [50] while 6, 7b, 9b, 11 are expressed in the early stage of nephrogenesis. In our study, we found Wnt2B was upregulated in db/db DN mice and in the high-glucose treated mouse mesangial cells (MMCs) as well as β-catenin and cyclin D1. In addition, Hottip knockdown decreased their expression while miR-455-3p inhibitor increased the expression of Wnt2B, β-catenin and cyclin D1. This means the canonical Wnt signaling pathway was involved in DN.
In conclusion, our present study demonstrated that high glucose (HG) treatment in mouse mesangial cells (MMCs, SV40-MES13) induced the expression of long non-coding RNA Hottip and inhibited miR-455-3p expression. HG treatment enhanced cell proliferation and inflammation, induced fibrosis-related proteins including Col. I, Col. IV, FN and PAI-1 expression, as well as activated the Wnt2B/β-catenin/cyclin D1 pathway. However, knockdown of Hottip partially reversed the effects caused by HG treatment in MMCs which provides a novel and promising target in DN treatment.
Disclosure of conflict of interest
None.
References
- 1.Barutta F, Mastrocola R, Bellini S, Bruno G, Gruden G. Cannabinoid receptors in diabetic kidney disease. Curr Diabetes Reports. 2018;18:9. doi: 10.1007/s11892-018-0975-7. [DOI] [PubMed] [Google Scholar]
- 2.Packham DK, Alves TP, Dwyer JP, Atkins R, de Zeeuw D, Cooper M, Shahinfar S, Lewis JB, Lambers Heerspink HJ. Relative incidence of ESRD versus cardiovascular mortality in proteinuric type 2 diabetes and nephropathy: results from the DIAMETRIC (diabetes mellitus treatment for renal insufficiency consortium) database. Am J Kidney Dis. 2012;59:75–83. doi: 10.1053/j.ajkd.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Afifi A, El Setouhy M, El Sharkawy M, Ali M, Ahmed H, El-Menshawy O, Masoud W. Diabetic nephropathy as a cause of end-stage renal disease in egypt: a six-year study. East Mediterr Health J. 2004;10:620–626. [PubMed] [Google Scholar]
- 4.Hadjadj S, Cariou B, Fumeron F, Gand E, Charpentier G, Roussel R, Kasmi AA, Gautier JF, Mohammedi K, Gourdy P. Death, end-stage renal disease and renal function decline in patients with diabetic nephropathy in French cohorts of type 1 and type 2 diabetes. Diabetologia. 2016;59:208–216. doi: 10.1007/s00125-015-3785-3. [DOI] [PubMed] [Google Scholar]
- 5.Chandie Shaw PK, Vandenbroucke JP, Tjandra YI, Rosendaal FR, Rosman JB, Geerlings W, de Charro FT, van Es LA. Increased end-stage diabetic nephropathy in indo-asian immigrants living in the netherlands. Diabetologia. 2002;45:337–341. doi: 10.1007/s00125-001-0758-5. [DOI] [PubMed] [Google Scholar]
- 6.Reddy MA, Park JT, Natarajan R. Epigenetic modifications in the pathogenesis of diabetic nephropathy. Semin Nephrol. 2013;33:341–353. doi: 10.1016/j.semnephrol.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hovind P, Rossing P, Tarnow L, Smidt UM, Parving HH. Progression of diabetic nephropathy. Kidney Int. 2001;59:702–709. doi: 10.1046/j.1523-1755.2001.059002702.x. [DOI] [PubMed] [Google Scholar]
- 8.Parving HH. Diabetic nephropathy: prevention and treatment. Kidney Int. 2001;60:2041–2055. doi: 10.1046/j.1523-1755.2001.00020.x. [DOI] [PubMed] [Google Scholar]
- 9.Wang YQ, Fan CC, Chen BP, Shi J. Resistin-like molecule beta (RELM-β) regulates proliferation of human diabetic nephropathy mesangial cells via mitogen-activated protein kinases (MAPK) signaling pathway. Med Sci Monit. 2017;23:3897–3903. doi: 10.12659/MSM.905381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Zhang X, Song S, Luo H. Regulation of podocyte lesions in diabetic nephropathy via miR-34a in the notch signaling pathway. Medicine. 2016;95:e5050. doi: 10.1097/MD.0000000000005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun YM, Su Y, Li J, Wang LF. Recent advances in understanding the biochemical and molecular mechanism of diabetic nephropathy. Biochem Biophys Res Commun. 2013;433:359–361. doi: 10.1016/j.bbrc.2013.02.120. [DOI] [PubMed] [Google Scholar]
- 12.Duransalgado MB, Rubioguerra AF. Diabetic nephropathy and inflammation. World J Diabetes. 2014;5:393. doi: 10.4239/wjd.v5.i3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanasaki K, Taduri G, Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol. 2013;4:7. doi: 10.3389/fendo.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Kanwar YS. Relevance of TNF-α in the context of other inflammatory cytokines in the progression of diabetic nephropathy. Kidney Int. 2015;88:662–665. doi: 10.1038/ki.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci. 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 16.Ivonne L, Gunter W. Epithelial-to-mesenchymal transition in diabetic nephropathy: fact or fiction? Cells. 2015;4:631–652. doi: 10.3390/cells4040631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat Rev Nephrol. 2014;10:369–378. doi: 10.1038/nrneph.2014.67. [DOI] [PubMed] [Google Scholar]
- 18.Cao AL, Wang L, Chen X, Wang YM, Guo HJ, Chu S, Liu C, Zhang XM, Peng W. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Investig. 2016;96:610. doi: 10.1038/labinvest.2016.44. [DOI] [PubMed] [Google Scholar]
- 19.Sapienza C, Lee J, Powell J, Erinle O, Yafai F, Reichert J, Siraj ES, Madaio M. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics. 2011;6:20–28. doi: 10.4161/epi.6.1.13362. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Li C, Sun G. Histone acetylation and its modifiers in the pathogenesis of diabetic nephropathy. J Diabetes Res. 2016;2016:1–11. doi: 10.1155/2016/4065382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun G, Cui W, Guo Q, Miao L. Histone lysine methylation in diabetic nephropathy. J Diabetes Res. 2015;2014:654148. doi: 10.1155/2014/654148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Badal SS, Yin W, Long J, Corcoran DL, Chang BH, Luan DT, Kanwar YS, Overbeek PA, Danesh FR. miR-93 regulates Msk2-mediated chromatin remodelling in diabetic nephropathy. Nat Commun. 2016;7:12076. doi: 10.1038/ncomms12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu L, Wang Q, Guo F, Ma X, Ji H, Liu F, Zhao Y, Qin G. MicroRNA-27a Induces mesangial cell injury by targeting of pPARγ, and its in vivo knockdown prevents progression of diabetic nephropathy. Sci Rep. 2016;6:26072. doi: 10.1038/srep26072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He F, Peng F, Xia X, Zhao C, Luo Q, Guan W, Li Z, Yu X, Huang F. MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia. 2014;57:1726–1736. doi: 10.1007/s00125-014-3282-0. [DOI] [PubMed] [Google Scholar]
- 25.Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015;19:1418. doi: 10.1111/jcmm.12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balakirev ES, Ayala FJ. Pseudogenes: are they “junk” or functional DNA? Ann Rev Genet. 2002;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- 27.Yang F, Yi F, Zheng Z, Ling Z, Ding J, Guo J, Mao W, Wang X, Wang X, Ding X. Characterization of a carcinogenesis-associated long non-coding RNA. Rna Biol. 2012;9:110–116. doi: 10.4161/rna.9.1.18332. [DOI] [PubMed] [Google Scholar]
- 28.Monika H, Tony G. Long non-coding RNAs in cancer and development: where do we go from here? Int J Mol Sci. 2015;16:1395–1405. doi: 10.3390/ijms16011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allison SJ. Diabetic nephropathy: a lncRNA and miRNA megacluster in diabetic nephropathy. Nat Rev Nephrol. 2016;12:713. doi: 10.1038/nrneph.2016.151. [DOI] [PubMed] [Google Scholar]
- 30.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med. 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 31.Cohen MP, Sharma K, Jin Y, Hud E, Wu VY, Tomaszewski J, Ziyadeh FN. Prevention of diabetic nephropathy in db/db mice with glycated albumin antagonists. A novel treatment strategy. J Clin Investig. 1995;95:2338–2345. doi: 10.1172/JCI117926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Chen G, Cheng X, Teng Z, Cai X, Yang J, Sun X, Lu W, Wang X, Yao Y. Therapeutic potential of digitoflavone on diabetic nephropathy: nuclear factor erythroid 2-related factor 2-dependent anti-oxidant and anti-inflammatory effect. Sci Rep. 2015;5:12377. doi: 10.1038/srep12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu M, Wang R, Li X, Fan M, Lin J, Zhen J, Chen L, Lv Z. Lnc RNA MALAT 1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with β-catenin. J Cell Mol Med. 2017;21:2732–2747. doi: 10.1111/jcmm.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min W, Wang S, Di Y, Qin Y, Lu W. A novel long non-coding RNA CYP4B1-PS1-001 regulates proliferation and fibrosis in diabetic nephropathy. Mol Cell Endocrinol. 2016;426:136–145. doi: 10.1016/j.mce.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Yao D, Wang S, Yan Q, Lu W. Long non-coding RNA ENSMUST00000147869 protects mesangial cells from proliferation and fibrosis induced by diabetic nephropathy. Endocrine. 2016;54:1–12. doi: 10.1007/s12020-016-0950-5. [DOI] [PubMed] [Google Scholar]
- 36.Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, Green NH, Chang BH, Overbeek PA, Danesh FR. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Investig. 2016;126:4205–4218. doi: 10.1172/JCI87927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duan LJ, Ding M, Hou LJ, Cui YT, Li CJ, Yu DM. Long noncoding RNA TUG1 alleviates extracellular matrix accumulation via mediating microRNA-377 targeting of PPARγ in diabetic nephropathy. Biochem Biophys Res Commun. 2017;484:598–604. doi: 10.1016/j.bbrc.2017.01.145. [DOI] [PubMed] [Google Scholar]
- 38.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- 39.Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012;60:976. doi: 10.1369/0022155412465073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bielesz B, Sirin Y, Han S, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Investig. 2010;120:4040–4054. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cha JJ, Hyun YY, Lee MH, Kim JE, Nam DH, Song HK, Kang YS, Lee JE, Kim HW, Han JY. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinol. 2013;154:2144–2155. doi: 10.1210/en.2012-2080. [DOI] [PubMed] [Google Scholar]
- 42.Hwang I, Seo EY, Ha H. Wnt/β-catenin signaling: a novel target for therapeutic intervention of fibrotic kidney disease. Arch Pharm Res. 2009;32:1653–1662. doi: 10.1007/s12272-009-2200-3. [DOI] [PubMed] [Google Scholar]
- 43.Cisternas P, Vio CP, Inestrosa NC. Role of wnt signaling in tissue fibrosis, lessons from skeletal muscle and kidney. Curr Mol Med. 2014;14:510–522. doi: 10.2174/1566524014666140414210346. [DOI] [PubMed] [Google Scholar]
- 44.He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/β-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:765–776. doi: 10.1681/ASN.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Macdonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao TP, Kühl M. An updated overview on wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 47.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 48.Berwick DC, Harvey K. The importance of wnt signalling for neurodegeneration in parkinson’s disease. Biochem Soc Trans. 2012;40:1123–1128. doi: 10.1042/BST20120122. [DOI] [PubMed] [Google Scholar]
- 49.Schinner S. Wnt-signalling and the metabolic syndrome. Horm Metab Res. 2009;41:159–163. doi: 10.1055/s-0028-1119408. [DOI] [PubMed] [Google Scholar]
- 50.Pulkkinen K, Murugan S, Vainio S. Wnt signaling in kidney development and disease. Organogenesis. 2008;4:55–59. doi: 10.4161/org.4.2.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]